Back to Journals » International Journal of Nanomedicine » Volume 18
Development of Diphenyl carbonate-Crosslinked Cyclodextrin Based Nanosponges for Oral Delivery of Baricitinib: Formulation, Characterization and Pharmacokinetic Studies
Authors Aldawsari MF, Alhowail AH ![]() , Anwer MK, Ahmed MM
, Anwer MK, Ahmed MM
Received 27 January 2023
Accepted for publication 19 April 2023
Published 27 April 2023 Volume 2023:18 Pages 2239—2251
DOI https://doi.org/10.2147/IJN.S405534
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Prof. Dr. Anderson Oliveira Lobo
Mohammed F Aldawsari,1 Ahmad H Alhowail,2 Md Khalid Anwer,1 Mohammed Muqtader Ahmed1
1Department of Pharmaceutics, College of Pharmacy, Prince Sattam Bin Abdulaziz University, Al-kharj, 11942, Saudi Arabia; 2Department of Pharmacology and Toxicology, College of Pharmacy, Qassim University, Buraydah, Al-Qassim, 51452, Saudi Arabia
Correspondence: Mohammed F Aldawsari, Department of Pharmaceutics, College of Pharmacy, Prince Sattam Bin Abdulaziz University, Al-kharj, 11942, Saudi Arabia, Tel +966-555101369, Email [email protected]
Background: The aim of the present investigation is to prepare baricitinib (BAR)-loaded diphenyl carbonate (DPC) β-cyclodextrin (βCD) based nanosponges (NSs) to improve the oral bioavailability.
Methods: BAR-loaded DPC-crosslinked βCD NSs (B-DCNs) were prepared prepared by varying the molar ratio of βCD: DPC (1:1.5 to 1:6). The developed B-DCNs loaded with BAR were characterized for particle size, polydispersity index (PDI), zeta potential (ZP), % yield and percent entrapment efficiency (%EE).
Results: Based on the above evaluations, BAR-loaded DPC βCD NSs (B-CDN3) was optimized with mean size (345.8± 4.7 nm), PDI (0.335± 0.005), Yield (91.46± 7.4%) and EE (79.1± 1.6%). The optimized NSs (B-CDN3) was further confirmed by SEM, spectral analysis, BET analysis, in vitro release and pharmacokinetic studies. The optimized NSs (B-CDN3) showed 2.13 times enhancement in bioavailability in comparison to pure BAR suspension.
Conclusion: It could be anticipated that NSs loaded with BAR as a promising tool for release and bioavailability for the treatment of rheumatic arthritis and Covid-19.
Keywords: baricitinib, cyclodextrin, diphenyl carbonate, crosslinker, bioavailability, BAR, DPC
Graphical Abstract:

Introduction
Baricitinib (BAR) is a azetidineacetonitrile, 1-(ethylsulfonyl)-3-[4-(7H-pyrrolo[2,3-d] pyrimidin-4-yl)-1H-pyrazol-1-yl]-, empirically represented as (C16H17N7O2S), with molar mass of 371.42 g/mol. The mechanism of action of BAR is as a Janus Kinase Inhibitor, there are four tyrosine receptor kinases Janus kinase (JAK1-3), and tyrosine kinase (TYK2), wherein JAK2 activation may be critical for tumour formation and progression, JAK3 is required for immune cell development. However, JAK1 and TYK2 indicated in disease conditions and immune suppression.1–3 BAR act against rheumatoid arthritis (RA) by disrupting the responses of cells due to the cytokines. Unrecognized proteins (cytokines) are inhibited that are actively involved in cells to cells communication, and excess of this cytokines may cause RA inflammation.4–6 Baricitinib (BAR) (Olumiant) relieve the symptoms of pain, stiffness and swelling in joints and slows the joint damage that RA can cause. Most people who benefit from this treatment will notice some improvement within the first 12 weeks.7,8 Recently, it was therapeutically used against the COVID-19 and SARS-CoV-2 by inhibiting the signalling pathways such as interleukin-2, interleukin-6, interleukin-10, interferon-γ, and impairing the AP2-associated protein kinase, respectively. It used as an antiarrhythmic agent, and JAK inhibitor with proven and effective anti-inflammatory and anti-viral effects, used during Covid-19 as a immunomodulatory,9,10 recently it got approved for treatment of alopecia.11,12 BAR is the first FDA-approved once-daily pill for the treatment of severe alopecia areata, this JAK inhibitor demonstrated excellent hair regrowth compared to the placebo.13 Moreover, it was also shown to decrease the proliferation of JAK1/JAK2 expression in mutated cells and causes caspase-mediated cell death.14
BAR is used as curative as well as palliative treatment purpose. BAR is not recommended in pregnancy and breastfeeding, therefore if anyone is under BAR treatment one should use contraceptive for at least one week after its treatment.15 Moreover, any person undergoing for operation or surgery must stop taking BAR for a week of before and after the surgical procedure. Treatment of BAR is recommended only for non-live vaccines and should be avoided with live vaccines. There is no therapeutic incompatibility between BAR and NSAIDs or other analgesics.16 However, there are a few side-effects observed with BAR treatments that include; nausea, vein embolisms, pulmonary embolism, breathlessness, affects on the immune system thereby causing cold sores, chest and urinary tract infections.17
BAR is reported to show poor aqueous solubility, which creates hindrance in developing formulations. Scientist have explored many formulations including nanocarriers to improve the solubility of this drug and to improve the efficacy.18–21 β-cyclodextrin (βCD) is a macrocyclic oligosaccharide cavity consisting of seven α-1,4-linked d-glucopyranosyl units, these are further crosslinked by using any cross linker; diphenyl carbonate (DPC), 1,1′-carbonyl diimidazole, 6-hexamethylene diisocyanate, pyromellitic dianhydride, 2,6-naphthalene dicarboxylic acid, 1,4-butanediol diglycidyl ether, in order to fabricate β-cyclodextrin nanosponges (β-CDNSs). In pharmaceutical industries, cyclodextrins (CD) usage for complexation, solubilization, stability, and improved dissolution and bioavailability is well-established and proven by many model drugs. CDs, due to their Lewis base character, can be able to accommodate guest supramolecular structures and facilitate the hydrophilicity. Moreover, the NSs formed by CDs has the hydrophilic, nanoporous, increases surface area, characteristics. β-CDNSs are stable at high temperatures up to 300°C, and over the pH range of 1 to 11. They can be easily fabricated by thermal desorption, and ultrasound techniques.21–25
The objective of the current study is to prepare the βCDNSs using DPC as cross linker and incorporate the BAR drug. The prepared DPC-crosslinked βCDNSs characterized for physicochemical, and pharmaceutical parameters. The optimized BAR-loaded DPC-crosslinked βCDNSs was then further evaluated for pharmacokinetic studies on rats.
Materials and Methods
Materials
BBAR) was purchased from “Mesochem Technology” Beijing, China. β-cyclodextrin (βCD), DPC and Dimethyl sulfoxide (DMSO) are procured from Sigma Aldrich, Germany and Alain PhrarmTech, India, respectively. All the other chemicals will be of analytical grades.
Synthesis of DPC Crosslinked β-CD-NSPs
Blank NSPs were synthesized by crosslinking the β-cyclodextrin (βCD) with DPC. Briefly, four batches of blank NSPs were prepared in the molar ratio (1:1.5, 1:3, 1:4.5 and 1:6, β-CD/DPC) by melting the dispersion at 90°C until to get liquefy, and added triethylamine (0.3 mL) that help in cross linking by proton exchange between DPC and βCD. The solution was solidified by cooling and then triturated in the mortar and pestle.25 The resultant mass was crushed, then rinsed repeatedly with ultrapure water to remove the phenol by product, extracted in triplicate with absolute acetone, and then left to air dry.
Phase Solubility Studies
The stability constant (K) and complexation efficiency (CE) of the combination including BAR/βCD were determined using a phase solubility analysis. Briefly, an excess quantity of BAR was dispersed into aqueous solution containing βCD (0–50 mM), and conical flasks were shaken at 25°C, 100 rpm for 72 h.26 The supernatant of the samples were carefully collected and filtered (0.45 µm), and the filtrate was diluted and analysed by UV- spectrophotometer at 224 nm (Jasco V-630 Made in Japan).27 The stability constant (Kc) was calculated from the phase solubility plot using following equations:26

Loading of BAR into NSPs
Four NSPs formulations (B-CDN1− B-CDN4) were developed by dispersing BAR with blank DPC crosslinked β-CD-NSPs in hydroalcoholic solvent (ethyl alcohol/water, 50:50 v/v) in the molar ratio (1:1, BAR:NSPs) ratio, using ultrasonicator “(Ultrasonic-Water Bath; Daihan Scientific, Model: WUG-D06H, Gangwon, Korea)” for 15 min. The BAR loaded β-CDNSs were then lyophilized, pulverized and stored in a glass-vial for further analysis.
Particle Characterization
Prepared BAR loaded β-CDNSs (B-CDN1-B-CDN4) were characterized for the particle analysis using Malvern particle analyser “Malvern zetasizer (ZEN-3600, Malvern Instruments Ltd., Malvern, UK)” worked on the principle of dynamic light scattering, where in the particles under Brownian motion scattered laser light and particle dimensions measured. The sample under investigation was diluted with Milli-Q water (1:200) and filled in the disposable polystyrene cuvettes (DTS0012), and for the zeta potential (ZP) sample liquid was filled in the cuvettes having two arms tubing fixed with copper strip to conduct current (DTS1070).28 All samples run in triplicate at temperature 25 °C.
DSC Studies
DSC curves of pure BAR, and DPC, β-CD and optimized NSPs (CDN3) were taken by thermoanalytical technique using Scinco N650 (made in Korea). The sample (5 mg) were cramped into the hemispherical aluminium pan, individually, placed beside reference (empty-pan) in the chamber supplied with nitrogen (20 mL/min) and heated at a rate of 20 °C in the temperature range of 50–250 °C.
FTIR Studies
The FTIR spectrums of pure BAR, β-CD, DPC and optimized NNs (CDN3) were taken by triturating these samples with KBr (FTIR grade) and compressed into a transparent pellet using die. The scanning was done in the range of wavenumber 4000 to 400 cm−1, each sample was passed through the IR intensity photons and fingerprints of functional groups present in the chemical entity was interpreted to propose the possible chemical interactions within the drug and β-CD-NS.
In vitro Drug Release Profile
In vitro drug release studies of pure BAR and optimized BAR loaded DPC cross linked βCD NSPs (B-CDN3) were conducted using dialysis bag (Hi-media Mol. 12,000 Dalton) shake flask method. The suspension of samples were sealed in the dialysis membrane and bag is put-up in the conical flask with phosphate buffer pH 6.8 kept at temperature 37 °C with continuous stirring at 100 rpm. At pre-determined time interval 1 mL sample is withdrawn from outer solution at each time interval and replaced by fresh PBS pH 6.8 medium. The aliquots were filtered and analyzed for drug release by using UV-spectrophotometer at 224 nm (Jasco V-630 Made in Japan).27 The experiment was done in triplicate. The drug release data were fitted to various kinetic models, including the zero order, first order, Higuchi, and Korsmeyer-Peppas models, by imposing relationships between the percentage of drug release per unit time, the percentage of log cumulative drug release per unit time, the percentage of log cumulative drug release per unit square root time, and the percentage of log cumulative drug release per unit log time, respectively. Based on the highest value of the correlation coefficient (R2), the best-fit model for the release of the medication was selected. The value of the release-exponent, which suggests the mechanism of drug release, was computed using the slope and R2 values of the plots.29,30
Morphology
The surface morphology of optimized NNs (B-DCN3) was examined by SEM “(Zeiss EVO LS10, Cambridge, UK)” by using the gold-sputter method. The sample was fixed in a stub and coated with gold and imaging was performed.
Brunauer-Emmett-Teller (BET) Analysis
BET analysis of CDN3 (optimized) β-CDNS was performed to determine the surface area, pore volume and pore radius by nitrogen sorption isotherm method. Briefly, CDN3 sample was filled into the glass bulb and heated overnight at 50 °C under negative pressure of 0.1 MPa to completely dry until constant weight was observed (Quantachrome Instruments-version 5.0, Anton Paar, FL, USA). Surface area (SA) of NSPs (CDN3) was calculated with BET theory using isotherm adsorption data at P/P0 from 1.003–5.047 range.
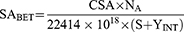
Where SABET is the BET surface area (m2/g), CSA is the analysis gas molecules cross sectional area (0.162 nm2 for nitrogen); NA stands for the Avogadro number; S is the slope (g/cm3); YInt is the intercept (g/cm3)
Moreover, the pore volume and pore size distribution was calculated by Barrett, Joyner and Halenda's (BJH) theory using the following equation.

Where rp is pore radius (Å)
Bio-Analytical Methods
By making a minor change to our previously published UPLC-MS/MS method,20,31 BAR was analyzed in rat plasma samples. BAR and an internal standard (rivaroxaban d-5) were quantified using the precursor to production ion transitions of 372.07 > 251.14 and 440.04 > 4144.9 (quantifier) in the multiple reactions monitoring mode for detection. The cone voltage and impact energy were set to 50 V and 30 eV for BAR and 46 V and 28 eV for IS, respectively, while the capillary voltage was set at 3.9 kV. All parameters were within the allowed range specified in the guideline for bioanalytical method validation before analysis, which followed the validation of the technique.
Pharmacokinetic Studies
Male rats were used in a comparative pharmacokinetic studies between a newly synthesized optimized formulation (NSPs) and a standard BAR solution. Twelve healthy adult male wistar albino rats (n = 6, weight 200–230 g) were used in the experiment. They were randomly assigned to one of two groups: CDN3 formulation (1 mg/kg, P.O.) or BAR suspension dispersed in 0.5% w/v carboxy methyl cellulose. The animals were housed in the suggested settings with access to food and water after being acquired from the “Animal Care Centre, College of Pharmacy, Prince Sattam Bin Abdulaziz University, Alkharj”. The Prince Sattam Bin Abdulaziz University’s Research Ethics Committee reviewed and approved the experimental protocol (approval code: BERC 003-03-21), and NIH Guideline for the Care and Use of Laboratory animals was followed for all animals used in the experiments during the conduct of the study. The animals were fasted overnight, and blood samples (0.5 mL) were taken at specific intervals (0, 0.25, 0.5, 1, 2, 4, 8, 12 and 24 h) after the administration of the appropriate forms in pre-heparinized tubes. The blood samples were safely maintained in deep freezers (−80±10 °C) until UPLC–MS/MS analysis after being centrifuged at 4500 × g for 5 minutes to separate the plasma from the blood. Using “WinNonlin software”, a product of Pharsight Co., Mountain View, California, the non-compartmental pharmacokinetic model was chosen to determine the various pharmacokinetic parameters. Every result was treated as mean standard deviation (SD). The P/K parameters were estimated to include the elimination half-life (T1/2), rate constant (kz), mean residence time (MRT), peak plasma concentration (Cmax), duration to achieve peak concentration (Tmax), area under the curve (AUC), [(AUC0–24) (AUC0–∞)]. Unpaired t-test was employed to compare the outcomes between the NSPs formulation and the standard BAR solution (p ˂ 0.05 was regarded as statistically significant).
Statistical Analysis
The generated data were treated statistically using one-way ANOVA followed by post hoc Tukey’s multiple comparison test using the GraphPad Prism program (version 4) (GraphPad Software, San Diego, CA, USA). p < 0.05 was considered as significant.
Results and Discussion
Phase Solubility Studies
The phase solubility investigation is basic requirement for optimizing the inclusion complex and determining a drug’s affinity with CDs. The molar ratio at which the drug forms a complex with CD has frequently been found using this method. The solubility of BAR increased linearly with increasing the concentration of βCD as shown in Figure 1, which confirmed AL-type system.25,32 The slope value was smaller than 1, which denotes an inclusion complex with molecules in a 1:1 molar ratio (BRA: βCD).26 The apparent stability constant, K1:1, was found to be 119.06 M−1. Therefore, it can be said that BAR and βCD formed a stable inclusion complex at a 1:1 molar ratio. These interaction with BAR were established by the cavity of βCD through intermolecular forces, confirming BAR’s incorporation inside the cavity of –CD.26
 |
Figure 1 Phase solubility plot of βCD and BAR. |
Particle Characterization
The BAR was encapsulated in β-CD NNs using DPC as a crosslinker. Four batches of BAR-loaded DPC-crosslinked βCD NNs (B-CDN1- B-CDN4) were prepared varying the molar concentration of (βCD: DPC; 1:1.5; 1:3; 1:4.5 and 1:6.0). The mean particles size, polydispersity index (PDI), ZP, yield and EE of the developed NNs (B-CDN1- B-CDN4) were determined in the range of 261.9–864.3 nm, 0.335–0.665, −15.8 to –25.2 mV, 89.15–91.46% and 28.7–82.6%, respectively (Table 1). The findings indicate that the size of the NN is significantly influenced by the ratio of βCD:DPC. The findings demonstrated a direct correlation between crosslinker concentration (DPC) and particle size, with the lowest crosslinker concentrations for NN formulations exhibiting the smallest particle size. High ZP values indicate that the NN would be stable because of stronger repulsive forces, which demonstrating that the supramolecular complexes will not assemble over time.33 Among these formulations, B-CDN3 with βCD/DPC molar ratio of 1:4.5 was having better physical characteristics with mean size (345.8±4.7 nm), PDI (0.335±0.005) (Figure 2), Yield (91.46±7.4%) and EE (79.1±1.6%) of B-CDN3 was smaller (345.8±4.7 nm), uniformly distributed with (0.335±0.005), which was further evaluated for spectral analysis, in vitro release and pharmacokinetic studies.
 |
Table 1 Particle Characterization of Developed BAR-Loaded NNs |
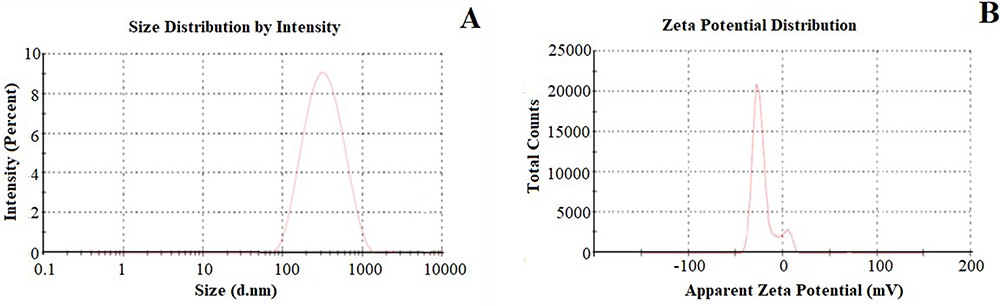 |
Figure 2 Particle size (A) and ZP (B) of optimized NSs (CDN3). |
DSC Studies
The comparative DSC spectra of pure BAR, βCD, DPC and optimized NNs (B-DCN3) were shown in Figure 3. Pure BAR showed an endothermic melting peak at 218.5 °C,27 whereas CDN3 reflects peak in the range of 80–90 °C, which could be due to the DPC, however BAR peak was completely disappeared in the CDN3, indicating the drug was molecularly dispersed in to the porous network of the NNs. Furthermore, this might show how BAR interacts with the NN structure through both the inclusion and non-inclusion phenomena.34 Crystalline drug was deformed to the amorphous nature inside the DPC crosslinked β-CD NN.
 |
Figure 3 Comparative DSC spectra of pure BAR, βCD, DPC and optimized NSs (CDN3). |
FTIR Studies
The comparative FTIR spectra of pure BAR, βCD, DPC and optimized NNs (B-DCN3) were presented in Figure 4. The FT-IR spectrum of pure BAR showed a number of strong absorption bands at wavenumbers of 3203.18 cm−1, 3116.40 cm−1, 2258.23 cm−1, 1873.51 cm−1, and 1442.49 cm−1, which correspond to N-H stretching, =C-H- stretching, -C≡N- stretching, -C=N- stretching, -C=C- (aromatic) stretching, respectively.20,27 The FTIR spectra of βCD exhibited a characteristic peaks at 3365.17 cm−1 and 2927.41 cm−1 for O-H group stretching and C-H stretching, respectively.35 The carbonate bond’s presence in the NNs was clearly verified using the FTIR spectra of optimized NNs (B-DCN3). This demonstrates the βCD tendency to crosslinked by DPC with one another which showed a peak at 1775.15 cm−1.36 The prominent broad peak of βCD could be also seen in optimized NNs (B-DCN3). However, BAR peaks were disappeared in optimized NNs (B-DCN3) that clearly evidenced complete encapsulation.
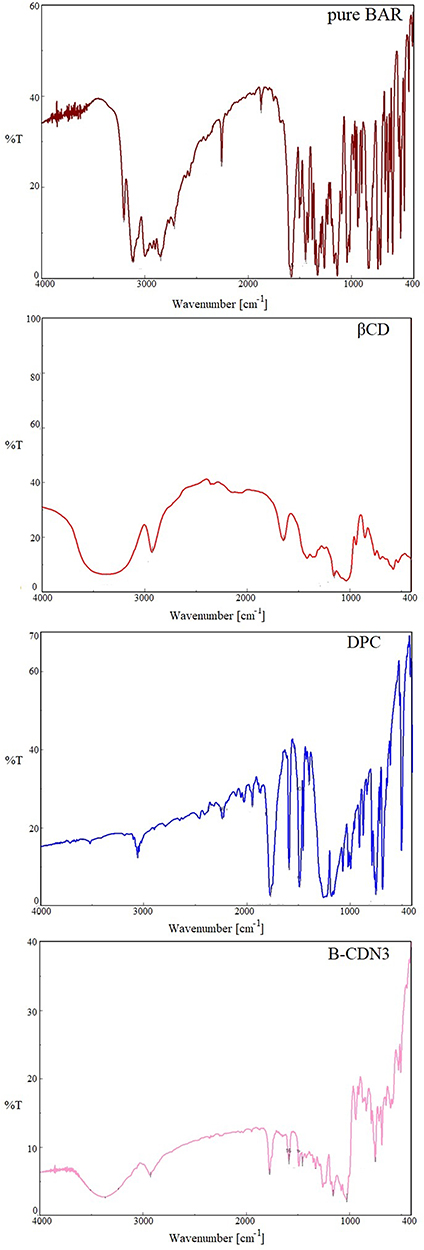 |
Figure 4 Comparative FTIR spectra of pure BAR, βCD, DPC and optimized NSs (CDN3). |
In vitro Drug Release Profile
The in vitro drug release study assesses the probable in vivo performance of dosage forms. The release data were presented as cumulative drug release vs time in hours. The release profile of pure BAR and optimized BAR loaded DPC crosslinked βCD NSPs (B-CDN3) are shown in Figure 5. A significant (p < 0.05) enhancement in release of BAR from optimized NSPs (B-CDN3) indicated as compared to the pure BAR (Figure 5). The optimized NSPs (B-CDN3) exhibited a quick release of drug (61±4.2%) within 3 h compared with pure BAR (40±6.4%). The free BAR showed complete release profile within 12 h of the study. The cross linked βCD present in the NSPs (B-CDN3) resulted in the production of porous, fluffy structure that has a greater capacity to speed up drug release and dissolution.37 The DPC crosslinked βCD NNs structure enhances CD's capacity to form particular complexes with guest molecules, which can be either strongly retained or released in a controlled manner.38,39 The regression coefficient for each of the four release kinetic models had values for the zero order of (0.9551), the first order of (0.9802), the Higuchi model of (0.9270), and the Korsmeyer Peppas model of (0.9912). The in vitro release data of the D-NSP3 formulation fit the Korsmeyer-Peppas kinetic model and had (n) value 0.517, indicating a non-Fickian (anomalous transport) and combination of chain relaxation and diffusion regulates release based on the greatest correlation coefficient (R2 = 0.9912).40–42
 |
Figure 5 Comparative release profile of pure BAR and optimized NSs (CDN3). |
Morphology
The SEM studies examined the morphology of optimized NNs (B-DCN3) (Figure 6). The image demonstrated the firm, rough surface and porous structure of the NSs formulation. This may be attributed to the BAR drug being encapsulated within the porous architecture of the NNs. In the past, reports with comparable morphological pictures and outcomes have also been reported.43
 |
Figure 6 SEM images of optimized NNs (B-DCN3). |
Brunauer-Emmett-Teller (BET) Analysis
The pore size, pore volume and surface area of optimized NN (B-CDN3) was determined by BET analysis using nitrogen adsorption/desorption technique (Figure 7). The B-CDN3 NNs exhibited a pore size (19.117 Å), pore volume (0.009 cc/g) and surface area (2.336 m²/g). This low value of pore size, pore volume and surface area was due to the fact BAR was adhered at the surface as as encapsulated within the pore of NN.31 These NSs release performance in the DPC-crosslinked βCDNSs can be significantly influenced by their particle size. In cases where the particle size is tiny, the system will have more surface area in comparison to samples with equal weights but higher particle sizes.4–46 The development of formulations will be aided by having a solid understanding of both particle size and surface area.
 |
Figure 7 Adsorption and desorption curves of optimized NSs (CDN3). |
In vivo Pharmacokinetic Study
Table 2 displays the pharmacokinetic parameters estimated following oral administration of the NSPs formulation and pure BAR suspension (as reported by a non-compartmental pharmacokinetic analysis). The Tmax, Cmax (**p ˂ 0.001), AUC0–24 (**p ˂ 0.001), and AUC0–∞ (**p ˂ 0.001) values of the optimized NSPs formulation were significantly greater at the administered dose than those of the pure BAR suspension (Figure 8). Because of this, the NSPs formulation had a high capacity for circulation, and as a result, its relative bioavailability was 2.13 times higher than that of pure BAR suspension, the result is so close to our previously prepared BAR-loaded Lipid/Polymer Hybrid Nanoparticles.20,46 Additionally, the T1/2 (6.83±1.1 h) and MRT (7.95±2.8 h) of the NSPs formulation were higher than that of the pure BAR suspension T1/2 (3.15±0.9 h) and MRT (3.77±2.1 h), showing that this formulation not only improves BAR’s bioavailability but also facilitates long-term retention, or sustain release performance. The BAR-loaded NSs exhibited long half-life compared to BAR suspension, indicating that formulation does indeed exert sustained release of BAR.47 However, compared to optimized NSPs formulation, the time to reach the maximum plasma concentration (Cmax) was lower for pure BAR suspension. Overall, the solubility and release of the BAR were improved by NNs carriers, in addition to the pharmacokinetics, which was also greatly improved.
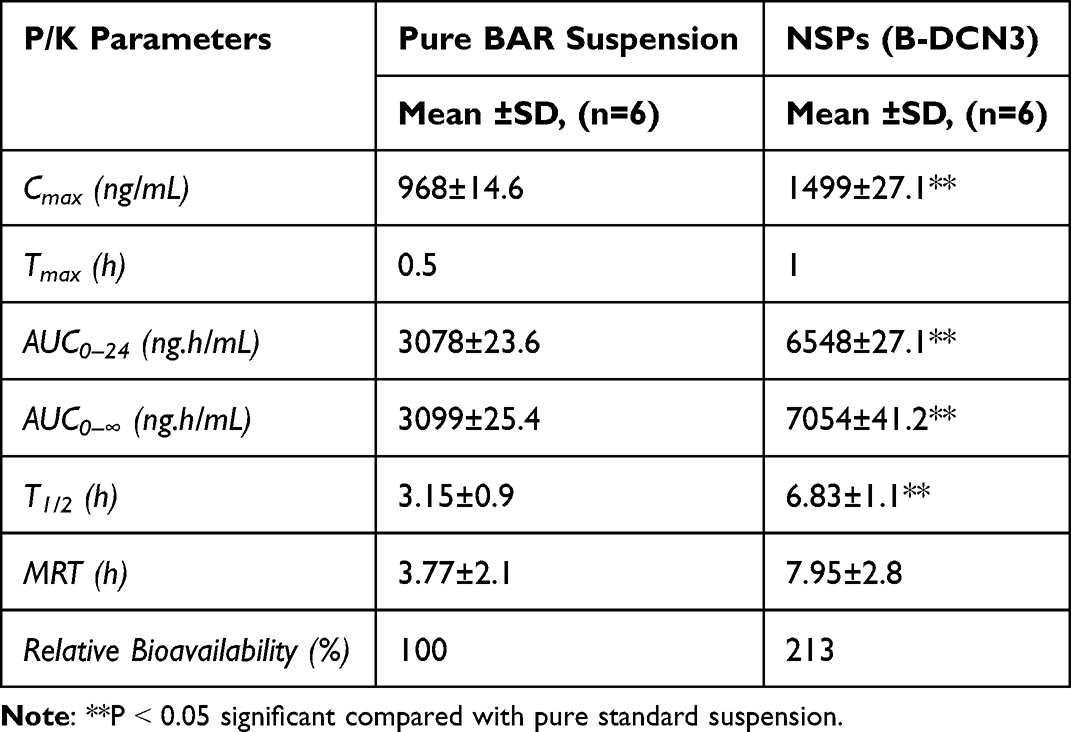 |
Table 2 P/K Parameters After Single Oral Dose of Pure BAR Suspension and NSPs Administration (1 mg/kg in Rats) |
 |
Figure 8 Comparative plasma concentration vs. time profile of pure BAR suspension and optimized NSPs formulation (CDN3). |
Conclusions
The current study aim was to BAR-loaded DPC-crosslinked β-CD NNs have been successfully developed to improve the bioavailability and sustain release the release of BAR. The optimized NNs (B-CDN3) had a suitable size, PDI, ZP and %EE to sustain the release of the drug, which was further confirmed by in vitro release and in vitro pharmacokinetic studies. The pharmacokinetic studies exhibited enhanced bioavailability compared to pure BAR. The optimized NNs (B-CDN3) was therefore shown to have superior therapeutic efficacy due to their porous, nanoscale structure. We conclude from our findings that optimized NNs (B-CDN3) is promising oral delivery for the treatment of arthritis.
Data Sharing Statement
The data presented in this study are available on request from the corresponding author.
Institutional Review Board Statement
The protocol for animal use was approved by the Standing Committee of Bioethics Research (SCBR) at Prince Sattam bin Abdulaziz University, Deanship of Scientific Research. (Approval No. SCBR-030-2022).
Author Contributions
All authors made a significant contribution to the work reported, including conception, study design, execution and acquisition of data, analysis, and interpretation. All authors took part in drafting, revising, or critically reviewing the article; gave final approval of the version to be published; have agreed on the journal to which the article has been submitted; and agree to be accountable for all aspects of the work.
Funding
This project was funded by the Deanship of Scientific Research at Prince Sattam Bin Abdulaziz University under the research project (PSAU-2022/03/20768).
Disclosure
The authors declare no conflicts of interest.
References
1. Nezamololama N, Fieldhouse K, Metzger K, Gooderham M. Emerging systemic JAK inhibitors in the treatment of atopic dermatitis: a review of abrocitinib, baricitinib, and upadacitinib. Drugs Context. 2020;9:8. doi:10.7573/dic.2020-8-5
2. Jorgensen SCJ, Tse CLY, Burry L, Dresser LD. Baricitinib: a review of pharmacology, safety, and emerging clinical experience in COVID‐19. Pharmacotherapy. J Hum Pharmacol Drug Ther. 2020;40:843–856. doi:10.1002/phar.2438
3. Nogueira M, Torres T. Janus kinase inhibitors for the treatment of atopic dermatitis: focus on abrocitinib, baricitinib, and upadacitinib. Dermatol Pract Concept. 2021;11:e2021145. doi:10.5826/dpc.1104a145
4. Tucci G, Garufi C, Pacella I, et al. Baricitinib therapy response in rheumatoid arthritis patients associates to STAT1 phosphorylation in monocytes. Front Immunol. 2022;13:932240. doi:10.3389/fimmu.2022.932240
5. Genovese MC, Kremer J, Zamani O, et al. Baricitinib in patients with refractory rheumatoid arthritis. N Engl J Med. 2016;374:1243–1252. doi:10.1056/NEJMoa1507247
6. Assadiasl S, Fatahi Y, Mosharmovahed B, Mohebbi B, Nicknam MH. Baricitinib: from rheumatoid arthritis to COVID-19. J Clin Pharmacol. 2021;61:1274–1285. doi:10.1002/jcph.1874
7. Taylor PC, Lee YC, Fleischmann R, et al. Achieving pain control in rheumatoid arthritis with baricitinib or adalimumab plus methotrexate: results from the RA-BEAM trial. J Clin Pharmacol. 2021;8:831. doi:10.3390/jcm8060831
8. Guo Q, Wang Y, Xu D, Nossent J, Pavlos NJ, Xu J. Rheumatoid arthritis: pathological mechanisms and modern pharmacologic therapies. Bone Res. 2018;6(1):15. doi:10.1038/s41413-018-0016-9
9. Favalli EG, Biggioggero M, Maioli G, Caporali R. Baricitinib for COVID-19: a suitable treatment? Lancet Infect Dis. 2020;20:1012–1013. doi:10.1016/S1473-3099(20)30262-0
10. Magro G. SARS-COV-2 and COVID-19: is interleukin-6 (IL-6) the ‘culprit lesion’ of ARDS onset? What is there besides tocilizumab? SGP130FC. Cytokine X. 2020;2:100029. doi:10.1016/j.cytox.2020.100029
11. Bewley A, Kwon O, Zlotogorski A, et al. P79: efficacy of baricitinib in adults with alopecia areata: patients’ perspectives on hair regrowth from two Phase III randomized controlled trials (brave‐Aa1 and brave‐Aa2). Br J Dermatol. 2022;187:70–71. doi:10.1111/bjd.21201
12. Mahmoud AM. Effectiveness and safety of baricitinib in patients with alopecia areata: a systematic review and meta-analysis of randomized controlled trials. Curr Med Res Opin. 2022;1–9. doi:10.1080/03007995.2022.2135838
13. Jabbari A, Dai Z, Xing L, et al. Reversal of alopecia areata following treatment with the JAK1/2 inhibitor baricitinib. EBioMedicine. 2015;2:351–355. doi:10.1016/j.ebiom.2015.02.015
14. Reddig A, Voss L, Guttek K, Roggenbuck D, Feist E, Reinhold D. Impact of different JAK inhibitors and methotrexate on lymphocyte proliferation and DNA damage. J Clin Med. 2021;10:1431. doi:10.3390/jcm10071431
15. Costanzo G, Firinu D, Losa F, Deidda M, Barca MP, Del Giacco S. Baricitinib exposure during pregnancy in rheumatoid arthritis. Ther Adv Musculoskelet Dis. 2020;12:1759720X19899296. doi:10.1177/1759720X19899296
16. Mucke J, Simon HU, Rüdiger Burmester G. The safety of antirheumatic drugs. Dtsch Arztebl Int. 2022;119:81–87. doi:10.3238/arztebl.m2022.0064
17. Ahmad A, Zaheer M, Balis FJ. Baricitinib. In: StatPearls. Treasure Island (FL): StatPearls Publishing; 2022. Available from: https://www.ncbi.nlm.nih.gov/books/NBK572064/.
18. Mohammadi-Meyabadi R, Beirampour N, Garrós N, et al. Assessing the solubility of baricitinib and drug uptake in different tissues using absorption and fluorescence spectroscopies. Pharmaceutics. 2022;14(12):2714. doi:10.3390/pharmaceutics14122714
19. Alshahrani SM, Shakeel F. Solubility data and computational modeling of baricitinib in various (DMSO + water) mixtures. Molecules. 2020;25:2124. doi:10.3390/molecules25092124
20. Anwer MK, Ali EA, Iqbal M, et al. Development of sustained release baricitinib loaded lipid-polymer hybrid nanoparticles with improved oral bioavailability. Molecules. 2022;27:168. doi:10.3390/molecules27010168
21. Garibyan A, Delyagina E, Agafonov M, Khodov I, Terekhova I. Effect of pH, temperature and native cyclodextrins on aqueous solubility of baricitinib. J Mol Liq. 2022;360:119548. doi:10.1016/j.molliq.2022.119548
22. Davis M, Brewster M. Cyclodextrin-based pharmaceutics: past, present and future. Nat Rev Drug Discov. 2004;3:1023–1035. doi:10.1038/nrd1576
23. Sherje AP, Dravyakar BR, Kadam D, Jadhav M. Cyclodextrin-based nanosponges: a critical review. Carbohydr Polym. 2017;173:37–49. doi:10.1016/j.carbpol.2017.05.086
24. Utzeri G, Matias PMC, Murtinho D, Valente AJM. Cyclodextrin-based nanosponges: overview and opportunities. Front Chem. 2022;10:859406. doi:10.3389/fchem.2022.859406
25. Anwer MK, Ahmed MM, Aldawsari MF, Iqbal M, Kumar V. Preparation and evaluation of diosmin-loaded diphenylcarbonate-cross-linked cyclodextrin nanosponges for breast cancer therapy. Pharmaceuticals. 2023;16(1):19. doi:10.3390/ph16010019
26. Ahad A, Jardan YAB, Raish M, Al-Mohizea AM, Al-Jenoobi FI. Ternary inclusion complex of sinapic acid with hydroxypropyl-β-cyclodextrin and hydrophilic polymer prepared by microwave technology. Processes. 2022;10(12):2637. doi:10.3390/pr10122637
27. Ansari MJ, Alshahrani SM. Nano-encapsulation and characterization of baricitinib using poly-lactic-glycolic acid co-polymer. Saudi Pharm J. 2019;27:491–501. doi:10.1016/j.jsps.2019.01.012
28. Ahmed MM, Fatima F, Anwer MK, Ansari MJ, Das SS, Alshahrani SM. Development and characterization of ethyl cellulose nanosponges for sustained release of brigatinib for the treatment of non-small cell lung cancer. J Polym Eng. 2020;40:823–832. doi:10.1515/polyeng-2019-0365
29. Ghose A, Nabi B, Rehman S, et al. Development and evaluation of polymeric nanosponge hydrogel for terbinafine hydrochloride: statistical optimization, in vitro and in vivo studies. Polymers. 2020;12(12):2903. doi:10.3390/polym12122903
30. Ahmed MM, Fatima F, Anwer MK, et al. Formulation and in vitro evaluation of topical nanosponge-based gel containing butenafine for the treatment of fungal skin infection. Saudi Pharm J. 2021;29(5):467–477. doi:10.1016/j.jsps.2021.04.010
31. He Y, Majid K, Maqbool M;, et al. Formulation and characterization of lornoxicam-loaded cellulosic-microsponge gel for possible applications in arthritis. Saudi Pharm J. 2020;28:994–1003. doi:10.1016/j.jsps.2020.06.021
32. Anwer MK, Jamil S, Ansari MJ, et al. Water soluble binary and ternary complexes of diosmin with β-cyclodextrin: spectroscopic characterization, release studies and anti-oxidant activity. J Mol Liq. 2014;199:35–41. doi:10.1016/j.molliq.2014.08.012
33. Salazar S, Yutronic N, Kogan MJ, Jara P. Cyclodextrin nanosponges inclusion compounds associated with gold nanoparticles for potential application in the photothermal release of melphalan and cytoxan. Int J Mol Sci. 2021;22:6446. doi:10.3390/ijms22126446
34. Ansari KA, Vavia PR, Trotta F, Cavalli R. Cyclodextrin-based nanosponges for delivery of resveratrol: in vitro characterisation, stability, cytotoxicity and permeation study. AAPS PharmSciTech. 2011;12:279–286. doi:10.1208/s12249-011-9584-3
35. Rachmawati H, Edityaningrum CA, Mauludin R. Molecular inclusion complex of curcumin-β-cyclodextrin nanoparticle to enhance curcumin skin permeability from hydrophilic matrix gel. AAPS PharmSciTech. 2013;14:1303–1312. doi:10.1208/s12249-013-0023-5
36. Pawan D, Sharma HK, Shah S, Tyagi CK, Chandekar AR, Jadon RS. Formulations and evaluation of cyclodextrin complexed ceadroxil loaded nanosponges. Int J Drug Deliv. 2017;9:84. doi:10.5138/09750215.2180
37. Swaminathan S, Vavia PR, Trotta F, et al. Structural evidence of differential forms of nanosponges of beta-cyclodextrin and its effect on solubilization of a model drug. J Incl Phenom Macrocycl Chem. 2013;76:201–211. doi:10.1007/s10847-012-0192-y
38. Trotta F, Zanetti M, Cavalli R. Cyclodextrin-based nanosponges as drug carriers. Beilstein J Org Chem. 2012;8:2091–2099. doi:10.3762/bjoc.8.235
39. Trotta F. Cyclodextrin nanosponges and their applications. In: Bilensoy E, editor. Cyclodextrins in Pharmaceutics, Cosmetics and Biomedicine: Current and Future Industrial Applications. Hoboken, NJ, USA: John Wiley & Sons; 2011:323–342.
40. Ritger PL, Peppas NA. A simple equation for description of solute release I. Fickian and non-fickian release from non-swellable devices in the form of slabs, spheres, cylinders or discs. J Controlled Release. 1987;5:23–36. doi:10.1016/0168-3659(87)90034-4
41. Ritger PL, Peppas NA. A simple equation for description of solute release II. Fickian and anomalous release from swellable devices. J Controlled Release. 1987;5:37–42. doi:10.1016/0168-3659(87)90035-6
42. Kalam MA, Iqbal M, Alshememry A, Alkholief M, Alshamsan A. Development and evaluation of chitosan nanoparticles for ocular delivery of tedizolid phosphate. Molecules. 2022;27(7):2326. doi:10.3390/molecules27072326
43. Ahmed MM, Anwer MK, Fatima F, et al. Development of ethylcellulose based nanosponges of apremilast: in vitro and in vivo pharmacokinetic evaluation. Lat Am J Pharm. 2020;39:1292–1299.
44. Martwong E, Chuetor S, Junthip J. Adsorption of paraquat by poly(vinyl alcohol)-cyclodextrin nanosponges. Polymers. 2021;13(23):4110. doi:10.3390/polym13234110
45. Almutairy BK, Alshetaili A, Alali AS, Ahmed MM, Anwer MK, Aboudzadeh MA. Design of olmesartan medoxomil-loaded nanosponges for hypertension and lung cancer treatments. Polymers. 2021;13(14):2272. doi:10.3390/polym13142272
46. Anwer MK, Ali EA, Iqbal M, et al. Development of chitosan-coated PLGA-based nanoparticles for improved oral olaparib delivery: in vitro characterization, and in vivo pharmacokinetic studies. Processes. 2022;10(7):1329. doi:10.3390/pr10071329
47. Kim YH, Ghim JL, Jung JA, et al. Pharmacokinetic comparison of sustained- and immediate-release formulations of cilostazol after multiple oral doses in fed healthy male Korean volunteers. Drug Des Devel Ther. 2015;9:3571–3577. doi:10.2147/DDDT.S86845
 © 2023 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2023 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.