Back to Journals » International Journal of Nanomedicine » Volume 11
Development of octreotide-conjugated polymeric prodrug of bufalin for targeted delivery to somatostatin receptor 2 overexpressing breast cancer in vitro and in vivo
Authors Liu T, Jia T, Yuan X, Liu C, Sun J, Ni Z, Xu J, Wang X, Yuan Y
Received 12 November 2015
Accepted for publication 1 February 2016
Published 23 May 2016 Volume 2016:11 Pages 2235—2250
DOI https://doi.org/10.2147/IJN.S100404
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Professor Lei Yang
Tao Liu,1,* Tingting Jia,2,* Xia Yuan,2 Cheng Liu,1 Jian Sun,1 Zhenhua Ni,1 Jian Xu,1 Xuhui Wang,2 Yi Yuan2
1Centralab, 2Department of Pharmacy, Putuo Hospital, Shanghai University of Traditional Chinese Medicine, Shanghai, People’s Republic of China
*These authors contributed equally to this work
Background: Development of polymeric prodrugs of small molecular anticancer drugs has become one of the most promising strategies to overcome the intrinsic shortcomings of small molecular anticancer drugs and improve their anticancer performance.
Materials and methods: In the current work, we fabricated a novel octreotide (Oct)-modified esterase-sensitive tumor-targeting polymeric prodrug of bufalin (BUF) and explored its anticancer performance against somatostatin receptor 2 overexpressing breast cancer.
Results: The obtained tumor-targeting polymeric prodrug of BUF, P(oligo[ethylene glycol] monomethyl ether methacrylate [OEGMA]-co-BUF-co-Oct), showed a nanosize dimension and controlled drug release features in the presence of esterase. It was demonstrated by in vitro experiment that P(OEGMA-co-BUF-co-Oct) showed enhanced cytotoxicity, cellular uptake, and apoptosis in comparison with those of free BUF. In vivo experiment further revealed the improved accumulation of drugs in tumor tissues and enhanced anticancer performance of P(OEGMA-co-BUF-co-Oct).
Conclusion: Taken together, this study indicated that polymeric prodrug of BUF holds promising potential toward the treatment of somatostatin receptor 2 overexpressing breast cancer.
Keywords: esterase responsive, controlled release, tumor targeting
Introduction
Breast cancer is now the most common cancer in women around the globe.1,2 For instance, >1.6 million people are being diagnosed and ~1.2 million die because of this disease every year in the People’s Republic of China.1 Thus, how to effectively prevent and combat this malignant disease is one of the most explored topics in cancer research.3 Chemotherapy has been widely employed in the treatment of different stages of breast cancer, and a series of anticancer drugs have been developed, such as paclitaxel (PTX), fluorouracil, and doxorubicin (DOX).4–6 In addition, traditional Chinese medicine receives more and more attention due to its promising potential in combating various cancers, including breast cancer. Some of the active components of traditional Chinese medicine have been introduced into clinical application, eg, PTX and camptothecin.7,8
Bufalin (BUF) extracted from the skin and parotid venom glands of the toad shows effective therapeutic activity against a broad spectrum of tumors, including breast cancer.9–15 However, its poor water solubility and severe adverse effects such as high cardiac toxicity, allergic shock, ardent fever, and sinus bradycardia limit its further clinical applications.16,17 In this regard, there is a need for the development of new formulations of BUF to overcome its side effects as well as to keep its original therapeutic activity.
Polymeric micelles constructed from polymeric prodrug of hydrophobic small molecular drugs have emerged as a very promising choice considering its good water solubility, easy to further modification, enhanced permeability and retention (EPR) effect, and tunable drug content.18–27 Moreover, it was reported that the introduction of tumor-targeting moieties onto polymeric prodrugs can effectively enhance the accumulation in tumor tissues and therapeutic efficacy of drugs.28
In this context, somatostatin receptors (SSTRs) were explored as potential targets because they are overexpressed in a variety of tumors and cancer cell lines, including breast cancer.29–31 SSTRs, which are members of the G protein-coupled receptor superfamily, consist of five subtypes (SSTR1–5) and are regarded as an important factor to influence series physiological functions.29–31 Octreotide (Oct) is an octapeptide analog of endogenous somatostatin and mainly binds to SSTRs, especially SSTR2 and SSTR5, in breast cancer; hence, it may be possible to employ Oct as a potential ligand in receptor-mediated nanodelivery system.32–36
Huang et al reported the fabrication of PTX–Oct conjugate and explored its anticancer performance against human breast MCF-7 carcinoma cells.4 It was showed that PTX–Oct exhibited less toxicity as well as comparable anticancer efficacy in comparison with that of free PTX. Zhang et al reported a novel Oct-modified sterically stabilized liposome loaded with anticancer drug DOX, Oct–SSL–DOX.37 Three cell lines, SSTR2-overexpressing NCI-H446 and MCF-7 and SSTR2-negative Chinese hamster ovary, were employed to evaluate the targeting capability and anticancer performance. It was demonstrated that compared with SSTR2-negative cells, increased cytotoxicity, enhanced drug accumulation in tumor tissues, and improved anticancer efficacy in SSTR2-positive tumors (NCI-H446 and MCF-7) were observed. They then synthesized Oct-modified PEG-b-poly(lactic acid; Oct-PEG-b-PLA) and physically encapsulated docetaxel to explore its anticancer application in NCI-H446 cells.38 After that they further developed a breast cancer and human embryonic lung fibroblast-targeting drug delivery system by using Oct–PEG–deoxycholic acid conjugate-modified N-deoxycholic acid-O,N-hydroxymethylation chitosan micelles, indicating promising potentials in anticancer drug delivery for targeted cancer therapy.36
It is worthy of noting that in addition to physically encapsulating drugs by nanoparticles, polymeric prodrug is another promising strategy owing to its intrinsic advantages such as passive targeting via the EPR effect, increased circulation time, improved water solubility, and tunable and well-defined drug content.18–23,39–44 Besides, premature/burst drug release, which is one of the most common shortcomings of encapsulating method, can be effectively addressed. By using responsive chemical bonds to link drugs and polymers, polymeric prodrug can effectively release drugs under specific stimuli when reached into tumor tissues or cancer cells but released little drug in the circulation period.40 Unfortunately, to the best of our knowledge, Oct-modified polymeric prodrugs against breast cancer were poorly explored, especially in responsive drug delivery systems. Huo et al fabricated Oct-modified PEGylated polymeric prodrug of PTX, Oct–PEG–PTX, by linking PTX with PEG via conventional ester bond.45 Anticancer evaluation against human small cell lung cancer cells NCI-H446 exhibited stronger antitumor efficacy and lower systemic toxicity than nontargeting polymeric prodrug, mPEG-PTX, and commercial Taxol.
In the present study, we intended to develop a novel type of synthetic polymer-based targeting delivery system by covalently linking anticancer drug (BUF) via esterase-responsive thioester bond and SSTR2-overexpressing breast cancer-targeted peptide, Oct, aiming to enhance their accumulation in breast cancer tissues and increase intracellular delivery of drugs as well as triggered drug release characteristic (Figure 1). 1H nuclear magnetic resonance (NMR), laser light scattering (LLS), gel permeation chromatography (GPC), and transmission electron microscopy (TEM) were employed to characterize the polymeric prodrugs and prodrug-based nanoparticles. Cell cytotoxicity, flow cytometry, and fluorescence imaging were employed to evaluate the anticancer performance of the polymeric prodrugs in vitro. Then in vivo fluorescence imaging was employed to evaluate the biodistribution of the polymeric prodrugs. In vivo anticancer experiments were conducted to probe the feasibilities and capabilities of this new type of polymeric prodrugs.
 | Figure 1 A schematic illustration for the fabrication of tumor-targeting micellar nanoparticles, P(OEGMA-co-BUF-co-Oct), covalently attached with targeting peptide (Oct) with stable chemical bond and anticancer drug (BUF) with esterase-responsive β-thioester. |
Materials and methods
Materials
Oligo(ethylene glycol) monomethyl ether methacrylate (OEGMA; Mn 300 g/mol, mean degree of polymerization [DP] 4–5) purchased from Aladdin was passed through a neutral alumina column to remove the inhibitor and then stored at −20°C prior to use. 2,2′-Azobis(2-methylpropionitrile) (AIBN) was recrystallized from 95% ethanol. BUF (99%) was purchased from Chengdu Puruifa Technology Development Co. Ltd. (Chengdu, People’s Republic of China) and used as received. 3-(4,5-Dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT), Oct (H2N-dPhe-Cys-Phe-dTrp-Lys-Thr-Cys-Thr-ol, molecular weight 1,019.26), and esterase solution (porcine liver, 5 kU) were purchased from Sigma-Aldrich Co. (St Louis, MO, USA) and used as received. Cyanine5 amine (Cy5; Lumiprobe, Hallandale Beach, Florida, USA), 4,6-diamidino-2-phenylindole (DAPI; Beyotime Biotech, Shanghai, People’s Republic of China), Annexin V fluorescein isothiocyanate (FITC)/propidium iodide (PI) apoptosis detection kit (BD Biosciences, San Jose, CA, USA), hematoxylin and eosin (H&E; Thermo Fisher Scientific, Waltham, MA, USA), and 4% paraformaldehyde solution (Beyotime Biotech) were used as received. Fetal bovine serum, penicillin, streptomycin, and Dulbecco’s Modified Eagle’s Medium (DMEM) were purchased from Thermo Fisher Scientific and used as received. The human breast cancer cell line MCF-7 was purchased from Cell Bank of Type Culture Collection of Chinese Academy of Sciences (Shanghai, People’s Republic of China). N-Hydroxysuccinimide (NHS; 99%), N, N′-dicyclohexylcarbodiimide (DCC), 4-dimethylaminopyridine (DMAP), and all other reagents were purchased from Sinopharm Chemical Reagent Co. Ltd. (Shanghai, People’s Republic of China), and used as received. Triethylamine, benzyl alcohol, and dichloromethane (CH2Cl2) were dried over CaH2 and distilled just prior to use. 2-Propylsulfanylthiocarbonyl sulfanyl-2-methyl propionic acid (PTPA) was synthesized according to literature procedures.46 The synthesis of 3-((2-(methacryloyloxy)ethyl)thio) propanoic acid (BSMA) is shown in Supplementary materials and the 1H NMR characterization of BSMA is shown in Figure S1.
Sample synthesis
Synthetic schemes employed for the fabrication of multifunctional polymeric prodrug, P(OEGMA-co-BUF-co-Oct), covalently linked with anticancer drug (BUF) and tumor-targeting peptide (Oct) are shown in Figure 2. All 1H NMR spectra were recorded on a Bruker AV300 NMR spectrometer (resonance frequency of 300 MHz for 1H NMR) operated in the Fourier transform mode. CDCl3 was used as the solvent. Molecular weights and molecular weight distributions were determined by GPC equipped with Waters 1,515 pump and Waters 2,414 differential refractive index detector (set at 30°C), employing a series of two linear Styragel columns (HR2 and HR4) at an oven temperature of 45°C. The eluent was N,N-dimethylformamide (DMF) at a flow rate of 1.0 mL/min.
 | Figure 2 Synthetic routes employed for the fabrication of multifunctional polymeric prodrug, P(OEGMA-co-BUF-co-Oct), covalently linked with anticancer drug BUF, breast cancer targeting peptide, and Oct. |
Synthesis of benzyl 2-methyl-2-(((propylthio)carbonothioyl)thio)propanoate
PTPA (6.67 g, 28 mmol), DCC (5.78 g, 28 mmol), and DMAP (289 mg) were dissolved in anhydrous CH2Cl2 (100 mL) and cooled to 0°C in an ice-water bath. Freshly distilled benzyl alcohol (2.0 g, 18.5 mmol) in anhydrous CH2Cl2 (20 mL) was then added dropwise over 20 minutes. The reaction mixture was stirred at 0°C for 2 hours and then 48 hours at room temperature. The mixture was filtered off, washed with saturated NaHCO3, and further purified by silica gel column chromatography using CH2Cl2/petroleum ether (1:1 v/v) as the eluent. After drying in a vacuum oven overnight, a yellowish oil was obtained (4.18 g, yield 48.2%), 1H NMR (CDCl3, δ, ppm, TMS; Figure S2): 7.30–7.40 (5H, aromatic protons), 5.15 (2H, ArCH2OOC–), 3.23–3.29 (2H, CH3CH2CH2S–), 1.73 (8H, –OC(=O)C(CH3)2S– and CH3CH2CH2S–), and 1.04 (3H, CH3CH2CH2S–).
Synthesis of P(OEGMA-co-BSMA)
β-Carboxyl group-containing copolymers, P(OEGMA-co-BSMA), were synthesized via reversible addition-fragmentation chain transfer (RAFT) polymerization. In a typical procedure, benzyl 2-methyl-2-(((propylthio)carbonothioyl)thio)propanoate (BPTPA; 33 mg, 0.1 mmol), OEGMA (2.70 g, 9.0 mmol), BSMA (218 mg, 1.0 mmol), and AIBN (1.6 mg, 0.01 mmol) were charged into a glass ampoule equipped with a magnetic stirring bar. The ampoule was degassed by three freeze–pump–thaw cycles and sealed under vacuum. After thermostating at 70°C in an oil bath and stirring for 5 hours, the reaction was terminated by quenching into liquid N2, exposed to air, and diluted with methanol. The mixture was then purified by dialysis (cellulose membrane; molecular weight cutoff [MWCO] 3,500 Da) against deionized water for 48 hours to afford P(OEGMA-co-BSMA). The obtained polymer was then treated at 70°C for 2 hours with an excess of AIBN (160 mg, 1.0 mmol) in 1,4-dioxane (5 mL) under nitrogen to remove the trithiocarbonate end-groups (1.74 g, yield 59.0%). The molecular weight and molecular weight distribution of P(OEGMA-co-BSMA) were determined by GPC using DMF as the eluent, revealing an Mn of 22.0 kDa and an Mw/Mn of 1.02 (Table 1). The conversion of OEGMA and BSMA was determined to be ~72% and ~89%, respectively, based on the 1H NMR analysis of the crude product of P(OEGMA-co-BSMA). The DP of P(OEGMA-co-BSMA) was determined to be ~74 by 1H NMR analysis in CDCl3 (Figure S3). Thus, the polymer was denoted as P(OEGMA0.88-co-BSMA0.12)74 and shortened as P(OEGMA-co-BSMA) in the subsequent sections.
 | Table 1 Molecular parameters of polymers used in this study |
Synthesis of P(OEGMA-co-BUF)
P(OEGMA-co-BSMA) (215 mg, 88.8 μmol COOH moieties), DCC (22 mg, 106 μmol), and DMAP (1 mg) were dissolved in anhydrous CH2Cl2 (15 mL) and cooled to 0°C in an ice-water bath. BUF (31 mg, 0.08 mmol) in anhydrous CH2Cl2 (5 mL) was then added dropwise over 10 minutes. The reaction mixture was stirred at 0°C for 2 hours and then 48 hours at room temperature. After filtration, the mixture was purified by dialysis (cellulose membrane; MWCO 3,500 Da) against methanol for 8 hours to afford P(OEGMA-co-BUF) (205 mg, yield 83.3%). The molecular weight and molecular weight distribution of P(OEGMA-co-BUF) were determined by GPC using DMF as the eluent, revealing an Mn of 24.5 kDa and an Mw/Mn of 1.15 (Table 1). BUF content in P(OEGMA-co-BUF) was determined to be ~10 mol% by UV–vis spectroscopy in ethanol by using BUF as the calibration standard (Unico UV/Vis 2802PCS). Thus, the polymer was denoted as P(OEGMA0.88-co-BUF0.1-co-BSMA0.02)74 and shortened as P(OEGMA-co-BUF) in the subsequent sections.
Synthesis of P(OEGMA-co-BUF-co-Oct)
P(OEGMA-co-BUF) (242 mg, 14.8 μmol COOH moieties), DCC (4.5 mg, 22 μmol), and DMAP (0.5 mg) were dissolved in anhydrous CH2Cl2 (10 mL) and cooled to 0°C in an ice-water bath. NHS (3.4 mg, 30 μmol) in anhydrous CH2Cl2 (5 mL) was then added dropwise over 20 minutes. The reaction mixture was stirred at 0°C for 2 hours and then 48 hours at room temperature. The insoluble salt was filtered off, washed with water, and evaporated to dryness. The obtained polymer and triethylamine (2 μL) were then dissolved in DMF (5 mL). Oct (8.4 mg, 7.4 μmol) was added, and the reaction mixture was stirred at ambient temperature overnight. The mixture was then purified by dialysis (cellulose membrane; MWCO 3,500 Da) against water for 8 hours to afford P(OEGMA-co-BUF-co-Oct) (213 mg, yield 85.1%). The molecular weight and molecular weight distribution of P(OEGMA-co-BUF-co-Oct) were determined by GPC using DMF as the eluent, revealing an Mn of 25.0 kDa and an Mw/Mn of 1.19 (Table 1). BUF content was determined to be ~11.5 wt% by UV–vis spectroscopy in ethanol by using BUF as the calibration standard. Thus, the polymer was denoted as P(OEGMA0.88-co-BUF0.1-co-Oct0.01-co-BSMA0.01)74 and shortened as P(OEGMA-co-BUF-co-Oct) in the subsequent sections. Following similar procedures, P(OEGMA0.88-co-BSMA0.11-co-Oct0.01)74, P(OEGMA0.88-co-BUF0.1-co-Cy50.01-co-BSMA0.01)74, and P(OEGMA0.88-co-BUF0.1-co-Oct0.01-co-Cy50.01)74 were obtained and shortened as P(OEGMA-co-Oct), P(OEGMA-co-BUF-co-Cy5), and P(OEGMA-co-BUF-co-Oct-co-Cy5), respectively, in the subsequent sections.
Preparation of micelles
Micelles assembled from polymeric prodrugs were prepared via the cosolvent approach. In a typical example, P(OEGMA-co-BUF-co-Oct) (10 mg) was dissolved in DMF (1 mL). The polymer solution was added into 9 mL deionized water under vigorous stirring. Then the solution was dialyzed against deionized water to remove organic solvent. Finally, the colloidal dispersion was diluted to the desired concentrations for further experiments.
Determination of hydrodynamic diameter (Dh) and zeta potential (ζ)
Malvern Zetasizer Nano ZS (Malvern Instruments, Malvern, UK) is used to characterize the zeta potentials and hydrodynamic dimensions of micellar nanoparticles with 632 nm set at a scattering angle of 173°. The solution was first sonicated for ~30 seconds, and then the measurements were performed in disposable sizing cuvettes or zeta-potential measurement cells. Each measurement was performed in triplicate. The micelles were characterized in phosphate-buffered saline (PBS) buffer (10 mM, pH 7.4) at a concentration of 0.5 g/L.
TEM characterization
TEM observation was conducted on a Hitachi H-800 electron microscope at an acceleration voltage of 200 kV. The sample for TEM observations was prepared by placing 10 mL of mixed micellar solution (0.2 g/L) onto copper grid coated with thin films of Formvar and carbon successively. The samples were stained with phosphotungstic acid (1%) for 2 minutes before observation.
In vitro drug release measurements
Approximately 100 μL of aqueous dispersion of drug-containing micelles (1.0 g/L) was transferred to a dialysis cell with MWCO of 2.0 kDa and then dialyzed against 3.4 mL of PBS buffer (pH 7.4) in the absence and presence of 10 U of esterase at 37°C or in the presence of serum and tumor homogenate at 37°C. The BUF concentration in the dialysate was quantified by high performance liquid chromatography (LC-20AD Series; Shimadzu Corporation, Kyoto, Japan) by measuring the absorption of BUF at 298 nm against a standard calibration curve.
In vitro cytotoxicity evaluation
Breast cancer cell line MCF-7 was employed for in vitro cytotoxicity evaluation via the MTT assay. Briefly, MCF-7 cells were seeded in a 96-well plate at an initial density of 5,000 cells/well in 200 μL of complete DMEM. After incubating for 24 hours, the medium was replaced with fresh medium, and the cells were treated with free BUF, P(OEGMA-co-BSMA), and P(OEGMA-co-BUF-co-Oct) at varying concentrations. After incubation for 24 hours further, MTT reagent (in 20 μL of PBS buffer, 5 mg/mL) was added to each well, and the cells were further incubated with 5% CO2 for 4 hours at 37°C. The culture medium in each well was removed and replaced by 150 μL of DMSO. The plate was gently agitated for 15 minutes before the absorbance at 490 nm was recorded by a microplate reader (Thermo Fisher Scientific). Each experiment condition was done in quadruple, and the data are shown as the mean value plus a standard deviation (±SD).
Cellular uptake of nanoparticles via flow cytometry
MCF-7 cells were seeded in six-well plates at ~105 cells per well in 1 mL supplemented DMEM. After 24-hour incubation, cells were stained by P(OEGMA-co-BUF-co-Oct-co-Cy5) and P(OEGMA-co-BUF-co-Cy5) in supplemented DMEM at a final Cy5 concentration of 3.0×10−6 M for 4 hours. Afterward, the culture medium was removed, and cells were washed three times with PBS and harvested with trypsin. The cells were resuspended in 500 μL of PBS. The quantitative uptake was recorded by a flow cytometry (Calibur; BD Biosciences).
Fluorescence imaging
MCF-7 cells were cultured in DMEM at 37°C in a CO2/air (5:95) incubator for 24 hours. Then, MCF-7 cells were stained in the presence of P(OEGMA-co-BUF-co-Oct-co-Cy5) ([Cy5] =3.0×10−6 M) and P(OEGMA-co-BUF-co-Cy5) ([Cy5] =3.0×10−6 M) for 4 hours and DAPI for 15 minutes at 37°C in a CO2/air (5:95) incubator. The concentration of DAPI was used according to the protocols provided by Beyotime Biotech. The cells were washed with PBS for three times and then fixed with 4% formaldehyde for 20 minutes at room temperature. DAPI and Cy5 were excited at 405 nm and 633 nm, respectively, and the fluorescence emissions were collected between 420 nm and 460 nm and 660 nm and 700 nm, respectively, by ZEISS710 confocal microscope (Carl Zeiss Meditec AG, Jena, Germany). Images were taken with a 63× objective lens.
Apoptosis assay via flow cytometry
To compare the apoptotic effects of free BUF, P(OEGMA-co-BUF), and P(OEGMA-co-BUF-co-Oct) in MCF-7 cells, an Annexin V FITC/PI apoptosis detection kit (BD Biosciences) was used. Loss of plasma membrane is one of the earliest features of apoptosis. In apoptotic cells, the membrane phospholipid phosphatidylserine (PS) is translocated from the inner to the outer leaflet of the plasma membrane, thereby exposing PS to the external cellular environment. Annexin V has a high affinity for PS and binds to cells with exposed PS. Annexin V may be conjugated to fluorochromes, including FITC. This format retains its high affinity for PS and thus serves as a sensitive probe for flow cytometric analysis of cells that are undergoing apoptosis. The red fluorescent PI dye enters cells through damaged membranes and binds to nucleic acids, but is excluded by the intact plasma membranes of live cells. Briefly, cells were seeded at a density of 5×105/well in six-well plates. After incubating for 24 hours, the medium with 2% fetal bovine serum was replaced with a fresh medium, and the cells were treated with 50 nM BUF-equivalent dosage of free BUF, P(OEGMA-co-BUF), and P(OEGMA-co-BUF-co-Oct) for 24 hours. The cells were then collected, washed twice with cold PBS, resuspended at a density of 1×106 cells/mL, and then stained with 5 μL of Annexin V FITC and 5 μL of PI for 15 minutes at room temperature in the dark according to the manufacturer’s instructions. Cell apoptosis was analyzed by using flow cytometry (Calibur).
Establishment of a tumor model
Subcutaneous tumors were established in nude mice (BALB/c, female, 4–6 weeks old) by injecting MCF-7 cells (5×106 cells in 0.2 mL PBS) into their left armpits. All experiments were carried out according to the Guidelines of the Laboratory Protocol of Animal Handling (affiliated Putuo Hospital of Shanghai University of Traditional Chinese Medicine) and the study protocols were approved by the Institutional Animal Care and Use Committee of Shanghai University of Traditional Chinese Medicine. Tumor sizes were measured using a caliper, and the tumor-bearing mice were randomized and classified into five groups (n=6): blank, free BUF, P(OEGMA-co-BSMA), P(OEGMA-co-BUF), and P(OEGMA-co-BUF-co-Oct).
Biodistribution of polymeric prodrug
Fluorescent dye Cy5-labeled polymeric micellar nanoparticles, P(OEGMA-co-BUF-co-Oct-co-Cy5) and P(OEGMA-co-BUF-co-Cy5), were employed to evaluate the tumor-targeting capability in MCF-7 breast cancer xenograft tumors. P(OEGMA-co-BUF-co-Oct-co-Cy5) (200 μL, 0.87 mg/mL, [Cy5] =2.5×10−5 M) and P(OEGMA-co-BUF-co-Cy5) (200 μL, 0.87 mg/mL, [Cy5] =2.5×10−5 M) were injected through the vena caudalis (tail vein) into the tumor-bearing xenografted mice. Fluorescence imaging of the tumor-bearing mice was performed at varying time intervals after injection by using a small animal in vivo fluorescence imaging system (LB 983; Berthold Technologies GmbH and Co KG, Bad Wildbad, Germany). After 48 hours, the nude mice were sacrificed. Tumor and organs, including heart, liver, spleen, lung, and kidney, were collected for ex vivo DOX distribution examination with an in vivo imaging system.
In vivo study of the therapeutic efficacy in mice
The mice were classified into five groups, normal saline group (13.5 mL/kg), BUF solution group (1 mg BUF/kg), P(OEGMA-co-BSMA) group (10 mg/kg), P(OEGMA-co-BUF) group (1 mg BUF/kg), and P(OEGMA-co-BUF-co-Oct) group (1 mg BUF/kg), on the basis of the solutions they were administered by intravenous injection through the vena caudalis every 2 days. The remaining mice in each group were killed, and the tumors were collected for examination.
Histological examination of tumor tissues
The removed xenograft tumors were fixed with 4% neutral buffered paraformaldehyde and embedded in paraffin before 4 μm tissue sections were prepared and stained with H&E for histological examination.
Statistical analysis
Unless specified otherwise, all experiments were performed in triplets, and data were reported as mean ± standard deviation. Statistical significance was determined using Student’s t-test.
Results and discussion
The focus of this study was to prepare and characterize the BUF prodrugs with the aim of enhancing its anticancer performance both in vitro and in vivo. SSTR2 overexpressing breast cancer cell line MCF-7 cells were used to examine their physiochemical properties, cellular uptake, ability to induce apoptosis and suppress proliferation of tumor cells, and in vivo anticancer effects.4,31,36,37 Throughout the studies, the effects of prodrugs were compared with those of native-free BUF.
Synthesis and characterization of polymeric prodrug
Polymeric prodrug of BUF, P(OEGMA-co-BUF-co-Oct), was fabricated via the combination of RAFT polymerization and polymer postfunctionalization (Figure 2).
Random copolymers, P(OEGMA-co-BSMA), were fabricated via the RAFT polymerization in the presence of BSMA (1H NMR characterization result is shown in Figure S1) and OEGMA using benzyl-containing small molecular RAFT agent (BPTPA; 1H NMR characterization of BPTPA is shown in Figure S2). GPC characterization revealed an Mn of 22.0 kDa and an Mw/Mn of 1.02 for P(OEGMA-co-BSMA) (Table 1). The DP was determined to be ~74 by 1H NMR analysis (Figure S3). P(OEGMA-co-BUF) was then obtained by the esterification reaction between the carboxyl groups of P(OEGMA-co-BSMA) and the 3α-hydroxyl group of BUF with an Mn of 24.5 kDa and Mw/Mn of 1.15 (Table 1). 1H NMR analysis further confirmed the successful linkage of BUF onto the polymers as shown in the appearance of characteristic peaks (peaks i, g, h) ascribed to BUF in Figure S4. Tumor-targeting polymeric prodrug, P(OEGMA-co-BUF-co-Oct), was finally obtained via the reaction of P(OEGMA-co-BUF) and Oct, which was confirmed by 1H NMR analysis (Figure S5). Mn and Mw/Mn were determined to be 25.0 kDa and 1.19, respectively (Table 1). BUF content was determined to be ~11.5 wt%. Following similar procedures, fluorescent dye Cy5-labeled polymeric prodrugs, P(OEGMA-co-BUF-co-Oct-co-Cy5) and P(OEGMA-co-BUF-co-Cy5), were obtained, exhibiting a strong infrared fluorescence emission peak at ~665 nm (Figure S6).
In aqueous media at pH 7.4, P(OEGMA-co-BUF-co-Oct) and its precursors tend to self-assemble into micellar nanoparticles. As shown in Figure 3a, the intensity-average hydrodynamic diameter (Dh) of P(OEGMA-co-BSMA) was determined to be ~70.3 (±1.38) nm with polydispersity index (μ2/G2) of 0.26. The zeta potential (ζ) was determined to be −17.7 (±0.15) mV, exhibiting negatively charged characteristics presumably due to the ionization of carboxyl groups. By introducing hydrophobic BUF onto the polymer backbone typical nanoparticles assembled from P(OEGAM-co-BUF) showed Dh of ~71.3 (±0.77) nm and μ2/G2 of 0.18 (Figure 3b), exhibiting little change compared with that of P(OEGMA-co-BSMA). However, the ζ dropped to −3.60 (±0.21) mV as a result of the fact that the carboxyl groups were reacted with BUF. The attachment of Oct onto P(OEGAM-co-BUF) showed slightly increased size with Dh of ~75.8 (±2.61) nm, μ2/G2 of 0.18, and ζ of −2.98 (±0.56) mV (Figure 3c). To further verify the presence of micellar nanoparticles in the star copolymer solution, TEM observations were conducted. The TEM image revealed the presence of robust and fairly narrow-distributed spherical nanoparticles with a diameter of ~30–40 nm. The dimension of nanoparticles determined by TEM reasonably agrees with that determined by dynamic LLS considering that the LLS technique reports the intensity-average dimensions in solution, whereas TEM determines nanoparticle dimensions in the dry state.
 | Figure 3 Typical hydrodynamic diameter distributions, f(Dh), recorded for 0.5 g/L aqueous solution of (a) P(OEGMA-co-BSMA), (b) P(OEGMA-co-BUF-co-BSMA), and (c) P(OEGMA-co-BUF-co-Oct). |
In vitro drug release
In vitro drug release of P(OEGMA-co-BUF-co-Oct) was then explored. As shown in Figure 4, under simulated physiological condition (PBS, pH 7.4, 37°C), only ~15% cumulative BUF release was observed over 24 hours, which was in accordance with the fact that β-thioester bonds are relatively stable at neutral conditions.40 This design can effectively circumvent burst and premature drug release after intravenous administration. While in the presence of esterase, which is abundant in cytosol and endo/lysosomes, cumulative ~86% BUF released from nanoparticles was obtained over the same period, showing controlled release characteristics. In addition, the introduction of tumor-targeting moiety Oct is assumed to enhance the accumulation of drugs in the cancerous tissue via receptor-mediated endocytosis.36 Considering the in vivo application of the prodrugs, in vitro drug release study in the presence of serum and tumor homogenate was also conducted to examine the real fate of the obtained prodrugs in circulation and tumor. As shown in Figure 4C, in the presence of serum, ~30.5% cumulative BUF release was observed over 24 hours, implying that a small part of BUF molecules might be released from the prodrugs in the circulation period. However, a majority of BUF molecules are still located in the nanoparticles. In addition, in the presence of tumor homogenate, cumulative ~82% BUF released from nanoparticles was obtained over the same period, indicating that nanoparticles reaching tumor tissues could effectively release BUF drugs.
 | Figure 4 In vitro BUF release profiles (37°C, 20 mM buffer solution) from polymeric micellar solution of P(OEGMA-co-BUF-co-Oct) in the (a) presence and (b) absence of 10 U esterase and in the presence of (c) serum and (d) tumor homogenate. |
In vitro cytotoxicity
In an effort to explore the in vitro anticancer efficacy, MCF-7 cells were employed as a model to evaluate the anticancer performance of the obtained polymeric prodrug in vitro. As shown in Figure 5, >90% MCF-7 cells are still alive even at a concentration as high as ~200 mg/L of P(OEGMA-co-BSMA), implying the good biocompatibility of the polymer. This is in agreement with the well-recognized biocompatibility of PEG-based polymers.39 In addition, the introduction of Oct onto the polymer backbone, P(OEGMA-co-Oct), exhibited little cytotoxicity change in comparison with that of P(OEGMA-co-BSMA; Figure S7), implying the good biocompatibility of P(OEGMA-co-Oct). However, it is encouraging to find that by treating MCF-7 cells with BUF-containing P(OEGMA-co-BUF) dramatically decreased cell viability, exhibiting much better anticancer performance than that of free BUF (Figure 6). The reason of improved anticancer efficacy of P(OEGMA-co-BUF) is still not quite clear and needs further exploration. One possible reason might be that more than one BUF molecule can be internalized simultaneously by cells with the help of the nanostructure of the polymeric prodrug. It was calculated that the BUF content in polymeric prodrug was ~11.5%, which means there were ~7.4 BUF molecules/polymer chain. Besides, it was reported that one polymeric micelle often consisted of several hundreds and even thousands of polymer chains.47 So, it was reasonable to assume that with the internalization of one single nanoparticle, >1,000 BUF molecules entered into cells. This might explain the higher anticancer efficacy of P(OEGMA-co-BUF) than free BUF.
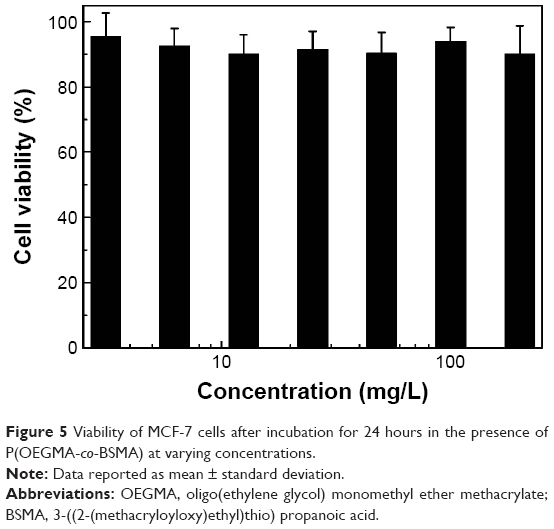 | Figure 5 Viability of MCF-7 cells after incubation for 24 hours in the presence of P(OEGMA-co-BSMA) at varying concentrations. |
 | Figure 6 In vitro cytotoxicity of (a) free BUF, (b) P(OEGMA-co-BUF), and (c) P(OEGMA-co-BUF-co-Oct) determined by the MTT assay against MCF-7 cells after incubating 24 hours at varying BUF concentrations. |
Moreover, the anticancer performance can be further enhanced by introducing tumor-targeting moieties, Oct in this case, to the polymeric prodrug. P(OEGMA-co-BUF-co-Oct) exhibits further improved cytotoxicity to MCF-7 cells in comparison with that of P(OEGMA-co-BUF), especially at lower BUF concentration. For example, at a BUF concentration of 100 nM, ~85.7% of cells are still alive for free BUF. While at the same BUF dosage, P(OEGMA-co-BUF) led to 53.9% cell death and P(OEGMA-co-BUF-co-Oct) caused 58.7% cell death. This result reveals that attaching Oct onto the polymer backbone might play an important role at lower BUF concentration in enhancing the cytotoxicity by promoting surface binding and cellular entry via receptor-mediated endocytosis mechanism (Figure 1).4,31,36,37 However, by increasing the BUF dosage, the anticancer difference between P(OEGMA-co-BUF) and P(OEGMA-co-BUF-co-Oct) decreased, although they still performed better than free BUF. The main reason might be related to the relative amount of SSTR2 on the membrane surface of MCF-7 cells and Oct peptide on the surface of nanoparticles. At relatively lower BUF concentrations, the amount of Oct was also small and the amount of SSTR2 was large enough to bind all the Oct-modified nanoparticles at the interface and mediated drugs into cells via receptor-mediated endocytosis. While, with the increase of BUF concentration, the amount of Oct increased and SSTR2 was saturated with increased Oct. At this point, receptor-mediated endocytosis reached its maximum. Further increasing BUF concentration cannot lead to more binds between Oct and SSTR2, and the surplus Oct cannot exert the role as effective as that at a lower concentration, which consequently led to the decreased anticancer difference between P(OEGMA-co-BUF) and P(OEGMA-co-BUF-co-Oct).
In vitro cellular uptake
The ability of selective delivery of drugs to tumor cells is an essential function of tumor-targeting nanocarriers, especially for prodrug systems. As described earlier, SSTR2 overexpressing breast cancer-targeting polymeric prodrug, P(OEGMA-co-BUF-co-Oct), showed dramatically enhanced antiproliferation effect, which was partly ascribed to the active targeting moieties to the MCF-7 cells. Currently, flow cytometry and confocal laser scanning microscopy were employed to explore the cellular uptake of polymeric prodrugs.
As shown in Figure 7A, the flow cytometry was first used to quantitatively evaluate the selective cellular uptake mechanism of P(OEGMA-co-BUF-co-Oct-co-Cy5) in MCF-7 cells. The results showed that P(OEGMA-co-BUF-co-Cy5) can effectively enter into cells as revealed by the strong fluorescence distribution in cells compared with control sample. However, the cellular uptake of Oct-modified P(OEGMA-co-BUF-co-Oct-co-Cy5) dramatically increased relative to the levels of P(OEGMA-co-BUF-co-Cy5). Considering that the only difference of P(OEGMA-co-BUF-co-Cy5) and P(OEGMA-co-BUF-co-Oct-co-Cy5) is the introduction of targeting peptide Oct, this implied the importance of Oct in the improvement of internationalization of prodrugs. To further examine the role of Oct in the internalization of prodrugs, SSTR2-negative MDA-MB-231 cells were employed to conduct flow cytometry experiment. As shown in Figure 7B, the cellular uptake of P(OEGMA-co-BUF-co-Cy5) and P(OEGMA-co-BUF-co-Oct-co-Cy5) exhibited negligible differences, indicating that the introduction of Oct impacts little to internalization for SSTR2-negative cells. In combination with the earlier results, it was reasonable to assume that the interaction of Oct peptide and SSTR2 on the cellular surface plays an important role in improving the internalization of prodrugs. Next, confocal laser scanning microscopy was applied to further investigate the intracellular distribution of Cy5-labeled prodrugs. As shown in Figure 8, P(OEGMA-co-BUF-co-Cy5) was effectively internalized and accumulated in the cytosol of cells as vividly revealed by the fluorescent dots distributed in cells. However, by staining cells with P(OEGMA-co-BUF-co-Oct-co-Cy5), substantially increased fluorescence can be observed, which further confirmed the importance of Oct. Correspondingly, fluorescence imaging for SSTR2-negative MDA-MB-231 cells was also conducted. As shown in Figure S8, both P(OEGMA-co-BUF-co-Cy5) and P(OEGMA-co-BUF-co-Oct-co-Cy5) can be effectively internalized by cells and little fluorescence intensity difference can be observed, which are in agreement with the results by flow cytometry.
 | Figure 7 Flow cytometry analysis of cellular uptake of (a) and (c) P(OEGMA-co-BUF-co-Cy5) and (b) and (d) P(OEGMA-co-BUF-co-Oct-co-Cy5) for 4 hours in (A) MCF-7 and (B) MDA-MB-231 cells. |
 | Figure 8 Typical confocal microscopy fluorescence images recorded for MCF-7 cells after incubating at 37°C with (top panels) P(OEGMA-co-BUF-co-Cy5) ([Cy5] =3.0×10−6 M) and (bottom panels) P(OEGMA-co-BUF-co-Oct-co-Cy5) ([Cy5] =3.0×10−6 M) for 4 hours. |
Apoptosis assay
As a programmed cell death, cell apoptosis could be featured by a series of morphological and biochemical changes. In the current work, MCF-7 cells were treated with free BUF, P(OEGMA-co-BUF), and P(OEGMA-co-BUF-co-Oct) to evaluate the apoptosis process. As shown in Figures 9 and S9, compared with the control sample, free BUF-treated cells exhibited an increase in early apoptosis and necrosis stages. On the other hand, P(OEGMA-co-BUF) led to little increase in early apoptosis but a dramatic increase in late apoptosis, and necrosis stages were observed. As expected, P(OEGMA-co-BUF-co-Oct) caused a further increase in late apoptosis and necrosis chambers in comparison with that of both free BUF and P(OEGMA-co-BUF). Besides, the apoptosis analysis via DAPI staining method also confirmed this. As shown in Figure S10, compared with untreated MCF-7 cells, free BUF led to condensed or fragmented nuclei, which is the characteristic of apoptosis, and polymeric prodrugs showed further condensed or fragmented nuclei.
 | Figure 9 Flow-cytometric analysis of the apoptosis of MCF-7 cells. |
In vivo biodistribution examination
As described previously, the introduction of Oct onto the prodrug led to enhanced internalization of drugs into tumor cells. Herein, Cy5-labeled targeting and nontargeting prodrugs, P(OEGMA-co-BUF-co-Oct-co-Cy5) and P(OEGMA-co-BUF-co-Cy5), were intravenously injected into MCF-7 tumor-bearing mice to evaluate the biodistribution of drugs. Figure 10 shows the real-time distribution and tumor accumulation of P(OEGMA-co-BUF-co-Oct-co-Cy5) and P(OEGMA-co-BUF-co-Cy5) at various time intervals. In P(OEGMA-co-BUF-co-Cy5) group, the fluorescence signals almost uniformly distributed around the body at first and then accumulated in kidney. This might be due to some free drugs cleaved and released from nanoparticles in the circulation period, considering the esterase activity of serum as revealed in Figure 4. Throughout the 48-hour period, only slight accumulation of signals in the tumor tissue can be observed. In contrast, the fluorescence intensity of the tumor region injected with P(OEGMA-co-BUF-co-Oct-co-Cy5) was significantly stronger, presumably owing to the modification of the active targeting peptide Oct. The fluorescence intensity in tumor was much stronger than other organs such as kidney and liver. Besides, the fluorescence intensity in tumor declined little even after 48-hour postinjection, implying the long retention of drugs in tumor tissues. Moreover, quantitative analysis of tissue distributions was also conducted. As shown in Figure 10C, the percentages of fluorescence intensities of heart, liver, spleen, and lung are significantly lower than those in tumors for both P(OEGMA-co-BUF-co-Cy5) and P(OEGMA-co-BUF-co-Oct-co-Cy5). For P(OEGMA-co-BUF-co-Cy5), ~25.8% of fluorescence intensity accumulated in tumor tissues and ~48.3% of fluorescence intensity accumulated in kidney. However, for P(OEGMA-co-BUF-co-Oct-co-Cy5), ~55.4% of fluorescence intensity accumulated in tumor tissues, while the amount in kidney was relatively smaller possibly due to the active targeting capability.
 | Figure 10 (A) In vivo imaging of tumor-bearing mice after IV administration with (a) P(OEGMA-co-BUF-co-Cy5) and (b) P(OEGMA-co-BUF-co-Oct-co-Cy5) for varying time. (B) Fluorescence images of organs, including heart, liver, spleen, lung, and kidney, and tumor collected at 48 h post injection of (c) P(OEGMA-co-BUF-co-Cy5) and (d) P(OEGMA-co-BUF-co-Oct-co-Cy5). (C) Bio-distributions of P(OEGMA-co-BUF-co-Cy5) and P(OEGMA-co-BUF-co-Oct-co-Cy5) in tumor issues and different organs. |
In vivo anticancer efficacy
Polymeric prodrugs of BUF showed significant improved antitumor activity in the MCF-7 tumor model. As illustrated in Figure 11, compared with saline control group, drug carrier, P(OEGMA-co-BSMA), showed no effect on the tumor in mice. When BUF was administered into the mice, the tumor growth was significantly inhibited as vividly revealed by the reduced tumor volume growth in comparison with that of control group. Furthermore, when the same dose of BUF was administered into the mice, the tumor inhibiting effect was much more obvious in the P(OEGMA-co-BUF) group than that in the BUF solution group. It was well documented that macromolecular drugs show much longer circulation time and more accumulation in tumor tissues than their small molecular counterparts based on EPR effects.18–23 Besides, in vitro cell cytotoxicity also demonstrated improved anticancer efficacy of P(OEGMA-co-BUF), possibly due to its nanostructure and prodrug design. The two factors might together lead to the improved tumor inhibition effect in in vivo experiment. P(OEGMA-co-BUF-co-Oct) further enhanced the inhibition of tumor growth, presumably due to increased accumulation of drug in the tumor tissue via Oct-mediated active targeting and macromolecular-based EPR effects. Moreover, no obvious toxicity was observed in the mice for all the groups, which was demonstrated by the well-maintained body weight of the mice. Finally, H&E staining results indicated that BUF-containing polymeric prodrugs led to much larger necrotic areas than free BUF and that there was no significant reaction observed in normal organs (Figure S11).
 | Figure 11 Polymeric prodrugs of BUF lead to enhanced antitumor efficacy in the BALB/c nude mice bearing MCF-7. |
Conclusion
In summary, we successfully developed a novel type of multifunctional polymeric prodrug of BUF, P(OEGMA-co-BUF-co-Oct), covalently modified with breast cancer-targeting peptide (Oct). The obtained polymeric prodrug exhibited improved anticancer efficacy against breast cancer both in vitro and in vivo in comparison with that of small molecular-free BUF. The employment of β-thioester bond to link BUF effectively circumvents burst/premature drug release, which often exists in the conventional physiologically encapsulation method. Meanwhile, in the presence of esterase, BUF can be effectively released. In addition, the introduction of Oct endows the polymeric prodrug good active targeting to tumor tissues in addition to passive targeting via EPR effect. In vivo fluorescence imaging and anticancer experiments indicated that P(OEGMA-co-BUF-co-Oct) exhibited excellent specific accumulation in tumor regions as well as dramatically improved antitumor efficacy. The reported tumor-targeting polymeric prodrug synergistically integrated with active targeting and triggered drug release augurs well for breast cancer therapy.
Acknowledgments
This work was supported by the “Budgeted Program” of Shanghai University of Traditional Chinese Medicine (No 2014YSN66), the “Doctoral Program” of Putuo Hospital of Shanghai University of Traditional Chinese Medicine (No 2015PT03), and the “Key Disciplines Program” of Putuo Hospital of Shanghai University of Traditional Chinese Medicine (No 2013XK151 I).
Disclosure
The authors report no conflicts of interest in this work.
References
Fan L, Strasser-Weippl K, Li JJ, et al. Breast cancer in China. Lancet Oncol. 2014;15:279–289. | ||
Siegel RL, Miller KD, Jemal A. Cancer statistics, 2015. CA Cancer J Clin. 2015;65:5–29. | ||
Harmer V. Breast cancer. Part 2: present and future treatment modalities. Br J Nurs. 2008;17:1028–1029. | ||
Huang CM, Wu YT, Chen ST. Targeting delivery of paclitaxel into tumor cells via somatostatin receptor endocytosis. Chem Biol. 2000;7:453–461. | ||
Duggan ST, Keating GM. Pegylated liposomal doxorubicin a review of its use in metastatic breast cancer, ovarian cancer, multiple myeloma and AIDS-related Kaposi’s sarcoma. Drugs. 2011;71:2531–2558. | ||
Qian Y, Liu Y, Yan Q, et al. Inhibition of Mus81 by siRNA enhances sensitivity to 5-FU in breast carcinoma cell lines. Onco Targets Ther. 2014;7:1883–1890. | ||
Luo C, Wang Y, Chen Q, et al. Advances of paclitaxel formulations based on nanosystem delivery technology. Mini Rev Med Chem. 2012;12: 434–444. | ||
Lu J, Liu C, Wang P, et al. The self-assembling camptothecin-tocopherol prodrug: an effective approach for formulating camptothecin. Biomaterials. 2015;62:176–187. | ||
Jiang Y, Zhang Y, Luan J, et al. Effects of bufalin on the proliferation of human lung cancer cells and its molecular mechanisms of action. Cytotechnology. 2010;62:573–583. | ||
Xie CM, Chan WY, Yu S, Zhao J, Cheng CH. Bufalin induces autophagy-mediated cell death in human colon cancer cells through reactive oxygen species generation and JNK activation. Free Radic Biol Med. 2011;51:1365–1375. | ||
Xie XB, Yin JQ, Wen LL, et al. Critical role of heat shock protein 27 in bufalin-induced apoptosis in human osteosarcomas: a proteomic-based research. PLoS One. 2012;7:e47375. | ||
Yin PH, Liu X, Qiu YY, et al. Anti-tumor activity and apoptosis-regulation mechanisms of bufalin in various cancers: new hope for cancer patients. Asian Pac J Cancer Prev. 2012;13:5339–5343. | ||
Qiu DZ, Zhang ZJ, Wu WZ, Yang YK. Bufalin, a component in Chansu, inhibits proliferation and invasion of hepatocellular carcinoma cells. BMC Complement Altern Med. 2013;13:185–195. | ||
Li MY, Yu XJ, Guo H, et al. Bufalin exerts antitumor effects by inducing cell cycle arrest and triggering apoptosis in pancreatic cancer cells. Tumour Biol. 2014;35:2461–2471. | ||
Qiu YY, Hu Q, Tang QF, et al. MicroRNA-497 and bufalin act synergistically to inhibit colorectal cancer metastasis. Tumour Biol. 2014;35:2599–2606. | ||
Yang Z, Teng Y, Wang H, Hou H. Enhancement of skin permeation of bufalin by limonene via reservoir type transdermal patch: formulation design and biopharmaceutical evaluation. Int J Pharm. 2013;447: 231–240. | ||
Hu Q, Liang B, Sun Y, et al. Preparation of bufalin-loaded pluronic polyetherimide nanoparticles, cellular uptake, distribution, and effect on colorectal cancer. Int J Nanomedicine. 2014;9:4035–4041. | ||
Matsumura Y, Maeda H. A new concept for macromolecular therapeutics in cancer chemotherapy: mechanism of tumoritropic accumulation of proteins and the antitumor agent smancs. Cancer Res. 1986;46: 6387–6392. | ||
Maeda H, Seymour LW, Miyamoto Y. Conjugates of anticancer agents and polymers: advantages of macromolecular therapeutics in vivo. Bioconjug Chem. 1992;3:351–362. | ||
Maeda H. SMANCS and polymer-conjugated macromolecular drugs: advantages in cancer chemotherapy. Adv Drug Deliv Rev. 2001;46:169–185. | ||
Maeda H, Sawa T, Konno T. Mechanism of tumor-targeted delivery of macromolecular drugs, including the EPR effect in solid tumor and clinical overview of the prototype polymeric drug SMANCS. J Control Release. 2001;74:47–61. | ||
Maeda H, Bharate GY, Daruwalla J. Polymeric drugs for efficient tumor-targeted drug delivery based on EPR-effect. Eur J Pharm Biopharm. 2009;71:409–419. | ||
Dozono H, Yanazume S, Nakamura H, et al. HPMA copolymer-conjugated pirarubicin in multimodal treatment of a patient with stage IV prostate cancer and extensive lung and bone metastases. Target Oncol. 2016;11:101–106. | ||
Fleige E, Quadir MA, Haag R. Stimuli-responsive polymeric nanocarriers for the controlled transport of active compounds: concepts and applications. Adv Drug Deliv Rev. 2012;64:866–884. | ||
Ren T, Xia W, Wu W, Li Y. Stimuli-responsive and polymeric prodrugs. Prog Chem. 2013;25:775–784. | ||
Oerlemans C, Bult W, Bos M, Storm G, Nijsen JF, Hennink WE. Polymeric micelles in anticancer therapy: targeting, imaging and triggered release. Pharm Res. 2010;27:2569–2589. | ||
Cabral H, Kataoka K. Progress of drug-loaded polymeric micelles into clinical studies. J Control Release. 2014;190:465–476. | ||
Zhang Y, Zhang H, Wang X, Wang J, Zhang X, Zhang Q. The eradication of breast cancer and cancer stem cells using octreotide modified paclitaxel active targeting micelles and salinomycin passive targeting micelles. Biomaterials. 2012;33:679–691. | ||
Virgolini I, Leimer M, Handmaker H, et al. Somatostatin receptor subtype specificity and in vivo binding of a novel tumor tracer, Tc-99m-P829. Cancer Res. 1998;58:1850–1859. | ||
Corleto VD, Nasoni S, Panzuto F, Cassetta S, Delle Fave G. Somatostatin receptor subtypes: basic pharmacology and tissue distribution. Dig Liver Dis. 2004;36:S8–S16. | ||
Watt HL, Kumar U. Colocalization of somatostatin receptors and epidermal growth factor receptors in breast cancer cells. Cancer Cell Int. 2006;6:1–19. | ||
Bruns C, Weckbecker G, Raulf F, et al. Molecular pharmacology of somatostatin-receptor subtypes. Ann N Y Acad Sci. 1994;733:138–146. | ||
Patel YC, Srikant CB. Subtype selectivity of peptide analogs for all five cloned human somatostatin receptors (hsstr 1–5). Endocrinology. 1994;135:2814–2817. | ||
Siehler S, Seuwen K, Hoyer D. [125I][Tyr3]octreotide labels human somatostatin sst2 and sst5 receptors. Eur J Pharmacol. 1998;348:311–320. | ||
Hashemi SH, Benjegård SA, Ahlman H, et al. 111In-labelled octreotide binding by the somatostatin receptor subtype 2 in neuroendocrine tumours. Br J Surg. 2003;90:549–554. | ||
Huo MR, Zou A, Yao C, et al. Somatostatin receptor-mediated tumor-targeting drug delivery using octreotide-PEG-deoxycholic acid conjugate-modified N-deoxycholic acid-O, N-hydroxyethylation chitosan micelles. Biomaterials. 2012;33:6393–6407. | ||
Zhang J, Jin W, Wang X, Wang J, Zhang X, Zhang Q. A novel octreotide modified lipid vesicle improved the anticancer efficacy of doxorubicin in somatostatin receptor 2 positive tumor models. Mol Pharm. 2010;7:1159–1168. | ||
Zhang Y, Wang X, Wang J, Zhang X, Zhang Q. Octreotide-modified polymeric micelles as potential carriers for targeted docetaxel delivery to somatostatin receptor overexpressing tumor cells. Pharm Res. 2011;28:1167–1178. | ||
Liu J, Zahedi P, Zeng F, Allen C. Nano-sized assemblies of a PEG-docetaxel conjugate as a formulation strategy for docetaxel. J Pharm Sci. 2008;97:3274–3290. | ||
Shen Y, Jin E, Zhang B, et al. Prodrugs forming high drug loading multifunctional nanocapsules for intracellular cancer drug delivery. J Am Chem Soc. 2010;132:4259–4265. | ||
Zhang G, Zhang M, He J, Ni P. Synthesis and characterization of a new multifunctional polymeric prodrug paclitaxel-polyphosphoester-folic acid for targeted drug delivery. Polym Chem. 2013;4:4515–4525. | ||
Dong C, Xia W, Li Y, Ren T. Self-assembled, redox-sensitive, H-shaped pegylated methotrexate conjugates with high drug-carrying capability for intracellular drug delivery. MedChemComm. 2014;5:147–152. | ||
Wang W, Li C, Zhang J, et al. Tailor-made gemcitabine prodrug nanoparticles from well-defined drug-polymer amphiphiles prepared by controlled living radical polymerization for cancer chemotherapy. J Mater Chem B. 2014;2:1891–1901. | ||
Zou J, Zhang F, Zhang S, et al. Poly(ethylene oxide)-block-polyphosphoester-graft-paclitaxel conjugates with acid-labile linkages as a pH-sensitive and functional nanoscopic platform for paclitaxel delivery. Adv Healthc Mater. 2014;3:441–448. | ||
Huo M, Zhu Q, Wu Q, et al. Somatostatin receptor-mediated specific delivery of paclitaxel prodrugs for efficient cancer therapy. J Pharm Sci. 2015;104:2018–2028. | ||
An Z, Tang W, Wu M, Jiao Z, Stucky GD. Heterofunctional polymers and core-shell nanoparticles via cascade aminolysis/Michael addition and alkyne-azide click reaction of RAFT polymers. Chem Commun (Camb). 2008;48:6501–6503. | ||
Liu T, Qian Y, Hu X, et al. Mixed polymeric micelles as multifunctional scaffold for combined magnetic resonance imaging contrast enhancement and targeted chemotherapeutic drug delivery. J Mater Chem. 2012;22:5020–5030. |
 © 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.