Back to Journals » Drug Design, Development and Therapy » Volume 11
Development of idursulfase therapy for mucopolysaccharidosis type II (Hunter syndrome): the past, the present and the future
Authors Whiteman DAH ![]() , Kimura A
, Kimura A
Received 13 April 2017
Accepted for publication 18 July 2017
Published 23 August 2017 Volume 2017:11 Pages 2467—2480
DOI https://doi.org/10.2147/DDDT.S139601
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 5
Editor who approved publication: Dr Qiongyu Guo
David AH Whiteman,* Alan Kimura*
Research & Development, Shire Human Genetic Therapies, Inc., Lexington, MA, USA
*These authors contributed equally to this work
Abstract: Mucopolysaccharidosis type II (MPS II; Hunter syndrome; OMIM 309900) is a rare, multisystemic, progressive lysosomal storage disease caused by deficient activity of the iduronate-2-sulfatase (I2S) enzyme. Accumulation of the glycosaminoglycans dermatan sulfate and heparan sulfate results in a broad range of disease manifestations that are highly variable in presentation and severity; notably, approximately two-thirds of individuals are affected by progressive central nervous system involvement. Historically, management of this disease was palliative; however, during the 1990s, I2S was purified to homogeneity for the first time, leading to cloning of the corresponding gene and offering a means of addressing the underlying cause of MPS II using enzyme replacement therapy (ERT). Recombinant I2S (idursulfase) was produced for ERT using a human cell line and was shown to be indistinguishable from endogenous I2S. Preclinical studies utilizing the intravenous route of administration provided valuable insights that informed the design of the subsequent clinical studies. The pivotal Phase II/III clinical trial of intravenous idursulfase (Elaprase®; Shire, Lexington, MA, USA) demonstrated improvements in a range of clinical parameters; based on these findings, intravenous idursulfase was approved for use in patients with MPS II in the USA in 2006 and in Europe and Japan in 2007. Evidence gained from post-approval programs has helped to improve our knowledge and understanding of management of patients with the disease; as a result, idursulfase is now available to young pediatric patients, and in some countries patients have the option to receive their infusions at home. Although ERT with idursulfase has been shown to improve somatic signs and symptoms of MPS II, the drug does not cross the blood–brain barrier and so treatment of neurological aspects of the disease remains challenging. A number of novel approaches are being investigated, and these may help to improve the care of patients with MPS II in the future.
Keywords: enzyme replacement therapy, Elaprase, idursulfase
Introduction
Mucopolysaccharidosis type II (MPS II; Hunter syndrome; OMIM 309900), one of a group of rare inherited disorders known as lysosomal storage diseases (LSDs), was first described in 1917.1 LSDs are characterized by defects in the functioning of lysosomal enzymes; in the case of MPS II, the underlying pathology is deficiency in iduronate-2-sulfatase (I2S; EC 3.1.6.13) activity, resulting in accumulation of the glycosaminoglycans (GAGs) heparan sulfate and dermatan sulfate within cells and tissues throughout the body.2 The disease is inherited in an X-linked manner and primarily affects males, although a small number of female patients have been described.3,4 The most common mechanism for symptomatic disease in females is skewed X-chromosome inactivation, but instances of X-chromosome rearrangement and 45,X karyotype have also been reported.3
Multiple organ systems are affected, with signs and symptoms usually emerging in the first few years of life. Disease presentation and rate of progression vary greatly, but the early manifestations of MPS II typically include recurrent respiratory infections, coarse facial features, joint stiffness, otitis media, umbilical/inguinal hernias, cardiomyopathies and hepatosplenomegaly;5,6 the characteristic short stature is usually apparent by ~8 years of age.7,8 Patients may also develop neurological symptoms, such as carpal tunnel syndrome, communicating hydrocephalus, spinal cord compression and hearing loss. For clinical purposes, patients are generally considered to fall into one of two categories according to the presence or absence of progressive central nervous system (CNS) involvement (typified by cognitive impairment, with behavioral difficulties and regression in developmental milestones), which occurs in as many as two-thirds of patients.2,9–12 All patients experience somatic signs and symptoms, although progression may be slower in individuals without cognitive impairment.2,9,13,14 Life expectancy is reduced in all patients; death usually occurs in the second decade of life in patients with cognitive impairment, whereas those without cognitive impairment may survive until their fifth or sixth decade.2,15
The possibility of treating LSDs by replacing the defective enzyme was put forward in the 1960s and the first such therapies completed clinical development in the early 1990s.16–21 Preclinical development of recombinant I2S (idursulfase; marketed as Elaprase® [Shire, Lexington, MA, USA]) as the first means of addressing the underlying enzyme deficiency in MPS II began in 1996; clinical studies commenced in 2001 and this phase of development was completed in 2005. At that time, MPS II was poorly characterized, with few data available on the natural history of the disease. The rarity of the condition was a significant factor in this paucity of knowledge: the estimated incidence is only 0.6–1.3 per 100,000 live male births.10,22–24 It is now a decade since intravenous enzyme replacement therapy (ERT) with idursulfase was approved for the treatment of patients with MPS II. In that time, our understanding of the disease and the management of patients with this condition has vastly increased, in large part owing to the clinical programs in place during this period. In this article, we provide an overview of the development of idursulfase for clinical use and the advances in patient care that have been made since marketing authorization was obtained. We also examine the unmet needs in patients with MPS II and look to the future and the novel therapeutic approaches that are being developed.
Development of idursulfase
Development of recombinant idursulfase
The first attempts to purify I2S from human tissues were made during the 1970s, but the low abundance of the protein, coupled with the potential for degradation and the presence of polypeptides of varying lengths, meant that purification to homogeneity was not achieved until 1990.21,25 Cloning of the iduronate-2-sulfatase (IDS) gene followed shortly afterward, paving the way for Shire (at that time, Transkaryotic Therapies, Inc.) to develop recombinant I2S (idursulfase) for therapeutic use in patients with MPS II.21,26 A human cell line was chosen for production of the recombinant enzyme (Box 1), ensuring that the therapeutic protein is indistinguishable from the endogenous form. Of particular note is the retention of the human pattern of posttranslational modifications, including the characteristic glycosylation profile. These modifications play an important role in targeting the enzyme to its site of action within the lysosomes of cells throughout the body via the mannose-6-phosphate (M6P) receptor-mediated uptake pathway (Figure 1).27–29 The likelihood of an immune response to the therapeutic protein is also believed to be reduced when the human posttranslational modification profile is retained.28
 | Box 1 Key points in the development of idursulfase |
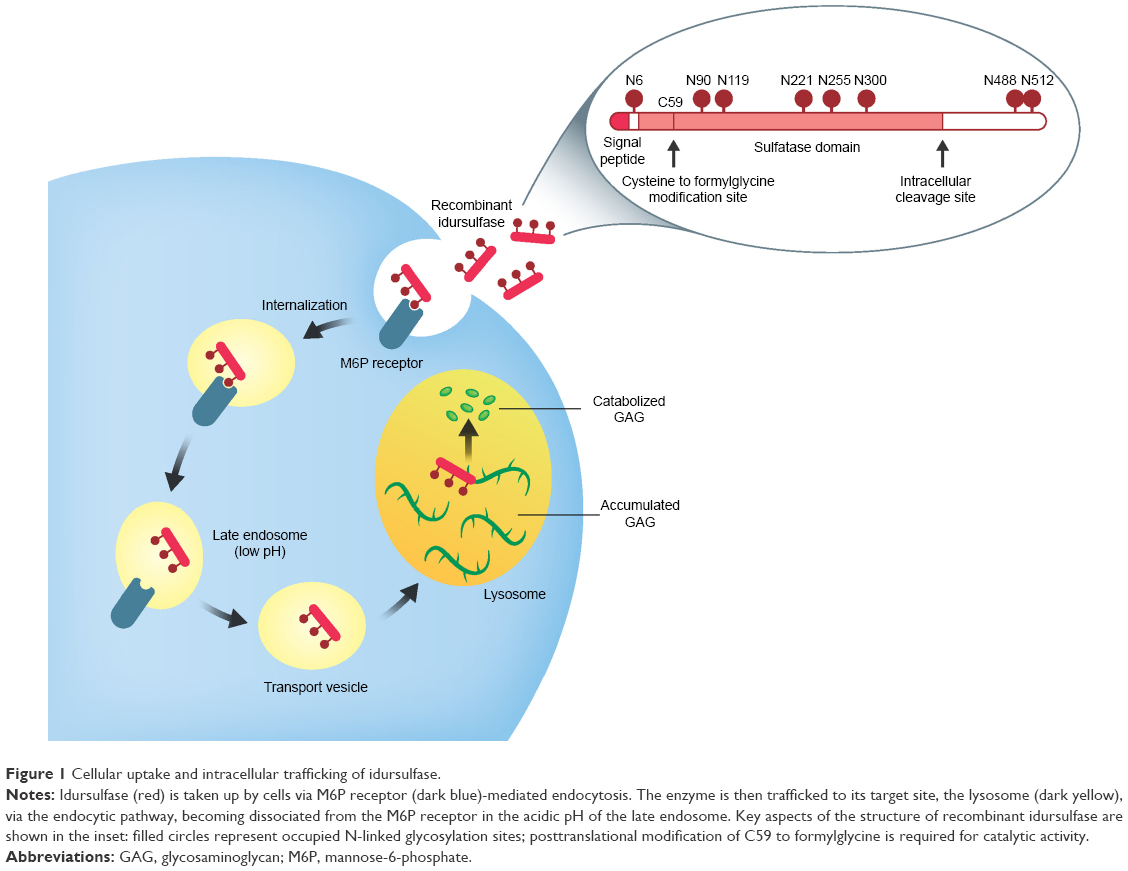 | Figure 1 Cellular uptake and intracellular trafficking of idursulfase. |
Preclinical development of idursulfase
Proof of principle for idursulfase therapy via intravenous infusion (formulation information is given in Box 2) was demonstrated in a series of animal studies, which were aided by the availability of an IDS-knockout mouse model of MPS II.21,30 This model not only has elevated levels of GAGs in urine and tissue (including in the liver, spleen, kidney and heart) but also exhibits many of the physical features of the disease.30 Intravenous idursulfase reduced levels of GAGs in urine and tissue, and the overall biodistribution of the infused enzyme was consistent with known M6P receptor distribution patterns. Together, these results indicate successful utilization of the intended uptake pathway, with the recombinant idursulfase reaching the lysosomes of cells in target organs to catabolize accumulated substrate.21,30
 | Box 2 Formulation of idursulfase for clinical use |
In addition to confirming the viability of the intended route of administration, the preclinical studies provided pivotal information for the design of the subsequent clinical program and the final choice of dosing regimen.21,31,32 For example, a weekly or every other week regimen was more effective at reducing levels of GAGs in tissue than monthly infusions, and degradation of GAGs in IDS-knockout mice was achieved with weekly administration of idursulfase at doses ranging from 0.1 mg/kg to 5.0 mg/kg.21,30 However, the tissue half-life for idursulfase of 1–2 days in both rats and mice indicated that the drug would be eliminated from the body by the second week after the infusion, and serum clearance mechanisms showed signs of saturation at doses of 0.5 mg/kg body weight or higher in cynomolgus monkeys.21 Together, these considerations were taken into account when making the decision to include the 0.5 mg/kg per week regimen in the Phase II/III study.21,30,32
Overall, the results of this series of preclinical studies, which included comprehensive repeat- and acute-dose toxicity studies, supported progression to clinical trials and laid a strong foundation for the design of the human clinical trial program.21
Idursulfase clinical development program
The development of a therapy for any rare disease poses significant challenges; these challenges are compounded in LSDs, and especially in MPS II, by the highly variable nature of disease presentation and progression, and an incomplete understanding of their natural history over time.21 In addition, at the time the idursulfase program was implemented, clinical development of therapies for rare genetic diseases was a field in its infancy; the rarity of the disease compared with the costs associated with the development of a new therapy was a considerable challenge. The design of the idursulfase clinical program built on experience gained in the development of ERTs for other LSDs (including Gaucher disease, Fabry disease and mucopolysaccharidosis type I [MPS I]) and comprised a Phase I/II study followed by a larger pivotal Phase II/III study designed to meet authorization requirements (Figure 2; Table 1).21 This approach aimed to achieve the necessary robustness with the small patient numbers available; the approach also reduced the time before the therapy could become available to patients by conducting only two studies in the clinical program.21
 | Figure 2 Overview of the idursulfase clinical development program. |
 | Table 1 Summary of completed intravenous idursulfase clinical studies |
The Phase I/II study investigated the safety of intravenous infusions of various doses of idursulfase administered according to an every other week regimen, examined pharmacodynamics and gathered exploratory efficacy data. Broad scope and rigorous design were key in ensuring that this single Phase I/II study with 12 patients enrolled enabled progression of idursulfase to the next phase of development.31 Overall, intravenous idursulfase was well tolerated, and infusion-related reactions (IRRs) were readily managed.31 Although the small number of patients and the variable nature of their disease at baseline made demonstration of clinical benefit challenging, exploratory efficacy outcomes indicated that the enzyme was biologically active and reached the lysosomes of cells in target tissues: GAG levels, which had been elevated at baseline, were reduced to near-normal levels in all patients; liver and spleen volumes were decreased; and walking distance, pulmonary function and left ventricular mass were improved in many patients.31
The choice of dose and the every other week regimen used in the Phase I/II study were based on the preclinical data; however, consideration of the tissue half-life of idursulfase in mice informed the decision that the Phase II/III study would investigate the 0.5 mg/kg weekly regimen, which had not been part of the Phase I/II study, in addition to the 0.5 mg/kg every other week regimen.21 At the time, with 96 patients enrolled and a duration of 1 year, the Phase II/III study was the largest and longest placebo-controlled trial performed for an LSD. Robust design of the Phase II/III study was essential to the success of the idursulfase clinical program, as regulatory approval would be based on the results of this single, relatively small, pivotal trial (study design is given in Table 1). Idursulfase was well tolerated, and no patient withdrew owing to an IRR.32 Compared with placebo, the greatest difference in the primary end point (a composite score) was with idursulfase 0.5 mg/kg weekly (P=0.0049); weekly infusions also resulted in significant improvements in 6-minute walk test distance (P=0.013) and forced vital capacity (FVC) compared with placebo (absolute FVC, P=0.001; % predicted FVC, P=0.065).32 In addition, urinary GAG levels and liver and spleen sizes were significantly reduced with idursulfase 0.5 mg/kg weekly (P<0.0001 for each). On the basis of the results of the Phase II/III study, idursulfase 0.5 mg/kg weekly was approved in the USA in 2006 and in Europe in 2007.32
Idursulfase post-approval programs
A wealth of knowledge has been accumulated since idursulfase was first approved, leading to expansion of the original indication as well as adding to our overall understanding of MPS II and management of patients with the disease. A large amount of data has been collected on idursulfase in both the clinical trial (reviewed by Scarpa33 and Sestito et al34) and the real-world settings,35,36 and more than 10,000 person-years of exposure to idursulfase have been accumulated since its approval in the USA in 2006 (Amanda Reynolds, Shire, personal communication, November 2016; exposure calculated using data as of July 2016).
An important backdrop to the post-approval activities is the additional long-term safety and effectiveness data required by the regulatory authorities in Europe, the USA, Canada and elsewhere as a condition of the marketing authorization for idursulfase. Requirements have changed over time, with the current focus being on long-term assessments of pulmonary and cardiovascular morbidity and mortality, urinary GAG levels and excretion patterns, and antibody levels. Post hoc analyses of data from the original clinical studies and continuation of the idursulfase clinical program have been key in addressing these requirements over the years, as have analyses of data collected in the Hunter Outcome Survey (HOS) registry. This registry was set up in 2005 to collect real-world data on the clinical presentation and progression of MPS II and the long-term safety and effectiveness of intravenous ERT with idursulfase.14,35
The HOS registry
HOS is a long-term, open-ended global registry designed to collect information from patients with MPS II; data are obtained during routine patient visits and assessments (Box 3).14,35 The registry was established in 2005 and has been used to address post-marketing commitments relating to long-term use of idursulfase. Although the multisystemic and variable nature of MPS II posed some challenges with designing the registry and continues to be a factor in efforts to collect comprehensive data, HOS gives us the ability to follow large numbers of patients for much longer than was possible in the original clinical trials.
 | Box 3 Overview of the HOS registry design and objectives |
Overall, the registry contains data from more than 1,000 patients from over 120 different clinics in 29 different countries (as of January 2016)35 and has resulted in 17 peer-reviewed journal articles8,14,15,35,37–49 and numerous congress abstracts and posters (more than 35 in the past 4 years alone). Our knowledge of the timing and prevalence of the many different manifestations of MPS II has been greatly increased as a consequence of the information collected in HOS.8,14,47 The registry was also instrumental in providing sufficient data to highlight and define the relatively hidden cardiac complications of the disease.43 The data collected have not only made a significant contribution to our knowledge of the natural history of this rare disease but are also an extremely valuable source of information on patient management and in particular the use of idursulfase: more than 750 patients being followed prospectively in HOS have received at least one infusion of the drug (data as of January 2016).35 Analyses of data from these patients have provided insight into compliance with treatment and long-term clinical outcomes following idursulfase therapy. The real-world experience captured by HOS also continues to provide an important contribution to our understanding of the profile of certain subsets of patients, such as those first receiving idursulfase very late in life, females with MPS II and those in whom therapy is initiated at a very young age.45
In addition to extending our knowledge of the effects of idursulfase,37,45 experience in HOS has influenced standards of care, with key areas of contribution being characterization and approaches to the management of IRRs, and the feasibility of home therapy.44,46
Immunological aspects
As with any protein-based therapy, there is the potential for an immune response to ERT; the nature and extent of this are dependent to a large extent on the therapeutic protein but also vary between patients.50 The overall picture for LSDs is reassuring: while some humoral responses are to be expected, the number of anaphylactic reactions is low.50 Nonetheless, it should be recognized that there is the potential for a severe reaction and so it is important that patients are monitored closely for hypersensitivity reactions, with the appropriate precautions taken where required.51
The immunological aspects of idursulfase therapy have been rigorously investigated both in the initial clinical trials and beyond, and the picture continues to indicate that in general there is no lessening of clinical response in the patients who develop antibodies to the therapeutic protein.31,32,51–53 It should be noted that the assays used to detect and characterize development of antibodies to idursulfase have been refined and improved over time, with the newer assays able to provide a clearer, more informative picture of immunological aspects; although it is not possible to compare the antibody titers obtained using the different assay methods directly, trends over time can still be assessed.
Formal post hoc analyses of the development of antibodies to idursulfase in patients receiving the drug were performed using data from the Phase II/III study and its extension and also from the young population (aged 1.4–7.5 years) in the HGT-ELA-038 study. These analyses clearly showed that although approximately one-half to two-thirds of patients developed immunoglobulin G (IgG) in response to idursulfase, with some showing persistent seropositivity, there was no association between antibody status and key clinical outcomes, including liver and spleen volumes, 6-minute walk test distance, predicted FVC or occurrence of serious adverse events. Urinary GAG levels in these patients decreased significantly with treatment, although to a lesser extent than in the seronegative individuals, but analysis of parameters such as 6-minute walk test distance and FVC did not indicate any impact on measures of functional clinical outcomes.52,53 Although there was generally little correlation between genotype and antibody status, a notable exception was those patients with a complete deletion/large rearrangement genotype, who almost invariably developed IgG in response to idursulfase, often at higher titers than other patients; neutralizing antibodies were also more common in this subgroup of patients. The limited data available on the impact of neutralizing antibodies on efficacy of treatment suggest that clinical benefit remains, perhaps with a slightly blunted response in terms of urinary GAG levels and liver size.53 Patients with the complete deletion/large rearrangement genotype are also more likely to experience treatment-emergent adverse events, including infusion-related events, and so are monitored carefully to ensure appropriate management when required.51,52
The extensive post-approval experience with idursulfase that we now have has enabled development of a clearly defined approach to the management of IRRs.44,54–56 The majority of these reactions are hypersensitivity reactions, also known as anaphylactoid reactions.50,57 These reactions are characterized by rash and fever and can typically be managed without interruption of treatment by slowing the rate of infusion and medication with antihistamines, antipyretics and/or low-dose corticosteroids.44,54–56 By contrast, true immunoglobulin E (IgE)-mediated anaphylaxis is dose dependent, and rechallenge typically results in a serious reaction or death.44,51,55 Although the distinction between these two types of reaction is not recognized by all regulatory agencies, it has important implications for patient management, with true anaphylaxis treated according to accepted standards of emergency care, but the commonly occurring cardiac complications of MPS II should be remembered if use of epinephrine is being considered. Importantly, no clinically relevant IgE-positive titers have been recorded in patients receiving idursulfase;31,32,44,45,52,58 the small number of patients who had a sample with a titer above the nominal threshold using the early assay methodology have all since been reinfused without issue, as have patients in a separate report analyzing IgE in patients receiving idursulfase.58
Availability of home therapy
In 2010, the idursulfase label was expanded to include provision for administration of infusions by a specialist nurse at home or in an alternative suitable nonclinic environment, where local health care regulations permit.51 Experience recorded in HOS provided considerable insight into the feasibility and safety of home therapy in routine practice and informed the development of initial recommendations for best management of the transition to receiving infusions at home.46 Individual institutions often build on these recommendations to construct their own more detailed guidelines to fit with local policies and infrastructure. Overall, experience with home therapy has been positive, with patients who receive infusions at home missing fewer infusions than patients receiving infusions in hospital, and with patients and families often reporting improved quality of life.59–61 Home therapy also offers the potential benefits of decreased pressure on resources at specialist centers and a reduction in the overall cost of treatment, including the lower cost in some countries for medications received in the home compared with those received in the hospital as well the reduced societal impact (for example, travel and caregiver time out of work).60,62
Experience with idursulfase in young pediatric patients
The idursulfase label was further expanded in 2013 to include pediatric patients younger than 5 years of age, with the HGT-ELA-038 study (ClinicalTrials.gov identifier: NCT00607386) providing important data to support the use of idursulfase in this group. This 1-year open-label study found that safety, tolerability and exploratory efficacy outcomes in children who started idursulfase therapy at 1.4–7.5 years of age were similar to those previously reported in male patients aged 5 years or older when they started ERT.31,32,63,64 In particular, patient age and body weight did not affect pharmacokinetic parameters, and idursulfase treatment reduced urinary GAG levels, liver size and spleen volume. A post hoc analysis examining immunogenicity aspects also found no reason to consider changes to patient management for this group.53 The clinical study data were complemented by an analysis of data in HOS, which found no new safety concerns in patients starting idursulfase when younger than 6 years of age and also demonstrated decreased urinary GAG levels and liver size with therapy.45 As experience with idursulfase accumulates, more is becoming known about ERT in very young patients, including those starting treatment when they are as young as 6 months of age.65 There is a growing focus on defining standards of care in these individuals, and the data being collected in HOS will continue to provide a valuable addition to our body of knowledge on this topic.63,65
Ongoing Phase IV clinical studies
In addition to the collection of data on use of idursulfase in routine clinical practice via the HOS registry, further Phase IV studies are planned or ongoing. One study (SHP-ELA-401; ClinicalTrials.gov identifier: NCT02455622), building on initial observations from analyses of data in HOS,15 focuses on prospective changes in height and weight over at least 5 years of treatment in patients who started idursulfase at a suitably young age. This study will also investigate cross-reactive immunological material (CRIM) status, with the ultimate goal of correlating it with clinical outcomes data.
Considerations for ERT for patients with MPS II
As experience with idursulfase has grown, guidelines and best practices have been developed providing region-specific perspectives and reflecting changes in the registered indications over time.56,66–69 Most aspects relating to the routine administration of the 3-hour idursulfase infusions are consistent across the various guidelines, with the emphasis being on supervision by a suitably experienced physician or other health care professional, and regular monitoring of vital signs and caution in certain subgroups of patients, for example, those with a febrile infection, airway abnormalities, respiratory distress or a history of allergies.56 Clinical experience, including the real-world data captured in HOS, has informed the development of a clear process for management of IRRs if they occur, and guidance has been produced on managing the transition to home therapy in countries where this is available.44,46 However, when to start and stop ERT remains a topic of debate.
It is generally agreed that early initiation of therapy is likely to be beneficial for somatic aspects of the disease, but this is not currently reflected by guidelines in all countries.56,66,70 Experience with idursulfase in very young children so far has been reassuring,65 but longer-term follow-up is needed to examine the impact of early initiation of therapy on disease progression.70–72 The approach for patients with severe neurological disease is not well defined; clinical experience indicates that somatic symptoms in such patients do improve with idursulfase therapy, and that patients and their families experience benefit as a result, but guidelines differ considerably between countries.65,67–69 Expert recommendations generally suggest that ERT initiation in patients exhibiting cognitive impairment should be at the clinician’s discretion in discussion with the child’s parents or caregivers, and note that ERT may not be appropriate in those expected to receive limited benefit, for example, patients with very severe neurological impairment or those in a vegetative state.56,65,68,69 Similarly, the decision to stop ERT may reflect progression of neurological involvement to a degree where the physician and family agree that the patient is no longer receiving benefit.56,65,68
It should be noted that the current data available on ERT with idursulfase suggest limited therapeutic effect on aspects of disease such as tracheal deformities, joint stiffness, bone deformities and hearing loss.34,64,73 In addition, rapid cessation of ERT has been reported to result in deterioration in clinical status,74 with some of the published guidelines providing an indication of the circumstances in which it is clinically appropriate for therapy to be discontinued.66,68
For all patients, it is clear that a strong, cohesive, multidisciplinary team approach is needed for them to receive the most effective care. Regular biochemical and clinical assessments, ideally every 6–12 months and more frequently in those with rapidly progressing disease, are generally recommended to monitor disease progression and inform treatment decisions.56 Individualized decisions on care continue to form a vital part of the management of this complex disease.
Future directions
We now have more than a decade of experience with idursulfase beyond the controlled environment of a clinical trial and have amassed more than 10,000 person-years of exposure to the drug among patients in a large number of countries since the initial approval in the USA in 2006 (Amanda Reynolds, Shire, personal communication, November 2016; exposure calculated using data as of July 2016). As we enter the next decade, it is important that we continue our efforts to improve the care of patients with MPS II. An area of focus is quantifying the true burden of disease and the impact of idursulfase therapy on the lives of patients and their families; as part of this, it is important that we find better means of assessing the severity of this complex disease and develop improved approaches to determining response to treatment.
Recently, a form of intravenous ERT, known as idursulfase beta, has been developed (Green Cross Corp., Yongin, Korea). This ERT, which is produced using a Chinese hamster ovary cell line rather than a human cell line, has followed a different development pathway from idursulfase and is available in some geographies on the basis of the 24-week Phase I/II study.75
A key area of unmet need is treatment of the CNS aspects of LSDs that have a neurological component; in MPS II, CNS disease affects approximately two-thirds of patients and has considerable impact on their lives and prognoses.2,15,56,68 Intravenously delivered proteins are generally not expected to cross the blood–brain barrier, and intravenous idursulfase did not reach the CNS in substantial quantities in animal studies.30 Therapy with intravenous idursulfase is therefore not able to mitigate the CNS symptoms of disease directly.56 Several approaches to delivering therapeutic enzymes, including idursulfase, to the CNS are therefore being investigated and are at varying stages of development.
One such approach is an investigational formulation of idursulfase that can be delivered via an intrathecal device directly into cerebrospinal fluid; this formulation is being evaluated by Shire for use in conjunction with intravenous idursulfase. Following a successful preclinical program,76,77 the Phase I/II dose-ranging study (HGT-HIT-045; ClinicalTrials.gov identifier: NCT00920647) demonstrated a decrease in cerebrospinal GAG levels.78 In addition, no serious adverse events were observed, supporting progression to further clinical development.78 An open-label extension of the Phase I/II study is ongoing and a 1-year Phase II/III study is underway (HGT-HIT-094; ClinicalTrials.gov identifier: NCT02055118), with expected completion in 2017.
An alternative approach employing a fusion protein technique is currently in Phase I clinical trials for patients with MPS II (AGT-182-101; ClinicalTrials.gov identifier: NCT02262338). Also known as the “Trojan Horse” approach, this noninvasive method utilizes endogenous receptor-mediated transport mechanisms to carry therapeutic proteins across the blood–brain barrier. In the case of therapy for patients with MPS II, a genetically engineered monoclonal antibody against the human insulin receptor is fused with I2S, with the antibody component carrying the therapeutic enzyme into the neuronal cells via the endogenous insulin receptor.79,80 The transferrin and low-density lipoprotein 1 pathways may also be used in this approach to transporting therapeutic agents across the blood–brain barrier, and the technology is evolving toward use of the appropriate peptide carriers, although this latter element is in its infancy.81,82
Bone marrow transplantation and hematopoietic stem cell transplantation have had some success in other mucopolysaccharidoses, especially MPS I, and international guidelines are now available.83,84 Although case reports and retrospective studies in patients with MPS II have found some improvement in somatic manifestations, significant morbidity and mortality have been reported and the impact of chronic graft-versus-host disease is of particular concern.56,85–94 In addition, no controlled clinical trials have yet been performed. Furthermore, there is no consistent evidence of overall clinical benefit, and expert guidelines suggest that these approaches are generally not advisable in the treatment of patients with MPS II.56,95
Gene therapy is also being investigated for many LSDs with a neurological component, including MPS II. Experimental approaches include viral vectors, such as adeno-associated virus 2/8 and retrovirus, stem cell gene therapy and nonviral technologies.96–99 Although viral vector gene therapy approaches have reached clinical trials for MPS I and mucopolysaccharidosis type III (MPS III), the technology remains at a preclinical stage for MPS II. Finally, various compounds that decrease accumulation of GAGs are being investigated at the laboratory level, including genistein and polypentosan sulfate;100,101 there is a preliminary report of genistein use in seven patients with MPS II,102 and a randomized clinical trial in patients with MPS III is ongoing (S Jones and B Bigger, Manchester, UK; EudraCT Number: 2013-001479-18). Additional gene therapy approaches, such as antisense technology, are also in the very early stages of evaluation;101,103–105 we look forward to seeing what the future holds with these technologies.
As we move toward an era in which it may be possible for the CNS manifestations of MPS II to be addressed, it is increasingly important that the clinical community rises to the challenge of detecting the onset of cognitive decline as early as possible. Our ultimate goal is to gain the ability to predict which patients will develop CNS involvement later in the course of their disease so that the appropriate treatment can be initiated as early as possible.
Summary and conclusion
The development of ERT with idursulfase for MPS II was a big step forward for patients and families living with this devastating disease. Idursulfase was the first therapy to specifically address the underlying cause of the disease and, in conjunction with the appropriate supportive care, remains a cornerstone of the management of patients with MPS II today. The idursulfase development program faced considerable challenges, including low patient numbers, variable disease presentation and progression and an incomplete knowledge of the natural history of MPS II. Despite this, the robust design of the clinical program enabled idursulfase to be approved on the basis of the pivotal Phase II/III study and thus become available to patients more rapidly than would have been possible under a standard clinical program. At the time that idursulfase was developed, there were few therapies for rare genetic diseases in development. Therefore, an important lasting legacy of this and other similar clinical programs from the same era is that therapies for other rare diseases are now being developed almost routinely, aided by the formalization of appropriate accelerated clinical development pathways.106
The post-approval clinical studies and ongoing HOS registry have added considerably to our understanding of MPS II and also augmented our knowledge of many aspects of the management of patients with the disease. As a result, idursulfase is now available to young pediatric patients, and home therapy is possible where local regulations permit. Our understanding of immunological aspects of idursulfase therapy is also vastly expanded. Treatment of the cognitive and neurological aspects of the disease remains a key challenge for the MPS II community, and we look forward to seeing what the future holds for therapies currently in development to address this aspect of the disease.
Acknowledgments
We would like to thank all those involved in the idursulfase preclinical and clinical programs, including physicians, site staff, patients and families. We are also grateful to Shire staff for their role in developing the scientific and clinical programs to investigate this important disease. In particular, we are grateful to Dr Michael Heartlein (Shire, Lexington, MA, USA) for his contribution to the early research on and preclinical development of idursulfase. We are also grateful to Amanda Reynolds (Shire, Basingstoke, UK) for the calculation of patient-years of exposure to idursulfase. We would also like to thank all those involved with the HOS registry mentioned in this article for their valuable contributions, in particular, the patients enrolled in HOS and their families, as well as the HOS Investigators and study coordinators. Data collection and analysis in the HOS registry are supported by Shire. Medical writing support was provided by Dr Helen Bremner (Oxford PharmaGenesis, Oxford, UK) and was funded by Shire.
Disclosure
David AH Whiteman is a full-time employee and shareholder of Shire. Alan Kimura was a full-time employee of Shire and a shareholder at the time this manuscript was drafted; he is now an employee of Translate Bio, Inc. (Lexington, MA, USA) and remains a shareholder of Shire. The authors report no other conflicts of interest in this work.
References
Hunter C. A rare disease in two brothers. Proc R Soc Med. 1917;10(Sect Study Dis Child):104–116. | ||
Neufeld EF, Muenzer J. The mucopolysaccharidoses. In: Scriver CR, Beaudet AL, Sly WS, et al, editors. The Metabolic and Molecular Bases of Inherited Disease. 8th ed. New York: McGraw-Hill; 2001:3421–3452. | ||
Pinto LL, Vieira TA, Giugliani R, Schwartz IV. Expression of the disease on female carriers of X-linked lysosomal disorders: a brief review. Orphanet J Rare Dis. 2010;5:14. | ||
Tuschl K, Gal A, Paschke E, Kircher S, Bodamer OA. Mucopolysaccharidosis type II in females: case report and review of literature. Pediatr Neurol. 2005;32(4):270–272. | ||
Martin R, Beck M, Eng C, et al. Recognition and diagnosis of mucopolysaccharidosis II (Hunter syndrome). Pediatrics. 2008;121(2):e377–e386. | ||
Burton BK, Giugliani R. Diagnosing Hunter syndrome in pediatric practice: practical considerations and common pitfalls. Eur J Pediatr. 2012;171(4):631–639. | ||
Rozdzynska A, Tylki-Szymanska A, Jurecka A, Cieslik J. Growth pattern and growth prediction of body height in children with mucopolysaccharidosis type II. Acta Paediatr. 2011;100(3):456–460. | ||
Parini R, Jones SA, Harmatz PR, Giugliani R, Mendelsohn NJ. The natural history of growth in patients with Hunter syndrome: data from the Hunter Outcome Survey (HOS). Mol Genet Metab. 2016;117(4):438–446. | ||
Schwartz IV, Ribeiro MG, Mota JG, et al. A clinical study of 77 patients with mucopolysaccharidosis type II. Acta Paediatr Suppl. 2007;96(455):63–70. | ||
Tylki-Szymańska A. Mucopolysaccharidosis type II, Hunter’s syndrome. Pediatr Endocrinol Rev. 2014;12(1):107–113. | ||
Young ID, Harper PS. Mild form of Hunter’s syndrome: clinical delineation based on 31 cases. Arch Dis Child. 1982;57(11):828–836. | ||
Young ID, Harper PS. The natural history of the severe form of Hunter’s syndrome: a study based on 52 cases. Dev Med Child Neurol. 1983;25(4):481–489. | ||
Wraith JE, Scarpa M, Beck M, et al. Mucopolysaccharidosis type II (Hunter syndrome): a clinical review and recommendations for treatment in the era of enzyme replacement therapy. Eur J Pediatr. 2008;167(3):267–277. | ||
Wraith JE, Beck M, Giugliani R, et al; HOS Investigators. Initial report from the Hunter Outcome Survey. Genet Med. 2008;10(7):508–516. | ||
Jones SA, Almassy Z, Beck M, et al. Mortality and cause of death in mucopolysaccharidosis type II-a historical review based on data from the Hunter Outcome Survey (HOS). J Inherit Metab Dis. 2009;32(4):534–543. | ||
Barton NW, Brady RO, Dambrosia JM, et al. Replacement therapy for inherited enzyme deficiency – macrophage-targeted glucocerebrosidase for Gaucher’s disease. N Engl J Med. 1991;324(21):1464–1470. | ||
Brady RO. The sphingolipidoses. N Engl J Med. 1966;275(6):312–318. | ||
Deduve C. From cytases to lysosomes. Fed Proc. 1964;23:1045–1049. | ||
Fratantoni JC, Hall CW, Neufeld EF. Hurler and Hunter syndromes: mutual correction of the defect in cultured fibroblasts. Science. 1968;162(3853):570–572. | ||
Grabowski GA, Barton NW, Pastores G, et al. Enzyme therapy in type 1 Gaucher disease: comparative efficacy of mannose-terminated glucocerebrosidase from natural and recombinant sources. Ann Intern Med. 1995;122(1):33–39. | ||
Heartlein MW, Kimura A. Discovery and clinical development of idursulfase (Elaprase®) for the treatment of mucopolysaccharidosis II (Hunter syndrome). In: David C, Pryde MJP, editors. Orphan Drugs and Rare Diseases. London: Royal Society of Chemistry; 2014:164–182. | ||
Baehner F, Schmiedeskamp C, Krummenauer F, et al. Cumulative incidence rates of the mucopolysaccharidoses in Germany. J Inherit Metab Dis. 2005;28(6):1011–1017. | ||
Meikle PJ, Hopwood JJ, Clague AE, Carey WF. Prevalence of lysosomal storage disorders. JAMA. 1999;281(3):249–254. | ||
Poorthuis BJ, Wevers RA, Kleijer WJ, et al. The frequency of lysosomal storage diseases in the Netherlands. Hum Genet. 1999;105(1–2):151–156. | ||
Bielicki J, Freeman C, Clements PR, Hopwood JJ. Human liver iduronate-2-sulphatase. Purification, characterization and catalytic properties. Biochem J. 1990;271(1):75–86. | ||
Wilson PJ, Morris CP, Anson DS, et al. Hunter syndrome: isolation of an iduronate-2-sulfatase cDNA clone and analysis of patient DNA. Proc Natl Acad Sci U S A. 1990;87(21):8531–8535. | ||
Baenziger JU. A major step on the road to understanding a unique posttranslational modification and its role in a genetic disease. Cell. 2003;113(4):421–422. | ||
Hille-Rehfeld A. Mannose 6-phosphate receptors in sorting and transport of lysosomal enzymes. Biochim Biophys Acta. 1995;1241(2):177–194. | ||
Walsh G, Jefferis R. Post-translational modifications in the context of therapeutic proteins. Nat Biotechnol. 2006;24(10):1241–1252. | ||
Garcia AR, DaCosta JM, Pan J, Muenzer J, Lamsa JC. Preclinical dose ranging studies for enzyme replacement therapy with idursulfase in a knock-out mouse model of MPS II. Mol Genet Metab. 2007;91(2):183–190. | ||
Muenzer J, Gucsavas-Calikoglu M, McCandless SE, Schuetz TJ, Kimura A. A phase I/II clinical trial of enzyme replacement therapy in mucopolysaccharidosis II (Hunter syndrome). Mol Genet Metab. 2007;90(3):329–337. | ||
Muenzer J, Wraith JE, Beck M, et al. A phase II/III clinical study of enzyme replacement therapy with idursulfase in mucopolysaccharidosis II (Hunter syndrome). Genet Med. 2006;8(8):465–473. | ||
Scarpa M. Evaluation of idursulfase for the treatment of mucopolysaccharidosis II (Hunter syndrome). Expert Opin Orphan Drugs. 2013;1(1):89–98. | ||
Sestito S, Ceravolo F, Grisolia M, Pascale E, Pensabene L, Concolino D. Profile of idursulfase for the treatment of Hunter syndrome. Res Rep Endocr Disord. 2015;5:79–90. | ||
Muenzer J, Jones SA, Tylki-Szymanska A, et al. Ten years of the Hunter Outcome Survey (HOS): insights, achievements, and lessons learned from a global patient registry. Orphanet J Rare Dis. 2017;12(1):82. | ||
Parini R, Rigoldi M, Tedesco L, et al. Enzymatic replacement therapy for Hunter disease: up to 9 years experience with 17 patients. Mol Genet Metab Rep. 2015;3:65–74. | ||
Jones SA, Parini R, Harmatz P, et al; HOS Natural History Working Group on behalf of HOS Investigators. The effect of idursulfase on growth in patients with Hunter syndrome: data from the Hunter Outcome Survey (HOS). Mol Genet Metab. 2013;109(1):41–48. | ||
del Toro-Riera M. Seguimiento de pacientes con síndrome de Hunter: el registro HOS (Hunter Outcome Survey) [Follow-up of patients with Hunter syndrome: the Hunter Outcome Survey (HOS) registry]. Rev Neurol. 2007;44(suppl 1):S13–S17. Spanish. | ||
del Toro-Riera M. World-wide experience in the treatment of mucopolysaccharidosis type II: the Hunter Outcome Survey (HOS) registry. Rev Neurol. 2008;47(S02):S3–S7. | ||
Mendelsohn NJ, Harmatz P, Bodamer O, et al. Importance of surgical history in diagnosing mucopolysaccharidosis type II (Hunter syndrome): data from the Hunter Outcome Survey. Genet Med. 2010;12(12):816–822. | ||
Cohn GM, Morin I, Whiteman DA; Hunter Outcome Survey Investigators. Development of a mnemonic screening tool for identifying subjects with Hunter syndrome. Eur J Pediatr. 2013;172(7):965–970. | ||
Keilmann A, Nakarat T, Bruce IA, Molter D, Malm G; HOS Investigators. Hearing loss in patients with mucopolysaccharidosis II: data from HOS – the Hunter Outcome Survey. J Inherit Metab Dis. 2012;35(2):343–353. | ||
Kampmann C, Beck M, Morin I, Loehr JP. Prevalence and characterization of cardiac involvement in Hunter syndrome. J Pediatr. 2011;159(2):327.e2–331.e2. | ||
Burton BK, Whiteman DA; HOS Investigators. Incidence and timing of infusion-related reactions in patients with mucopolysaccharidosis type II (Hunter syndrome) on idursulfase therapy in the real-world setting: a perspective from the Hunter Outcome Survey (HOS). Mol Genet Metab. 2011;103(2):113–120. | ||
Muenzer J, Beck M, Giugliani R, et al. Idursulfase treatment of Hunter syndrome in children younger than 6 years: results from the Hunter Outcome Survey. Genet Med. 2011;13(2):102–109. | ||
Burton BK, Guffon N, Roberts J, van der Ploeg AT, Jones SA. Home treatment with intravenous enzyme replacement therapy with idursulfase for mucopolysaccharidosis type II – data from the Hunter Outcome Survey. Mol Genet Metab. 2010;101(2–3):123–129. | ||
Link B, de Camargo Pinto LL, Giugliani R, et al. Orthopedic manifestations in patients with mucopolysaccharidosis type II (Hunter syndrome) enrolled in the Hunter Outcome Survey. Orthop Rev (Pavia). 2010;2(2):e16. | ||
Alcalde-Martin C, Muro-Tudelilla JM, Cancho-Candela R, et al. First experience of enzyme replacement therapy with idursulfase in Spanish patients with Hunter syndrome under 5 years of age: case observations from the Hunter Outcome Survey (HOS). Eur J Med Genet. 2010;53(6):371–377. | ||
Bodamer O, Scarpa M, Hung C, Pulles T, Giugliani R. Birth weight in patients with mucopolysaccharidosis type II: data from the Hunter Outcome Survey (HOS). Mol Genet Metab Rep. 2017;11:62–64. | ||
Brooks DA, Kakavanos R, Hopwood JJ. Significance of immune response to enzyme-replacement therapy for patients with a lysosomal storage disorder. Trends Mol Med. 2003;9(10):450–453. | ||
Elaprase Summary of Product Characteristics. 2016. Available from: http://www.ema.europa.eu/docs/en_GB/document_library/EPAR_-_Product_Information/human/000700/WC500023008.pdf. Accessed July 24, 2016. | ||
Barbier AJ, Bielefeld B, Whiteman DA, Natarajan M, Pano A, Amato DA. The relationship between anti-idursulfase antibody status and safety and efficacy outcomes in attenuated mucopolysaccharidosis II patients aged 5 years and older treated with intravenous idursulfase. Mol Genet Metab. 2013;110(3):303–310. | ||
Pano A, Barbier AJ, Bielefeld B, Whiteman DA, Amato DA. Immunogenicity of idursulfase and clinical outcomes in very young patients (16 months to 7.5 years) with mucopolysaccharidosis II (Hunter syndrome). Orphanet J Rare Dis. 2015;10:50. | ||
Miebach E, Schulze-Frenking G, Knoche K, Mengel E, Beck M. Management of hypersensitivity reactions in mucopolysaccharidosis type II. Acta Paediatr. 2008;97(suppl 457):98. | ||
Miebach E. Management of infusion-related reactions to enzyme replacement therapy in a cohort of patients with mucopolysaccharidosis disorders. Int J Clin Pharmacol Ther. 2009;47(suppl 1):S100–S106. | ||
Scarpa M, Almassy Z, Beck M, et al. Mucopolysaccharidosis type II: European recommendations for the diagnosis and multidisciplinary management of a rare disease. Orphanet J Rare Dis. 2011;6:72. | ||
Sampson HA, Munoz-Furlong A, Campbell RL, et al. Second symposium on the definition and management of anaphylaxis: summary report – Second National Institute of Allergy and Infectious Disease/Food Allergy and Anaphylaxis Network Symposium. J Allergy Clin Immunol. 2006;117(2):391–397. | ||
Kim J, Park MR, Kim DS, et al. IgE-mediated anaphylaxis and allergic reactions to idursulfase in patients with Hunter syndrome. Allergy. 2013;68(6):796–802. | ||
Burton BK, Wiesman C, Paras A, Kim K, Katz R. Home infusion therapy is safe and enhances compliance in patients with mucopolysaccharidoses. Mol Genet Metab. 2009;97(3):234–236. | ||
Milligan A, Hughes D, Goodwin S, Richfield L, Mehta A. Intravenous enzyme replacement therapy: better in home or hospital? Br J Nurs. 2006;15(6):330–333. | ||
Hughes DA, Mlilligan A, Mehta A. Home therapy for lysosomal storage disorders. Br J Nurs. 2007;16(22):1384, 1386–1389. | ||
Bagewadi S, Roberts J, Mercer J, Jones S, Stephenson J, Wraith JE. Home treatment with Elaprase and Naglazyme is safe in patients with mucopolysaccharidoses types II and VI, respectively. J Inherit Metab Dis. 2008;31(6):733–737. | ||
Giugliani R, Hwu WL, Tylki-Szymanska A, Whiteman DA, Pano A. A multicenter, open-label study evaluating safety and clinical outcomes in children (1.4–7.5 years) with Hunter syndrome receiving idursulfase enzyme replacement therapy. Genet Med. 2014;16(6):435–441. | ||
Muenzer J, Beck M, Eng CM, et al. Long-term, open-labeled extension study of idursulfase in the treatment of Hunter syndrome. Genet Med. 2011;13(2):95–101. | ||
Lampe C, Atherton A, Burton BK, et al. Enzyme replacement therapy in mucopolysaccharidosis II patients under 1 year of age. JIMD Rep. 2014;14:99–113. | ||
Giugliani R, Villarreal ML, Valdez CA, et al. Guidelines for diagnosis and treatment of Hunter syndrome for clinicians in Latin America. Genet Mol Biol. 2014;37(2):315–329. | ||
McGill J, Sillence D, Ketteridge D, Peters H. Guidelines for the Eligibility to Receive Treatment for Mucopolysaccharidosis Type II (MPS-II) with Idursulfase (Elaprase®) through the Life Saving Drugs Programme. MPS Advisory Committee, Australian Government Department of Health and Aging; 2008. Available from: http://www.health.gov.au/internet/main/publishing.nsf/Content/lsdp-info/$File/MPS-II-060515.pdf. Accessed August 7, 2017. | ||
Muenzer J, Bodamer O, Burton B, et al. The role of enzyme replacement therapy in severe Hunter syndrome – an expert panel consensus. Eur J Pediatr. 2012;171(1):181–188. | ||
Vellodi A, Wraith JE, Cleary MA, Ramaswami U, Lavery C, Jessop E. Guidelines for the Investigation and Management of Mucopolysaccharidosis Type II. Department of Health National Specialist Commissioning Group (NSCAG); 2007. Available from: http://www.ufrgs.br/redempsbrasil/artigos/MPSII4.pdf. Accessed August 3, 2017. | ||
Muenzer J. Early initiation of enzyme replacement therapy for the mucopolysaccharidoses. Mol Genet Metab. 2014;111(2):63–72. | ||
Tajima G, Sakura N, Kosuga M, Okuyama T, Kobayashi M. Effects of idursulfase enzyme replacement therapy for mucopolysaccharidosis type II when started in early infancy: comparison in two siblings. Mol Genet Metab. 2013;108(3):172–177. | ||
Tylki-Szymańska A, Jurecka A, Zuber Z, Rozdzynska A, Marucha J, Czartoryska B. Enzyme replacement therapy for mucopolysaccharidosis II from 3 months of age: a 3-year follow-up. Acta Paediatr. 2012;101(1):e42–e47. | ||
Okuyama T, Tanaka A, Suzuki Y, et al. Japan Elaprase Treatment (JET) study: idursulfase enzyme replacement therapy in adult patients with attenuated Hunter syndrome (Mucopolysaccharidosis II, MPS II). Mol Genet Metab. 2010;99(1):18–25. | ||
Jurecka A, Zuberuber Z, Opoka-Winiarska V, Wegrzyn G, Tylki-Szymanska A. Effect of rapid cessation of enzyme replacement therapy: a report of 5 cases and a review of the literature. Mol Genet Metab. 2012;107(3):508–512. | ||
Sohn YB, Cho SY, Park SW, et al. Phase I/II clinical trial of enzyme replacement therapy with idursulfase beta in patients with mucopolysaccharidosis II (Hunter Syndrome). Orphanet J Rare Dis. 2013;8:42. | ||
Felice BR, Wright TL, Boyd RB, et al. Safety evaluation of chronic intrathecal administration of idursulfase-IT in cynomolgus monkeys. Toxicol Pathol. 2011;39(5):879–892. | ||
Xie H, Chung JK, Mascelli MA, McCauley TG. Pharmacokinetics and bioavailability of a therapeutic enzyme (idursulfase) in cynomolgus monkeys after intrathecal and intravenous administration. PLoS One. 2015;10(4):e0122453. | ||
Muenzer J, Hendriksz CJ, Fan Z, et al. A phase I/II study of intrathecal idursulfase-IT in children with severe mucopolysaccharidosis II. Genet Med. 2016;18(1):73–81. | ||
Boado RJ, Ka-Wai Hui E, Zhiqiang Lu J, Pardridge WM. Insulin receptor antibody-iduronate 2-sulfatase fusion protein: pharmacokinetics, anti-drug antibody, and safety pharmacology in Rhesus monkeys. Biotechnol Bioeng. 2014;111(11):2317–2325. | ||
Lu JZ, Boado RJ, Hui EK, Zhou QH, Pardridge WM. Expression in CHO cells and pharmacokinetics and brain uptake in the Rhesus monkey of an IgG-iduronate-2-sulfatase fusion protein. Biotechnol Bioeng. 2011;108(8):1954–1964. | ||
Karkan D, Pfeifer C, Vitalis TZ, et al. A unique carrier for delivery of therapeutic compounds beyond the blood-brain barrier. PLoS One. 2008;3(6):e2469. | ||
Nounou MI, Adkins CE, Rubinchik E, et al. Anti-cancer antibody trastuzumab-melanotransferrin conjugate (BT2111) for the treatment of metastatic HER2+ breast cancer tumors in the brain: an in-vivo study. Pharm Res. 2016;33(12):2930–2942. | ||
Aldenhoven M, Jones SA, Bonney D, et al. Hematopoietic cell transplantation for mucopolysaccharidosis patients is safe and effective: results after implementation of international guidelines. Biol Blood Marrow Transplant. 2015;21(6):1106–1109. | ||
Boelens JJ, Wynn RF, O’Meara A, et al. Outcomes of hematopoietic stem cell transplantation for Hurler’s syndrome in Europe: a risk factor analysis for graft failure. Bone Marrow Transplant. 2007;40(3):225–233. | ||
Beck M. Mucopolysaccharidosis type II (Hunter syndrome): clinical picture and treatment. Curr Pharm Biotechnol. 2011;12(6):861–866. | ||
Bergstrom SK, Quinn JJ, Greenstein R, Ascensao J. Long-term follow-up of a patient transplanted for Hunter’s disease type IIB: a case report and literature review. Bone Marrow Transplant. 1994;14(4):653–658. | ||
Coppa GV, Gabrielli O, Zampini L, et al. Bone marrow transplantation in Hunter syndrome (mucopolysaccharidosis type II): two-year follow-up of the first Italian patient and review of the literature. Pediatr Med Chir. 1995;17(3):227–235. | ||
Coppa GV, Gabrielli O, Zampini L, et al. Bone marrow transplantation in Hunter syndrome. J Inherit Metab Dis. 1995;18(1):91–92. | ||
Guffon N, Bertrand Y, Forest I, Fouilhoux A, Froissart R. Bone marrow transplantation in children with Hunter syndrome: outcome after 7 to 17 years. J Pediatr. 2009;154(5):733–737. | ||
Li P, Thompson JN, Hug G, Huffman P, Chuck G. Biochemical and molecular analysis in a patient with the severe form of Hunter syndrome after bone marrow transplantation. Am J Med Genet. 1996;64(4):531–535. | ||
Muenzer J, Beck M, Eng CM, et al. Multidisciplinary management of Hunter syndrome. Pediatrics. 2009;124(6):e1228–e1239. | ||
Mullen CA, Thompson JN, Richard LA, Chan KW. Unrelated umbilical cord blood transplantation in infancy for mucopolysaccharidosis type IIB (Hunter syndrome) complicated by autoimmune hemolytic anemia. Bone Marrow Transplant. 2000;25(10):1093–1097. | ||
Peters C, Krivit W. Hematopoietic cell transplantation for mucopolysaccharidosis IIB (Hunter syndrome). Bone Marrow Transplant. 2000;25(10):1097–1099. | ||
Vellodi A, Young E, Cooper A, Lidchi V, Winchester B, Wraith JE. Long-term follow-up following bone marrow transplantation for Hunter disease. J Inherit Metab Dis. 1999;22(5):638–648. | ||
Giugliani R, Federhen A, Rojas MV, et al. Mucopolysaccharidosis I, II, and VI: brief review and guidelines for treatment. Genet Mol Biol. 2010;33(4):589–604. | ||
Braun SE, Aronovich EL, Anderson RA, Crotty PL, McIvor RS, Whitley CB. Metabolic correction and cross-correction of mucopolysaccharidosis type II (Hunter syndrome) by retroviral-mediated gene transfer and expression of human iduronate-2-sulfatase. Proc Natl Acad Sci U S A. 1993;90(24):11830–11834. | ||
Cardone M, Polito VA, Pepe S, et al. Correction of Hunter syndrome in the MPSII mouse model by AAV2/8-mediated gene delivery. Hum Mol Genet. 2006;15(7):1225–1236. | ||
Friso A, Tomanin R, Zanetti A, et al. Gene therapy of Hunter syndrome: evaluation of the efficiency of muscle electro gene transfer for the production and release of recombinant iduronate-2-sulfatase (IDS). Biochim Biophys Acta. 2008;1782(10):574–580. | ||
Tomanin R, Friso A, Alba S, et al. Non-viral transfer approaches for the gene therapy of mucopolysaccharidosis type II (Hunter syndrome). Acta Paediatr Suppl. 2002;91(439):100–104. | ||
Schuchman EH, Ge Y, Lai A, et al. Pentosan polysulfate: a novel therapy for the mucopolysaccharidoses. PLoS One. 2013;8(1): e54459. | ||
Friso A, Tomanin R, Salvalaio M, Scarpa M. Genistein reduces glycosaminoglycan levels in a mouse model of mucopolysaccharidosis type II. Br J Pharmacol. 2010;159(5):1082–1091. | ||
Marucha J, Tylki-Szymanska A, Jakobkiewicz-Banecka J, et al. Improvement in the range of joint motion in seven patients with mucopolysaccharidosis type II during experimental gene expression-targeted isoflavone therapy (GET IT). Am J Med Genet A. 2011;155A(9):2257–2262. | ||
Matos L, Goncalves V, Pinto E, et al. Data in support of a functional analysis of splicing mutations in the IDS gene and the use of antisense oligonucleotides to exploit an alternative therapy for MPS II. Data Brief. 2015;5:810–817. | ||
Matos L, Goncalves V, Pinto E, et al. Functional analysis of splicing mutations in the IDS gene and the use of antisense oligonucleotides to exploit an alternative therapy for MPS II. Biochim Biophys Acta. 2015;1852(12):2712–2721. | ||
Moskot M, Gabig-Ciminska M, Jakobkiewicz-Banecka J, Wesierska M, Bochenska K, Wegrzyn G. Cell cycle is disturbed in mucopolysaccharidosis type II fibroblasts, and can be improved by genistein. Gene. 2016;585(1):100–103. | ||
Griggs RC, Batshaw M, Dunkle M, et al. Clinical research for rare disease: opportunities, challenges, and solutions. Mol Genet Metab. 2009;96(1):20–26. | ||
Paterson H, Reeves B, Brown R, et al. Activated N-ras controls the transformed phenotype of HT1080 human fibrosarcoma cells. Cell. 1987;51(5):803–812. | ||
Rasheed S, Nelson-Rees WA, Toth EM, Arnstein P, Gardner MB. Characterization of a newly derived human sarcoma cell line (HT-1080). Cancer. 1974;33(4):1027–1033. | ||
Strauss DM, Lute S, Brorson K, Blank GS, Chen Q, Yang B. Removal of endogenous retrovirus-like particles from CHO-cell derived products using Q sepharose fast flow chromatography. Biotechnol Prog. 2009;25(4):1194–1197. | ||
Wheatley DN. Pericentriolar virus-like particles in Chinese hamster ovary cells. J Gen Virol. 1974;24(2):395–399. | ||
Shahrokh Z, Royle L, Saldova R, et al. Erythropoietin produced in a human cell line (Dynepo) has significant differences in glycosylation compared with erythropoietins produced in CHO cell lines. Mol Pharm. 2011;8(1):286–296. | ||
Kariya Y, Kawamura C, Tabei T, Gu J. Bisecting GlcNAc residues on laminin-332 down-regulate galectin-3-dependent keratinocyte motility. J Biol Chem. 2010;285(5):3330–3340. | ||
Ghaderi D, Taylor RE, Padler-Karavani V, Diaz S, Varki A. Implications of the presence of N-glycolylneuraminic acid in recombinant therapeutic glycoproteins. Nat Biotechnol. 2010;28(8):863–867. | ||
Andrews EB, Arellano FM, Avorn J, et al. Guidelines for Good Pharmacoepidemiology Practices (GPP). International Society for Pharmacoepidemiology; 2015. Available from: https://www.pharmacoepi.org/resources/guidelines_08027.cfm. Accessed August 2, 2017. | ||
Dreyer NA, Schneeweiss S, McNeil B, et al. GRACE principles: recognizing high-quality observational studies of comparative effectiveness. Am J Manag Care. 2010;16(6):467–471. | ||
ICH Harmonised Tripartite Guideline for Good Clinical Practice E6(R1). International Council for Harmonisation of Technical Requirements for Pharmaceuticals for Human Use; 1996. Available from: https://www.ich.org/fileadmin/Public_Web_Site/ICH_Products/Guidelines/Efficacy/E6/E6_R1_Guideline.pdf. Accessed August 2, 2017. |
 © 2017 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2017 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.