Back to Journals » Drug Design, Development and Therapy » Volume 11
Development of novel ultrashort antimicrobial peptide nanoparticles with potent antimicrobial and antibiofilm activities against multidrug-resistant bacteria
Authors Almaaytah A, Mohammed GK ![]() , Abualhaijaa A, Al-Balas Q
, Abualhaijaa A, Al-Balas Q ![]()
Received 27 July 2017
Accepted for publication 5 October 2017
Published 3 November 2017 Volume 2017:11 Pages 3159—3170
DOI https://doi.org/10.2147/DDDT.S147450
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Dr Sukesh Voruganti
Ammar Almaaytah,1 Gubran Khalil Mohammed,1 Ahmad Abualhaijaa,2 Qosay Al-Balas3
1Department of Pharmaceutical Technology, Faculty of Pharmacy, 2Department of Applied Biological Sciences, Faculty of Science and Arts, 3Department of Medicinal Chemistry, Faculty of Pharmacy, Jordan University of Science and Technology, Irbid, Jordan
Abstract: Conventional antibiotics are facing strong microbial resistance that has recently reached critical levels. This situation is leading to significantly reduced therapeutic potential of a huge proportion of antimicrobial agents currently used in clinical settings. Antimicrobial peptides (AMPs) could provide the medical community with an alternative strategy to traditional antibiotics for combating microbial resistance. However, the development of AMPs into clinically useful antibiotics is hampered by their relatively low stability, toxicity, and high manufacturing costs. In this study, a novel in-house-designed potent ultrashort AMP named RBRBR was encapsulated into chitosan-based nanoparticles (CS-NPs) based on the ionotropic gelation method. The encapsulation efficacy reported for RBRBR into CS-NPs was 51.33%, with a loading capacity of 10.17%. The release kinetics of RBRBR from the nanocarrier exhibited slow release followed by progressive linear release for 14 days. The antibacterial kinetics of RBRBR-CS-NPs was tested against four strains of Staphylococcus aureus for 4 days, and the developed RBRBR-CS-NPs exhibited a 3-log decrease in the number of colonies when compared to CS-NP and a 5-log decrease when compared to control bacteria. The encapsulated peptide NP formulation managed to limit the toxicity of the free peptide against both mammalian cells and human erythrocytes. Additionally, the peptide NPs demonstrated up to 98% inhibition of biofilm formation when tested against biofilm-forming bacteria. Loading RBRBR into CS-NPs could represent an innovative approach to develop delivery systems based on NP technology for achieving potent antimicrobial effects against multidrug-resistant and biofilm-forming bacteria, with negligible systemic toxicity and reduced synthetic costs, thereby overcoming the obstructions to clinical development of AMPs.
Keywords: ultrashort antimicrobial peptides, nanoparticles, drug delivery, antibiofilm
Introduction
Recent decades have witnessed a significant surge in bacterial resistance to antibiotics due to the tremendous overuse and misuse of antibiotics that was facilitated by the medical community.1 The escalating problem of microbial resistance has been correlated with a sharp decrease in the number of antibacterial drugs currently being developed by pharmaceutical companies, which are experiencing dried research pipelines in regard to the development of novel antimicrobial agents.2 Resistance to antibiotics has reached critical levels with reports of the emergence of panresistant bacteria, consequently setting the stage for humanity to experience the fear of an anticipated postantibiotic era.3 Therefore, a strong need to develop new classes of antimicrobial compounds to limit the problems of microbial resistance is urgently needed.4
Antimicrobial peptides (AMPs) have gained great interest recently to serve as potential alternatives to conventional antibiotics.5,6 Due to their unique properties such as unique structures, modes of action, and low probability of inducing microbial resistance, AMPs are considered as excellent alternatives to conventional antibiotics. AMPs are amphiphilic cationic peptides and a member of the innate host-defense system of most living organisms, such as plants, bacteria, fungi, and yeast.7–9 AMPs are small molecules (<10 kDa), with variable length and amino-acid sequence (10–50 amino acids).10 It is hypothesized that when AMPs come in contact with the cell membranes, AMPs adopt amphiphilic structures that allow them to induce pore formation in target membranes and consequently cause cell death.11 AMPs exhibit a low tendency to induce bacterial resistance, with rapid and potent wide-spectrum antimicrobial activities against a wide range of organisms involving multiresistant Gram-negative and Gram-positive bacteria, parasites, fungi, and viruses.12–14 The broad-spectrum activity, fast-killing kinetics, and hydrophobicity of these peptides allow AMPs to gain attractive properties with enhanced selectivity and activity toward pathogens.15
Unfortunately, and despite the initial enthusiasm regarding AMPs as candidate alternatives to antibiotics, many obstacles have limited their development and clinical use. These limitations are attributed to poor stability in biological fluids, due to inactivation by lipoproteins and anionic albumins. Additionally, AMPs have limited antimicrobial target selectivity, which leads to undesirable interactions with host macromolecules and high systemic toxicity.16–18
Due to these limitations, most research efforts are currently focused on improving AMP stability and reducing systemic toxicity. In this regard, the encapsulation of AMPs by nanostructures might represent an innovative approach to overcome some problems related to the limited use of AMPs clinically.19,20
Nanotechnology has witnessed rapid development in many different fields, including infectious diseases and applied microbiology.21,22 Nanotechnology involves structures or materials sized 1–100 nm that exhibit powerful potential leverage for drug-delivery system purposes when compared to traditional delivery systems.23 Nanoparticles (NPs) comprise various biodegradable materials in the nanometric size range, such as natural and synthetic polymers and lipids. NPs can incorporate both hydrophilic and lipophilic drugs, due to their high degree of biocompatibility.
Chitosan (CS) is a biodegradable and biocompatible polycationic polysaccharide with high molecular weight.24,25 CS consists of copolymers of β-1,4-linked D-glucosamine and N-acetyl-D-glucosamine obtained by the deacetylation of chitin, a natural polymer found in the enormous number of invertebrates and in the cell walls of fungi.25 Many studies have reported the use of CS in both microparticle and NP formulations in novel potential drug-delivery systems. CS is nontoxic and a safe excipient that is constantly employed in drug formulation, and exhibits intrinsic antimicrobial activity against many microorganisms, such as fungi, yeast, and Gram-positive and Gram-negative bacteria.26,27 The encapsulation of AMPs into CS would develop innovative nanotherapeutics in the treatment of microbial infections and possibly provide alternative solutions to the limitations currently hindering the clinical development of AMPs.
Few studies have reported on the use of CS-NPs as a delivery system for AMPs. The use of CS-NPs for delivery of renin substrate I, the cationic amphiphilic peptide that shares the physicochemical properties of many AMPs, resulted in 100% encapsulation efficacy, low burst, and continuous linear kinetic release.28 Another AMP loaded in CS-NP is temporin B (TB). The results indicate that CS-NP encapsulation is correlated with reduced cytotoxicity against mammalian cells.29
Cationic ultrashort AMPs (USAMPs) consisting of fewer than eight residues are a promising group of AMPs that meet the required criteria of novel antimicrobial drug development due to their unparalleled mode of action, which is a result of their high diversity in regard to peptide length, amino-acid sequence, and structure.30,31 Additionally, USAMPs display high antimicrobial activity, with negligible or very low toxicity against mammalian host cells.32 Developing USAMPs is also attractive because of low production costs, low probability of developing resistance, and low or negligible hemolytic toxicity when compared with the conventional AMPs reported in literature. The production cost is also an advantage of these molecules, due to the short sequence of the peptides.
There are no reported studies demonstrating the application of USAMPs or AMPs into CS-NPs. The present study aimed to evaluate CS’s ability to encapsulate USAMPs and evaluate the efficacy of incorporating USAMPs in CS-NPs as molecule carriers for maintaining USAMP activity while reducing mammalian cell toxicity. Additionally, the study aimed to explore the antibiofilm activity of the NP-USAMP combination against strains of biofilm-forming Gram-positive bacteria.
Materials and methods
Materials
The AMP employed in the present study was the novel in-house-designed potent USAMP named RBRBR. The peptide was rationally designed in house, and resembles no other peptide deposited in official protein databases. It consists of five amino acids (RBRBR). Arginine (R) was chosen to represent the charged moieties, while L-4-phenyl-phenylalanine (B) was incorporated to represent the hydrophobic residues.
The peptide was synthesized using the solid-phase method and fluorenylmethyloxycarbonyl (Fmoc) chemistry, and purified by reverse-phase high-performance liquid chromatography using an acetonitrile–H2O–trifluoroacetic acid gradient. The identity of the peptide was confirmed by electrospray-ionization mass spectrometry (GL Biochem, Shanghai, China). Medium-molecular-weight CS (deacetylation ~92%) and sodium tripolyphosphate (TPP) were purchased from Sigma Aldrich (St Louis, MO, USA).
Microorganisms
The bacterial strains used for the determination and testing of the RBRBR-CS-NP formulation were acquired from the American Type Culture Collection (ATCC). These included Staphylococcus aureus (ATCC 29213), which was used as a control strain, and methicillin-resistant S. aureus (MRSA; ATCC 33591, 43300). Clinically isolated MRSA (ATCC BAA41) was also employed in this study.
Preparation of blank CS-NPs and RBRBR-CS-NPs
CS-NPs and RBRBR-CS-NPs were prepared with a simple ionic gelation method, as described previously, with slight modifications.28,29 Briefly, CS was dissolved in 1.75% (v:v) acetic acid (1 mg/mL, 5 pH), while TPP was dissolved in water (1 mg/mL). For RBRBR-CS-NP preparation, 500 μg of RBRBR was added to the CS solution. After the addition of 1 mL TPP aqueous solution to 5 mL of the CS solution, the NPs were formed. Formation took place under magnetic stirring, and the mixture was stirred at room temperature for 2 hours. NP suspensions were purified by centrifugation at 2,980 g at 4°C for 2 hours.
Characterization of NPs
Dynamic light scattering was used to measure the size distribution of the NPs. Developed NP ζ-potential was evaluated at 25°C in 10 mM phosphate buffered saline (PBS), pH 7.4.
In vitro release kinetics of NPs and evaluation of RBRBR-loading capacity
After RBRBR-CS-NP purification, the RBRBR found in the supernatant was evaluated at 562 nm using a Micro BCA protein-assay kit (Thermo Fisher Scientific, Waltham, MA, USA):
|
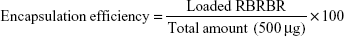
|
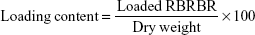
Purified RBRBR-CS-NPs were redispersed in 1 mL PBS (pH 7.4) and stirred at 37°C in a shaking incubator. The sample was centrifuged at 2,980 g 4°C for 2 hours at different intervals. Fresh medium (1 mL) was added to replace the collected supernatant. The Micro BCA protein assay was used to determine the amount of peptide released from the NP formula.
Bacterial susceptibility assay
The number of colony-forming units per milliliter (CFU/mL) of the bacterial strains used in this study after treatment with peptide loaded in CS-NPs was determined to evaluate the efficacy and antimicrobial activity of the peptide-NP formula. Briefly, tryptic soy broth (TSB) was used as the culture medium for bacterial growth, and bacterial strains were diluted to 106 CFU/mL. RBRBR-CS-NPs concentrations of 2.5, 5, and 10 mg/mL were prepared accordingly. The antibacterial activity of each concentration of developed RBRBR-CS-NPs was compared to activity of CS-NPs.
The bacterial suspension of 106 CFU/mL was added to buffer assay at a volume ratio of 1:9. The buffer assay containing both RBRBR-CS-NPs and CS-NPs and bacterial strains was incubated for 4 continuous days at 37°C. After 1-, 2-, 3-, and 4-day intervals, 10 μL from each sample tube were withdrawn, serially diluted, and cultured on sterilized Müller–Hinton agar plates for 24 or 48 hours at 37°C, followed by colony counting. Results are expressed as log10 CFU/mL.
Antibiofilm-activity formation
Biofilm formation was performed as reported in earlier studies33–37 employing the Calgary biofilm device (Innovotech, Edmonton, Canada). Gram-positive S. aureus (33591) was left to grow in TSB at 37°C for 20 hours. Then, a concentration of 107 CFU/mL was prepared by diluting the cultures in the same medium. Using 96-peg lids on which the cells of biofilm can build up, 150 μL of that bacteria culture was added to each peg lid to allow the formation of biofilm on the purposed designed pegs, followed by incubating the pegs for 20 hours at 37°C under 125 rpm rotation. Blank lanes were prepared by adding 150 μL TSB to six wells. To discard planktonic cells after biofilm formation, PBS was used to wash pegs three times.
Viable bacterial cell counts after treatment
After washing, each peg lid containing 200 μL of 5 and 10 mg/mL RBRBR-CS-NPs was transferred into a “challenge 96-well microtiter plate”. The biofilms in the peg lids were allowed to grow by incubating the plates at 37°C for 9 hours. Peg lids containing only PBS were used as control and PBS only as a blank. After incubation, the pegs were transferred into sterilized Eppendorf tubes containing 500 μL PBS, sonicated for 20 minutes, and 5 μL from each Eppendorf tube was transferred to sterilized labeled agar plates and incubated for 48 hours at 37°C. The number of bacterial colonies was counted, and results are expressed as CFU/mL (%).
Cytotoxicity assays
The cell line employed in the present study was the mammalian Vero cell line, acquired commercially form ATCC (ATCC CCL81). The Vero cell line represents immortalized kidney epithelial cells extracted from the African green monkey, formerly called Cercopithecus aethiops. Roswell Park Memorial Institute (RPMI) media were employed as culture media for cell growth supplemented with 1% streptomycin–ampicillin and 10% fetal bovine serum (Sigma-Aldrich). The MTT cell-proliferation assay was used to measure the cell-proliferation rate of the peptide-CS-NP-treated cell line.38 For the MTT assay, cells were seeded at 5×103 cells per well in flat-bottomed 96-well plates, and the plates were incubated for 18–24 hours at 37°C under 5% CO2. The following day, different concentrations of RBRBR-CS-NPs (2.5, 5, and 10 mg/mL, loaded with 255, 510, and 1,020 μg/mL RBRBR, respectively) were prepared using RPMI as the dissolving media and added to the cells in the plates.
Plates were incubated for 24 hours at 37°C under 5% CO2. After 24 hours of treatment, 20 μL MTT solution (2.5 mg/mL) was added to all wells and plates incubated for 2–5 hours at 37°C, 5% CO2. After this incubation, the MTT–peptide solution was removed using a 21 gauge needle and a 10 mL syringe. Dimethyl sulfoxide (100 μL) was added to each well and mixed thoroughly by pipetting to dissolve the formazan crystals at the bottom of the wells until a clear purple color was achieved. The plates were then placed on an absorbance microplate reader (BioTek, Winooski, VT, USA) and absorbance measured at λ=550 nm.
Erythrocyte-hemolysis assay
For determination of the ability of CS-NPs and RBRBR-CS-NPs to induce hemolysis in human erythrocytes, assays were performed as reported earlier.39 Briefly, PBS containing 2.5, 5, and 10 mg/mL RBRBR-CS-NPs were prepared and compared to free RBRBR (255, 510, and 1,020 μg/mL). Then, 1 mL of each concentration was added to 1 mL of 4% erythrocyte suspension.
Controls were prepared by adding 5 μL Triton X-100 to 1 mL red blood cell (RBC) suspension (positive control), blanks were prepared by adding 1 mL RBC suspension to PBS, and suspensions were incubated with CS-NPs, RBRBR-CS-NPs, and free RBRBR for 60 minutes at 37°C. After the incubation step, tubes were gently vortexed, 1 mL of each sample removed and placed into sterilized Eppendorf tubes, centrifuged for 5 minutes at 3,000 g, and 100 μL of each supernatant placed into a well in a 96-well plate. Absorbance was measured at λ=450 nm with the aid of the microplate reader. Percentage hemolysis was calculated thus:
|
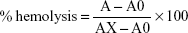
where A is OD450 with the peptide solution, A0 the OD450 of blank (PBS), and AX the OD450 of control (0.1% Triton X-100).
Statistical analysis
All data-generating experiments involved in this study were performed in triplicate. The values generated in all experiments were compared and analyzed by one-way analysis of variance using the least significant-difference multiple-comparison tests on the means. Differences were reported at the 95% confidence level (P<0.05). Data analysis was conducted using SPSS software version 21.
Results
Preparation and characterization of CS-NPs and RBRBR-CS-NPs
Preparation of CS-NPs and RBRBR-CS-NPs was performed using simple ionic gelation. NPs were analyzed by dynamic light scattering to determine their average diameter. This showed an increase in average NP diameter due to RBRBR loading. The average diameter increased from 104.27±0.5 nm for CS-NPs to 121.13±1.01 nm for RBRBR-CS-NPs (Table 1). Size-distribution figures of the CS-NPs and RBRBR-CS-NPs NPs formulated in the study as they were acquired by dynamic light scattering are represented in Figures S1 and S2. Both CS-NPs and RBRBR-CS-NPs exhibited positive ζ-potential values in PBS at pH 7.4 (Table 1), which was probably attributable to the cationic nature of CS. Loaded RBRBR significantly decreased the ζ-potential value of the NP formulation (Table 1).
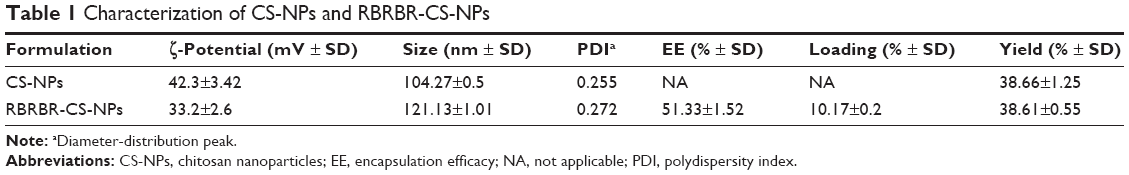 | Table 1 Characterization of CS-NPs and RBRBR-CS-NPs |
Evaluation of RBRBR-loading capacity
Encapsulation efficacy and loading capacity of RBRBR into CS-NPs were evaluated at 562 nm using the Micro BCA protein assay. The results showed an encapsulation efficacy of 51.33% and loading capacity of 10.17%, for RBRBR (Table 1).
In vitro release kinetics of NPs
Release kinetics of RBRBR from RBRBR-CS-NPs were studied in PBS pH 7.4 for 14 days (Figure 1). According to the DDSolver software program, the best release model that mathematically fitted with this profile was Korsmeyer–Peppas.40 The system exhibited a slow-release pattern followed by progressive linear release for 14 days.
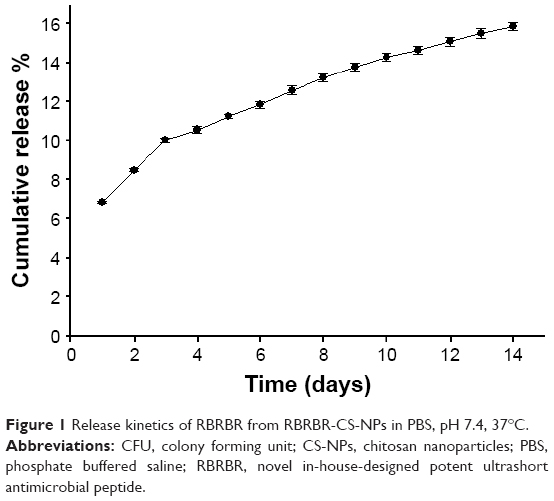 | Figure 1 Release kinetics of RBRBR from RBRBR-CS-NPs in PBS, pH 7.4, 37°C. |
Bacterial susceptibility assay
Preparatory experiments were performed to optimize long-term interval antibacterial activity for CS-NPs and RBRBR-CS-NPs. The optimum concentration of tryptic soy broth (TSB) in PBS for sustaining bacterial growth for 4 days was 1.25% (v:v). Additionally, 5 mg/mL RBRBR-CS-NPs was found to be the optimum concentration to determine the activity of the formula for 4 days.
The 5 mg/mL RBRBR-CS-NP formulation displayed significant antimicrobial activity against all studied bacterial strains. Encapsulated RBRBR-NPs were able to inhibit the growth of all studied bacterial strains employed in this study. Antibacterial activity of RBRBR-CS-NP was assessed by measuring cell viability, expressed as log10 CFU/mL for each of the tested bacterial strains. Then, this was compared to 5 mg/mL CS-NPs and to the control.
Figure 2 and Table 2 show the kinetics of the antibacterial activity against the control strain S. aureus (ATCC 29213). After incubation, bacteria CFU/mL when exposed to the RBRBR-CS-NPs was significantly reduced for both CS-NPs and the control formulation. At 4 days of exposure, the CFU/mL of RBRBR-CS-NPs showed 3-log reduction when compared to the CS-NP formulation and 5-log reduction when compared to control bacteria.
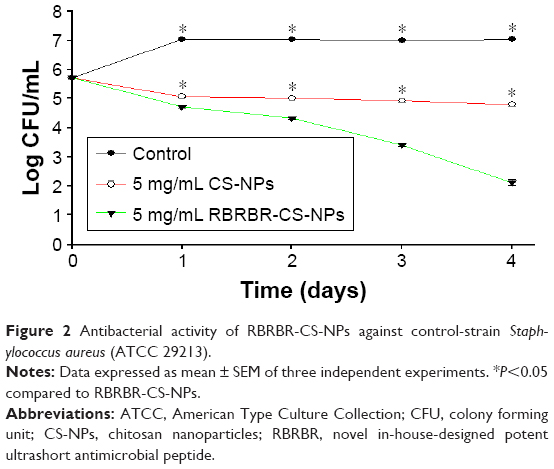 | Figure 2 Antibacterial activity of RBRBR-CS-NPs against control-strain Staphylococcus aureus (ATCC 29213). |
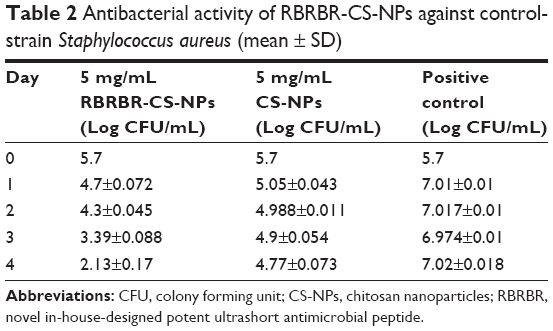 | Table 2 Antibacterial activity of RBRBR-CS-NPs against control-strain Staphylococcus aureus (mean ± SD) |
Other comparisons of the antibacterial activity of RBRBR-CS-NPs to CS-NPs and control were performed against two MRSA strains (ATCC 33591, 43300). Additionally, clinically isolated MRSA (ATCC BAA41) was employed in antimicrobial susceptibility assays. Figure 3 and Table 3 clearly display that all three bacterial strains showed a statistically significant difference in cell viability between RBRBR-loaded CS-NPs and CS-NPs.
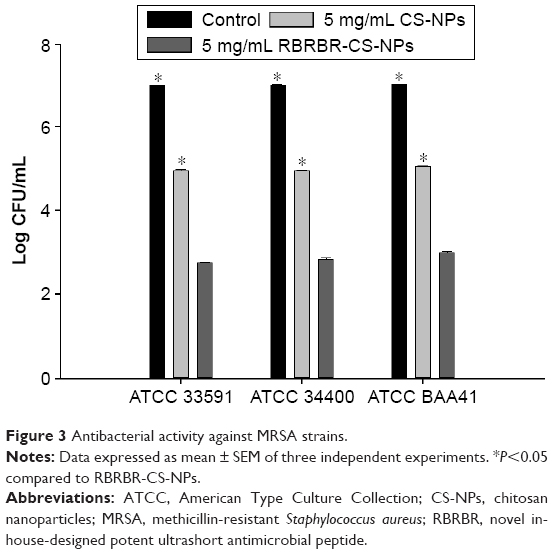 | Figure 3 Antibacterial activity against MRSA strains. |
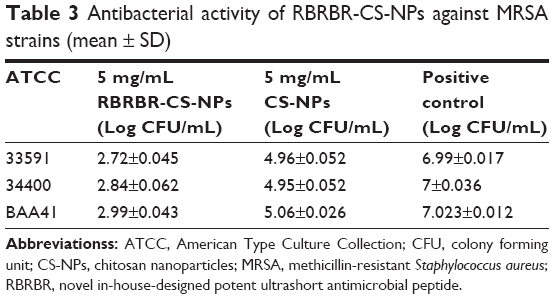 | Table 3 Antibacterial activity of RBRBR-CS-NPs against MRSA strains (mean ± SD) |
Antibiofilm activity
The antibiofilm activity of the RBRBR-CS-NP was assessed by evaluating the reduction in viable bacterial cell counts after peptide treatment using the colony-count method. The RBEBR-CS-NP formulation was able to reduce the number of viable bacterial cells compared to the control (Table 4). CFU/mL was reduced by 86.5% and 98% for 5 and 10 mg/mL formulations, respectively. Both formulations of RBRBR-CS-NP showed statistically significant differences in cell viability when compared to the control. Percentages of biofilm reduction as CFU/mL are depicted in Figure 4.
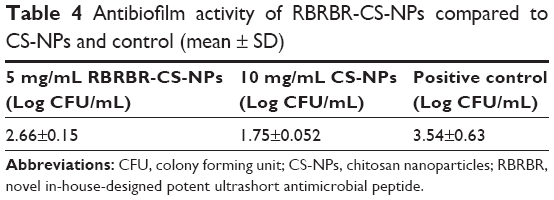 | Table 4 Antibiofilm activity of RBRBR-CS-NPs compared to CS-NPs and control (mean ± SD) |
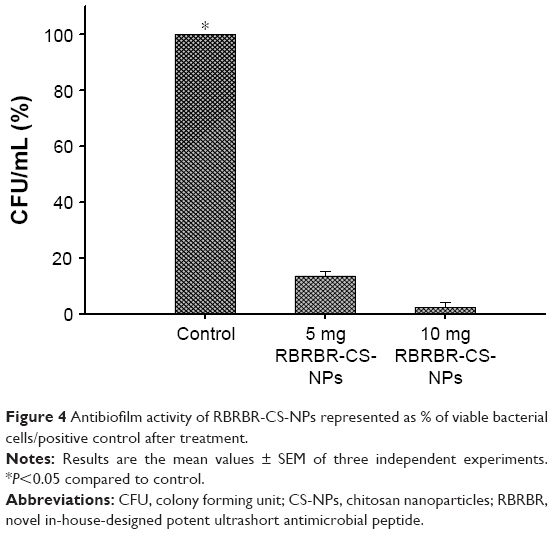 | Figure 4 Antibiofilm activity of RBRBR-CS-NPs represented as % of viable bacterial cells/positive control after treatment. |
Hemolysis assay
The hemolytic activity of RBRBR-CS-NPs against human erythrocytes (RBCs) was determined to evaluate the peptide–NP formula’s toxicity toward normal mammalian cells. In this study, CS-NPs and RBRBR-CS-NPs (2.5, 5 and 10 mg/mL) were incubated with a 4% human erythrocyte suspension and compared to free RBRBR (255, 510, and 1,020 μg/mL).
There was negligible hemolysis for both the CS-NPs and RBRBR-CS-NPs at the concentrations employed in the hemolytic assays (Table 5). Figure 5 displays the results of the hemolytic assays for all formulations. The percentage hemolysis reported for the RBRBR-CS-NP formulation at the highest concentration employed in the study was 3.6%, while the percentage hemolysis reported for the free peptide was 13.1%. The results of the hemolytic assays clearly indicate that the encapsulation of RBRBR with the NP carrier managed to significantly decrease the hemolytic and toxic activity of the free peptide against mammalian erythrocytes.
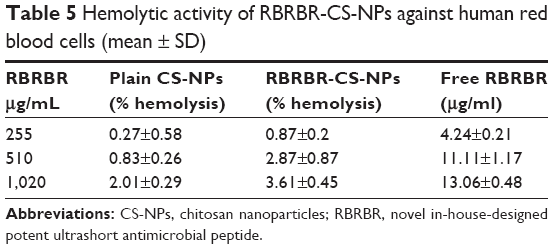 | Table 5 Hemolytic activity of RBRBR-CS-NPs against human red blood cells (mean ± SD) |
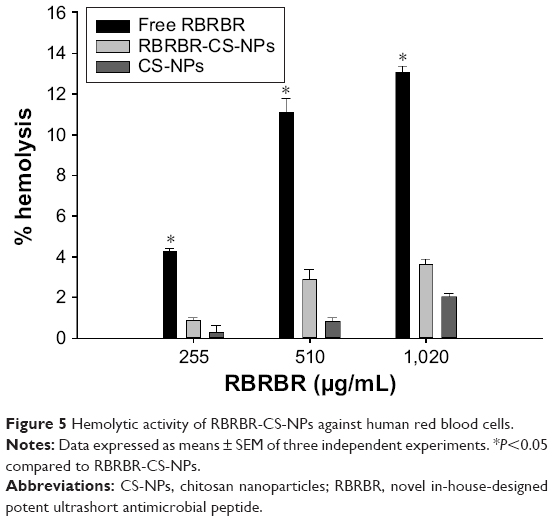 | Figure 5 Hemolytic activity of RBRBR-CS-NPs against human red blood cells. |
MTT cell-proliferation assay
Cell-proliferation assays were performed to measure the antiproliferative activity of RBRBR-CS-NPs against mammalian cells (Vero), in order to measure the selectivity and toxicity of the developed nanocarrier. Developed NPs managed to limit the toxicity of the same amount of loaded RBRBR against normal cell lines. The half maximal inhibitory concentration (IC50) of free RBRBR reported was 187.2 μg/mL (Figure 6). The IC50 reported for the RBRBR-CS-NP was 6.62 mg/mL. The data generated from the cell-proliferation assays showed that the cytotoxicity of the RBRBR was reduced significantly when encapsulated in the nanocarrier and the CS nanocarrier managed to shield the cytotoxic activity of the free RBRBR on the growth and proliferation of mammalian cell lines, confirming results from the hemolytic assays (Figure 7).
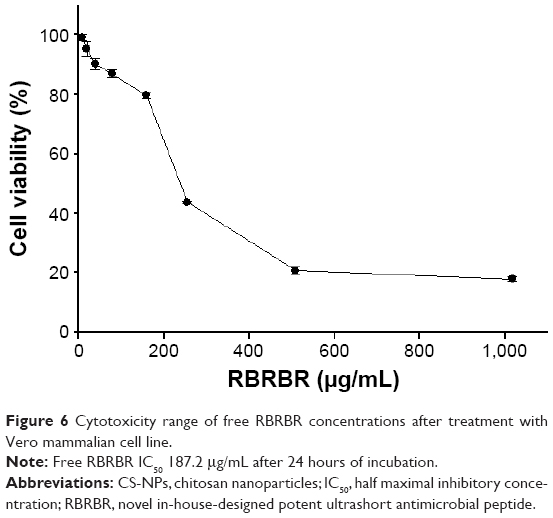 | Figure 6 Cytotoxicity range of free RBRBR concentrations after treatment with Vero mammalian cell line. |
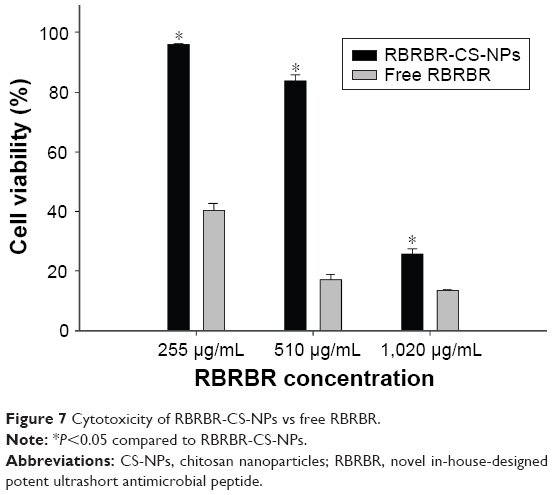 | Figure 7 Cytotoxicity of RBRBR-CS-NPs vs free RBRBR. |
Discussion
Although the number of resistant bacterial strains and infectious diseases is on the rise, the number of antibiotics being developed and reaching the clinic in the last few decades has declined significantly.41 Due to their potent antimicrobial activity, rapid-killing kinetics, and broad spectrum of activity, AMPs are considered promising alternative agents for forming a new class of antimicrobial agents.
One of the most significant obstacles to the application of AMPs in the treatment of microbial infections is related to their lack of microbial target selectivity and their toxicity against normal cells. Several AMPs display potent antimicrobial activity, but suffer from high toxicity and hemolytic activity against normal cells. To overcome the problems of stability and selectivity of AMPs, research efforts are currently focused on developing technologies to reduce AMP toxicity. One of the promising technologies being explored recently is the use of nanotechnology for the loading and entrapment of AMPs into nanocarriers. The encapsulation of AMPs into nanocarriers could enhance therapeutic activity and selectivity by forming a shield around AMPs and causing equilibrium-mediated sustained release of active molecules directly to bacterial cells.
Recently, several AMPs have been encapsulated into various forms of nanostructures, including nanofibers, nanovesicle-coated metallic NPs, and self-assembled structures. Among these, polymeric NPs represent the most common application for the delivery of AMPs, and are considered as the most promising approach for the therapeutic application of AMPs.42
Several studies have explored the potential delivery of AMPs using polymeric NPs based on synthetic or natural polymers. However, among the several polymers investigated, CS has been shown to be highly attractive for drug delivery. Due to its bioadhesive property, biodegradation, safety, biocompatibility, and attractive encapsulation of several drugs, CS is studied widely for drug delivery. Ionic gelation represents the most reliable and commonly used method for the preparation of CS-NPs. The method is based on the reversible physical electrostatic cross-linking between positively charged CS and the polyanion TPP.43
The present work intended to employ recent advances in NP formulation along with the advantage of USAMPs by encapsulating an in-house-designed novel USAMP named RBRBR into CS-NPs to identify if the incorporation of RBRBR into CS-NPs might retain the antimicrobial activity of the peptide while reducing its toxicity, and consequently determine the therapeutic potential of this approach. The formulated RBRBR-CS-NPs were prepared by ionic gelation and evaluated afterward by studying the physicochemical characterization, release kinetics, bacterial susceptibility, antibiofilm formation, and cytotoxicity. Due to the positive charge of CS in acidic media, electrostatic repulsion with the cationic RBRBR was expected to occur. To overcome this anticipated repulsion, the pH was adjusted to 5, in order to reduce the positive charge of CS and enhance the encapsulation process. For better understanding of the interaction among the charged molecule and to evaluate the stability of the nanosystem, ζ-potential was evaluated. The expected positive value of ζ-potentials was observed, due to the cationic charge of CS. This value was significantly decreased (P<0.05) by loading RBRBR into CS-NPs, which was attributed to conformation and rearrangement of the CS charge.
The encapsulation and development of the RBRBR-CS-NPs clearly exhibited nanoscale dimensions accompanied by a statistically significant enhancement in the average diameter of the system when compared to the blank NPs, which confirmed the incorporation of RBRBR into CS-NPs. Interestingly, the nanosystem displayed progressive linear release kinetics that were continuous for a long period. Few studies have evaluated the effectiveness of encapsulating AMPs NPs. For instance, polylactic acid NPs were investigated to incorporate nisin for sustained release and inhibition of Lactobacillus delbrueckii growth.44 Poly(lactic-co-glycolic acid) (PLGA) NPs have been used to encapsulate LL37 for wound healing and inhibition of Escherichia coli growth. The nanosystem showed low loading of LL37 into NPs and high burst release in the first 24 hours of an amount equal to about 40% of the total peptide. Nevertheless, the free LL37 displayed higher antibacterial activity than PLGA-LL37-NPs against E. coli.45
Another AMP that has been loaded into CS-NPs is TB. The results displayed that the encapsulation ensured a burst effect with long-lasting antibacterial effect to prevent the regrowth of residual bacterial cells. The peptide’s cytotoxicity against mammalian cells was significantly reduced after encapsulation of TB in CS-NPs.29 Despite the low amount of TB compared to RBRBR into the nanocarrier (116.5 vs 255, 233 vs 510, and 466 vs 1,020 μg/mL in 2.5, 5 and 10 mg/mL CS-NPs, respectively), the RBRBR-CS-NP formulation exhibited significantly lower cell toxicity when compared with TB, which was probably due to the intrinsic low cytotoxicity of USAMPs.
The smallness and low hydrophobicity of USAMPs play a curative role in lowering the toxicity of AMPs in general. These features ensure higher selectivity against bacteria due to the abundance of anionic phospholipids, rather than zwitterion ones that are present in general in erythrocytes and mammalian cells.46 In the present study, the novel RBRBR was loaded into CS-NPs that displayed high-quality features of an improved nanoscale system, including a stable formula, linear release kinetics, and low cytotoxicity.
The antibacterial susceptibility assays carried out in this study showed that RBRBR-CS-NPs were active against wild-type and the multidrug-resistant clinical isolated strains of Gram-positive bacteria. Compared to the peptide-unloaded CS-NPs, RBRBR-CS-NPs demonstrated broader-spectrum and more potent antimicrobial effects against all tested bacterial strains, including the resistant isolates.
Plain CS-NPs were able to inhibit the growth of control strains of Gram-positive bacteria, with a 2.25-log reduction observed at day 4. On the other hand, the developed RBRBR-CS-NPs displayed a 5-log reduction compared to untreated bacteria. It is noteworthy that CS-NPs ensured a burst effect followed by a further 3-log reduction as a result of the released peptide. According to the release pattern, the peptide concentration released from the nanocarrier was responsible for the strong reduction in bacterial cell viability. Therefore, the developed nanosystem employed in this study might possibly provide a synergistic mode of action with regard to antimicrobial effects lasting 4 days. Additionally, RBRBR-CS-NPs significantly inhibited the growth of the multidrug-resistant and clinical isolates of Gram-positive strains for all bacteria employed in the present study.
To the best of our knowledge, there are no reports in the literature that have evaluated an AMP-NP formula for inhibition of biofilm formation. The antibiofilm activity of RBRBR-CS-NPs was determined using viable bacterial cell counts after treatment. The nanoscale system displayed potent antibiofilm activity against the formed biofilms of bacterial strains that were employed in this study. Used at concentrations of 5 and 10 mg/mL, RBRBR-CS-NPs were able to inhibit viable cell growth within 9 hours by 87% and 98%, respectively.
With regard to the role of the nanosystem in toxicity management of free RBRBR, cytotoxicity assays, MTT cell proliferation, and erythrocyte hemolysis achieved promising results. The toxicity of RBRBR against the normal mammalian cell line and human erythrocytes was reduced significantly by peptide incorporation into the nanocarrier system. The improvement in toxicity profile and selectivity index in the nanosystem formulation can be attributed to the positive charge and improved binding to microbial membranes. The positive charge influences the electrostatic binding of the nanosystem to its target cell, and consequently facilitates the killing mechanism.
CS-NPs were acting here as nanoscale carrier to deliver the RBRBR directly to its target surface. In the present work, the encapsulation not only protected RBRBR from the external environment but also ensured a high abundance of peptide concentration at bacterial surfaces. As such, RBRBR-CS-NPs exhibit potent antibacterial activity with negligible toxicity.
In conclusion, our results demonstrate that the use of nanotechnology represented by the nanocarrier system employed in this study could be employed as an innovative therapeutic essentially based on CS-NPs loaded with novel USAMP to inhibit bacterial growth and proliferation. The results of the antimicrobial studies performed here indicate that RBRBR-CS-NPs have potent selective and long-acting activities against a wide range of Gram-positive bacteria, including the clinical isolates of the resistant strains of S. aureus. The results obtained from the hemolytic and MTT assays indicate that RBRBR loaded into nanocarriers exerts minimal toxicity against human erythrocytes and normal mammalian cell lines and has significant selectivity against microbial cells when compared to free RBRBR. The encapsulation of AMPs in NPs would provide an innovative and promising approach in AMP-delivery application.
Acknowledgment
This study was funded by the Deanship of Research at the Jordan University of Science and Technology. The authors are extremely grateful and sincerely acknowledge the guidance, encouragement, and help of Dr Nid’’a Alshraiedeh and Dr Rana Obeidat at the Faculty of Pharmacy, Jordan University of Science and Technology, during the course of this study.
Disclosure
The authors report no conflicts of interest in this work.
References
Carlet J, Jarlier V, Harbarth S, Voss A, Goossens H, Pittet D. Ready for a world without antibiotics? The Pensières Antibiotic Resistance Call to Action. Antimicrob Resist Infect Control. 2012;1:11. | ||
Fair RJ, Tor Y. Antibiotics and bacterial resistance in the 21st century. Perspect Medicin Chem. 2014;6:25–64. | ||
Hey SP, Kesselheim AS. Reprioritizing research activity for the post-antibiotic era: ethical, legal, and social considerations. Hastings Cent Rep. 2017;47:16–20. | ||
Carlet J, Pulcini C, Piddock LJV. Antibiotic resistance: a geopolitical issue. Clin Microbiol Infect. 2014;20:949–953. | ||
Aoki W, Ueda M. Characterization of antimicrobial peptides toward the development of novel antibiotics. Pharmaceuticals (Basel). 2013;6:1055–1081. | ||
Roscia G, Falciani C, Bracci L, Pini A. The development of antimicrobial peptides as new antibacterial drugs. Curr Protein Pept Sci. 2013;14(8):641–649. | ||
Hancock R, Brown K, Mookherjee N. Host defence peptides from invertebrates: emerging antimicrobial strategies. Immunobiology. 2006;211:315–322. | ||
Wiesner J, Vilcinskas A. Antimicrobial peptides: the ancient arm of the human immune system. Virulence. 2010;1:440–464. | ||
Zasloff M. Antimicrobial peptides of multicellular organisms. Nature. 2002;415:389–395. | ||
Hancock R. Expert opinion on investigational drugs cationic antimicrobial peptides: towards clinical applications. Expert Opin Investig Drugs. 2000;9:1723–1729. | ||
Giuliani A, Pirri G, Nicoletto SF. Antimicrobial peptides: an overview of a promising class of therapeutics. Cent Eur J Biol. 2007;2:1–33. | ||
Fox JL. Antimicrobial peptides stage a comeback. Nat Biotechnol. 2013;31:379–382. | ||
Maisetta G, Batoni G, Esin S, et al. In vitro bactericidal activity of human β-defensin 3 against multidrug-resistant nosocomial strains. Antimicrob Agents Chemother. 2006;50:806–809. | ||
Del Gaudio G, Lombardi L, Maisetta G, et al. Antifungal activity of the noncytotoxic human peptide hepcidin 20 against fluconazole-resistant Candida glabrata in human vaginal fluid. Antimicrob Agents Chemother. 2013;57:4314–4321. | ||
Henriksen JR, Etzerodt T, Gjetting T, Andresen TL. Side chain hydrophobicity modulates therapeutic activity and membrane selectivity of antimicrobial peptide Mastoparan-X. PloS One. 2014;9:e91007. | ||
Hancock R, Yeung A, Gellatly S. Multifunctional cationic host defence peptides and their clinical applications. Cell Mol Life Sci. 2011;68:2161–2176. | ||
Marr A, Gooderham W, Hancock R. Antibacterial peptides for therapeutic use: obstacles and realistic outlook. Curr Opin Pharmacol. 2006;6:468–472. | ||
Kang SJ, Park SJ, Mishig-Ochir T, Lee BJ. Antimicrobial peptides: therapeutic potentials. Expert Rev Anti Infect Ther. 2014;12:1477–1486. | ||
Yadav SC, Kumari A, Yadav R. Development of peptide and protein nanotherapeutics by nanoencapsulation and nanobioconjugation. Peptides. 2011;32:173–187. | ||
Brandelli A. Nanostructures as promising tools for delivery of antimicrobial peptides. Mini Rev Med Chem. 2012;12:731–741. | ||
Seil JT, Webster TJ. Antimicrobial applications of nanotechnology: methods and literature. Int J Nanomedicine. 2012;7:2767–2781. | ||
Shrivastava S, Dash D. Applying nanotechnology to human health: revolution in biomedical sciences. J Nanotechnol. 2009;2009:184702. | ||
De Jong WH, Borm PJ. Drug delivery and nanoparticles: applications and hazards. Int J Nanomedicine. 2008;3(2):133–149. | ||
Denkbaş EB, Ottenbrite RM. Perspectives on: chitosan drug delivery systems based on their geometries. J Bioact Compat Polym. 2006;21:351–368. | ||
Duttagupta DS, Jadhav VM, Kadam VJ. Chitosan: a propitious biopolymer for drug delivery. Curr Drug Deliv. 2015;12:369–381. | ||
Kong M, Chen XG, Xing K, Park HJ. Antimicrobial properties of chitosan and mode of action: a state of the art review. Int J Food Microbiol. 2010;144:51–63. | ||
Thanou M, Verhoef JC, Junginger HE. Oral drug absorption enhancement by chitosan and its derivatives. Adv Drug Deliv Rev. 2001;52:117–126. | ||
Piras AM, Sandreschi S, Maisetta G, Esin S, Batoni G, Chiellini F. Chitosan nanoparticles for the linear release of model cationic peptide. Pharm Res. 2015;32:2259–2265. | ||
Piras A, Maisetta G, Sandreschi S, et al. Chitosan nanoparticles loaded with the antimicrobial peptide temporin B exert a long-term antibacterial activity in vitro against clinical isolates of Staphylococcus epidermidis. Front Microbiol. 2015;6:372. | ||
Strøm MB, Rekdal O, Svendsen JS. Antimicrobial activity of short arginine- and tryptophan-rich peptides. J Pept Sci. 2002;8:431–437. | ||
Strøm M, Haug B, Skar M, Stensen W, Stiberg T, Svendsen J. The pharmacophore of short cationic antibacterial peptides. J Med Chem. 2003;46:1567–1570. | ||
Verkleij A, Zwaal R, Roelofsen B, Comfurius P, Kastelijn D, van Deenen L. The asymmetric distribution of phospholipids in the human red cell membrane: a combined study using phospholipases and freeze-etch electron microscopy. Biochim Biophys Acta. 1973;323:178–193. | ||
Ceri H, Olson M, Morck D, et al. The MBEC assay system: multiple equivalent biofilms for antibiotic and biocide susceptibility testing. Methods Enzymol. 2001;337:377–385. | ||
Falciani C, Lozzi L, Pollini S, et al. Isomerization of an antimicrobial peptide broadens antimicrobial spectrum to Gram-positive bacterial pathogens. PLoS One. 2012;7:e46259. | ||
Feng X, Sambanthamoorthy K, Palys T, Paranavitana C. The human antimicrobial peptide LL-37 and its fragments possess both antimicrobial and antibiofilm activities against multidrug-resistant Acinetobacter baumannii. Peptides. 2013;49:131–137. | ||
Luca V, Stringaro A, Colone M, Pini A, Mangoni ML. Esculentin(1–21), an amphibian skin membrane-active peptide with potent activity on both planktonic and biofilm cells of the bacterial pathogen Pseudomonas aeruginosa. Cell Mol Life Sci. 2013;70:2773–2786. | ||
Almaaytah A, Tarazi S, Alsheyab F, Al-Balas Q, Mukattash T. Antimicrobial and antibiofilm activity of mauriporin, a multifunctional scorpion venom peptide. Int J Pept Res Ther. 2014;20:397–408. | ||
Slater B. Epidemiology of congenital malformations. Dev Med Child Neurol. 1963;5:351–354. | ||
Almaaytah A, Zhou M, Wang L, Chen T, Walker B, Shaw C. Antimicrobial/cytolytic peptides from the venom of the North African scorpion, Androctonus amoreuxi: biochemical and functional characterization of natural peptides and a single site-substituted analog. Peptides. 2012;35:291–299. | ||
Zhang Y, Huo M, Zhou J, et al. DDSolver: an add-in program for modeling and comparison of drug dissolution profiles. AAPS J. 2010;12(3):263–271. | ||
Zhang Y, Liu Y, Sun Y, et al. In vitro synergistic activities of antimicrobial peptide brevinin-2CE with five kinds of antibiotics against multidrug-resistant clinical isolates. Curr Microbiol. 2014;68:685–692. | ||
Sandreschi S, Piras AM, Batoni G, Chiellini F. Perspectives on polymeric nanostructures for the therapeutic application of antimicrobial peptides. Nanomedicine (Lond). 2016;11:1729–1744. | ||
Nagavarma B, Yadav H, Ayaz A, Vasudha L, Shivakumar H. Different techniques for preparation of polymeric nanoparticles: a review. Asian J Pharm Clin Res. 2012;5:16–23. | ||
Salmaso S, Elvassore N, Bertucco A, Lante A, Caliceti P. Nisin-loaded poly-L-lactide nano-particles produced by CO2 anti-solvent precipitation for sustained antimicrobial activity. Int J Pharm. 2004;287:163–173. | ||
Chereddy K, Her C, Comune M, et al. PLGA nanoparticles loaded with host defense peptide LL37 promote wound healing. J Control Release. 2014;194:138–147. | ||
Mangoni ML, Shai Y. Short native antimicrobial peptides and engineered ultrashort lipopeptides: similarities and differences in cell specificities and modes of action. Cell Mol Life Sci. 2011;68:2267–2280. |
Supplementary materials
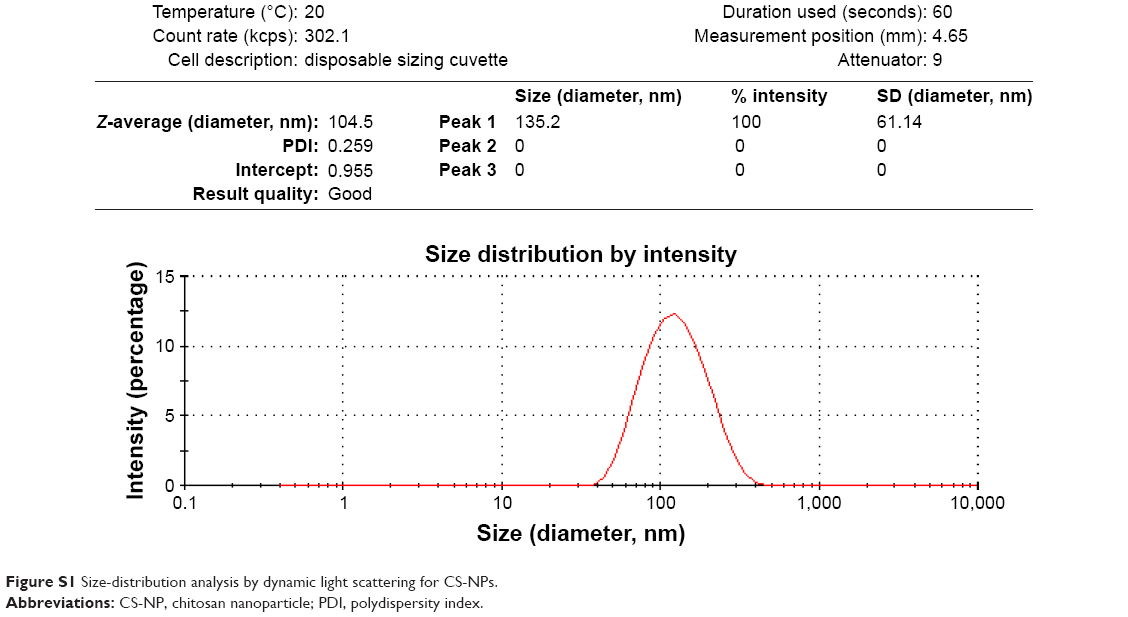 | Figure S1 Size-distribution analysis by dynamic light scattering for CS-NPs. |
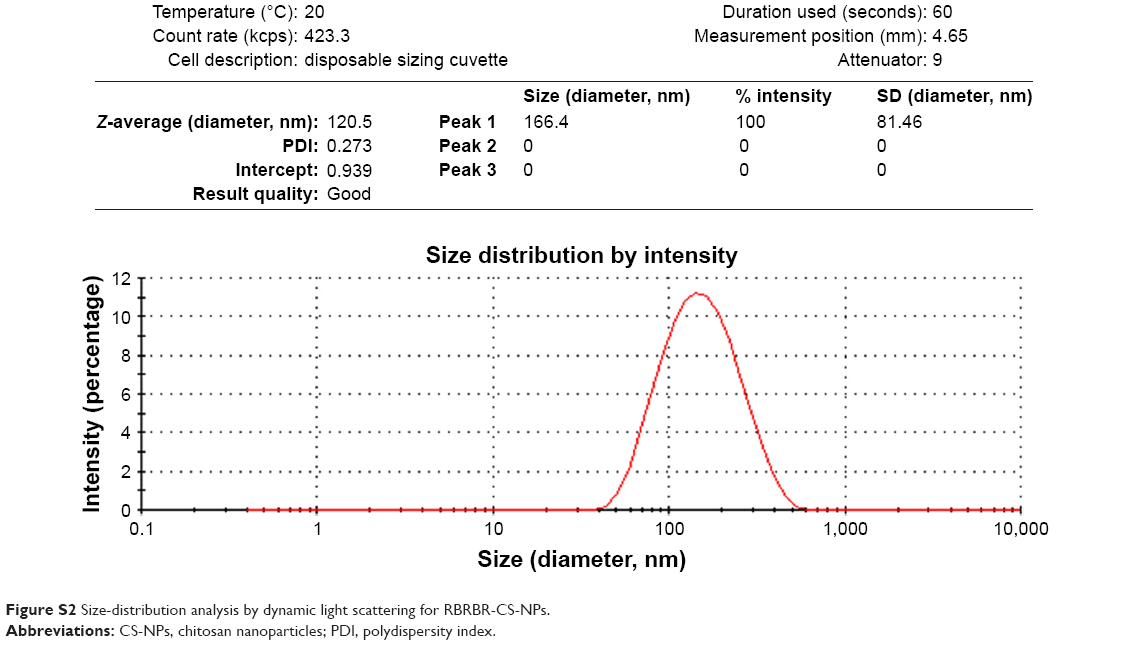 | Figure S2 Size-distribution analysis by dynamic light scattering for RBRBR-CS-NPs. |
 © 2017 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2017 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.