Back to Journals » International Journal of Nanomedicine » Volume 10 » Issue 1
Development of polymeric–cationic peptide composite nanoparticles, a nanoparticle-in-nanoparticle system for controlled gene delivery
Authors Jain AK ![]() , Massey A, Yusuf H, McDonald D, McCarthy H, Kett VL
, Massey A, Yusuf H, McDonald D, McCarthy H, Kett VL
Received 27 August 2015
Accepted for publication 7 October 2015
Published 24 November 2015 Volume 2015:10(1) Pages 7183—7196
DOI https://doi.org/10.2147/IJN.S95245
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Thomas Webster
Arvind K Jain,1,2 Ashley Massey,1 Helmy Yusuf,1 Denise M McDonald,3 Helen O McCarthy,1 Vicky L Kett1
1School of Pharmacy, Medical Biology Centre, Queen’s University Belfast, Belfast, Northern Ireland, UK, 2Weatherall Institute of Molecular Medicine, MRC Molecular Haematology Unit, University of Oxford, John Radcliffe Hospital, Oxford, UK, 3Centre for Vision & Vascular Science, Queen’s University Belfast, Belfast, Northern Ireland, UK
Abstract: We report the formulation of novel composite nanoparticles that combine the high transfection efficiency of cationic peptide-DNA nanoparticles with the biocompatibility and prolonged delivery of polylactic acid–polyethylene glycol (PLA-PEG). The cationic cell-penetrating peptide RALA was used to condense DNA into nanoparticles that were encapsulated within a range of PLA-PEG copolymers. The composite nanoparticles produced exhibited excellent physicochemical properties including size <200 nm and encapsulation efficiency >80%. Images of the composite nanoparticles obtained with a new transmission electron microscopy staining method revealed the peptide-DNA nanoparticles within the PLA-PEG matrix. Varying the copolymers modulated the DNA release rate >6 weeks in vitro. The best formulation was selected and was able to transfect cells while maintaining viability. The effect of transferrin-appended composite nanoparticles was also studied. Thus, we have demonstrated the manufacture of composite nanoparticles for the controlled delivery of DNA.
Keywords: PLA-PEG, cationic peptide, gene delivery, composite nanoparticles, DNA, transfection
Introduction
Gene therapy involves the introduction of genes, commonly in the form of DNA or RNA, into a cell either to rectify a missing or defective gene or to express a protein of therapeutic interest. The use of DNA for gene therapy is a challenge because of its inability to cross the cell membrane barrier in its native solution form and its instability in the extracellular matrix. Application of a suitable delivery vector can compact the DNA providing stability as well as efficient translocation inside the cell through attachment to the target cells, cell membrane passage, escape from the endolysosome to reach into the cytosol, and, if possible, transport to the nucleus for the expression of the desired protein.1,2 Numerous viral and nonviral carriers have shown in vitro efficacy, including cationic nanoparticles.3–5 But their major limitation is their high toxicity, nonspecificity, and short life span in the blood circulation.1,6–8
PEGylation of particles increases their blood circulation half-life9 leading to enhanced bioavailability, also helping them to cross physical and physiological barriers.10,11 Polylactic acid–polyethylene glycol (PLA-PEG) nanoparticles have been widely used for the safe and prolonged delivery of therapeutics owing to their versatile and defined degradation profile that can be customized according to their molecular composition and chain lengths. When the block copolymer PLA-PEG is formulated into nanoparticles, PLA forms the hydrophobic core and the PEG chains form a corona around the nanoparticles giving a brush-like topography,12,13 and the degradation profile of the PLA defines the rate of drug delivery, while PEG determines the biological fate of the particles.
In this paper, composite nanoparticles were developed to exploit the advantages of both cationic cell-penetrating peptides and PLA-PEG nanoparticles. Cell-penetrating peptides have emerged as a specialized tool for the efficient intracellular delivery of the therapeutic cargo.14 For gene delivery, cell-penetrating peptides that include positively charged amphiphilic peptides ≥30 amino acid in length15 exhibit enhanced cell compatibility when compared with other available options.16–18 The cationic cell-penetrating peptide RALA19 was chosen to condense the DNA and form the core of the nanoparticles for efficient transfection of the cells. RALA exhibits an alpha helical structure in a hydrophilic environment with hydrophobic amino acids on one face and hydrophilic on the other, making it a suitable candidate for DNA condensation and efficient membrane perturbation. The sequence and predicted secondary structure of the peptide were generated using PEPstr server,20 and the surface was generated using UCSF chimera (Version 1.9) software (Figure 1).21
 | Figure 1 Sequence and structure of RALA. Overview of composition of the composite nanoparticles. |
We have found that DNA condensed with RALA to form cationic RALA nanoparticles (RNPs) can be encapsulated into PLA-PEG nanoparticles to form a composite DNA:RNP termed here as “polymeric–cationic peptide composite nanoparticles”. This enables the possibility of prolonging the rate of DNA release along with the potential for surface modification for its targeted delivery. The electrostatic interaction between the DNA and cationic peptide was optimized to achieve a cationic nanoparticle formulation close to monodispersity in order to allow their encapsulation within the PLA-PEG nanoparticles. A series of PLA-PEG polymers were synthesized to evaluate the optimum copolymer composition for the encapsulation of RNPs and controlled and safe delivery of the DNA. On the basis of their use in various biological applications, two PEG chain lengths, 2 and 5 kDa, were selected and various PLA chain lengths were polymerized with them.22–25 Thus, this paper describes the manufacture of a composite nanoparticulate system to offer the advantages of PLA-PEG copolymer in terms of safe, controlled, and targeted delivery26,27 in combination with the high encapsulation and transfection efficiency of the DNA payload offered by cationic RNPs.19
Experimental section
Materials
The RALA peptide was purchased from Biomatik, USA. The reporter plasmid pEGFP-N1 was purchased from Clontech (USA), cloned in DHF5-∝ (Thermo Fisher Scientific, Waltham, MA, USA), and purified using a Maxi-prep PureLink™ HiPure Plasmid Purification Kit (Thermo Fisher Scientific). For the formulation of RNPs, DNAase/RNAase-free distilled water (USP water for injection) purchased from Thermo Fisher Scientific was used; for all other applications, HPLC ultrapure Type 1 (ASTM, CAP, NCCLS standards) water processed by PURELAB Prima and PURELAB Maxima HPLC (ELGA LabWater) was used. D,L-Lactide (3,6-dimethyl-1,4-dioaxane-2,5-dione), polyethylene glycol methyl ether (mPEG 2,000/5,000), stannous octoate (Tin(II) 2-ethylhexanoate, ~95%), chloroform-d (100%, 0.03% TMS as standard) PVA (polyvinyl alcohol, average Mw 30,000–70,000, degree of hydrolysis 87–90%), Ethidium bromide (EtBr), proteinase K (BioUltra, ≥30 units/mg protein), were purchased from Sigma-Aldrich Co. ∝,∝-Trehalose dehydrate (high purity, endotoxin free) was procured from Ferro Pfanstiehl Laboratories Inc., USA. Roswell Park Memorial Institute (RPMI) 1640, 2.5% Trypsin (10×), 1 kb plus DNA ladder, and Quant-iT™ PicoGreen® ds DNA assay kit were purchased from Thermo Fisher Scientifc. Fetal calf serum was purchased from PAA, Austria. WST-1 cell proliferation reagent was bought from Roche Diagnostics Ltd, UK.
Cationic RNPs preparation
RNPs were formed by electrostatic interaction between the cationic RALA peptide and the negatively charged phosphate backbone of the DNA.19 The charge ratio of the components was calculated as N:P ratio, which is the molar ratio of the amine groups in the RALA to the phosphates in the DNA. RNPs were prepared at various N:P ratios to investigate and optimize the condensation behavior. One microgram of DNA was mixed with a calculated amount of RALA peptide and incubated at room temperature for 20 minutes before further experimentation. For the preparation of composite nanoparticles, RNPs in higher DNA concentration were prepared in 50 mM MOPS buffer at 50°C.
Gel retardation assay was performed by electrophoresis through a 0.8% agarose gel containing EtBr with Tris acetate-ethylenediaminetetraacetic acid running buffer at 80 V for 1 hour. Gels were imaged using a Biochemi® Multispectrum imaging system (UVP, UK). Images are representative of a minimum of three independent studies.
PLA-PEG copolymer synthesis and characterization
Copolymers were synthesized by the ring opening polymerization of D,L-lactide in the presence of prepolymerized mPEG 2,000/5,000 (monomethyl ether of polyethylene glycol) using stannous octoate as a catalyst.28 Reactants and catalyst solutions were azeotropically distilled independently before carrying out the polymerization reaction. The crude product was obtained by removing toluene and was dissolved in dichloromethane (DCM) to precipitate, twice, by cold ethyl ether (−80°C) for the purification of the product. The purified copolymers were then dried overnight in a vacuum desiccator. 1H-NMR spectroscopy was performed to assess the ratio of PLA and PEG chain lengths (LA/EG) in the synthesized block copolymers using a Bruker Ultrashield 400 plus instrument. The LA/EG ratio was calculated using the integration value of the peaks corresponding to methine protons (–CH) of the lactide at ∂ 5.1 ppm and methylene protons (–CH2) of PEG at ∂ 3.7 ppm.28 Molecular weight and polydispersity index (PDI) of the synthesized copolymers were determined by gel permeation chromatography (GPC) using Varian/Polymer Laboratories GPC-50 instrument equipped with refractive index detector. For molecular weight determination, chloroform was the mobile phase (40°C and flow rate of 1 mL/min). Polymers were dissolved in chloroform, filtered, and then injected into a column of 3 μm Resipore Mixed-B, 300 cm ×7.5 cm (Agilent Technologies, Santa Clara, CA, USA). Average molecular weights were calculated using a series of polystyrene standards. Products are abbreviated to PLAx-PEGy, where x and y represent the molecular weight of the respective block in kilodaltons.
Thermal analysis of the copolymers and the composite nanoparticles
Modulated temperature differential scanning calorimetry was performed using a QA 100 (TA Instruments, Newcastle, DE, USA) calibrated for temperature, enthalpy, and heat capacity. Samples in hermetically sealed aluminum pans were heated at 3°C/min from −20°C to 150°C with modulation of ±1°C for >60 seconds using a heat–cool–heat method to remove the effect of the accompanying relaxation endotherm from the glass transition (Tg). Values were determined from the midpoint of the signal step change in the reversing heat flow and shown as a “+” sign in the figures. Measurements were made in triplicate from three independently prepared formulations, and results are presented as mean ± standard deviation (SD).
Formulation of composite nanoparticles by double emulsion solvent evaporation method
Composite nanoparticles were made with various PLA-PEG copolymers using a modified double emulsion solvent evaporation method.28 Briefly, an aqueous RNP suspension was added to 4% w/v copolymeric solution in DCM (1:5 v/v) under vortex and probe sonicated (120 Sonic Dismembrator with 3 mm probe; Thermo Fisher Scientific) for 60 seconds. The water-in-oil (w/o) emulsion was added to 5% w/v polyvinyl alcohol solution in distilled water (1:5 v/v) under vortex and sonicated again. The resultant w/o/w emulsion was stirred overnight to evaporate the organic phase. The composite nanoparticles were collected by centrifugation at 30,000× g for 30 minutes (3K30; Sigma Centrifuge, UK) and washed twice with distilled water before re-suspending in 5% w/v aqueous trehalose solution and freeze-dried (Advantage, VirTis, Gardiner, NY, USA) using a protocol modified from Jain et al.29
To investigate the effect of surface modification on transfection efficiency, Tf-appended composite nanoparticles were also prepared. Transferrin was adsorbed on the surface of nanoparticles using modifications of reported methods30,31 suspending the nanoparticles in 10 mM 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid buffer, pH 7.4, with 150 mM NaCl containing transferrin. The resultant nanoparticles were collected by centrifugation at 25,000× g for 30 minutes.
Further nanoparticle characterization
Particle size and zeta potential values were determined by dynamic light scattering using a Malvern zetasizer (Nano ZS; Malvern Instruments, Malvern, UK). Freeze-dried composite nanoparticles were re-suspended in distilled water before analysis. For size determination, the average of five readings (at least ten runs each) was taken of each sample; data are presented as mean ± SD (n=10) of ten samples from different batches.
DNA was quantified using a proteinase K-assisted PicoGreen assay according to our previously developed protocol.32 Calibration plots were prepared in the range of 50–1,000 ng/mL for both DNA and RNPs (having equivalent DNA concentrations) in Tris buffer (pH 8.0, 20 mM).
Transmission electron microscopy was performed (JEOL JEM1400 transmission electron microscope at an accelerating voltage of 80 kV) to image the RNPs within the composite nanoparticles. Composite nanoparticles were prepared with the addition of 0.1% w/v osmium tetroxide in the polymer containing organic phase to generate the contrast between the RNPs and the outer polymer matrix of the composite nanoparticles. Samples were loaded over the copper grid (Formvar/Carbon 200 mesh, Agar Scientific) by putting a drop of sample onto a wax sheet then covering with the grid for 1 hour. Excess water was removed from the grid with tissue paper before air-drying overnight. For RNPs, the grids were negatively stained with 4% ammonium molybdate.
In-process stability study
In-process stability studies were performed to determine the effect of probe sonication, DCM, and emulsification on the RNPs and condensed plasmid DNA (pDNA) during the preparation of composite nanoparticles. To investigate the effect of sonication, samples were sonicated at amplitudes of 40%, 50%, and 60% for durations of 30, 60, and 120 seconds. As a control, the pDNA alone was sonicated at the lowest amplitude setting (40%) for 30 seconds. Samples were loaded onto an agarose gel with and without digestion with proteinase K. To investigate the effect of DCM, RNPs were vortexed with DCM for different time intervals. To investigate the effect of emulsification, the double emulsion solvent evaporation method was performed without the addition of polymer to the organic phase, thus removing the requirement of the polymeric nanoparticle disruption step. Samples were collected after 1 and 2 minutes sonication of the secondary emulsion and then loaded onto a gel with and without proteinase K digestion.
In vitro release study
Freeze-dried powders were re-suspended in 1 mL of Tris acetate-ethylenediaminetetraacetic acid buffer (40 mM, pH adjusted to 7.2 with HCl) with 0.02% sodium azide and centrifuged to quantify the RNP concentration in the supernatants, which corresponds to the amount of free or surface adsorbed RNPs present in the composite nanoparticles. The composite nanoparticles were re-suspended and incubated at 37°C in a shaking incubator to perform the in vitro release study >6 weeks. At each time point, the composite nanoparticles were centrifuged to collect the supernatant and re-suspended with the fresh buffer. DNA quantification was as described earlier.
Cell culture studies
Cell culture studies were performed using ZR-75-1 breast cancer cells from the American Tissue Culture Collection maintained in RPMI 1640 medium supplemented with 10% fetal calf serum. ZR-75-1 cells were seeded at a density of 1×105 cells/well onto 24-well tissue culture plates (VWR, UK) for 24 hours prior to transfection. Cells were conditioned for 2 hours in Opti-MEM (Thermo Fisher Scientific), which was then supplemented with either RNPs or composite nanoparticles. Following incubation for 6 hours (RNPs) or 24 hours (with composite nanoparticles), the media were removed and replaced with the serum-supplemented culture media and further incubated for 24 hours. Transfection efficiency was qualitatively evaluated using an inverted fluorescence microscope (Nikon Eclipse TE300; Nikon Instruments, Melville, NY, USA) with an epifluorescence source (Nikon Instruments). Images were captured using a DXM1200 (Nikon Instruments) digital camera at ×100 magnification. After imaging, cells were harvested and transfection efficiency was quantitatively measured using flow cytometry (FACSCalibur system; BD Biosciences, San Jose, CA, USA). The data were analyzed using Cyflogic software (Version 1.2.1; CyFlo, Ltd., Turku, Finland), and percent fluorescent cell values were reported as 4% gating of the controlled untreated cells.
Cell viability after treatment with nanoparticles was determined by washing the cells with phosphate-buffered saline followed by addition of 100 μL of 10% WST-1 reagent in serum-free media and incubation in standard CO2 incubator for 2 hours. The plate was read (EL808 plate-reader; Biotek, USA), and values are presented as percentage compared to untreated cells as a positive control.
Statistical analysis
Statistical analysis was performed using GraphPad prism 6 and GraphPad InStat 3 (GraphPad Software, Inc., La Jolla, CA, USA). One-way analysis of variance followed by Dunnett’s post hoc test was used to compare the data set (more than three groups). A P-value <0.05 was considered to indicate statistical significance for comparison. All data are presented as mean ± SD of three independently performed repeats unless otherwise specified.
Results and discussion
Formulation of cationic peptide-DNA nanoparticles (RNPs)
Figure 2A shows a representative image of the gel retardation assay performed to determine the N:P ratio required to condense DNA into the RNPs. Lanes corresponding to nanoparticles prepared with N:P ratios of 0.5–2.5 exhibit a DNA band at the same distance as pDNA (labeled D), indicating that in these formulations, the amount of RALA peptide was not sufficient to condense the DNA completely. At N:P ratio ≥3, no migration of DNA into the gel was seen indicating that beyond this ratio the DNA was condensed with the peptide. Lanes corresponding to N:P ratios of 3–4 showed some fluorescence in the well indicating formation of DNA-peptide aggregates, while at higher N:P ratios no DNA fluorescence was seen, confirming that in these lanes, the DNA is buried within the RNPs.
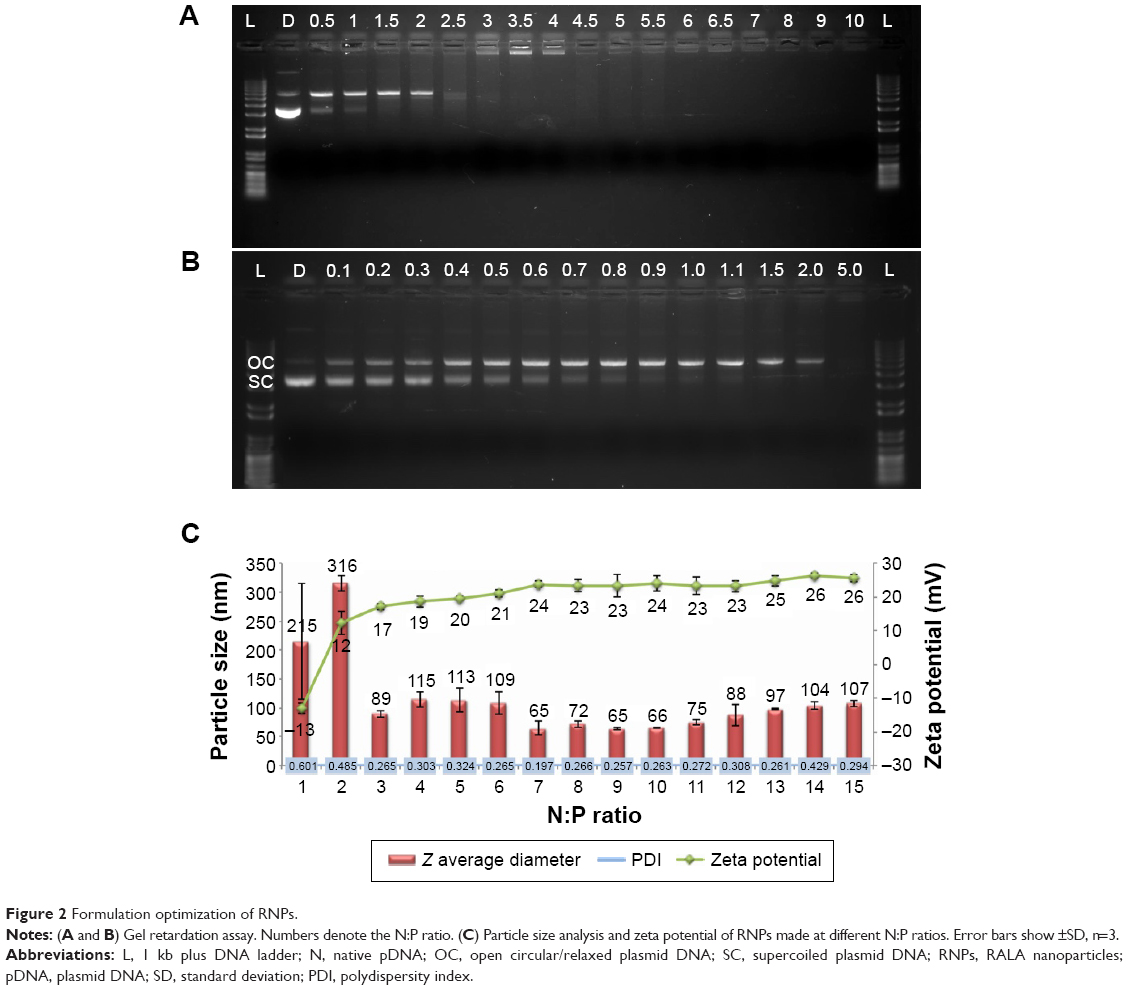 | Figure 2 Formulation optimization of RNPs. |
These findings indicate that the condensation of pDNA by RALA peptide progresses in a similar manner to that described previously for multivalent cations; this spontaneous collapse of DNA is defined as condensation33,34 and explained by an all-or-none transition phenomenon.35–37 At low concentrations, RALA disrupts the supercoiled structure of the pDNA, but beyond the critical condensing concentration (NP >2), it causes the collapse of the DNA and forms nanoparticles (Figure 2B). These observations are in-line with the explanation provided by Porschke,38 where they found that spermine (a tetravalent cation) forms a sheath around DNA until its concentration is raised to a threshold concentration enabling condensation of the DNA. We also observed that the zeta potential increased with N:P ratio and exhibited a sudden charge inversion at N:P ratio of 2 (Figure 2C). This effect and net positive charge on the surface of the nanoparticles even after the complete neutralization of DNA molecule can be explained by the “tails and arches” theory.39,40 Further, the zeta potential remained consistently >20 mV for the formulations prepared with N:P ratio ≥4 indicating the complete condensation of DNA, while the particle diameter was in the sub-100 nm range at N:P ratios >6. On the basis of these findings, a N:P ratio of 10 was considered to be optimum for the efficient production of RNPs and used for further studies; the Z-average diameter of ten independently prepared batches was determined to be 70.6±7.5 nm with a PDI of 0.240±0.054. Preparation of the composite nanoparticles required RNPs at a higher concentration where these conditions failed to control the condensation giving rise to polydispersed nanoparticles with a bimodal size distribution. After further optimizing the pH and thermodynamic conditions for the controlled condensation, these high concentration RNPs were prepared in 50 mM MOPS buffer at 50°C (optimization data are not shown). The Z-average particle size of the nanoparticles produced under these conditions was 55.2±10.6 nm with a PDI of 0.232±0.050 (n=16).
Characterization of the PLA-PEG copolymers
NMR indicated that the calculated chain length ratio of the synthesized polymers was close to the desired chain length ratio (Table 1) and GPC showed monomodal distribution curves with no peaks corresponding to the molecular weights of the reactants. The polydispersity values (Mw/Mn) of the synthesized copolymers were in the range 1.2–1.4 indicating their suitability for the manufacture of composite nanoparticles.
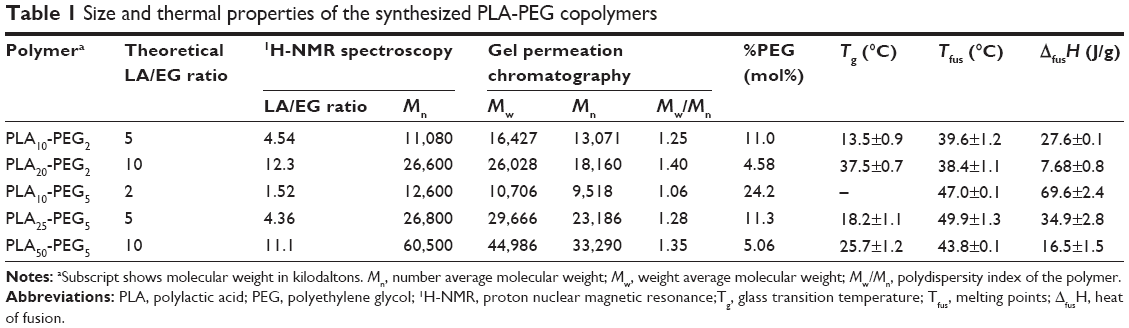 | Table 1 Size and thermal properties of the synthesized PLA-PEG copolymers |
Thermal properties of the polymers
Melting points (Tfus) values for copolymers containing PEG 2000 were lower than those containing PEG 5000 because of the higher Tfus value of PEG 5000. Increasing the PEG content lowered the Tg and increased the heat of fusion (ΔfusH) of the copolymers (Table 1; Figure S1). For example, the copolymers PLA20-PEG2 and PLA25-PEG5 have similar PLA chain lengths, but the latter has a higher PEG content that leads to its higher ΔfusH and lower Tg, whereas PLA10-PEG2 and PLA25-PEG5 exhibited closer values for ΔfusH and lower Tg because of their similar %PEG contents irrespective of the higher PLA and PEG chain lengths of PLA25-PEG5. These results indicate that the total PEG content has the largest influence on the thermal properties of the bulk copolymers.
Formulation of composite nanoparticles
Composite nanoparticles were prepared using the modified double emulsion solvent evaporation method with optimization for the synthesis of copolymeric nanoparticles as follows.
Optimization of DNA/polymer ratio
The formulation method was optimized for RNP content by adjusting the DNA/polymer ratio (μg/mg) using PLA25-PEG5 copolymer in the range of 0–1.0 μg/mg (Figure S2). Particle size and zeta potential of the copolymer nanoparticles were not affected by loading with the RNPs. Similarly, constant zeta potential values were observed across the range of loadings studied, indicating that in all cases, the RNPs were incorporated within the core of the composite nanoparticles and that the surface charge was solely determined by the copolymer. A small but statistically insignificant decrease in the particle size was observed in composite nanoparticles compared to empty polymeric nanoparticles (Figure S2). This size decrease might be attributed to the charge–charge interaction between the cationic RNPs and the negative charge of the polymer chains. The composite nanoparticles prepared with 0.25 μg/mg DNA/polymer ratio were selected for use in further studies.
Characterization of composite nanoparticles
Composite nanoparticles were characterized for their size, size distribution, surface charge, and encapsulation efficiency (Table 2). The smallest nanoparticles were formed by PLA10-PEG2 and PLA10-PEG5 with particle size of 145 nm that was attributed to their low molecular weight. The yields of these nanoparticles were very low; furthermore, the nanoparticles made with PLA10-PEG5 exhibited a low encapsulation efficiency indicating that they failed to form a stable emulsion during preparation. These nanoparticles were therefore considered unsuitable for further characterization. Increasing the PLA molecular weight increased the particle size, for example, PLA20-PEG2 > PLA10-PEG2 (P<0.001) and PLA25-PEG5 > PLA10-PEG5 (P<0.001). PLA50-PEG5 also showed a smaller but statistically significant increase in the particle size when compared with PLA25-PEG5 (P<0.01). These observations indicate that PLA forms the core of the nanoparticles that increases in size with increase in PLA chain length.28
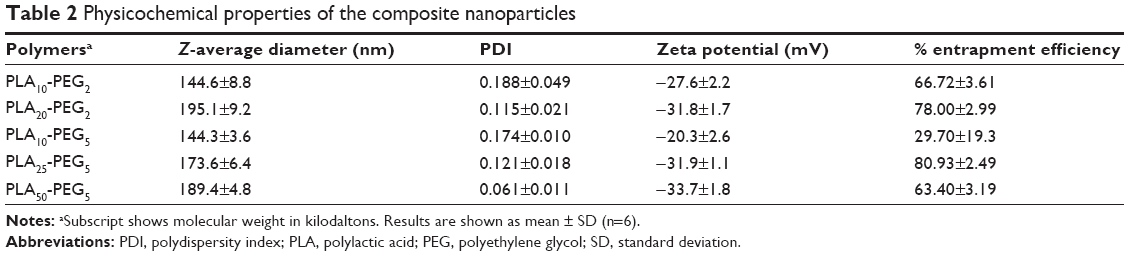 | Table 2 Physicochemical properties of the composite nanoparticles |
Composite nanoparticles prepared with similar PLA molecular weight but longer PEG chains produced a smaller particle size, for example, PLA25-PEG5 < PLA20-PEG2 (P<0.001). We suggest that the increased amphiphilicity conferred by the longer chain length stabilizes the globules during emulsification. This in turn helps to produce uniform and smaller nanoparticles by avoiding coalescence.
All nanoparticles exhibited encapsulation efficiency >60% except PLA10-PEG5 as described earlier. Further, PLA20-PEG2 and PLA25-PEG5 produced nanoparticles with exceptionally high entrapment efficiency, which is attributed to the similar PLA chain length used in their formation that helped to produce a stable o/w/o emulsion enabling maximum engulfment of the RNPs. The negative zeta potential exhibited by all the composite nanoparticles indicates that all the positively charged RNPs were encapsulated into the core. On the basis of these results, PLA20-PEG2, PLA25-PEG5, and PLA50-PEG5 were selected for further studies, with PLA25-PEG5 considered the best formulation because of its small size and high encapsulation efficiency.
Thermal properties of the composite nanoparticles
All products exhibited a Tg at 120°C, indicating that the trehalose was present in the amorphous state (Figure 3A),41 which is desirable since crystallization of protectants in nanoparticle formulations has been shown to cause instability.42 The Tg values of composite nanoparticles were found to be higher than those of their respective copolymers (Figure 3B), which was attributed to the more confined architecture of the polymer chains within the composite nanoparticles than in the bulk polymer.43,44 A relatively small Tg increase was seen for the Tg value of PLA20-PEG2 nanoparticles compared with the bulk polymer indicating that other factors are also involved in determining the value of Tg, which could be interesting to explore in future studies.
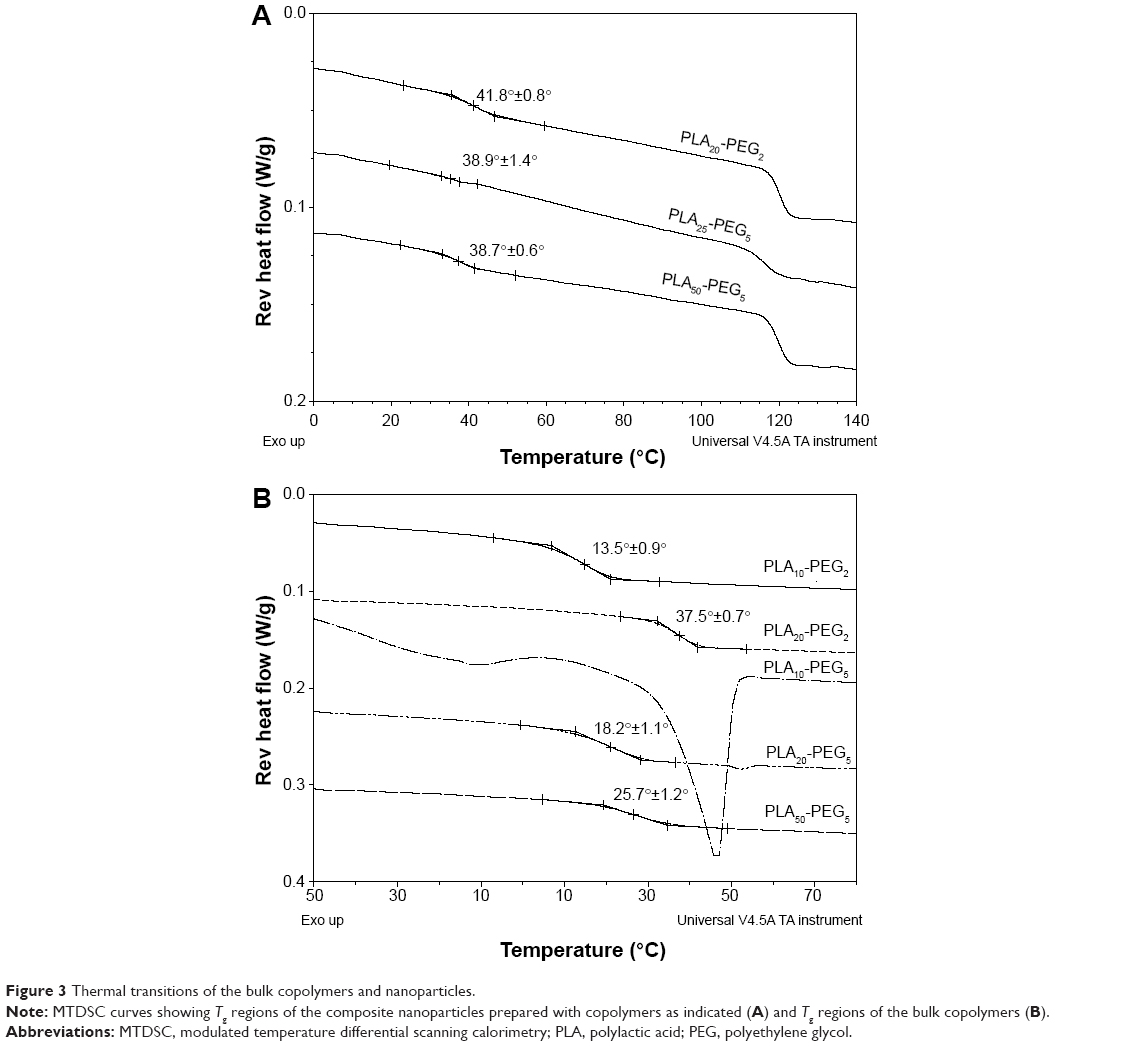 | Figure 3 Thermal transitions of the bulk copolymers and nanoparticles. |
The Tg values of the nanoparticles were found to be unaffected by the overall molecular weight of the polymer as exemplified by the similar values of PLA25-PEG5 and PLA50-PEG5 (Figure 3A). The PEG molecular weight had a greater plasticizing effect on Tg values than the PEG content. For example, PLA25-PEG5 and PLA50-PEG5 exhibited similar Tg values and contained 18 mol% and 8 mol% PEG, respectively. All the formulations exhibited a Tg value >38°C indicating their structural stability at room temperature.
In-process stability study
Figure 4 shows representative images of the three different kinds of experiments performed to assess stability of the RNPs and the pDNA condensed in it during the formulation of composite nanoparticles by double emulsion solvent evaporation method.
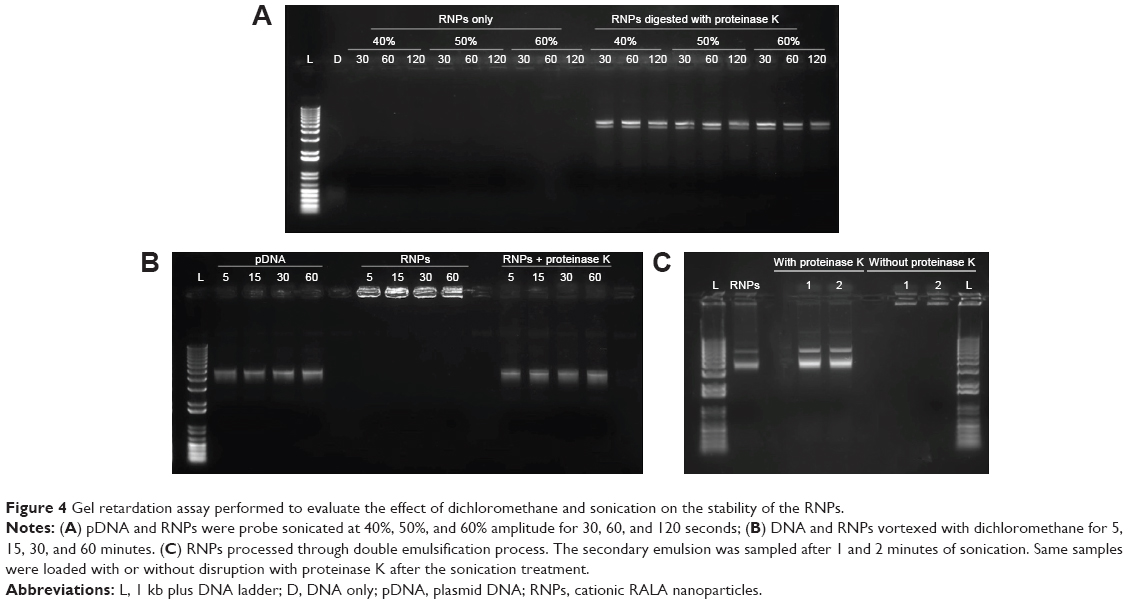 | Figure 4 Gel retardation assay performed to evaluate the effect of dichloromethane and sonication on the stability of the RNPs. |
Gel electrophoresis was used to analyze the effect of probe sonication on the stability of the RNPs and the entrapped DNA (Figure 4A). No DNA band was observed in the control lane D, which contained pDNA alone sonicated at 40% for 30 seconds. A faint DNA smear at the bottom of the lane shows that sonication degraded the DNA. In the lanes containing RNPs, no DNA was observed, indicating that it remained inside the RNPs during sonication. Sonicated RNPs predigested with proteinase K before loading to the gel released the DNA to migrate into the gel confirming that the DNA was stable inside the RNPs.
Data presented in Figure 4B indicate that both free DNA and RNPs are stable when vortexed with DCM. Samples digested with proteinase K exhibited DNA bands at the expected height confirming stability of DNA inside RNPs, whereas undigested samples did not show any fluorescence in the lane indicating the stability of RNPs with DCM. The data presented in Figure 4C show that the RNPs remained stable during the complete double emulsification process and also protected the encapsulated DNA from the shear stress posed by DCM and sonications.
Transmission electron microscopy
A new approach was developed whereby the hydrophobic dye osmium tetroxide was added to the polymeric organic phase during the preparation of the composite nanoparticles. The dye becomes molecularly dispersed in the polymer matrix only, giving a contrast background to the RNPs that makes them visible inside the composite nanoparticles (Figure 5). To our knowledge, this is the first time that this preparation method has been reported. The images show that the RNPs were spherical and ≤50 nm (Figure 5A). RNPs are the bright areas in the center of the composite nanoparticles. These images give visual confirmation of RNP encapsulation inside the composite nanoparticles. Depending on the size, single or multiple RNPs are encapsulated within the composite nanoparticles.
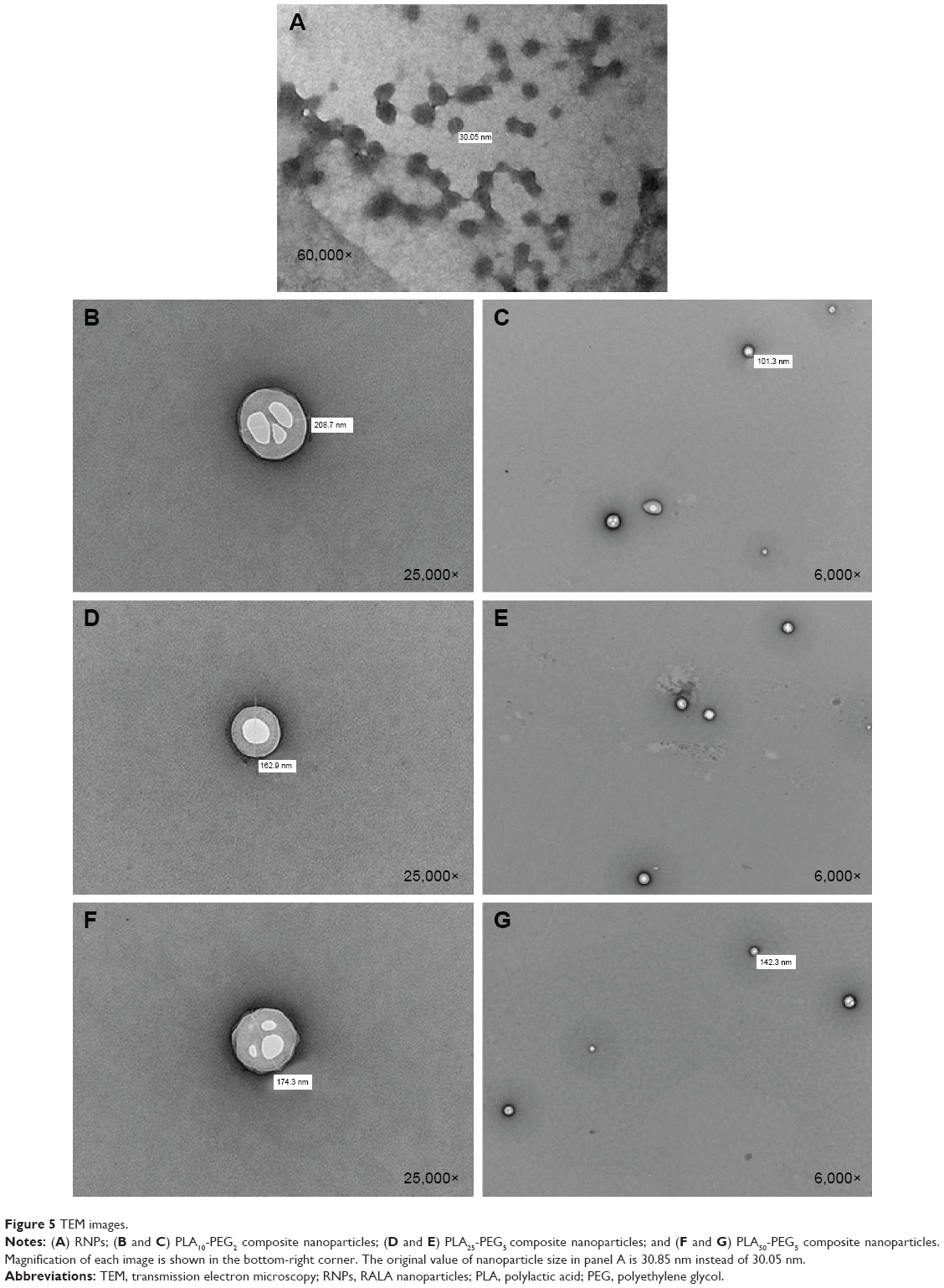 | Figure 5 TEM images. |
In vitro release study
All the composite nanoparticles exhibited an initial burst release that accounted for the 8%–10% DNA released in first 24 hours, which is good for the priming of a therapeutic response (Figure 6). Four to five percent of this was DNA that had been released during freeze-drying as determined by centrifugation. The PLAx-PEG5 copolymers exhibited a larger burst release than PLA20-PEG2, which could be attributed to the longer PEG chains of the PLAx-PEG5 formulations since it is known that higher contact area with water enhances degradation rate.45 PLA25-PEG5 showed the highest amount of DNA release at every time point, which was attributed to a number of factors, longer PEG chains, shorter PLA chains, and the low Tg, which was closest to the temperature of the release medium. The longer PEG chains might have caused a higher degradation rate in this system, and increased PEG content has been correlated with increase in cargo release.46 The other two formulations have a higher ratio of PLA chain length to PEG, which slows the degradation profile. All three composite nanoparticles were deemed acceptable. The PLA25-PEG5 was taken forward for the transfection study because of its higher release rate.
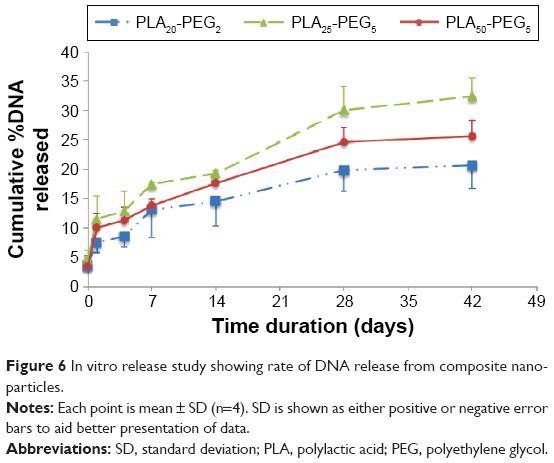 | Figure 6 In vitro release study showing rate of DNA release from composite nanoparticles. |
These data also demonstrate the stability of the released DNA over the time course of the study since the PicoGreen reagent binds specifically to the double-stranded DNA; therefore, the concentrations are of stable double-stranded DNAs only.
In vitro transfection and cell viability study
In vitro transfection studies were performed to evaluate the transfection efficiency of composite nanoparticles manufactured with PLA25-PEG5. Transferrin-appended PLA25-PEG5 composite nanoparticles were also studied as previous reports have shown that transferrin-adsorbed nanoparticles can exhibit enhance cellular uptake with good stability in vivo.31 Transfection was compared with un-encapsulated RNPs. Figure 7A shows the particle size and zeta potential data obtained for the nanoparticles used in the study and indicate that transferrin adsorption caused a large increase in zeta potential, confirming the adsorption of the transferrin on the surface of composite nanoparticles. In the current study, transferrin-adsorbed nanoparticles were collected by high-speed centrifugation and vigorous vortexing; the presence of transferrin on the surface of the nanoparticles even after these shear forces indicates their physical stability of the system.
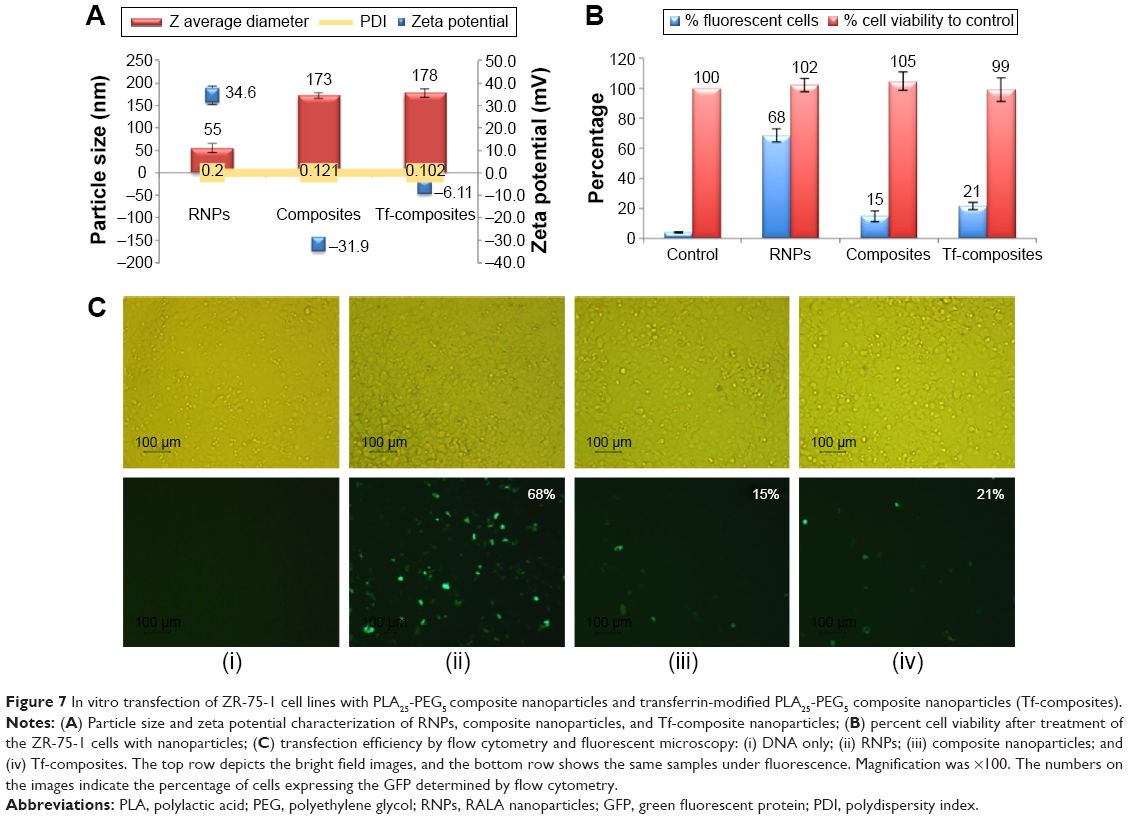 | Figure 7 In vitro transfection of ZR-75-1 cell lines with PLA25-PEG5 composite nanoparticles and transferrin-modified PLA25-PEG5 composite nanoparticles (Tf-composites). |
The cell viability assay indicated that the nanoparticles were all nontoxic (Figure 7B). The RNPs exhibited 68% transfection, and expression of the green fluorescent protein was analyzed by flow cytometry (Figure 7C), which is in line with our previous findings,19 and confirmed the suitability of using these RNPs for efficient translocation of the DNA into the cells. In contrast, composite nanoparticles showed a much lower expression. Transferrin-appended composite nanoparticles exhibited a significantly (P<0.05) higher fluorescence that could be attributed to transferrin receptor-mediated uptake.30,47,48 These results indicate that RNPs are superior over composite nanoparticles over the 24-hour period of the study, even with the additional advantage conferred by Tf-conjugation. This apparent low-transection efficiency is attributed to the slow release of the RNPs from composite nanoparticles and could be advantageous in the in vivo setup.
Conclusion
This study has successfully demonstrated the manufacture of composite nanoparticle systems based on cationic peptide-DNA nanoparticles (RNPs) encapsulated within a PLA-PEG nanoparticle. Formulation of RNPs was studied at various N:P ratios in order to obtain a reproducible Z-average diameter <100 nm. RNPs were further optimized with more stringent conditions such as type of buffer, buffer capacity, ionic strength, buffer pH, and microenvironment to control this electrostatic interaction and obtain RNPs, with the suitable characteristics required for formulation of composite NPs. Finally, RNPs of 55 nm (Z-average diameter) were prepared in 50 mM MOPS buffer at 50°C with DNA concentration of 400 μg/mL and were shown to be stable during the emulsification-based encapsulation process. For the second component, various compositions of PLA-PEG block copolymers were explored. On the basis of the thermal characterization of the copolymers and their efficiency to encapsulate RNPs, three copolymers were selected for the further studies, such as PLA20-PEG2, PLA25-PEG5, and PLA50-PEG5. Composite nanoparticles based on these copolymers exhibited a range of acceptable DNA release rates over a 6-week in vitro study. The continuous slow release indicates that these systems might be able to exhibit prolonged delivery of DNA after administration that could avoid the requirement for frequent dosing, which is known to enhance patient compliance, while a transfection study showed their ability to transfect cells without compromising cell viability. It is beyond the scope of the short in vitro transfection study to show the impact of controlled release over a longer time period to evaluate the efficacy of the composite nanoparticles as a whole system. A dedicated in vivo gene delivery study would be useful to further explore the potential of this new type of composite nanoparticle system. Thus, this study has shown the potential to design composite nanoparticles for prolonged and safe gene delivery and demonstrates that such systems are promising candidates for effective gene therapy.
Acknowledgments
AK Jain acknowledges Queen’s University of Belfast for providing University International Research Studentship. We also thank Dr P Redpath and Dr M Migaud for help with the polymer synthesis, Prof C Alexander, Dr KD Bansal, and Dr S Spain (School of Pharmacy, University of Nottingham, Nottingham, UK) for support for gel permeation chromatography, and Dr Gary Lyons and Ms Deborah Moffett (Agri-Food and Biosciences Institute for Northern Ireland, Belfast, Northern Ireland) for the transmission electron microscopy analysis.
Disclosure
The authors report no conflicts of interest in this work.
References
Pouton CW, Seymour LW. Key issues in non-viral gene delivery. Adv Drug Deliv Rev. 2001;46(1–3):187–203. | ||
Spain SG, Yasayan G, Soliman M, Heath F, Saeed AO, Alexander C. 4.424 – nanoparticles for nucleic acid delivery. In: Paul D, editor. Comprehensive Biomaterials. Oxford: Elsevier; 2011:389–410. | ||
Park TG, Jeong JH, Kim SW. Current status of polymeric gene delivery systems. Adv Drug Deliv Rev. 2006;58(4):467–486. | ||
Cho YW, Kim JÄ, Park K. Polycation gene delivery systems: escape from endosomes to cytosol. J Pharm Pharmacol. 2003;55(6):721–734. | ||
Li W, Nicol F, Szoka FC Jr. GALA: a designed synthetic pH-responsive amphipathic peptide with applications in drug and gene delivery. Adv Drug Deliv Rev. 2004;56(7):967. | ||
Bexiga MG, Varela JA, Wang F, et al. Cationic nanoparticles induce caspase 3-, 7- and 9-mediated cytotoxicity in a human astrocytoma cell line. Nanotoxicology. 2011;5(4):557–567. | ||
Oh Y-K, Park TG. siRNA delivery systems for cancer treatment. Adv Drug Deliv Rev. 2009;61(10):850–862. | ||
Howard KA. Delivery of RNA interference therapeutics using polycation-based nanoparticles. Adv Drug Deliv Rev. 2009;61(9):710–720. | ||
Owens DE, Peppas NA. Opsonization, biodistribution, and pharmacokinetics of polymeric nanoparticles. Int J Pharm. 2006;307(1):93–102. | ||
Vila A, Gill H, McCallion O, Alonso MJ. Transport of PLA-PEG particles across the nasal mucosa: effect of particle size and PEG coating density. J Control Release. 2004;98(2):231–244. | ||
Suh J, Choy KL, Lai SK, et al. PEGylation of nanoparticles improves their cytoplasmic transport. Int J Nanomedicine. 2007;2(4):735. | ||
Gref R, Domb A, Quellec P, et al. The controlled intravenous delivery of drugs using PEG-coated sterically stabilized nanospheres. Adv Drug Deliv Rev. 2012;16:215–233. | ||
Riley T, Heald C, Stolnik S, et al. Core-shell structure of PLA-PEG nanoparticles used for drug delivery. Langmuir. 2003;19(20):8428–8435. | ||
Koren E, Torchilin VP. Cell-penetrating peptides: breaking through to the other side. Trends Mol Med. 2012;18(7):385–393. | ||
Deshayes S, Morris M, Divita G, Heitz F. Cell-penetrating peptides: tools for intracellular delivery of therapeutics. Cell Mol Life Sci. 2005;62(16):1839–1849. | ||
Saccardo P, Villaverde A, González-Montalbán N. Peptide-mediated DNA condensation for non-viral gene therapy. Biotechnol Adv. 2009;27(4):432–438. | ||
Fominaya J, Gasset M, García R, Roncal F, Pablo Albar J, Bernad A. An optimized amphiphilic cationic peptide as an efficient non-viral gene delivery vector. J Gene Med. 2000;2(6):455–464. | ||
Wyman TB, Nicol F, Zelphati O, Scaria PV, Plank C, Szoka FC Jr. Design, synthesis, and characterization of a cationic peptide that binds to nucleic acids and permeabilizes bilayers. Biochemistry. 1997;36(10):3008–3017. | ||
McCarthy HO, McCaffrey J, McCrudden CM, et al. Development and characterization of self-assembling nanoparticles using a bio-inspired amphipathic peptide for gene delivery. J Control Release. 2014;189(0):141–149. | ||
Harpreet K, Aarti G, Raghava GPS. PEPstr: a de novo method for tertiary structure prediction of small bioactive peptides. Protein Pept Lett. 2007;14(7):626–631. | ||
Pettersen EF, Goddard TD, Huang CC, et al. UCSF Chimera – a visualization system for exploratory research and analysis. J Comput Chem. 2004;25(13):1605–1612. | ||
Göpferich A, Gref R, Minamitake Y, et al. Drug Delivery from Bioerodible Polymers: Systemic and Intravenous Administration. Washington: American Chemical Sciety; 1994. | ||
Zhang Y, Zhang Q, Zha L, et al. Preparation, characterization and application of pyrene-loaded methoxy poly (ethylene glycol), Äìpoly (lactic acid) copolymer nanoparticles. Colloid Polym Sci. 2004;282(12):1323–1328. | ||
Farokhzad OC, Cheng J, Teply BA, et al. Targeted nanoparticle-aptamer bioconjugates for cancer chemotherapy in vivo. Proc Natl Acad Sci U S A. 2006;103(16):6315–6320. | ||
Nguyen CA, Allémann E, Schwach G, Doelker E, Gurny R. Cell interaction studies of PLA-MePEG nanoparticles. Int J Pharm. 2003;254(1):69–72. | ||
Panyam J, Labhasetwar V. Biodegradable nanoparticles for drug and gene delivery to cells and tissue. Adv Drug Deliv Rev. 2012;64:61–71. | ||
Ravi Kumar M, Hellermann G, Lockey RF, Mohapatra SS. Nanoparticle-mediated gene delivery: state of the art. Expert Opin Biol Ther. 2004;4(8):1213–1224. | ||
Jain AK, Goyal AK, Gupta PN, et al. Synthesis, characterization and evaluation of novel triblock copolymer based nanoparticles for vaccine delivery against hepatitis B. J Control Release. 2009;136(2):161–169. | ||
Jain AK, Swarnakar NK, Godugu C, Singh RP, Jain S. The effect of the oral administration of polymeric nanoparticles on the efficacy and toxicity of tamoxifen. Biomaterials. 2011;32(2):503–515. | ||
Gan CW, Feng S-S. Transferrin-conjugated nanoparticles of poly (lactide)-D-α-tocopheryl polyethylene glycol succinate diblock copolymer for targeted drug delivery across the blood–brain barrier. Biomaterials. 2010;31(30):7748–7757. | ||
Chang J, Paillard A, Passirani C, et al. Transferrin adsorption onto PLGA nanoparticles governs their interaction with biological systems from blood circulation to brain cancer cells. Pharm Res. 2012;29(6):1495–1505. | ||
Jain AK, Yusuf H, Pattani A, McCarthy HO, McDonald DM, Kett VL. Development of a method to quantify the DNA content in cationic peptide–DNA nanoparticles. J Pharm Biomed Anal. 2014;100:236–242. | ||
Bloomfield VA. Condensation of DNA by multivalent cations: considerations on mechanism. Biopolymers. 1991;31(13):1471–1481. | ||
Bloomfield VA. DNA condensation. Curr Opin Struct Biol. 1996;6(3):334–341. | ||
Hirano K, Ichikawa M, Ishido T, Ishikawa M, Baba Y, Yoshikawa K. How environmental solution conditions determine the compaction velocity of single DNA molecules. Nucleic Acids Res. 2012;40(1):284–289. | ||
Murayama H, Yoshikawa K. Thermodynamics of the collapsing phase transition in a single duplex DNA molecule. J Phys Chem B. 1999;103(47):10517–10523. | ||
Yoshikawa Y, Suzuki Y, Yamada K, et al. Critical behavior of megabase-size DNA toward the transition into a compact state. J Chem Phys. 2011;135(22):225101–225107. | ||
Porschke D. Dynamics of DNA condensation. Biochemistry. 1984;23(21):4821–4828. | ||
Nguyen TT, Shklovskii BI. Inversion of DNA charge by a positive polymer via fractionalization of the polymer charge. Physica A. 2002;310(1–2):197–211. | ||
Nguyen TT, Shklovskii BI. Model of inversion of DNA charge by a positive polymer: fractionalization of the polymer charge. Phys rev let. 2002;89(1):018101. | ||
McGarvey OS, Kett VL, Craig DQM. An investigation into the crystallization of α,α-trehalose from the amorphous state. J Phys Chem B. 2003;107(27):6614–6620. | ||
Abdelwahed W, Degobert G, Fessi H. Investigation of nanocapsules stabilization by amorphous excipients during freeze-drying and storage. Eur J Pharm Biopharm. 2006;63(2):87–94. | ||
Martínez-Tong DE, Soccio M, Sanz A, García C, Ezquerra TA, Nogales A. Chain arrangement and glass transition temperature variations in polymer nanoparticles under 3D-confinement. Macromolecules. 2013;46(11):4698–4705. | ||
Guo Y, Morozov A, Schneider D, et al. Ultrastable nanostructured polymer glasses. Nat Mater. 2012;11(4):337–343. | ||
Huang Y-Y, Chung T-W, Tzeng T-W. Drug release from PLA/PEG microparticulates. Int J Pharm. 1997;156(1):9–15. | ||
Ren J, Yu X, Ren T, Hong H. Preparation and characterization of fenofibrate-loaded PLA-PEG microspheres. J Mater Sci Mater Med. 2007;18(8):1481–1487. | ||
Chang J, Jallouli Y, Kroubi M, et al. Characterization of endocytosis of transferrin-coated PLGA nanoparticles by the blood–brain barrier. Int J Pharm. 2009;379(2):285–292. | ||
Cui Y, Xu Q, Chow PK-H, Wang D, Wang C-H. Transferrin-conjugated magnetic silica PLGA nanoparticles loaded with doxorubicin and paclitaxel for brain glioma treatment. Biomaterials. 2013;34(33):8511–8520. |
Supplementary materials
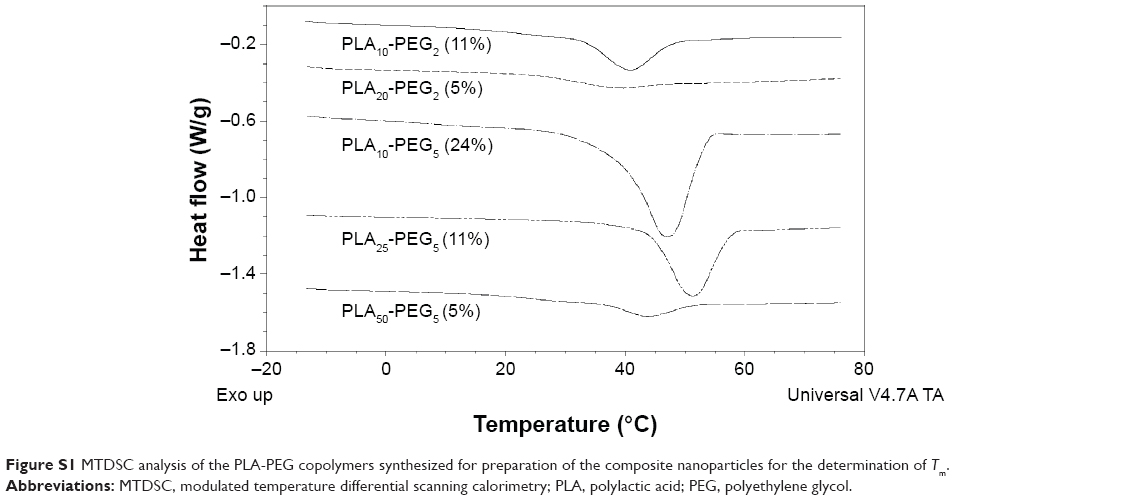 | Figure S1 MTDSC analysis of the PLA-PEG copolymers synthesized for preparation of the composite nanoparticles for the determination of Tm. |
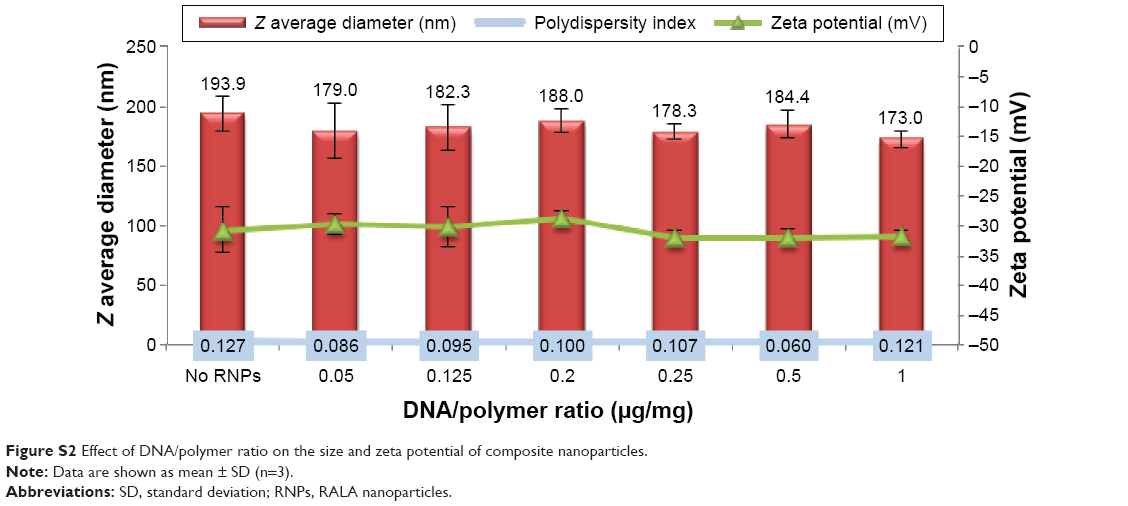 | Figure S2 Effect of DNA/polymer ratio on the size and zeta potential of composite nanoparticles. |
 © 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.