")
Back to Journals » International Journal of Nanomedicine » Volume 9 » Issue 1
Development of nucleic acid vaccines: use of self-amplifying RNA in lipid nanoparticles
Authors Rodríguez-Gascón A, del Pozo-Rodríguez A, Solinís MA
Received 18 December 2013
Accepted for publication 15 February 2014
Published 10 April 2014 Volume 2014:9(1) Pages 1833—1843
DOI https://doi.org/10.2147/IJN.S39810
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 5
Alicia Rodríguez-Gascón, Ana del Pozo-Rodríguez, María Ángeles Solinís
Pharmacokinetic, Nanotechnology and Gene Therapy Group (PharmaNanoGene), Faculty of Pharmacy, Centro de Investigación Lascaray Ikergunea, University of the Basque Country UPV/EHU, Vitoria-Gasteiz, Spain
Abstract: Self-amplifying RNA or RNA replicon is a form of nucleic acid-based vaccine derived from either positive-strand or negative-strand RNA viruses. The gene sequences encoding structural proteins in these RNA viruses are replaced by mRNA encoding antigens of interest as well as by RNA polymerase for replication and transcription. This kind of vaccine has been successfully assayed with many different antigens as vaccines candidates, and has been shown to be potent in several animal species, including mice, nonhuman primates, and humans. A key challenge to realizing the broad potential of self-amplifying vaccines is the need for safe and effective delivery methods. Ideally, an RNA nanocarrier should provide protection from blood nucleases and extended blood circulation, which ultimately would increase the possibility of reaching the target tissue. The delivery system must then be internalized by the target cell and, upon receptor-mediated endocytosis, must be able to escape from the endosomal compartment into the cell cytoplasm, where the RNA machinery is located, while avoiding degradation by lysosomal enzymes. Further, delivery systems for systemic administration ought to be well tolerated upon administration. They should be safe, enabling the multiadministration treatment modalities required for improved clinical outcomes and, from a developmental point of view, production of large batches with reproducible specifications is also desirable. In this review, the concept of self-amplifying RNA vaccines and the most promising lipid-based delivery systems are discussed.
Keywords: self-amplifying RNA vaccine, RNA replicon, liposomes, solid lipid nanoparticles, nucleic acid vaccines
Nucleic acid vaccines
The application of new technologies to identify protective antigens, to optimally present antigens to the immune system, and to manufacture vaccines using highly characterized, synthetic methods of production has greatly facilitated the discovery and development of new and improved vaccines.1 Genetic vaccines involve direct immunization with RNA or DNA encoding the antigen(s) of interest and have as one of their main advantages the simplicity and purity with which they can be produced.2 The use of nucleic acid-based vaccines to combine the benefits of in situ expression of antigens with the safety of inactivated and subunit vaccines has been a key advancement. Upon their discovery more than 20 years ago, nucleic acid vaccines promised to be a safe and effective means to mimic immunization with a live organism vaccine, particularly for induction of T-cell immunity.3 Gene-based vaccines are under development for a broad variety of applications, ranging from vaccines to immunotherapies for infectious diseases, cancer, autoimmune diseases, and allergy. Nucleic acid vaccines have significant advantages over conventional vaccines,4 including: safety, since living organisms and potent adjuvants are not required; effectiveness, since after immunization they express antigens in situ, mimicking a true infection; induction of both B and T-cell responses (including cytotoxic T lymphocytes); specificity, since they induce the immune response only to the antigen of interest; and relatively low production cost, high stability, ease of manipulation, and the possibility of expressing complex antigens such as transmembrane proteins.5
DNA vaccines are bacterial plasmids constructed to express an encoded protein following in vivo administration and subsequent transfection of cells.6 Initially, these plasmids were thought to function simply as a shuttle system for the gene, resulting in the in situ production of antigen (for vaccines) or therapeutic protein (for gene therapy applications). Since the initial demonstration that such a plasmid encoding a viral protein could result in both cellular and antibody immunity, as well as protection from viral challenge (including cross-strain challenge by a strain of virus quite different from the strain from which the gene was cloned), the technology has been used for wide-ranging applications, from a laboratory tool to licensed veterinary vaccines, and is under development for a variety of human biomedical applications.
Besides DNA, the naturally transient and cytosolically active messenger RNA (mRNA) molecules are seen as a possibly safer and more potent alternative to DNA for gene vaccination. Optimized mRNA was demonstrated to be a potent gene vaccination vehicle when delivered naked, in liposomes, or coated on particles. mRNA can also be utilized for transfecting dendritic cells ex vivo, which are then transferred back to the patient or directly administered in vivo. For instance, in a Phase I clinical trial, it was shown that tumor-associated antigen-mRNA-electroporated mature dendritic cells are capable of inducing tumor-associated antigen-T-cell responses in multiple myeloma patients after stem cell transplantation. Moreover, vaccination was well tolerated with limited toxicity.7
The proof of concept of the utility of RNA in vaccination was demonstrated when intramuscular injection of mRNA in mice resulted in local production of an encoded protein8 and induction of immune responses against an encoded antigen.9 A large body of nonclinical data and a considerable number of clinical studies have demonstrated the excellent safety profile of mRNA-based immunotherapeutic strategies, mainly in tumor vaccination.10 Moreover, induction of target-specific immune responses has been established in vaccinated patients, albeit at weak levels in most cases.
The use of RNA in vaccination has been studied mainly with mRNA or nonamplifying RNA, and to a lesser extent, with self-amplifying RNA generated with modified RNA from virus. Moreover, RNA interference (RNAi) has also been studied as a possible strategy in vaccination. RNAi is a natural process based on complementarity between RNA and its target mRNA to cause destruction of the target.11 In a previous study, RNAi-mediated chemokine receptor 5 (CCR5) silencing prevented human immunodeficiency virus (HIV) infection in BLT mice and, therefore, it may be useful as anti-HIV prophylaxis.12 The blockage of negative regulatory molecules on dendritic cells with RNAi has also been studied to prevent and treat chronic virus infections.13 One of the highly potential applications for the clinical use of RNAi is cancer vaccination; the suppression of gene expression (for instance, certain cytokines) in dendritic cells has been shown to enhance their immunostimulatory capacity and to result in an enhanced antitumor response.14
Gene targets for vaccination with RNA include reporter genes,15 tumor antigens,16 viral antigens,17 and allergens.18 Table 1 presents several diseases that can potentially be treated with RNA vaccines. Regardless of the encoded antigen, viruses with an RNA genome are able to induce the innate immune response of the host.19 This means that certain RNA molecules may cause a strong innate response that leads to induction of a broad and efficient adaptative immune response.20 For instance, double-stranded RNA, normally not present in cells but synthesized during viral replication, is recognized by host cells as a signal to mount a strong immune response.21 It has been shown that induction of antigen-specific immunity can be achieved by administering RNA vaccines through several routes, ie, intravenous, intradermal, subcutaneous, intranodal, and intrasplenic.1 Additionally, mRNA has also been used via intradermal application to induce its uptake by Langerhans cells and dermal dendritic cells for further transport to the lymph node.
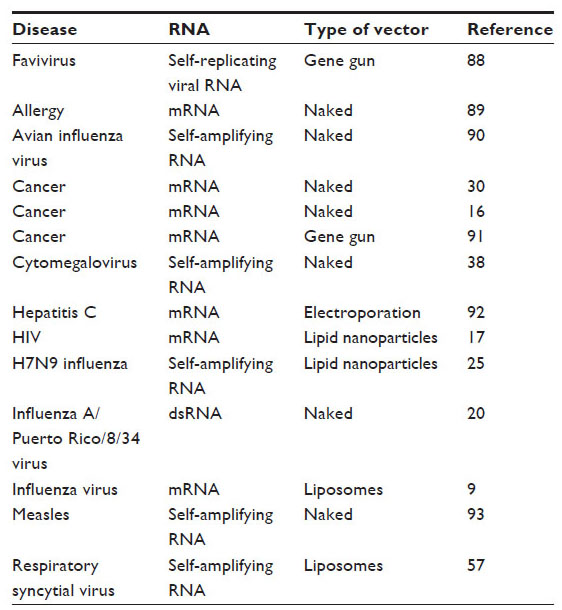 | Table 1 Diseases potentially treated with RNA vaccines |
Although attention has been mainly focused on plasmid-based DNA vaccines, use of RNA also has advantages.5 First, the theoretical risk of vector integration into the host genome and subsequent malignant cell transformation is omitted. Second, due to the relatively short half-life of the RNA molecule, its expression is transient and, therefore, exposure to the antigen is more controlled. Consequently, the risk when using tumor-associated antigen genes such as proto-oncogenes for immunization decreases. Moreover, this transient expression minimizes the risk of induction of tolerance.17 Third, RNA must be released into the cytoplasm to be transcribed into the protein, rendering its application much safer.22 However, DNA must be delivered into the nucleus for transcription into mRNA, which is transported back to the cytoplasm to be translated; therefore, DNA must cross the nuclear membrane, a limiting step that is avoided with RNA. Several studies have shown that microinjections of plasmid DNA into the cytoplasm of nondividing cells resulted in very low levels of gene expression, but direct intranuclear injection of the same number of plasmid DNA copies led to efficient transfection.23 Therefore, an additional advantage of RNA-based vaccines is the possibility to transfect slow or nondividing cells, the nuclear membrane of which is much more difficult to cross than rapidly dividing cells.24
RNA can be produced by a cell-free enzymatic transcription reaction, thus avoiding the use of micro-organisms or cultured cells in manufacturing, with associated quality and safety issues. This method enables simple downstream purification and very rapid and cost-effective manufacturing.25 Lyophilization studies have shown that RNA vaccines are not less stable than conventional vaccines that require a cold chain to be effective.26 Therefore, mRNA can be produced in large amounts and with good manufacturing practice quality, thus allowing further development of mRNA-based therapies.
In Europe, all mRNA-based therapies fall under the scope of the regulation for advanced therapy medicinal products.27,28 According to the European regulation,28 a “gene therapy medicinal product is an active substance which contains or consists of a recombinant nucleic acid used in or administered to human beings”; therefore, mRNA is categorized as gene therapy. In contrast, in the USA, mRNA-based vaccination therapies are not considered as gene therapy since they do not imply the same risks as they are not capable of integrating into the genome.10 This means that, at least in the USA, clinical translation of mRNA-based medicinal products is simpler and faster than that of DNA-based therapies.
In spite of the advantages of RNA vaccines in comparison with those that are DNA-based, few studies have explored approaches to increase the delivery and effectiveness of mRNA vaccines in vivo. As a result, knowledge concerning the potency of nonviral delivery of mRNA vaccines is scarce and data describing their immunological properties are largely lacking.17 Clinical experience with RNA vaccines is limited. There are some experiences in Phase I/II clinical vaccination trials with mRNA that codes for tumor antigens;29,30 these studies have shown that the treatment is feasible and safe, although the clinical effectiveness for antitumor immunotherapy must be evaluated in further trials.
Self-amplifying RNA vaccines
Self-replicating RNA vectors (termed RNA replicons) have been developed based on advancements in the molecular virology of single-stranded RNA viruses. They are a form of a nucleic acid-based vaccine derived from either positive-strand or negative-strand RNA viruses. The gene sequences encoding structural proteins in these RNA viruses are replaced by mRNA encoding antigens of interest as well as by the RNA polymerase for RNA replicon replication and transcription.31 The foreign genes inserted in place of the structural protein gene region, when introduced into cells, will replicate and express the heterologous genes, inducing synthesis of large amounts of the foreign gene product within the cell, reaching levels of 15%–20% of total cell protein.32 Self-amplifying RNA can be regarded as “disabled” virus vectors that are capable of amplifying within the cytoplasm of host cells for a prolonged period but are unable to produce infectious progeny.33 They may be potentially more potent than mRNA vaccines. Figure 1 shows a schematic representation of a self-amplifying RNA. As RNA replicons encode gene sequences of structural proteins and an RNA transcript form, their size is larger than that of conventional nonamplifying mRNA.4
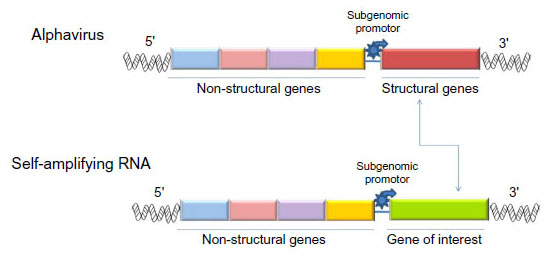 | Figure 1 Scheme showing a self-amplifying RNA derived from an alphavirus in which structural genes have been replaced by the gene of interest. |
For vaccine application, the genes encoding the structural proteins are replaced by the antigen or antigens of interest. Their production under special conditions (eg, packaging cell lines) leads to formation of single-round infectious particles carrying RNAs encoding the antigens.34 In this way, transient high levels of antigen production can be achieved without the use of a “live” spreading viral infection.
The most studied RNA molecules of this class are derived from alphavirus,4 such as Sindbis, Semliki Forest, and Venezuelan equine encephalitis viruses, and from flaviviruses35 (for instance, Kunjin virus and Yellow Fever virus). A major distinction between the alphavirus and flavivirus replicons is their cytopathic nature; alphavirus replicons are generally cytopathic and flavivirus replicons are generally noncytopathic, although whether cytopathicity is an advantage or disadvantage still remains unclear. In a previous study,35 Semliki Forest virus (alphavirus) and Kunjin virus (flavivirus), when delivered as naked RNAs, elicited comparable CD8+ T-cell responses, but the Semliki Forest virus vectors elicited greater humoral responses to an encoded cytoplasmic antigen, ß-galactosidase. Studies in major histocompatibility complex class II-deficient mice revealed that neither vector could overcome the dependence of CD4+ T-cell help in development of humoral and cellular responses following immunization. These studies indicate that the distinct biology of the two replicon systems may differentially impact the adaptive immune response, and this may need to be considered when designing vaccination strategies. Therefore, replicons derived from different RNA viruses differ with regard to levels and duration of heterologous gene expression, allowing generation of a versatile toolbox for vaccine or gene therapy applications.36
As mRNA, RNA replicons are effective at eliciting antigen-specific humoral and cellular immune responses in animal models of infectious and noninfectious diseases.4 Like DNA vaccines and viral vectors, the RNA vaccine candidate induces both CD4+ and CD8+ T-cell responses, as transfection of host cells can facilitate antigen presentation by major histocompatibility complex molecules and priming of naive T lymphocytes. Self-amplifying RNA vaccines have been successfully assayed with many different antigens as vaccine candidates, and have been shown to be potent in several animal species, including mice,5 nonhuman primates,37 and humans.38 In a Phase I trial of an alphavirus replicon vaccine for cytomegalovirus in cytomegalovirus-seronegative adult volunteers, the vaccine was safe and induced neutralizing antibody and multifunctional T-cell responses against three cytomegalovirus antigens that are important targets for protective immunity.38 To the best of our knowledge, this has been the only clinical trial of self-amplifying mRNA for vaccination.
Although the possibility of an infection is eliminated because of removal of the genes encoding structural proteins, more research on the potential for induction of a harmful infection is needed. On the other hand, this kind of vaccine is more complex than nonamplifying mRNA vaccines, and their greater size and complexity make it difficult to design an efficient delivery system.
Delivery systems for self-amplifying RNA
A key challenge in realizing the broad potential of RNA-based therapeutics is finding safe and effective delivery methods. Unmodified RNA is unstable in the bloodstream, can be immunogenic, and does not readily cross membranes to enter cells. Therefore, chemical modifications and/or delivery material are required to bring RNA to its site of action without adverse effects.39 In some applications, effective delivery of naked RNAs, without a carrier, may be possible.40 However, systemic delivery to many tissues, including the liver, requires a vehicle to provide protection and to transport the RNA to the cells of interest. Therefore, the most important, and most difficult challenge in therapy using nucleic acids, including self-amplifying RNA vaccines, is the issue of delivery.41 Lately, there has been an important development of nonviral delivery systems for RNAi,39 but the experience with mRNA or self-amplifying RNA is very limited. However, most of the systems studied for delivery of the former can be applied to the latter.
Ideally, an RNA nanocarrier should provide protection from blood nucleases and extended blood circulation, which ultimately would increase the possibility of reaching the target tissue.42 The delivery system containing the RNA must then be internalized by the target cell and, upon receptor-mediated endocytosis, must be able to escape from the endosomal compartment into the cell cytoplasm where the RNA machinery is located, while avoiding degradation by lysosomal enzymes.43 Further, delivery systems for the systemic administration of RNA ought to be well tolerated upon administration. They should be safe, enabling the multiadministration treatment modalities required for improved clinical outcomes.44 From a manufacturing point of view, production of large batches with reproducible specifications is also desirable. A broad diversity of materials is under exploration to address the challenges of in vivo delivery, including viral and nonviral vectors. Although viral vectors have been extensively studied for delivery of nucleic acids, safety (immunogenicity and oncogenicity) concerns limit their clinical application. Nonviral vectors include inorganic particles, polymeric-based, cationic lipid-based, and peptide-based vectors, and physical methods such as electroporation, sonoporation, and magnetofection.41 Despite the delivery success achieved by some of these carriers, advances are necessary to allow the fullest application of RNA at the clinical level.
Self-amplifying RNA vaccines have been delivered mainly as naked RNA or with viral vectors. The practical utility of viral vectors, however, is limited by manufacturing considerations, cost-effectiveness, and potential adverse health effects.45 Very limited data have been published on nonviral delivery of RNA replicons, although it is a highly attractive approach. Moreover, and as mentioned in the section “Self-amplifying RNA vaccines”, RNA replicons usually have very large sizes (≈9 kb) compared with mRNA (≈2 kb), which adds an additional challenge to the development of drug delivery vehicles.
Lipid-based formulations as vehicles for self-amplifying RNA
Lipid-based formulations have been increasingly recognized as one of the most promising delivery systems for RNA due to their biocompatibility and their ease of large-scale production.45,46 Cationic lipids have been widely studied as synthetic materials for delivery of RNA. After mixing together, nucleic acids are condensed by cationic lipids to form lipid/nucleic acid complexes known as lipoplexes. These lipid complexes are able to protect genetic material from the action of nucleases and deliver it into cells by interacting with the negatively charged cell membrane.47 Many research groups focus their work on the design of new cationic lipids as candidates for gene delivery,48 and some of them have been assayed in clinical trials.49 Lipoplexes can be prepared by directly mixing positively charged lipids at physiological pH with negatively charged nucleic acids, so their preparation is simple. More complex lipid-based formulations including liposomes, nanoemulsions, and solid lipid nanoparticles, have also been studied in depth for nucleic acid delivery.47
To deliver RNA efficiently, several limiting steps must be overcome, including dissociation from the nanocarrier, cellular uptake, endosomal escape, and appropriate intracellular disassembly.50 The relationship between the physicochemical properties of the nanocarrier (particle size, RNA entrapment, acid dissociation constant) and biological barriers (for instance, cellular uptake), and that between biological barriers and RNA activity remain unclear.51 This lack of clarity is one of the reasons for poor in vitro–in vivo translation. This type of analysis aids in the identification of meaningful structure–function activity relationships, improves the in vitro screening process before in vivo use, and facilitates the future design of potent nanocarriers. In spite of that, significant progress has been made toward developing lipid-based systems for nucleic acids, such as immunostimulatory oligonucleotides, plasmid DNA containing therapeutic genes, and antisense oligonucleotides and RNA.41–43
Cationic lipids used in vitro and in vivo to prepare lipid-based formulations to deliver nucleic acids may have toxic effects, although there is little to no effect on organ function or tissue architecture if they are used at relatively low doses. However, acute inflammation and tissue damage may occur at higher doses.52 The toxic effect is due mainly to the cationic nature of the vector, although the biodegradability of the compound is also important.47 Upon intravenous administration, positively charged nanoparticles interact with negatively charged serum proteins, forming aggregates that accumulate mainly in the lungs, liver, and spleen. Moreover, lipoplexes often induce pulmonary toxicity associated with complement activation and inflammation.42 Therefore, formulations for RNA delivery must have adequate properties to minimize these effects.
Despite tremendous efforts to develop lipid-based formulations for drug delivery, only a few formulations are approved for clinical use, and none of them has included nucleic acids. Efforts must be focused primarily on two areas:53 1) technical aspects, such as manufacturing strategies, development of techniques for reproducible formulations, large-scale production, and conjugation of targeting molecules; and 2) novel concepts and approaches to accomplish on-demand release of active molecules from the lipid nanocarrier (based on the unique properties of the assembly components of lipid-based nanocarriers). Additionally, the delivery systems should be low-cost and accessible. Another factor that limits clinical application is the lack of well defined mandatory preclinical investigations for RNA-based approaches.
Liposomes
Liposomes are colloidal lipid-based and surfactant-based delivery systems composed of a phospholipid bilayer surrounding an aqueous compartment. They may present as spherical vesicles and can range in size from 20 nm to a few microns. Cationic lipid-based liposomes are able to complex with negatively charged nucleic acids via electrostatic interactions, resulting in complexes that offer biocompatibility, low toxicity, and the possibility of the large-scale production required for in vivo clinical applications.54 The lipid to RNA ratio, and overall lipid concentration used in forming these complexes, are very important for efficient gene delivery and vary depending on applications. Liposomes can fuse with the plasma membrane for uptake; once inside the cell, the liposomes are processed via the endocytic pathway and the genetic material is then released from the endosome/carrier into the cytoplasm. Compared with polymeric nanoparticles, liposomes have long been perceived as better drug delivery vehicles because of their superior biocompatibility, given that liposomes are basically analogs of biological membranes, and can be prepared from both natural and synthetic phospholipids.54
Several studies have shown the usefulness of liposomes in delivery of mRNA for vaccination. For instance, liposomes containing mRNA encoding the influenza virus nucleoprotein induced anti-influenza cytotoxic T lymphocytes, which were identical to those obtained in vivo with infectious virus in terms of specificity, lysing both peptide-sensitized and virus-infected targets.9 Intravenous delivery of mRNA encapsulated into liposomes has been shown to be the most efficient route of vaccination, although intradermal and eventually subcutaneous injections could also induce immunity.22
In a recent study, a liposomal formulation loaded with mRNA encoding tumor-associated antigens induced a dendritic cell-mediated antitumor response in vivo and a 3–5-fold decrease in the number of lung metastases when compared with a control group. These pulsed dendritic cells caused a slight proinflammatory response in mice which was balanced by a positive effect of the dendritic cell-induced antitumor cytokine response.55
Liposomes can be combined with polymers to form lipopolyplexes, which are also useful for vaccination with nucleic acids. In a recent study, mice receiving systemic injections of a new tumor antigen mRNA (MART1) formulated in a polyethylene glycol (PEG)ylated derivative of histidylated polylysine and L-histidine-(N,N-di-n-hexadecylamine)ethylamide liposomes were specifically and significantly protected against melanoma tumor progression.56
Although many studies using liposomes for gene therapy have been published, it was not until recently that Geall et al57 confirmed the delivery of self-amplifying RNA via liposomes. In a model of respiratory syncytial virus infection, the authors showed that a self-amplifying RNA vaccine formulated in liposomes potently induced neutralizing antibodies in cotton rats, as well as antigen-specific interferon-γ-producing CD4+ and CD8+ T-cells in mice. These responses were comparable with or exceeded those elicited by RNA delivered via viral particles or electroporation of plasmid DNA and provided protection against subsequent respiratory syncytial virus infection.
Even though cationic lipid-based liposomes have shown good gene transfection ability and biocompatibility in vitro, and although a few therapeutic clinical trials are underway, the clinical applications of liposomes have been limited because of in vivo instability.58 It has been reported that intravenously injected liposome/DNA complexes form large aggregates with blood components and that these aggregates become entrapped in the lung capillary bed.59 Therefore, cationic lipid-based liposomes usually require addition of components to enhance their in vivo stability after exposure to blood components. For example, hydrophobic cholesterol, nonionic surfactants, or PEG have been used to increase the physical stability of cationic lipid-based liposomes under such conditions.58 It has been reported that such stability-enhanced liposomes have much better transfection efficiency, especially under in vivo conditions.60 Despite these drawbacks, liposomes are very promising systems for nonviral gene delivery. In fact, several liposome-based vectors have been assayed in clinical trials. For example, Allovectin-7® (Vical, San Diego, CA, USA), a plasmid DNA carrying major histocompatibility complex, class 1, B (HLA-B) and ß2-microglobulin genes complexed with 1,2-dimyristyloxy-propyl-3-dimethyl-hydroxy ethyl ammonium bromide/dioleoylphosphatidylethanolamine (DMRIE/DOPE) liposomes, has been assessed for safety and efficacy in Phase I and II clinical trials.61,62 However, further research is necessary to improve their shape, size, cytotoxicity, efficiency, and biocompatibility, which may yield an effective method of gene delivery in the future.
Nanoemulsions
Nanoemulsions are thermodynamically stable isotropic systems in which two immiscible liquids (water and oil) are mixed to form a single phase by means of appropriate surfactants, with a droplet diameter in the approximate range of 0.5–100 μm.63 Depending on the nature of the core, ie, water or oil, the emulsions can be water-in-oil or oil-in-water systems, respectively. Since a nanoemulsion is a stable isotropic system, careful balance of the three phases is essential to achieve a thermodynamically stable state. Nanoemulsions often have a negatively surfaced charge, so are not suitable for gene therapy. However, by using cationic surfactants or cationic lipids, it is possible to prepare cationic nanoemulsions. The extent of the electrostatic interaction between the nucleic acid and the nanoemulsion depends on the cationic lipid (nature of the cationic polar head group and the acyl chains) and the nucleic acid structures (lengths and base composition).64 Stearylamine, 1,2-dioleoyl-sn-glycero-3-trimethylammonium-propane (DOTAP), and 3β-[N-(N′,N′-dimethylaminoethane)-carbamoyl]cholesterol (DC-cholesterol) are examples of cationic surfactants used to prepare cationic nanoemulsions able to interact electrostatically with negatively charged nucleic acids.63 Nevertheless, these cationic surfactants were also noted to exhibit toxicity. New cationic lipids, which have an aspartate or glutamate backbone, a lysine head-group, and two alkyl tails, yield efficient gene expression with lower cytotoxicity. In addition, amino acid-modified fatty acids, which mimic natural lipoamino acids, have been developed. For example, lauroyl-arginine methyl ester was reported to be a cationic surfactant with rich self-aggregation properties, biodegradability, and low toxicity, and can be used in food, pharmaceutical, and cosmetic applications.65 The colipid DOPE is extensively used because of its fusogenic properties to improve the ability of emulsions and cationic liposomes to transfect cells.66 This is partially explained by the fact that the amine group of DOPE interacts with DNA phosphate groups, thus weakening the binding affinity between cationic lipids and DNA.
Cationic nanoemulsions have been described as a promising strategy to improve transfection of nucleic acids to mammalian cells, protecting them against nuclease attack. For instance, Bruxel et al67 prepared a cationic nanoemulsion with DOTAP as a delivery system for antisense oligonucleotides targeting malarial topoisomerase II. The physical characteristics and serum-resistant properties of the nanoemulsion complexes indicate that cationic nanoemulsions could be a more efficient carrier system for nucleic acids and/or immunogene delivery than liposomes.66 The fact that transfection is efficient in the presence of serum is an important advantage. One of the reasons for the serum-resistant properties of the cationic lipid nanoemulsion may be the stability of the nanoemulsion/nucleic acid complex. In several in vivo studies, cationic nanoemulsions were found to be more suitable for gene delivery than liposomes because of the higher transfection and lower toxicity. Compared with liposomes, emulsions also have the advantages of easy processing and low production costs.68 Additionally, large-scale production of emulsions can be performed in a cost-effective and simple way using high-pressure homogenization.69
The introduction of nonionic surfactants with a branched PEG head group, such as Tween 80®, increments nanoemulsion stability, probably because of their steric hindrance and generation of a hydrophilic surface, which prevents physical aggregation of the nucleic acid/nanoemulsion complexes. Moreover, due to their hydrophilicity and lack of aggregates, emulsions decorated with PEG avoid protein adsorption. Therefore, incorporation of PEG derivatives into the cationic lipid nanoemulsions may prevent them from enzymatic degradation, resulting in prolonged circulation in the blood. Further, nanoemulsions with a hydrophilic surfactant are taken up slowly by phagocytic cells.66
In spite of the advantages of nanoemulsions for the delivery of nucleic acids, only a few attempts have been made to use this new delivery system for RNA, and all of them have been made with RNAi. For instance, Kaneda et al70 showed the potential application of cationic nanoemulsion prepared with DOTAP, DOPE, and cholesterol for delivery of small interfering RNA. Transfection complexes with a mean particle diameter of approximately 300 nm were able to suppress expression of upregulated vascular adhesion molecules by endothelial cells. To the best of our knowledge, no studies with mRNA or self-amplifying RNA have been published.
Solid lipid nanoparticles
Solid lipid nanoparticles (SLN) comprise a variety of systems with a particle diameter of 50–1,000 nm. They are colloidal particles made up of a relatively rigid biocompatible and biodegradable matrix of hydrophobic lipids that are solid at room and body temperatures. SLN were developed in the early 1990s in an attempt to combine the advantages of solid particles, emulsions, and liposomes. On the one hand, solid particles protect their incorporated active compounds and are more flexible in modulating the release of these compounds. On the other hand, as with liposomes and emulsions, they are composed of nontoxic excipients, and their large-scale production may be easily accomplished.71 SLN can be prepared by using high-pressure homogenization, so use of organic solvents is avoided; this is an important advantage from the point of view of industrial production.72 Moreover, SLN possess very good stability,73 and they can be lyophilized.74 In a previous study reported by our group,75 we showed long permanence of the lipid nanocarriers in blood and tissues after intravenous administration to rats. The particle size, surface charge, and surfactant content of the nanoparticles affected their tissue biodistribution profile.
Although SLN have undergone constant development over recent years as drug delivery systems, there is not much literature concerning their application in gene therapy. However, their capacity to transfect different cell lines has been demonstrated in vitro76,77 and in vivo.78,79 Their ability to condense and protect genetic material, and their efficiency when entering cells, and once inside, to release DNA or RNA, make this nanoparticulate system an interesting vector for gene therapy.80,81 The suitability of SLN for gene therapy depends on the ratio between the nanoparticles and the nucleic acid; an equilibrium between the binding forces to achieve protection without hampering posterior release at the site of action is necessary.82 Figure 2 shows an SLN prepared by our group using Precirol® ATO5 as the solid lipid and DOTAP and Tween as surfactants. This image was captured by atomic force microscopy.
 | Figure 2 Photograph of solid lipid nanoparticles captured by atomic force microscopy. |
After intravenous administration, positively charged nanoparticles interact with negatively charged serum proteins, forming aggregates that accumulate mainly in the lungs, liver, and spleen.83 In order to avoid this problem, as with liposomes and emulsions, significant advances have been seen in the development of different types of PEG-grafted SLN.43 The hydrophilic nature of PEG provides an aqueous shield around the nanoparticle surface, thus decreasing the extent of opsonization and subsequent recognition by macrophages of the mononuclear phagocytic system, that ultimately leads to an increase in nanoparticle blood residence time.43 PEG has been used extensively by the pharmaceutical industry to improve the pharmacokinetic properties of different therapeutic agents and drug nanocarriers. In order to prolong circulation of the nanoparticles in blood, other strategies can be considered. For instance, in a previous study, SLN were prepared with dextran and protamine, and their in vivo transfection capacity improved.78 The improved transfection profile was explained by the presence of negatively charged dextran on the nanoparticle surface. This polyanion-biocompatible polysaccharide hampers strong interactions with other components such as serum proteins,84 so may induce a longer circulation time of the vector in blood. Moreover, the high condensation of the nucleic acid due to protamine, which contributes to nuclease resistance, may also result in an extended stay of the plasmid in the organism. Peptides may also favor other limiting steps for transfection, such as cellular internalization. For instance, SLN can incorporate cell-penetrating peptides, such as the dimeric HIV-1 transactivator of transcription (TAT) peptide85 or the synthetic sweet arrow peptide.86
Cationic SLN produced by microemulsion with Compritol® ATO 888 as matrix lipid, Pluronic® F68 as surfactant, and dimethyldioctadecylammonium bromide as cationic lipid were able to protect mRNA against enzymatic degradation. Moreover, toxicity studies in a living organism (eggs of the sea urchin Paracentrotus lividus, an organism very sensitive to a number of chemical and environmental agents) revealed that SLN allows normal embryonic development, high viability, a regular cell cycle, and correct morphogenesis. The authors observed correct biodistribution of the RNA, suggesting that the complexes allow an efficient transfer of mRNA into the cell, thus maintaining its functionality.87
Conclusion
Vaccines based on nucleic acids (both DNA and RNA) have been broadly investigated for several years, but have not yet resulted in a commercial product for human use. Self-amplifying RNA vaccines have been shown to induce both humoral and cellular immune responses. However, their clinical application depends on the delivery system. Ideally, the delivery vectors should be specific, effective, long-lasting, and safe. Lipid nanoparticles, including liposomes and SLN, are two of the most promising delivery systems for self-amplifying RNA vaccines due to their biocompatibility and the ease of large-scale production. Therefore, this approach could provide a potential generic platform for rapid development of potent and versatile vaccines.
Acknowledgment
This work was supported by the Basque Government’s Department of Education, Universities and Investigation (IT-341-10).
Disclosure
The authors report no conflicts of interest in this work.
References
Ulmer JB, Mason PW, Geall A, Mandl CW. RNA-based vaccines. Vaccine. 2012;30(30):4414–4418. | |
Liu MA. DNA vaccines: a review. J Intern Med. 2003;253(4):402–410. | |
Ulmer JB, Donnelly JJ, Parker SE, et al. Heterologous protection against influenza by injection of DNA encoding a viral protein. Science. 1993;259(5102):1745–1749. | |
Geall AJ, Mandl CW, Ulmer JB. RNA: the new revolution in nucleic acid vaccines. Semin Immunol. 2013;25(2):152–159. | |
Johansson DX, Ljungberg K, Kakoulidou M, Liljeström P. Intradermal electroporation of naked replicon RNA elicits strong immune responses. PLoS One. 2012;7(1):e29732. | |
Liu MA. DNA vaccines: an historical perspective and view to the future. Inmunol Rev. 2011;239(1):62–84. | |
Hobo W, Novobrantseva TI, Fredrix H, et al. Improving dendritic cell vaccine immunogenicity by silencing PD-1 ligands using siRNA-lipid nanoparticles combined with antigen mRNA electroporation. Cancer Immunol Immunother. 2013;62(2):285–297. | |
Wolff JA, Malone RW, Williams P, et al. Direct gene transfer into mouse muscle in vivo. Science. 1990;247(4949 Pt 1):1465–1468. | |
Martinon F, Krishnan S, Lenzen G, et al. Induction of virus-specific cytotoxic T lymphocytes in vivo by liposome-entrapped mRNA. Eur J Immunol. 1993;23(7):1719–1722. | |
Kreiter S, Diken M, Selmi A, Türeci ö, Sahin U. Tumor vaccination using messenger RNA: prospects of a future therapy. Curr Opin Immunol. 2011;23(3):399–406. | |
Rayburn ER, Zhang R. Antisense, RNAi, and gene silencing strategies for therapy: mission possible or impossible? Drug Discov Today. 2008;13(11–12):513–521. | |
Kim SS, Peer D, Kumar P, et al. RNAi-mediated CCR5 silencing by LFA-1-targeted nanoparticles prevents HIV infection in BLT mice. Mol Ther. 2010;18(2):370–376. | |
Jiang W. Blockade of B7-H1 enhances dendritic cell-mediated T-cell response and antiviral immunity in HBV transgenic mice. Vaccine. 2012;30(4):758–799. | |
Akita H, Kogure K, Moriguchi R, et al. Nanoparticles for ex vivo siRNA delivery to dendritic cells for cancer vaccines: programmed endosomal escape and dissociation. J Control Release. 2010;143(3):311–317. | |
Piggott JM, Sheahan BJ, Soden DM, O’Sullivan GC, Atkins GJ. Electroporation of RNA stimulates immunity to an encoded reporter gene in mice. Mol Med Rep. 2009;2(5):753–736. | |
Diken M, Kreiter S, Selmi A, Türeci O, Sahin U. Antitumor vaccination with synthetic mRNA: strategies for in vitro and in vivo preclinical studies. Methods Mol Biol. 2013;969:235–246. | |
Pollard C, Rejman J, De Haes W, et al. Type I IFN counteracts the induction of antigen-specific immune responses by lipid-based delivery of mRNA vaccines. Mol Ther. 2013;21(1):251–259. | |
Weiss R, Scheiblhofer S, Roesler E, Weinberger E, Thalhamer J. mRNA vaccination as a safe approach for specific protection from type I allergy. Expert Rev Vaccines. 2012;11(1):55–67. | |
Jensen S, Thomsen AR. Sensing of RNA viruses: a review of innate immune receptors involved in recognizing RNA virus invasion. J Virol. 2012;86(6):2900–2910. | |
Martínez-Gil L, Goff PH, Hai R, García-Sastre A, Shaw ML, Palese P. A Sendai virus-derived RNA agonist of RIG-I as a virus vaccine adjuvant. J Virol. 2013;87(3):1290–1300. | |
Jin B, Sun T, Yu XH, et al. Immunomodulatory effects of dsRNA and its potential as vaccine adjuvant. J Biomed Biotechnol. 2010;2010:690438. | |
Pascolo S. Vaccination with messenger RNA (mRNA). Handb Exp Pharmacol. 2008;183:221–235. | |
Zabner J, Fasbender AJ, Moninger T, Poellinger KA, Welsh MJ. Cellular and molecular barriers to gene transfer by a cationic lipid. J Biol Chem. 1995;270(32):18997–19007. | |
Bettinger T, Carlisle RC, Read ML, Ogris M, Seymour LW. Peptide-mediated RNA delivery: a novel approach for enhanced transfection of primary and post-mitotic cells. Nucleic Acids Res. 2001;29(18):3882–3891. | |
Hekele A, Bertholet S, Archer J, et al. Rapidly produced SAM® vaccine against H7N9 influenza is immunogenic in mice. Emerging Microbes and Infections. 2013;2:e52. | |
Jones KL, Drane D, Gowans EJ. Long-term storage of DNA-free RNA for use in vaccine studies. Biotechniques. 2007;43(5):675–681. | |
European Medicines Agency. Guideline on human cell-based medicinal products. London, UK: European Medicines Agency; 2007. Available from: http://www.ema.europa.eu/docs/en_GB/document_library/Scientific_guideline/2009/09/WC500003898.pdf. | |
European Commission. Directive 2001/83/EC of the European Parliament and of the Council of 6 November 2001 on the community code relating to medicinal products for human use: Brussels, Belgium: European Commission; 2001. Available from: http://www.ema.europa.eu/docs/en_GB/document_library/Regulatory_and_procedural_guideline/2009/10/WC500004481.pdf. | |
Weide B, Carralot JP, Reese A, et al. Results of the first phase I/II clinical vaccination trial with direct injection of mRNA. J Immunother. 2008;31(2):180–188. | |
Weide B, Pascolo S, Scheel B, et al. Direct injection of protamine-protected mRNA: results of a phase 1/2 vaccination trial in metastatic melanoma patients. J Immunother. 2009;32(5):498–507. | |
Xu J, Luft JC, Yi X, et al. RNA replicon delivery via lipid-complexed PRINT protein particles. Mol Pharm. 2013;10(9):3366–3374. | |
Kamrud KI, Coffield VM, Owens G, et al. In vitro and in vivo characterization of microRNA-targeted alphavirus replicon and helper RNAs. J Virol. 2010;84(15):7713–7725. | |
Diken M, Kreiter S, Selmi A, et al. Selective uptake of naked vaccine RNA by dendritic cells is driven by macropinocytosis and abrogated upon DC maturation. Gene Ther. 2011;18(7):702–708. | |
Widman DG, Frolov I, Mason PW. Third-generation flavivirus vaccines based on single-cycle, encapsidation-defective viruses. Adv Virus Res. 2008;72:77–126. | |
Tannis LL, Gauthier A, Evelegh C, et al. Semliki forest virus and Kunjin virus RNA replicons elicit comparable cellular immunity but distinct humoral immunity. Vaccine. 2005;23(33):4189–4194. | |
Gehrke R, Heinz FX, Davis NL, Mandl CW. Heterologous gene expression by infectious and replicon vectors derived from tick-borne encephalitis virus and direct comparison of this flavivirus system with an alphavirus replicon. J Gen Virol. 2005;86 Pt 4:1045–1053. | |
Carroll TD, Matzinger SR, Barro M, et al. Alphavirus replicon-based adjuvants enhance the immunogenicity and effectiveness of Fluzone® in rhesus macaques. Vaccine. 2011;29(5):931–940. | |
Bernstein DI, Reap EA, Katen K, et al. Randomized, double-blind, Phase 1 trial of an alphavirus replicon vaccine for cytomegalovirus in CMV seronegative adult volunteers. Vaccine. 2009;28(2):484–493. | |
Kanasty R, Dorkin JR, Vegas A, Anderson D. Delivery materials for siRNA therapeutics. Nat Mater. 2013;12(11):967–977. | |
Kleinman ME, Yamada K, Takeda A, et al. Sequence- and target-independent angiogenesis suppression by siRNA via TLR3. Nature. 2008;452(7187):591–597. | |
Rodríguez-Gascón AR, Solinis MA, del Pozo-Rodríguez A. Non-viral delivery systems in gene therapy. In: Martin F, editor. Gene Therapy – Tools and Potential Applications. Rijeka, Croatia: Intech; 2013. | |
Gomes-da-Silva LC, Fonseca NA, Moura V, Pedroso de Lima MC, Simões S, Moreira JN. Lipid-based nanoparticles for siRNA delivery in cancer therapy: paradigms and challenges. Acc Chem Res. 2012;45(7):1163–1171. | |
Moreira JN, Santos A, Moura V, Pedroso de Lima MC, Simões S. Non-viral lipid-based nanoparticles for targeted cancer systemic gene silencing. J Nanosci Nanotechnol. 2008;8(5):2187–2204. | |
Vaishnaw AK, Gollob J, Gamba-Vitalo C, et al. A status report on RNAi therapeutics. Silence. 2010;1(1):14. | |
Zimmer G. RNA replicons – a new approach for influenza virus immunoprophylaxis. Viruses. 2010;2(2):413–434. | |
McCaskill J, Singhania R, Burgess M, et al. Efficient biodistribution and gene silencing in the lung epithelium via intravenous liposomal delivery of siRNA. Mol Ther Nucleic Acids. 2013;2:e96. | |
Gascón AR, Pedraz JL. Cationic lipids as gene transfer agents: a patent review. Expert Opin Ther Patent. 2008;18(5):515–524. | |
Mahato RI, Kim SW, editors. Pharmaceutical Perspectives of Nucleic Acid-Based Therapeutics. London, UK: Taylor and Francis; 2002. | |
Edelstein ML, Abedi MR, Wixon J. Gene therapy clinical trials worldwide to 2007 – an update. J Gene Med. 2007;9(10):833–842. | |
Alabi CA, Love KT, Sahay G, et al. Multiparametric approach for the evaluation of lipid nanoparticles for siRNA delivery. Proc Natl Acad Sci U S A. 2013;110(32):12881–12886. | |
Lin PJ, Tam YY, Hafez I, et al. Influence of cationic lipid composition on uptake and intracellular processing of lipid nanoparticle formulations of siRNA. Nanomedicine. 2013;9(2):233–246. | |
Yew NS, Scheule RK. Toxicity of cationic lipid-DNA complexes. Adv Genet. 2005;53P:189–214. | |
Puri A, Loomis K, Smith B, et al. Lipid-based nanoparticles as pharmaceutical drug carriers: from concepts to clinic. Crit Rev Ther Drug Carrier Syst. 2009;26(6):523–580. | |
Torchilin VP. Recent advances with liposomes as pharmaceutical carriers. Nat Rev Drug Discov. 2005;4(2):145–160. | |
Markov OO, Mironova NL, Maslov MA, et al. Novel cationic liposomes provide highly efficient delivery of DNA and RNA into dendritic cell progenitors and their immature offsets. J Control Release. 2012;160(2):200–210. | |
Mockey M, Bourseau E, Chandrashekhar V, et al. mRNA-based cancer vaccine: prevention of B16 melanoma progression and metastasis by systemic injection of MART1 mRNA histidylated lipopolyplexes. Cancer Gene Ther. 2007;14(9):802–814. | |
Geall AJ, Verma A, Otten GR, et al. Nonviral delivery of self-amplifying RNA vaccines. Proc Natl Acad Sci U S A. 2012;109(36):14604–14609. | |
Kwon SM, Nam HY, Nam T, et al. In vivo time-dependent gene expression of cationic lipid-based emulsion as a stable and biocompatible non-viral gene carrier. J Control Release. 2008;128(1):89–97. | |
Lizinger DC, Brown JM, Wala I, et al. Fate of cationic liposomes and their complex with oligonucleotide in vivo. Biochim Biophys Acta. 1996;1281(2):139–149. | |
Hyvonen Z, Ronkko S, Toppinen MR, Jaaskeainen I, Plotniece A, Urtti A. Dioleoyl phosphatidylethnolamine and PEG-lipid conjugates modify DNA delivery medicated by 1,4-dihydropyridine amphiphiles. J Control Release. 2004;99(1):177–190. | |
Stopeck AT, Hersh EM, Akporiaye ET, et al. Phase I study of direct gene transfer of an allogeneic histocompatibility antigen, HLA-B7, in patients with metastatic melanoma. J Clin Oncol. 1997;15(1):341–349. | |
Stopeck AT, Jones A, Hersh EM, et al. Phase II study of direct intralesional gene transfer of allovectin-7, an HLA-B7/beta2-microglobulin DNA-liposome complex, in patients with metastatic melanoma. Clin Cancer Res. 2001;7(8):2285–2291. | |
Souto EB, Nayak AP, Murthy RS. Lipid nanoemulsions for anti-cancer drug therapy. Pharmazie. 2011;66(7):473–478. | |
Bruxel F, Vilela JM, Andrade MS, et al. Investigation of the structural organization of cationic nanoemulsion/antisense oligonucleotide complexes. Colloids Surf B Biointerfaces. 2013;112:530–536. | |
Liu CH, Yu SY. Cationic nanoemulsions as non-viral vectors for plasmid DNA delivery. Colloids Surf B Biointerfaces. 2010;79(2):509–515. | |
Verissimo LM, Lima LF, Egito LC, de Oliveira AG, do Egito ES. Pharmaceutical emulsions: a new approach for gene therapy. J Drug Target. 2010;18(5):333–342. | |
Bruxel F, Cojean S, Bochot A, et al. Cationic nanoemulsion as a delivery system for oligonucleotides targeting malarial topoisomerase II. Int J Pharm. 2011;416(2):402–409. | |
Nam HY, Park JH, Kim K, Kwon IC, Jeong SY. Lipid-based emulsion system as non-viral gene carriers. Arch Pharm Res. 2009;32(5):639–646. | |
Choi WJ, Kim JK, Choi SH, Park JS, Ahn WS, Kim CK. Low toxicity of cationic lipid-based emulsion for gene transfer. Biomaterials. 2004;25(27):5893–5903. | |
Kaneda MM, Sasaki Y, Lanza GM, Milbrandt J, Wickline SA. Mechanisms of nucleotide trafficking during siRNA delivery to endothelial cells using perfluorocarbon nanoemulsions. Biomaterials. 2010;31(11):3079–3086. | |
del Pozo-Rodríguez A, Delgado D, Solinís MA, Gascón AR. Lipid nanoparticles as vehicles for macromolecules: nucleic acids and peptides. Recent Pat Drug Deliv Formul. 2011;5(3):214–226. | |
Teeranachaideekul V, Müller RH, Junyaprasert VB. Encapsulation of ascorbyl palmitate in nanostructured lipid carriers (NLC) – effects of formulation parameters on physicochemical stability. Int J Pharm. 2007;340(1–2):198–206. | |
Müller R, Mäder K, Gohla S. Solid lipid nanoparticles (SLN) for controlled drug delivery – a review of the state of the art. Eur J Pharm Biopharm. 2000;50(1):161–177. | |
del Pozo-Rodríguez A, Solinís MA, Gascón AR, Pedraz JL. Short- and long-term stability study of lyophilized solid lipid nanoparticles for gene therapy. Eur J Pharm Biopharm. 2009;71(2):181–189. | |
Beloqui A, Solinís MA, Delgado A, Evora C, del Pozo-Rodríguez A, Rodríguez-Gascón A. Biodistribution of nanostructured lipid carriers (NLCs) after intravenous administration to rats: influence of technological factors. Eur J Pharm Biopharm. 2013;84(2):309–314. | |
del Pozo-Rodríguez A, Delgado D, Solinís MA, Gascón AR, Pedraz JL. Solid lipid nanoparticles: formulation factors affecting cell transfection capacity. Int J Pharm. 2007;339(1–2):261–268. | |
Delgado D, del Pozo-Rodríguez A, Solinís MÁ, Rodríguez-Gascón A. Understanding the mechanism of protamine in solid lipid nanoparticle-based lipofection: the importance of the entry pathway. Eur J Pharm Biopharm. 2011;79(3):495–502. | |
Delgado D, Gascón AR, del Pozo-Rodríguez A, et al. Dextran-protamine-solid lipid nanoparticles as a non-viral vector for gene therapy: in vitro characterization and in vivo transfection after intravenous administration to mice. Int J Pharm. 2012;425(1–2):35–43. | |
Delgado D, del Pozo-Rodríguez A, Solinís MA, Bartkowiak A, Rodríguez-Gascón A. New gene delivery system based on oligochitosan and solid lipid nanoparticles: ‘in vitro’ and ‘in vivo’ evaluation. Eur J Pharm Sci. 2013;50(3–4):484–491. | |
Rodríguez-Gascón A, Solinis MA, del Pozo-Rodríguez A, Delgado D, Pedraz JL, inventors. University of the Basque Country UPV/EHU. Lipid nanoparticles for gene therapy. US 20120183589 A1. 2012. | |
Rodríguez-Gascón A, Solinis MA, del Pozo-Rodríguez A, Delgado D, Fernandez E, inventors. Lipid nanoparticles for treating ocular diseases. University of the Basque Country UPV/EHU and Universidad Miguel Hernández de Elche. WO 2012085318 A1. 2012. | |
Ruiz de Garibay AP, Delgado D, Del Pozo-Rodríguez A, Solinís MÁ, Gascón AR. Multicomponent nanoparticles as nonviral vectors for the treatment of Fabry disease by gene therapy. Drug Des Devel Ther. 2012;6:303–310. | |
Simões S, Filipe A, Faneca H, et al. Cationic liposomes for gene delivery. Expert Opin Drug Deliv. 2005;2(2):237–254. | |
Maruyama K, Iwasaki F, Takizawa T, et al. Novel receptor-mediated gene delivery system comprising plasmid/protamine/sugar-containing polyanion ternary complex. Biomaterials. 2004;25(16):3267–3273. | |
Rudolph C, Schillinger U, Ortiz A, et al. Application of novel solid lipid nanoparticle (SLN)-gene vector formulations based on a dimeric HIV-1 TAT-peptide in vitro and in vivo. Pharm Res. 2004;21(9):1662–1669. | |
del Pozo-Rodríguez A, Pujals S, Delgado D, et al. A proline-rich peptide improves cell transfection of solid lipid nanoparticle-based non-viral vectors. J Control Release. 2009;133(1):52–59. | |
Montana G, Bondì ML, Carrotta R, et al. Employment of cationic solid-lipid nanoparticles as RNA carriers. Bioconjug Chem. 2007;18(2):302–308. | |
Kofler RM, Aberle JH, Aberle SW, Allison SL, Heinz FX, Mandl CW. Mimicking live flavivirus immunization with a noninfectious RNA vaccine. Proc Natl Acad Sci U S A. 2004;101(7):1951–1956. | |
Weiss R, Scheiblhofer S, Roesler E, Ferreira F, Thalhamer J. Prophylactic mRNA vaccination against allergy. Curr Opin Allergy Clin Immunol. 2010;10(6):567–574. | |
Kalhoro NH, Veits J, Rautenschlein S, Zimmer G. A recombinant vesicular stomatitis virus replicon vaccine protects chickens from highly pathogenic avian influenza virus (H7N1). Vaccine. 2009;27(8):1174–1183. | |
Steitz J, Britten CM, Wölfel T, Tüting T. Effective induction of anti-melanoma immunity following genetic vaccination with synthetic mRNA coding for the fusion protein EGFP.TRP2. Cancer Immunol Immunother. 2006;55(3):246–253. | |
Yu H, Babiuk LA, van Drunen Littel-van den Hurk S. Immunity and protection by adoptive transfer of dendritic cells transfected with hepatitis C NS3/4A mRNA. Vaccine. 2007;25(10):1701–1711. | |
Pan CH, Greer CE, Hauer D, et al. A chimeric alphavirus replicon particle vaccine expressing the hemagglutinin and fusion proteins protects juvenile and infant rhesus macaques from measles. J Virol. 2010;84(8):3798–3807. |
 © 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms.php
and incorporate the Creative Commons Attribution
- Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms.php
and incorporate the Creative Commons Attribution
- Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.