Back to Journals » International Journal of Nanomedicine » Volume 14
Development Of Novel Liposome-Encapsulated Combretastatin A4 Acylated Derivatives: Prodrug Approach For Improving Antitumor Efficacy
Authors Gu Y, Ma J, Fu Z ![]() , Xu Y, Gao B, Yao J, Xu W
, Xu Y, Gao B, Yao J, Xu W ![]() , Chu K, Chen J
, Chu K, Chen J
Received 2 April 2019
Accepted for publication 21 October 2019
Published 7 November 2019 Volume 2019:14 Pages 8805—8818
DOI https://doi.org/10.2147/IJN.S210938
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Dr Mian Wang
Yongwei Gu,1,2,* Juanjuan Ma,2,* Zhiqin Fu,1,2,* Youfa Xu,1,2 Baoan Gao,1,2 Jianzhong Yao,3 Wei Xu,1 Kedan Chu,1 Jianming Chen1,2
1Department of Pharmacy, Fujian University of Traditional Chinese Medicine, Fujian 350108, People’s Republic of China; 2Shanghai Wei Er Biopharmaceutical Technology Co., Ltd., Shanghai 201707, People’s Republic of China; 3School of Pharmacy, Second Military Medical University, Shanghai 200433, People’s Republic of China
*These authors contributed equally to this work
Correspondence: Wei Xu; Jianming Chen
Department of Pharmacy, Fujian University of Traditional Chinese Medicine, Fujian 350108, People’s Republic of China
Tel/fax +86-591-22861693
; +86-21-31198900-8949
Email [email protected]; [email protected]
Purpose: The objective of the present study was to develop a liposomal drug delivery system based on combretastatin A4 (CA4) prodrugs modified with varying alkyl chains and investigate the in vitro drug conversion from prodrug and in vivo antitumor effect.
Methods: The prodrug of CA4 was synthesized with stearyl chloride (18-carbon chain), palmitoyl chloride (16-carbon chain), myristoyl chloride (14-carbon chain), decanoyl chloride (10-carbon chain), and hexanoyl chloride (6-carbon chain) at the 3′-position of the CA4. Subsequently, it was encapsulated with liposomes through the thin-film evaporation method. Furthermore, the characteristics of prodrug-liposome were evaluated using in vitro drug release, conversion, and cytotoxicity assays, as well as in vivo pharmacokinetic, antitumor, and biodistribution studies.
Results: The liposome system with loaded CA4 derivatives was successfully developed with nano-size and electronegative particles. The rate of in vitro drug release and conversion was reduced as the fatty acid carbon chain lengthened. On the contrary, in vivo antitumor effects were improved with the enlargement of the fatty acid carbon chain. The results of the in vivo pharmacokinetic and tissue distribution studies indicated that the reduced rate of CA4 release with a long carbon chain could prolong the circulation time and increase the drug concentration in the tumor tissue.
Conclusion: These results suggested that the release or hydrolysis of the parent drug from the prodrug was closely related with the in vitro and in vivo properties. The slow drug release of CA4 modified with longer acyl chain could prolong the circulation time and increase the concentration of the drug in the tumor tissue. These effects play a critical role in increasing the antitumor efficacy.
Keywords: combretastatin A4, liposome, prodrug, drug release, anticancer
Introduction
Cancer is a multifaceted disease, representing one of the leading causes of disease-associated death worldwide.1,2 Common characteristics among traditional small molecule anticancer drugs are their poor solubility in aqueous solutions, serious toxicity, and side effects.3 The “prodrug” strategy was proposed to alleviate or even suppress these drawbacks. Prodrugs are bioreversible derivatives of pharmacologically active agents that can be either chemically or enzymatically degraded into the parent active drug inside the body to elicit its desired pharmacological effect.4,5 They have received considerable attention for their ability to improve insufficient solubility and permeability, poor chemical stability, rapid metabolism, low blood–brain barrier transport, and toxicity of drugs.6 Currently, approximately 5–7% of the drugs approved can be classified as prodrugs.7
For prodrugs, drug release plays an important role in the delivery process,8 which may alter the efficacy, pharmacokinetics, tissue distribution, and toxicity of the parent drug.9 Ideally, the parent drug should be released from the prodrug at the desired anatomical site either by specific enzymes or through chemical methods and maintain the drug concentration within the therapeutic range for the desired duration of time.10,11 The druggability of some drugs can be markedly increased after being designed as prodrugs. However, the active parent drugs are usually rapidly released from prodrugs prior to reaching tumor sites,12 leading to low activity, toxicity, and undesired side effects.13 Thus, drug release should be considered prior to prodrug design.
The aim of this study was to explain the relationship between the release or hydrolysis of the parent drug from the prodrug and its efficacy, pharmacokinetics, and tissue distribution by studying a series of combretastatin A4 (CA4) prodrugs (Figure 1).
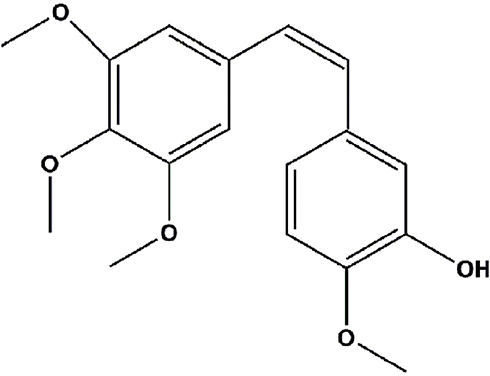 |
Figure 1 Molecular structure of CA4. Abbreviation: CA4, combretastatin A4. |
CA4 is a novel vascular disrupting agent isolated from the South African shrub Combretum caffrum. It is a cis-styrene compound with a similar structure to colchicine,14 and similar action target. CA4 can effectively bind at the colchicine site of tubulin and inhibit polymerization, resulting in the arrest of cell proliferation and death.15–17 Moreover, CA4 – a promising anticancer drug – shows strong cytotoxicity against a broad spectrum of cancer cell lines, including murine melanoma, human breast carcinoma colon, and ovarian cancer cells.18 Unfortunately, it is insoluble in water and has low bioavailability, which impairs its antitumor activity and limits its application in the clinical setting.19 CA4 phosphate (CA4P), the water-soluble prodrug of CA4, was developed to overcome these obstacles. CA4P, which is currently being investigated in phase II/III clinical trials, is clinically more favorable than CA4 owing to its significantly greater solubility in water.20,21 However, the short half-life (T1/2) and wide distribution of the prodrug in vivo may influence its therapeutic efficacy and alter blood flow in numerous normal tissues, which can cause undesirable side effects (eg, cardiotoxicity and ataxia) and limit its clinical application.18,20,22 In addition, CA4P readily isomerizes to the more stable trans-isomer which is inactive under external stimuli (eg, heat or light), resulting in complete abolition of cytotoxicity.23
Lipophilic prodrugs and liposomes are promising strategies for prolonging the effect time in vivo and improving the other shortcomings of CA4. The use of lipophilic prodrugs can improve the absorption, distribution, metabolism, and excretion profiles for parenteral administration; lipophilic therapeutics also exhibit greater half-lives.9,24 Liposomes are nano-agents composed of phospholipids with excellent biodegradability, biocompatibility, and low toxicity; they are an ideal carrier system for drug delivery.25 Especially, liposomes with size ranging from 70 nm to 200 nm are preferentially accumulated in tumors through the enhanced permeability and retention (EPR) effect.26 Therefore, the liposome is a suitable drug delivery for lipophilic CA4 prodrugs.
In this study, we synthesized a series of CA4 prodrugs with fatty chains attached at the 3′-position of the CA4 B-ring varying in length (ie, 6, 10, 14, 16, and 18 carbons) as a model drug, and prepared the prodrugs as liposomes to study the release of the parent drug. To the best of our knowledge, this was the first study to investigate the in vitro release and conversion of CA4-prodrug lipid in plasma with regard to the length of the fatty chain.24,27–29 In addition, the in vitro experiments were designed to investigate the relationship between the release trend and in vivo pharmacokinetics, biodistribution, and antitumor effect. This approach evaluates a suitable preparation for CA4 prodrug and provides a reference for other drugs.
Materials And Methods
Materials
CA4 and CA4P were purchased from Hangzhou Rui Shu Biochemical Co., Ltd (Hangzhou, China). Fatty acyl chloride, stearyl chloride, palmitoyl chloride, myristoyl chloride, decanoyl chloride, hexanoyl chloride, and N, N-diisopropylethylamine were purchased from Adamas Reagent Co., Ltd (Shanghai, China). Egg yolk phospholipids (PC-98T) and DSPE-PEG2000 were obtained from Shanghai Advanced Vehicle Technology Pharmaceutical Co., Ltd (Shanghai, China). Cholesterol, potassium dihydrogen phosphate, and sucrose were sourced from Sinopharm Chemical Reagent Co., Ltd (Shanghai, China). Acetonitrile and methanol were purchased from Merck (Darmstadt, Germany). Dichloromethane and formic acid were provided by Shanghai Titan Scientific Co., Ltd. (Shanghai, China).
Human breast adenocarcinoma MCF-7 cells, mouse sarcoma S180 cells, and human hepatocellular liver carcinoma HepG2 cells were purchased from the Chinese Academy of Sciences Shanghai Institute of Cell Bank (Shanghai, China). Kunming (KM) mice and Sprague–Dawley (SD) rats were provided by Shanghai SLAC Laboratory Animal Co. Ltd (Shanghai, China). All animal protocols complied with the Guide for the Care and Use of Laboratory Animals and Institute of Laboratory Animal Resources and were approved by the Institutional Animal Care and Use Committee of Second Military Medical University (Shanghai, China).
Methods
Synthesis And Identification Of CA4 Prodrugs
CA4 derivatives were synthesized by conjugating the CA4 with varying fatty acid chains of stearyl chloride (CA4-18, (Z)-2-methoxy-5-(3,4, 5-trimethoxystyryl) phenyl stearate), palmitoyl chloride (CA4-16, (Z)-2-methoxy-5-(3, 4, 5-trimethoxystyryl) phenyl palmitate), myristoyl chloride (CA4-14, (Z)-2-methoxy-5-(3, 4, 5-trimethoxystyryl) phenyl myristate), decanoyl chloride (CA4-10, (Z)-2-methoxy-5-(3, 4, 5-trimethoxystyryl) phenyl decanoate), and hexanoyl chloride (CA4-6, (Z)-2-methoxy-5-(3, 4, 5-trimethoxystyryl) phenyl hexanoate). Acylate CA4 prodrugs were synthesized in a one-step process, in which CA4 was reacted with the acyl chloride using N, N-diisopropylethylamine (DIPEA) as acid-binding agent. The synthetic route of CA4 prodrugs is shown in Figure 2. Briefly, CA4 (500 mg, 1.58 mmol) was dissolved in anhydrous dichloromethane (20 mL) under stirring in an ice-water bath. DIPEA (0.306 mL, 1.75 mmol) was added to the reaction mixture. After the completion of addition, the reaction mixture was stirred for an additional 20 min. The fatty acid chain solution (0.642 mL, 1.90 mmol) was added dropwise to the reaction mixture while maintaining the internal temperature of the reaction mixture between 0°C and 5°C. The reaction mixture was stirred for an additional 15 min at 0°C, followed by stirring at room temperature for another 2 h. The reaction mixture was extracted thrice with ethyl acetate (25 mL). The combined organic extracts were washed with brine (10 mL), dried with anhydrous sodium sulfate, and concentrated under reduced pressure. Subsequently, silica gel (2 g) was added and the solvent was evaporated under reduced pressure. A column with ethyl acetate and methanol (20:1) was used to analyze the yield of the eluted prodrugs. Also, the melting points, nuclear magnetic resonance (NMR) hydrogen spectra, NMR carbon spectra, and mass spectrometry data were analyzed to verify the structures of the prodrugs, and high-performance liquid chromatography (HPLC) was applied to evaluate the purity of the prodrugs.
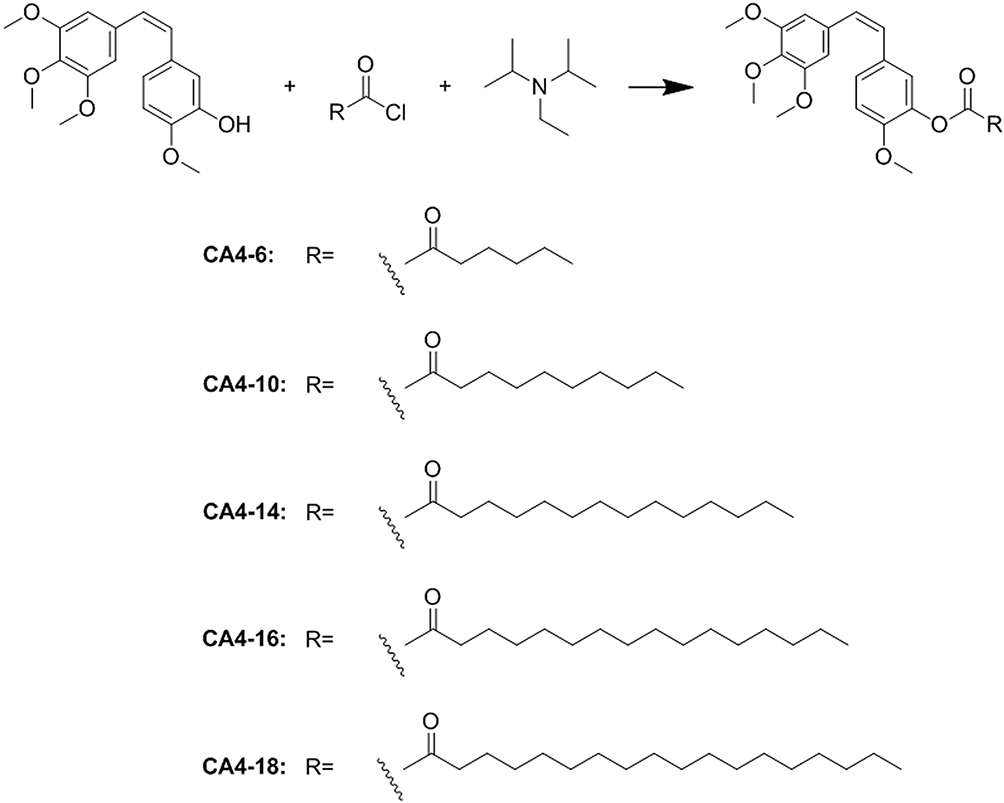 |
Figure 2 Synthesis of CA4 prodrugs with fatty chains attached to the 3′-position of the CA4 B-ring. Abbreviation: CA4, combretastatin A4. |
Preparation Of Liposomes
CA4-18 liposomes (CA4-18-L) were prepared using the conventional thin-film evaporation method.30,31 Briefly, CA4-18 (40 mg), PC-98T (200 mg), DSPE-PEG2000 (40 mg), and cholesterol (20 mg) were co-dissolved in 10 mL dichloromethane in a round-bottom flask. Subsequently, the organic solvent was removed from the mixture at reduced pressure and 45°C using a rotatory evaporator to form a thin lipid film. The film was hydrated with 10 mL deionized water at 70°C. The resulting colloidal solution was sonicated for 3 min using a probe ultrasonic processor (JY92-IIN, Xinzhi, Zhejiang, China). The system was filtered through a 0.22 μm membrane to obtain liposomes with uniform particle size, and sucrose (8%) was added into the liposomes to adjust the osmotic pressure. CA4-6 liposomes (CA4-6-L), CA4-10 liposomes (CA4-10-L), CA4-14 liposomes (CA4-14-L), and CA4-16 liposomes (CA4-16-L) were prepared through the same phospholipid composition and method.
Particle Size, Zeta Potential, And Morphology
The particle size and zeta potential of liposomes were measured using Zetasizer Nano-ZS 90 (Malvern Instruments, Malvern, UK) at 25°C. The surface morphology of liposomes was visualized through transmission electron microscopy (JEM-100CXII, Japan). Prior to measurement, the liposomes were diluted 20-fold in distilled water. Data were obtained from triplicate samples.
Encapsulation Efficiency
The encapsulation efficiency (EE%) was determined using the Sephadex G-50 gel (Pharmcia) mini-column centrifugation-HPLC method.32 A total of 100 μL CA4-18-L (100 μL) was added into the Sephadex G-50 gel mini-column. The first two tubes of eluent were collected and diluted with methanol (5 mL) to destroy the liposome. The solution was filtered through a 0.22 μm membrane and the filtrate was analyzed through HPLC. CA4-6/10/14/16-L were assayed using the same method. The following formula was employed to calculate the EE% of liposomes.
(1)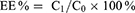
C0 represents the drug content prior to elution and C1 represents the drug content after elution.
Stability Of Prodrugs Loaded In The Liposome
The short-term and accelerated physical stability of the prepared CA4 prodrugs loaded in the liposome were studied by evaluating the particle size and polydispersity index (PDI). In the short-term physical stability, CA4-6/10/14/16/18-L were placed in a desiccator with relative humidity at 50±5% at 25°C for 2 weeks. The samples were analyzed at predetermined time points (days 0, 7, and 14). For the accelerated physical stability, the formulations were centrifuged at 1000 rpm for 30 min, and the particle size and PDI were measured.33
In Vitro Release
In vitro release was evaluated using the bulk-equilibrium reverse dialysis bag technique.33 The acylate CA4 are hydrophobic compounds; hence, the dissolution medium was composed of 20% ethanol, 5% Tween 80, and phosphate-buffered saline (pH 7.4). The medium was incubated in a 37±1°C water bath at a shaking speed of 100 rpm. Each of the CA4-6/10/14/16/18-L was directly placed into 250 mL of stirred sink solution. The dialysis bags (molecular weight cut-off: 12,000 kDa) containing 1 mL of the same sink solution were equilibrated with the sink solution for 12 h prior to conducting the experiments. At predetermined time points (2, 6, 12, 24, and 48 h), the dialysate was removed and replaced with an equal amount of fresh medium to maintain the sink condition. The samples were filtered through 0.22 μm membrane and analyzed through HPLC.
In Vitro Conversion Assay
This experiment was performed to assay the percent conversion of prodrug-liposomes in rat plasma. Blank blood was collected from the orbital vein of SD rats and centrifuged for 10 min at 4,000 rpm to obtain the blank plasma. CA4-6/10/14/16/18-L and CA4P (200 μL) were separately added into 2 mL blank plasma and incubated in a 37±1°C water bath at a shaking speed of 180 rpm. At the designated time points, 100 μL samples were withdrawn and analyzed using HPLC.
In Vitro Cytotoxicity Assay
The Cell Counting Kit-8 assay was used to evaluate the cytotoxicity of CA4-6/10/14/16/18-L, free CA4P solution, and free CA4 in dimethyl sulfoxide solution in human breast adenocarcinoma MCF-7 cells, mouse sarcoma S180 cells, and human hepatocellular liver carcinoma HepG2 cells. The cells were seeded in 96-well plates (5×103 cells per well) in Dulbecco’s modified Eagle’s medium (containing 10% fetal bovine serum, 100 μg/mL streptomycin, and 100 IU/mL penicillin). Subsequently, the cells were incubated for 24 h in a humidified atmosphere of 5% CO2 at 37°C. The medium was replaced with fresh growth medium with different concentrations (0.0025, 0.025, 0.25, 2.5, 25, 125, and 250 μM) of CA4.
After 48 h of incubation, the medium was replaced with fresh growth medium containing 10% Cell Counting Kit-8. Following incubation for 1.5 h at 37°C, the absorbance was measured using a SpectraMaxplus microplate reader (Molecular Devices, Sunnyvale, CA, USA) at 450 nm. The half-maximal inhibitory concentration (IC50) was calculated using the SPSS (19.0) software. Subsequently, the relative cell viability (%) was calculated using the following Equation (2).
(2)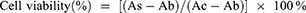
As, Ac, and Ab represent the absorbances of the treatment, control, and blank groups, respectively.
In Vivo Pharmacokinetics
SD rats (male, weight: 180–200 g) were used to evaluate the pharmacokinetics of CA4P and CA4-6/10/18-L, which were used as positive control, as well as the representative of the short-chain acylate CA4, the medium-chain acylate CA4, and the long-chain acylate CA4. SD rats were fasted for 12 h prior to tail vein injection at drug-equivalent doses of 28 mg/kg CA4P. Blood samples were collected from the orbital vein at 2 min, 5 min,10 min, 15 min, 20 min, 30 min, 45 min, 1 h, 1.5 h, 2 h, 4 h, 6 h, and 10 h after injection. The blood samples were put in heparinized vials and centrifuged for 10 min at 4,000 rpm to obtain the plasma samples. Subsequently, the plasma samples were stored at −20°C until HPLC analysis.
In Vivo Antitumor Studies
S180 cells (2×106 per mouse) were intraperitoneally inoculated into the KM mice to evaluate the therapeutic antitumor effects. The tumor volume was measured on days 1, 3, 5, 7, and 9. The volume was calculated using Equation (2). Nine days later, the mice were sacrificed through cervical dislocation, and the S180 cells in mouse ascites were excised and diluted to a cell suspension of 106 cells/mL using sterile saline. Subsequently, 0.2 mL cell suspension was subcutaneously injected in the right flank of male KM mice (aged 6–8 weeks). Post inoculation, tumor-bearing KM mice were randomly divided into four groups (n=9), namely CA4P, CA4-10-L, CA4-18-L, and normal saline. CA4P was dissolved in sterile saline prior to the experiment. Saline, CA4P, CA4-10-L, and CA4-18-L were intravenously administered through the tail vein after 24 h of inoculation at drug-equivalent doses of 20 mg/kg of CA4. The drug was administered for 7 consecutive days. Animal weight was monitored every 2 days, and the mice were sacrificed through cervical dislocation after drug withdrawal for 2 days. The excised tumors were weighed and the tumor inhibition rate (TIR) was determined using Equation (3).
(3)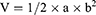
V, a, and b represent the tumor volume, longer tumor size, and shorter tumor size, respectively.
(4)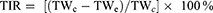
TWc and TWe represent the tumor weights of the saline and experimental groups, respectively.
In Vivo Biodistribution
S180 tumor-bearing mice were used to investigate the biodistribution of CA4P, CA4-10-L, and CA4-18-L. The animal models were prepared as described in the section titled “In vivo antitumor studies”. Tumor-bearing mice for 1 week were randomly divided into three groups (n=24) and received intravenous CA4P, CA4-10-L, and CA4-18-L at drug-equivalent doses of 28 mg/kg CA4P. At the designated time points (2 min, 15 min, 30 min, 1 h, 2 h, 4 h, 8 h, and 24 h post injection; three mice per time point), tumors and major organs (hearts, kidneys, livers, spleens, and lungs) were excised and washed with saline. Subsequently, 0.1 g of each organ was homogenized with saline at a ratio of 1:4 (weight:volume) and the CA4 concentration in the different groups was assayed using HPLC.
Statistical Analysis
Data are presented as mean ± standard deviation. Student’s t-test was used for comparisons between two groups, while one-way analysis of variance analysis (Student–Newman–Keuls q) was used for comparisons between multiple groups. All analyses were performed using the SPSS (19.0) (IBM Corporation, Armonk, NY, USA). A P<0.05 denoted statistical significance, while P<0.01 and P<0.001 denoted highly significant differences.
Results And Discussion
Synthesis And Identification Of CA4 Prodrugs
The physical properties of the synthesized CA4-6/10/14/16/18 are shown in Table 1. The results of mass spectrometry, proton-NMR, and carbon-13-NMR analyses for the prodrugs are illustrated in supplementary information (Tables 1S–5S in supplementary). The data indicated that CA4 was successfully modified by the 18-carbon chain, 16-carbon chain, 10-carbon chain, and six-carbon chain to CA4-6/10/14/16/18.
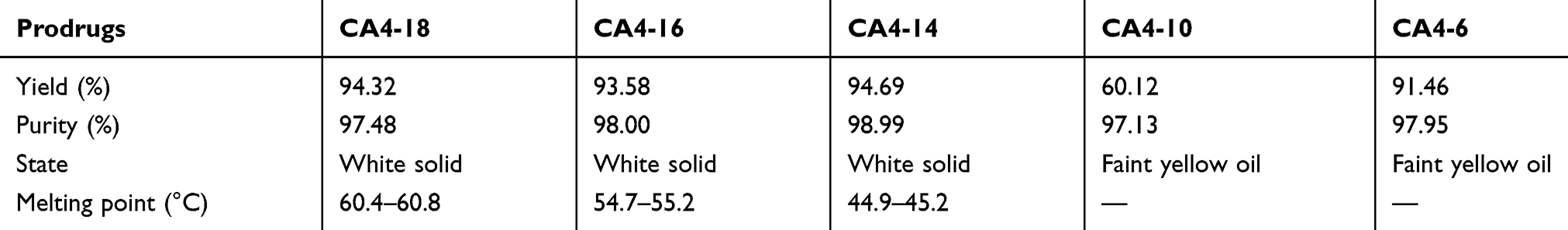 |
Table 1 Physical Properties Of The Synthesized CA4-18, CA4-16, CA4-14, CA4-10, And CA4-6 |
Preparation And Characterization Of Liposomes
After modification with fatty acid, acylate CA4 exhibited better lipid solubility than CA4. Thus, the thin-film evaporation method was used to prepare liposomes, and the drugs were encapsulated in the liposome bilayer. The size distribution, zeta potentials, and encapsulation efficiency are presented in Table 2. CA4-6/10/14/16/18-L all demonstrated high encapsulation efficiency (>95%). Hence, this design successfully increases the encapsulation efficiency of drugs compared with that noted for CA4 loaded in nano-agents.34 The particle sizes of liposomes were 75–120 nm with an acceptable PDI.35,36 Nanoparticles with sizes ranging 70–200 nm demonstrated longer circulation time than free drug, and can accumulate in pathological sites with affected and leaky vasculature (ie, tumors, inflammations, and infarcted areas) via the EPR effect.37 In addition, the surface charge can influence the stability of the liposome.38 CA4-6/10/14/16/18-L had a negative charge (approximately −50 mV), which indicated good stability of the liposomes. Furthermore, the shapes of liposomes were spherical, and uniform as shown in Figure 3.
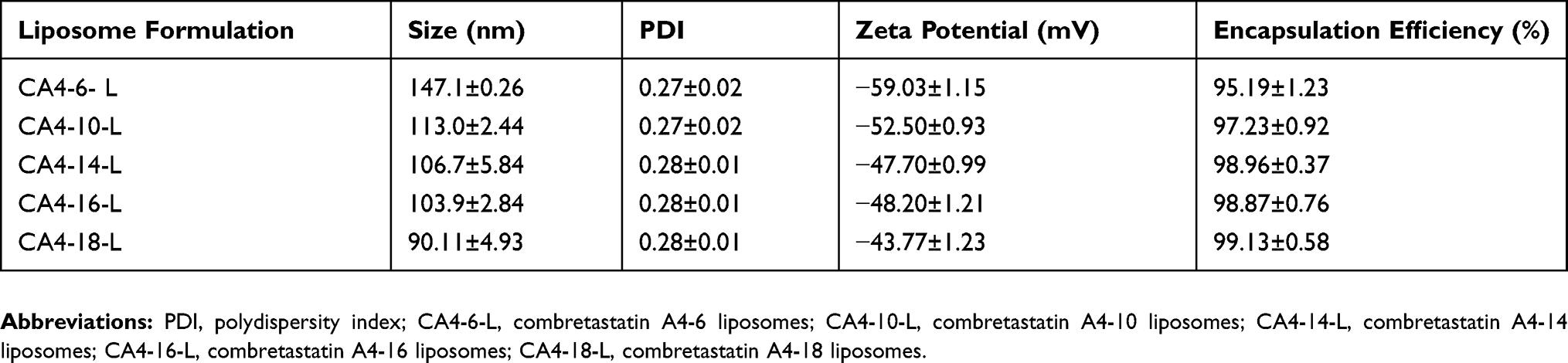 |
Table 2 Sizes, Zeta Potentials, And Encapsulation Efficiencies Of Liposomes |
 |
Figure 3 TEM micrograph of CA4-L (A), CA4-6-L (B), CA4-10-L (C), CA4-14-L (D), CA4-16-L (E), and CA4-18-L (F), respectively. Abbreviations: TEM, transmission electron microscopy; CA4-L, combretastatin A4-liposome; CA4-6-L, combretastatin A4-6 liposomes; CA4-10-L, combretastatin A4-10 liposomes; CA4-14-L, combretastatin A4-14 liposomes; CA4-16-L, combretastatin A4-16 liposomes; CA4-18-L, combretastatin A4-18 liposomes. |
Stability Of Prodrugs Loaded In Liposomes
The short-term stability of prodrugs loaded in liposomes was evaluated for 2 weeks. The particle size and PDI at days 0, 7, and 14 are presented in Table 3. The formulations were homogeneous and there was no significant difference (P<0.05) in the particle size and PDI at days 0, 7, and 14, and after centrifugation. These results indicated that the CA4-6/10/14/16/18-L were stable.39
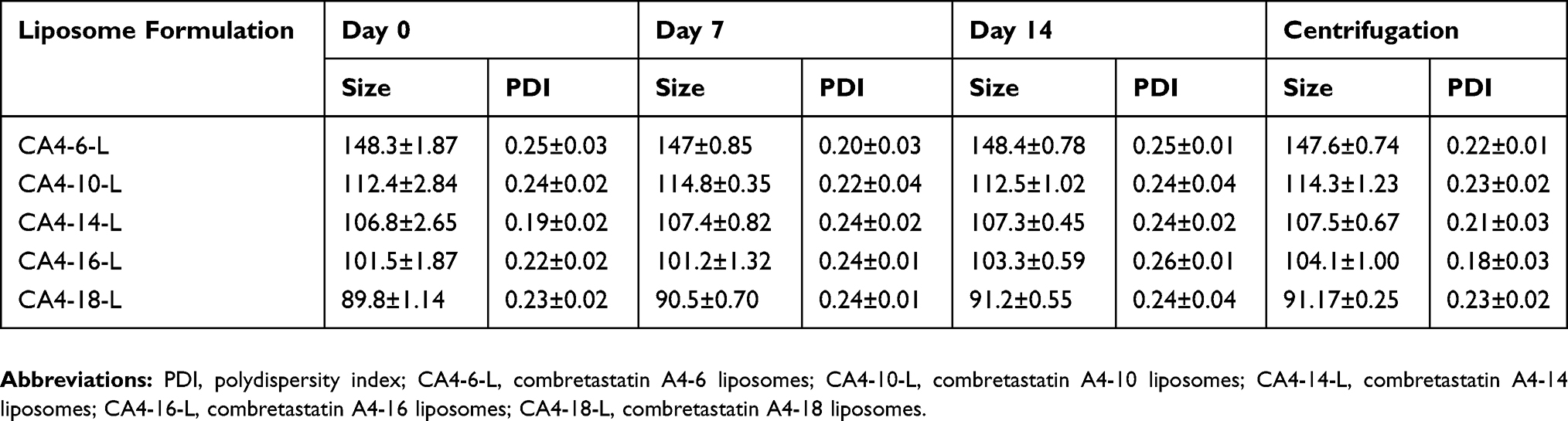 |
Table 3 Sizes And PDI Of Liposomes In The Stability Experiments |
Drug Release And Conversion Behavior Assay
The release profiles of CA4-6/10/14/16/18 from CA4-6/10/14/16/18-L are represented in Figure 4A. As shown in the graph, there was no burst release during the first 4 h, and all drugs were released slowly. After 48 h, CA4-6 was almost completely released from CA4-6-L, while only 46.34%, 39.47%, 33.15%, and 25.96% of CA4-10/14/16/18, respectively, were released from liposomes. Obviously, the release rate was as follows: CA4-6-L>CA4-10-L>CA4-14-L>CA4-16-L>CA4-18-L. The results suggest that longer modified acyl chains for CA4 were associated with slower release rates in vitro. The results are consistent with those previously reported.29 This may be attributed to the increased lipid solubility of the obtained compound in parallel with the increase in the modified fatty acid chain for CA4. Of note, higher lipid solubility indicates more challenging separation from the liposome. This may indicate that CA4-18-L can remain longer in vivo than other liposomes.
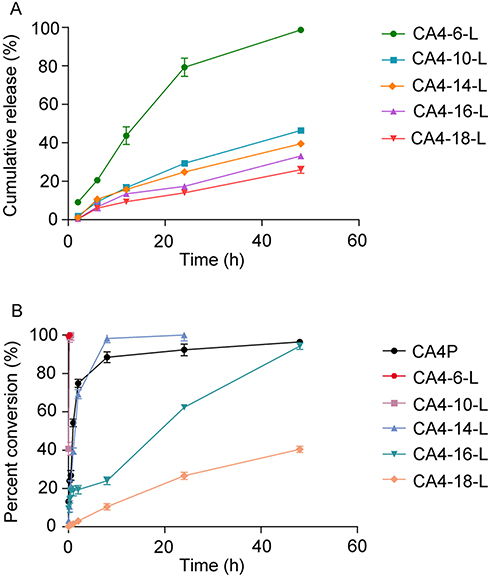 |
Figure 4 (A) Cumulative release profile of CA4-6, CA4-10, CA4-14, CA4-16, and CA4-18 from CA4-6-L, CA4-10-L, CA4-14-L, CA4-16-L, and CA4-18-L, respectively. (B) The percent conversion of CA4P, CA4-6-L, CA4-10-L, CA4-14-L, CA4-16-L, and CA4-18-L after incubation with the rat plasma (n=3). Abbreviations: CA4, combretastatin A4; CA4P, combretastatin A4 phosphate; CA4-6-L, combretastatin A4-6 liposomes; CA4-10-L, combretastatin A4-10 liposomes; CA4-14-L, combretastatin A4-14 liposomes; CA4-16-L, combretastatin A4-16 liposomes; CA4-18-L, combretastatin A4-18 liposomes. |
CA4P and CA4-6/10/14/16/18 are all prodrugs of CA4, which need to be converted into the active compound CA4. Hence, we evaluated the percent conversion of CA4P and CA4-6/10/14/16/18-L in rat plasma. As shown in Figure 4B and supplementary data (Figure 1S and Table 6S in supplementary), CA4-6-L and CA4-10-L were almost completely converted into CA4 after incubation in plasma for 15 min, whereas merely 40.41% of the CA4-18-L was converted after 48 h. This experiment indicates that all could be converted into CA4 in vivo. The rate of conversion in rat plasma was as follows: CA4-6-L>CA4-10-L>CA4-14-L>CA4P>CA4-16-L>CA4-18-L. These results were consistent with those of the in vitro release, indicating that longer modified acyl chains for CA4 were linked to slower conversion rates. Thus, in agreement with the above hypothesis, CA4-18-L may remain longer in vivo than other liposomes.
In Vitro Cytotoxicity
The cytotoxicity results for CA4-6/10/14/16/18-L, CA4P solution, and free CA4 solution are presented in Table 4 and Figure 5. After 48 h, the IC50 values of free CA4 against MCF-7, S180, and HepG2 cells were 0.004±0.002 μM, <0.001 μM, and 0.064±0.006 μM, respectively. These findings indicated that S180 cells are more sensitive to CA4 than MCF-7 and HepG2 cells. Obviously, all the CA4 derivatives were less active than CA4 against MCF-7, S180, and HepG2 cells. This may be attributed to the fact that prodrug derivatives are not activated until they are hydrolyzed into CA4. The IC50 values were as follows: CA4<CA4-6-L<CA4-10-L<CA4-14-L<CA4-16-L<CA4-18-L. Figure 5A–C show the relative cell viability (%) of MCF-7, S180, and HepG2 at the CA4 concentration of 0.0025 μM. We observed a trend of increase in relative cell viability (%) with a decrease in the modified acyl chain. The results suggest that the shorter-chain acylate CA4 exhibited greater cytotoxicity than longer-chain acylate CA4 at the same concentration. This may be due to the easier release from the liposome and hydrolysis into the active parent drug CA4 of the former versus the latter. The data indicated that the release of the active drug could significantly influence the in vitro cytotoxicity. The results were consistent with those of the prodrug release and conversion studies, demonstrating that the slower rate of drug release and conversion was associated with lower cytotoxicity.
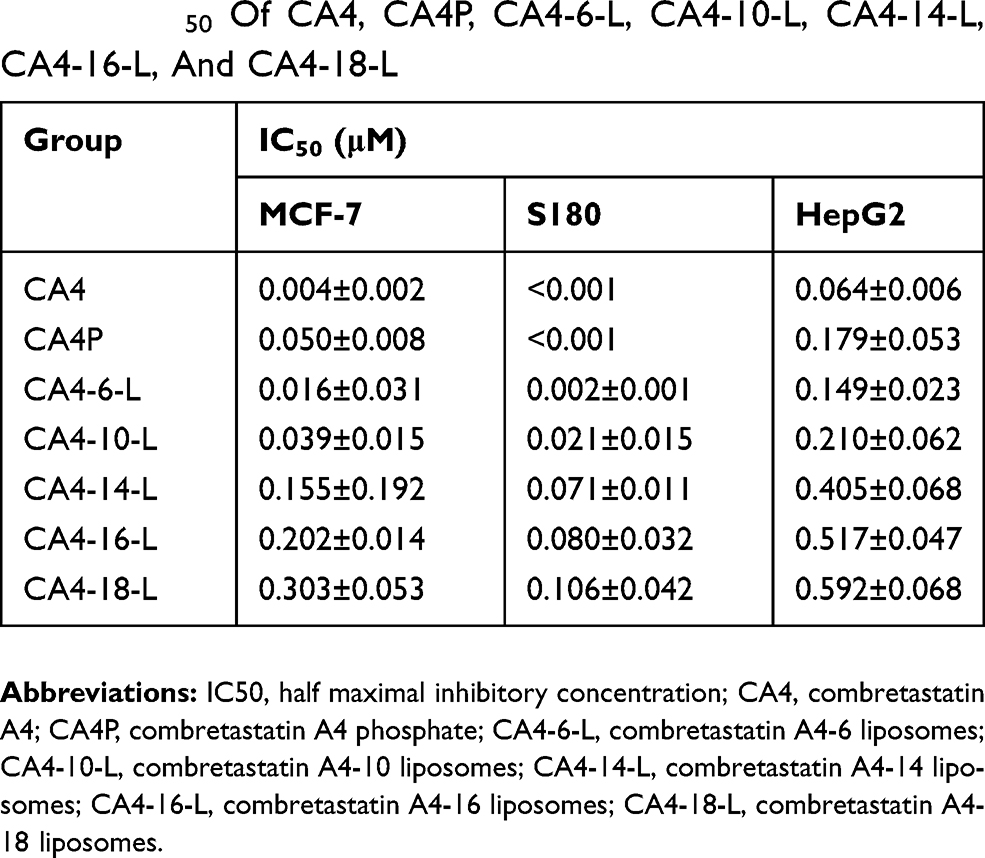 |
Table 4 IC50 Of CA4, CA4P, CA4-6-L, CA4-10-L, CA4-14-L, CA4-16-L, And CA4-18-L |
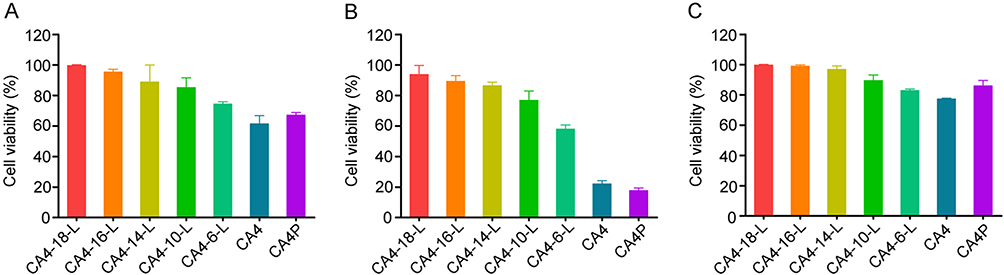 |
Figure 5 Cell viability of MCF-7 (A), S180 (B), and HepG2 (C) after treatment with CA4, CA4P, CA4-6-L, CA4-10-L, CA4-14-L, CA4-16-L, and CA4-18-L for 48 h. Data are presented as mean±SD (n=3). Abbreviations: CA4, combretastatin A4; CA4P, combretastatin A4 phosphate; CA4-6-L, combretastatin A4-6 liposomes; CA4-10-L, combretastatin A4-10 liposomes; CA4-14-L, combretastatin A4-14 liposomes; CA4-16-L, combretastatin A4-16 liposomes; CA4-18-L, combretastatin A4-18 liposomes. |
In Vivo Pharmacokinetics
Figure 6 shows the plasma concentration–time curve of drugs after treatment with CA4P and CA4-6/10/18-L. The results showed that the conversion of the short-chain CA4-6-L and the medium-chain CA4-10-L into CA4 was excessively rapid to be detected. However, the long-chain CA4-18-L remained longer in vivo and was slowly converted into CA4 in comparison with the other groups. Following injection (2 h), CA4P could not be detected, whereas CA4-18-L with slow drug release could be detected until 10 h later (Figure 6A). The obtained results were consistent with those of the in vitro drug release and conversion behavior studies, which indicated that slow drug release and conversion could induce a longer circulation time. Compared with CA4P, the drug - time area under curve (AUC) of CA4-18 was increased by 43-fold (P<0.001), and the clearance (CL) and V of CA4-18-L were markedly lower (P<0.01) (Table 5). As mentioned above, CA4P is linked to undesired side effects, including the relatively short T1/2 and wide distribution of the drug in vivo. The use of CA4-18-L significantly improves this pharmacokinetic parameters and characteristics.
 |
Table 5 Pharmacokinetic Parameters Of CA4P And CA4-18 After Intravenous Injection Of CA4P, CA4-6-L, CA4-10-L, And CA4-18-L To SD Rats Via The Tail Vein |
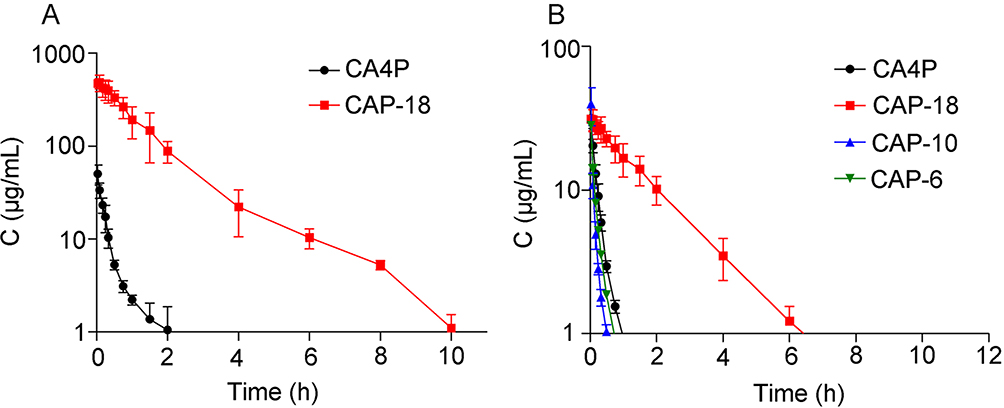 |
Figure 6 Plasma concentration–time curve of CA4P and CA4-18-L (A) and CA4 converted from CA4P, CA4-6-L, CA4-10-L, and CA4-18-L (B) after intravenous injection to SD rats via the tail vein (n=6). Abbreviations: CA4, combretastatin A4; CA4P, combretastatin A4 phosphate; CA4-6-L, combretastatin A4-6 liposomes; CA4-10-L, combretastatin A4-10 liposomes; CA4-18-L, combretastatin A4-18 liposomes. |
Figure 6B and Table 6 show the circulation time of CA4 converted from CA4-18-L, demonstrating a remarkable improvement compared with the other groups. Moreover, the AUC of the CA4-18-L group was 8.35-fold (P<0.001), 15.94-fold (P<0.001), and 15.64-fold (P<0.001) than those of the CA4P, CA4-6-L, and CA4-10-L groups, respectively. The CL and V of the CA4-18-L group were markedly lower than those of the other groups (P<0.01). The T1/2 and mean residence time were significantly prolonged compared with those of the other groups (P<0.05), including the long average time of CA4 residence in the body.40 The results revealed that slow release can increase the T1/2, mean residence time, and AUC0-τ, and decrease CL and V of the parent drug. This indicates that the slower rates of drug release and conversion induce a significantly longer circulation time in vivo.
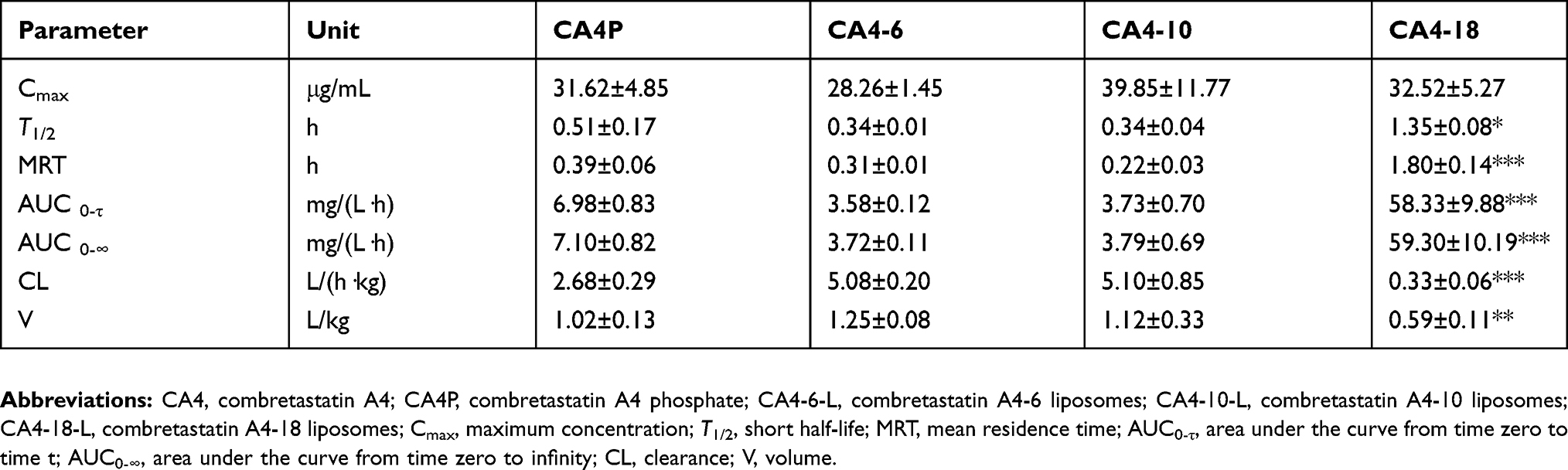 |
Table 6 Pharmacokinetic Parameters Of CA4 Converted From CA4P, CA4-6, CA4-10, And CA4-18 After Intravenous Injection Of CA4P, CA4-6-L, CA4-10-L, And CA4-18-L To SD Rats Via The Tail Vein |
In Vivo Antitumor Studies
As shown in Table 4, S180 cells are sensitive to CA4 with the lowest IC50. Hence, S180 tumor-bearing mice were established to evaluate the in vivo antitumor effects of the different CA4 derivatives. As demonstrated in the pharmacokinetics studies, there was no statistically significant difference in CA4-6-L and CA4-10-L after administration. Therefore, CA4P, CA4-10-L, and CA4-18-L were evaluated in this study. Table 7 displays the dose and TIR. The TIR of CA4P, CA4-10-L, and CA4-18-L (equal dosage) were 57.87%, 43.06%, and 94.44%, respectively. Figure 7 shows the excised S180 tumor (Figure 7A) and tumor weight (Figure 7B) on the last day of the experiment, as well as the tumor growth curves (Figure 7C). As shown in Figure 7 and Table 7, all the treatment groups could significantly inhibit tumor growth (P<0.001) compared with the negative control group (saline). The CA4-18-L group exhibited favorable tumor suppression compared with the CA4-10-L and CA4P groups (both P<0.001). There was no statistically significant difference between the CA4-10-L and CA4P groups in terms of tumor suppression (P>0.05). As shown in Table 7, it was obvious that the slow-drug-release CA4-18-L exerted a greater antitumor effect than the fast-drug-release CA4-10-L.
 |
Table 7 Antitumor Effects Of CA4P, CA4-10-L, And CA4-18-L On S180 Tumor-Bearing Mice |
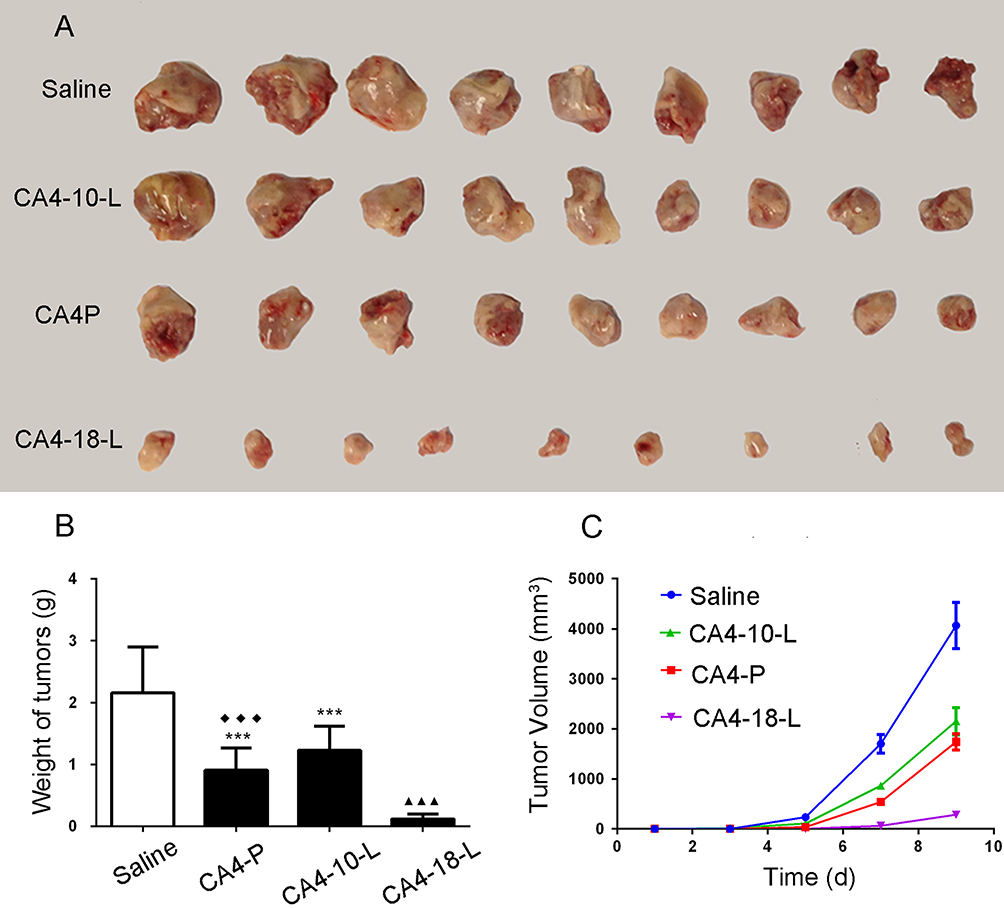 |
Figure 7 Antitumor activity of CA4P, CA4-10-L, and CA4-18-L in S180 tumor-bearing mice. (A) Excised S180 tumor. (B) Tumor weights and volumes (C) of different treatment groups. Data are presented as mean±SD (n=9). ***P<0.001 compared with saline. ▲▲▲P<0.001 compared with CA4P. ◆◆◆P<0.001 compared with CA4-18-L. Abbreviations: CA4P, combretastatin A4 phosphate; CA4-10-L, combretastatin A4-10 liposomes; CA4-18-L, combretastatin A4-18 liposomes. |
The results indicate that the rate of drug release and hydrolysis are related to the antitumor effect in vitro and in vivo. As the modified acyl chain for CA4 increases, the rates of prodrug release and the activated CA4 hydrolyzed from the prodrug derivatives are reduced. Therefore, the longer-chain acylate CA4 exhibits lower cytotoxicity in vitro. On the contrary, they exert a greater antitumor effect in vivo, which may be attributed to the long-chain acylate CA4 was lipophilic compound and hydrolyzed slowly and has a longer circulation time in vivo. The short-chain acylate CA4 would be hydrolyzed into CA4 immediately after injection and remain as CA4 with lower lipid solubility, which is easy to eliminate from the body. Thus, the slow rate of drug release and conversion can prolong the circulation time in vivo, and consequently enhance the antitumor effect in vivo.
In Vivo Biodistribution
S180 tumor-bearing mice were used to investigate the biodistribution of CA4P, CA4-10-L, and CA4-18-L. Figure 8 shows the tissue distributions of CA4P in the CA4P group and CA4 converted from CA4P, CA4-10-L, and CA4-18-L at different times post administration. Figure 8A shows that CA4P could be detected in the heart, liver, spleen, lung, kidney, and tumor, with a higher concentration recorded in the liver, spleen, and kidney. The results indicate that CA4P has a wide distribution in vivo and higher reticuloendothelial system uptake. The high level of CA4P in the kidney implied that CA4P can be rapidly cleared from the body.41 Not surprisingly, CA4-10 could not be detected in the tissue due to the fast release and conversion. Of note, CA4-18 was detected only in the liver, which strongly correlated with its lower apparent volume of distribution (0.08±0.02 L/kg). Figure 8B shows the concentration of CA4 converted from CA4-18-L. CA4 was mostly distributed in the liver, spleen, and lung during the first 2 h, while the concentration of CA4 in the tumor demonstrated an increasing trend. CA4 had a higher concentration in the tumor at approximately 2 h, which was perhaps due to the slow drug release and the EPR effect of the liposome.
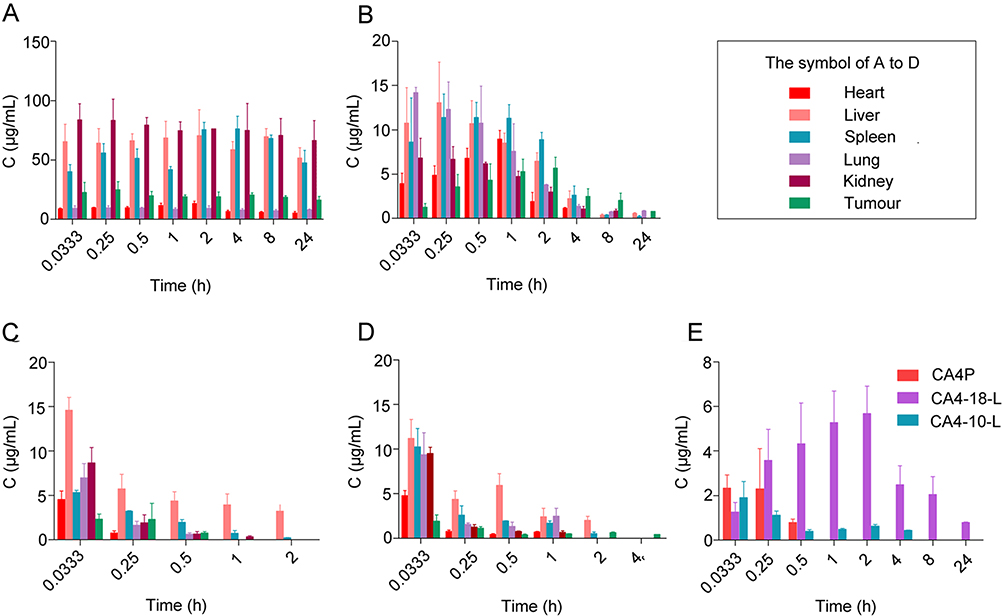 |
Figure 8 Concentration of CA4P, CA4-10-L, and CA4-18-L in tissues of S180 tumor-bearing mice after intravenous administration (n=3). (A) Concentration of CA4P. (B) Concentration of CA4 converted from CA4-18-L. (C) Concentration of CA4 converted from CA4P. (D) Concentration of CA4 converted from CA4-10-L. (E) Concentration of CA4 converted from CA4P, CA4-18-L, and CA4-10-L in tumor. Abbreviations: CA4P, combretastatin A4 phosphate; CA4-10-L, combretastatin A4-10 liposomes; CA4-18-L, combretastatin A4-18 liposomes. |
Figure 8C and D show the similar distribution of CA4 converted from CA4P and CA4-10-L. CA4 was mostly distributed in the liver and could be rapidly cleared from the body. Figure 8E shows the concentration of CA4 converted from CA4P, CA4-10-L, and CA4-18-L in tumor tissue at different time points. The EPR effect of the long-circulating liposome may explain the slow elimination of CA4 converted from CA4-10-L and CA4-18-L versus that of CA4P in tumor tissue.42 Furthermore, the AUC tumor value of CA4 converted from CA4-18-L (49.36±11.10 μg/(g·h)) was significantly higher than that obtained for the CA4-10-L group (2.24±0.41 μg/(g·h)) (P<0.001). This finding showed that the slow drug release increased the accumulation of active drug in tumor tissue and enhanced the antitumor effect.41 Overall, these results demonstrated that CA4-18-L can increase the concentration of CA4 in tumor tissue at the equal dosage with the other groups owing to the slow drug release.
CA4P, a vascular disrupting agent, effectively destroys the structural integrity of tumor vasculature.43 However, its poor solubility in water limits the clinical application of this agent. CA4P was developed as the water-soluble prodrug of CA4 and is currently being investigated in phase II/III clinical trials.21 Unfortunately, the limitations of this prodrug (ie, short T1/2 and undesirable side effects) in normal organs need to be resolved.18,20,22 The results of this research study indicated that the lipophilic prodrug could change the tissue distribution of the drug, prolong the biological T1/2, and improve the antitumor effect. Moreover, certain lipophilic prodrugs (eg, dexamethasone palmitate injection and clindamycin palmitate hydrochloride formulations) are commercially available.44,45 It can be concluded that the development of lipophilic prodrugs provides a promising platform for the use of CA4, and the CA4 lipophilic prodrug loaded in liposome carries potential for clinical application.
Conclusion
In this study, a series of CA4 prodrugs were used as model drugs to determine the influence of drug release on the pharmacokinetics, biodistribution, and antitumor effect. Lipophilic acylate CA4 prodrugs were synthesized and prepared as liposomes with high EE% (ie, small particle size and negative charge of approximately −50 mV). The results of the prodrug release and conversion behavior studies indicated that longer modified acyl chains for CA4 were associated with slower rates of drug release and conversion. The results of the in vitro cytotoxicity analysis on MCF-7, S180, and HepG2 cells indicate that CA4-18-L with the longest carbon chain exhibited less cytotoxicity than the other acylate CA4 prodrugs. On the contrary, in vivo, the long-chain acylate CA4 was more effective than the other acylate CA4 prodrugs. The in vitro and in vivo findings further support the hypothesis that the parent drug released or hydrolyzed from the prodrug could influence its efficacy, pharmacokinetics, and tissue distribution. Slow drug release prolongs the circulation time and increases the concentration of the drug in the tumor tissue. These effects play a critical role in enhancing antitumor efficacy. Particularly, it was found that CA4-18-L exhibited higher antitumor efficacy than CA4P which is currently being investigated in clinical trials. In summary, the synthesis of prodrugs with poor solubility and high toxicity is an effective strategy to improve their antitumor effect.
Acknowledgments
This study was supported by the National Natural Science Foundation of China (No. 81772749), the Shanghai Rising-Star Program (No. 18QB1400400), Shanghai Qingpu District Industry-University-Research Cooperation Development Fund Project (QIUR 2019-5), and Shanghai Science and Technology Project of Little Giant (1902HX76600).
Disclosure
The authors report no conflicts of interest in this work.
References
1. Bray F, Ferlay J, Soerjomataram I, Siegel RL, Torre LA, Jemal A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin. 2018;68(6):394–424. doi:10.3322/caac.21492
2. Siegel RL, Miller KD, Jemal A. Cancer statistics, 2019. CA Cancer J Clin. 2019;69(1):7–34. doi:10.3322/caac.21551
3. Wang Y, Cheetham AG, Angacian G, Su H, Xie L, Cui H. Peptide-drug conjugates as effective prodrug strategies for targeted delivery. Adv Drug Deliv Rev. 2017;110-111:112-126.
4. Fang JY, Al-Suwayeh SA. Nanoparticles as delivery carriers for anticancer prodrugs. Expert Opin Drug Deliv. 2012;9(6):657–669. doi:10.1517/17425247.2012.679927
5. Vig BS, Huttunen KM, Laine K, Rautio J. Amino acids as promoieties in prodrug design and development. Adv Drug Deliv Rev. 2013;65(10):1370–1385. doi:10.1016/j.addr.2012.10.001
6. Markovic M, Ben-Shabat S, Keinan S, Aponick A, Zimmermann EM, Dahan A. Lipidic prodrug approach for improved oral drug delivery and therapy. Med Res Rev. 2019;39(2):579–607.
7. Bala V, Rao S, Boyd BJ, Prestidge CA. Prodrug and nanomedicine approaches for the delivery of the camptothecin analogue SN38. J Control Release. 2013;172(1):48–61. doi:10.1016/j.jconrel.2013.07.022
8. Ulmschneider MB, Searson PC. Mathematical models of the steps involved in the systemic delivery of a chemotherapeutic to a solid tumor: from circulation to survival. J Control Release. 2015;212:78–84. doi:10.1016/j.jconrel.2015.06.026
9. Hinna AH, Hupfeld S, Kuntsche J, Bauer-Brandl A, Brandl M. Mechanism and kinetics of the loss of poorly soluble drugs from liposomal carriers studied by a novel flow field-flow fractionation-based drug release-/transfer-assay. J Control Release. 2016;232:228–237. doi:10.1016/j.jconrel.2016.04.031
10. Tam YT, Shin DH, Chen KE, Kwon GS. Poly(ethylene glycol)-block-poly(d,l-lactic acid) micelles containing oligo(lactic acid)-paclitaxel prodrug: in vivo conversion and antitumor efficacy. J Control Release. 2019;298:186–193. doi:10.1016/j.jconrel.2019.02.017
11. Böttger R, Knappe D, Hoffmann R. Readily adaptable release kinetics of prodrugs using protease-dependent reversible PEGylation. J Control Release. 2016;230:88–94. doi:10.1016/j.jconrel.2016.04.010
12. Arosio D, Casagrande C. Advancement in integrin facilitated drug delivery. Adv Drug Deliv Rev. 2016;97:111–143. doi:10.1016/j.addr.2015.12.001
13. Mura S, Bui DT, Couvreur P, Nicolas J. Lipid prodrug nanocarriers in cancer therapy. J Control Release. 2015;208:25–41. doi:10.1016/j.jconrel.2015.01.021
14. Han F, Wang P, Zhang W, et al. CA-1H, a novel oxazole bearing analogue of combretastatin A-4, disrupts the tumor vasculatures and inhibits the tumor growth via inhibiting tubulin polymerization. Biomed Pharmacother. 2016;80:151–161. doi:10.1016/j.biopha.2016.03.016
15. Colliez F, Fruytier AC, Magat J, et al. Monitoring Combretastatin A4-induced tumor hypoxia and hemodynamic changes using endogenous MR contrast and DCE-MRI. Magn Reson Med. 2016;75(2):866–872. doi:10.1002/mrm.25642
16. Daswani VP, Ayesa U, Venegas B, Chong. PL. Concentration-induced J-aggregate formation causes a biphasic change in the release of trans-combretastatin A4 disodium phosphate from archaeosomes and the subsequent cytotoxicity on mammary cancer cells. Mol Pharm. 2015;12(10):3724–3734. doi:10.1021/acs.molpharmaceut.5b00500
17. Su M, Huang J, Liu S, et al. The anti-angiogenic effect and novel mechanisms of action of Combretastatin A-4. Sci Rep. 2016;6:28139. doi:10.1038/srep28139
18. Liu H, Wang J. Loading IR820 using multifunctional dendrimers with enhanced stability and specificity. Pharmaceutics. 2018;10(3):77. doi:10.3390/pharmaceutics10030077
19. Mahal K, Biersack B, Caysa H, Schobert R, Mueller. T. Combretastatin A-4 derived imidazoles show cytotoxic, antivascular, and antimetastatic effects based on cytoskeletal reorganisation. Invest New Drugs. 2015;33(3):541–554. doi:10.1007/s10637-015-0215-9
20. Venegas B, Zhu W, Haloupek NB, et al. Cholesterol superlattice modulates CA4P release from liposomes and CA4P cytotoxicity on mammary cancer cells. Biophys J. 2012;102(9):2086–2094. doi:10.1016/j.bpj.2012.03.063
21. Zheng S, Zhong Q, Mottamal M, et al. Design, synthesis, and biological evaluation of novel pyridine-bridged analogues of combretastatin-A4 as anticancer agents. J Med Chem. 2014;57(8):3369–3381. doi:10.1021/jm500002k
22. Li G, Song YZ, Huang ZJ, Chen K, Chen DW, Deng YH. Novel, nano-sized, liposome-encapsulated polyamidoamine dendrimer derivatives facilitate tumour targeting by overcoming the polyethylene glycol dilemma and integrin saturation obstacle. J Drug Target. 2017;25(8):734–746. doi:10.1080/1061186X.2017.1324860
23. Xu Q, Qi H, Sun M, et al. Synthesis and biological evaluation of 3-Alkyl-1,5-Diaryl-1H-Pyrazoles as rigid analogues of Combretastatin A-4 with potent antiproliferative activity. PLoS One. 2015;10(6):e0128710. doi:10.1371/journal.pone.0128710
24. Lorscheider M, Tsapis N, Simón-Vázquez R, et al. Nanoscale lipophilic prodrugs of dexamethasone with enhanced pharmacokinetics. Mol Pharm. 2019;16(7):2999–3010. doi:10.1021/acs.molpharmaceut.9b00237
25. Allen TM, Cullis PR. Liposomal drug delivery systems: from concept to clinical applications. Adv Drug Deliv Rev. 2013;65(1):36–48. doi:10.1016/j.addr.2012.09.037
26. Zhang C, An T, Wang D, et al. Stepwise pH-responsive nanoparticles containing charge-reversible pullulan-based shells and poly(β-amino ester)/poly(lactic-co-glycolic acid) cores as carriers of anticancer drugs for combination therapy on hepatocellular carcinoma. J Control Release. 2016;226:193–204. doi:10.1016/j.jconrel.2016.02.030
27. Hu L, Luo X, Zhou S, et al. Neutrophil-mediated delivery of dexamethasone palmitate-loaded liposomes decorated with a sialic acid conjugate for rheumatoid arthritis treatment. Pharm Res. 2019;36(7):97. doi:10.1007/s11095-019-2609-4
28. N’Guessan A, Fattal E, Chapron D, Gueutin C, Koffi A, Tsapis N. Dexamethasone palmitate large porous particles: a controlled release formulation for lung delivery of corticosteroids. Eur J Pharm Sci. 2018;113:185–192. doi:10.1016/j.ejps.2017.09.013
29. Gaekens T, Guillaume M, Borghys H, et al. Lipophilic nalmefene prodrugs to achieve a one-month sustained release. J Control Release. 2016;232:196–202. doi:10.1016/j.jconrel.2016.04.029
30. Terakawa MS, Lin Y, Kinoshita M, et al. Impact of membrane curvature on amyloid aggregation. Biochim Biophys Acta Biomembr. 2018;1860:1741–1764. doi:10.1016/j.bbamem.2018.04.012
31. Zhang W, Song Y, Eldi P, et al. Targeting prostate cancer cells with hybrid elastin-like polypeptide/liposome nanoparticles. Int J Nanomedicine. 2018;13:293–305. doi:10.2147/IJN.S152485
32. Mishra D, Garg M, Dubey V, Jain S, Jain NK. Elastic liposomes mediated transdermal delivery of an anti-hypertensive agent: propranolol hydrochloride. J Pharm Sci. 2007;96(1):145–155. doi:10.1002/jps.20737
33. Gu Y, Tang X, Yang M, Yang D, Liu J. Transdermal drug delivery of triptolide-loaded nanostructured lipid carriers: preparation, pharmacokinetic, and evaluation for rheumatoid arthritis. Int J Pharm. 2019;554:235–244. doi:10.1016/j.ijpharm.2018.11.024
34. Su T, Long Y, Deng C, et al. Construction of a two-in-one liposomal system (TWOLips) for tumor-targeted combination therapy. Int J Pharm. 2014;476:241–252. doi:10.1016/j.ijpharm.2014.09.055
35. Monteiro LOF, Fernandes RS, Castro L, et al. Paclitaxel-loaded folate-coated pH-sensitive liposomes enhance cellular uptake and antitumor activity. Mol Pharm. 2019;16:3477–3488. doi:10.1021/acs.molpharmaceut.9b00329
36. Lakkadwala S, Dos Santos Rodrigues B, Sun C, Singh J. Dual functionalized liposomes for efficient co-delivery of anti-cancer chemotherapeutics for the treatment of glioblastoma. J Control Release. 2019;307:247–260. doi:10.1016/j.jconrel.2019.06.033
37. Ravar F, Saadat E, Gholami M, et al. Hyaluronic acid-coated liposomes for targeted delivery of paclitaxel, in-vitro characterization and in-vivo evaluation. J Control Release. 2016;229:10–22. doi:10.1016/j.jconrel.2016.03.012
38. Yuba E, Kanda Y, Yoshizaki Y, et al. pH-sensitive polymer-liposome-based antigen delivery systems potentiated with interferon-γ gene lipoplex for efficient cancer immunotherapy. Biomaterials. 2015;67:214–224. doi:10.1016/j.biomaterials.2015.07.031
39. Yang Q. Liu S., Gu Y., et al. Development of sulconazole-loaded nanoemulsions for enhancement of transdermal permeation and antifungal activity. Int J Nanomedicine. 2019;14:3955–3966. doi:10.2147/IJN.S206657
40. Gaspar DP, Serra C, Lino PR, et al. Microencapsulated SLN: an innovative strategy for pulmonary protein delivery. Int J Pharm. 2017;516(1–2):231–246. doi:10.1016/j.ijpharm.2016.11.037
41. Liu M, Du H, Khan AR, Ji J, Yu A, Zhai G. Redox/enzyme sensitive chondroitin sulfate-based self-assembled nanoparticles loading docetaxel for the inhibition of metastasis and growth of melanoma. Carbohydr Polym. 2018;184:82–93. doi:10.1016/j.carbpol.2017.12.047
42. Liu M, Khan AR, Ji J, Lin G, Zhao X, Zhai G. Crosslinked self-assembled nanoparticles for chemo-sonodynamic combination therapy favoring antitumor, antimetastasis management and immune responses. J Control Release. 2018;290:150–164. doi:10.1016/j.jconrel.2018.10.007
43. Nagaiah G, Remick SC. Combretastatin A4 phosphate: a novel vascular disrupting agent. Future Oncol. 2010;6(8):1219–1228. doi:10.2217/fon.10.90
44. China Food And Drug Administration [homepage on the Internet]. Available from: http://app1.sfda.gov.cn/datasearch/face3/base.jsp?tableId=36&tableName=TABLE36&title=%BD%F8%BF%DA%D2%A9%C6%B7&bcId=124356651564146415214424405468.
45. China Food And Drug Administration [homepage on the Internet]. Available from: http://app1.sfda.gov.cn/datasearch/face3/base.jsp?tableId=25&tableName=TABLE25&title=%B9%FA%B2%FA%D2%A9%C6%B7&bcId=124356560303886909015737447882.
 © 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.