")
Back to Journals » International Journal of Nanomedicine » Volume 12
Development of coated liposomes loaded with ghrelin for nose-to-brain delivery for the treatment of cachexia
Authors Salade L, Wauthoz N , Deleu M, Vermeersch M, De Vriese C, Amighi K, Goole J
Received 29 July 2017
Accepted for publication 9 September 2017
Published 28 November 2017 Volume 2017:12 Pages 8531—8543
DOI https://doi.org/10.2147/IJN.S147650
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Dr Thomas Webster
Laurent Salade,1 Nathalie Wauthoz,1 Magali Deleu,2 Marjorie Vermeersch,3 Carine De Vriese,1 Karim Amighi,1 Jonathan Goole1
1Laboratoire de Pharmacie Galénique et de Biopharmacie, Université libre de Bruxelles (ULB), Brussels, 2Laboratoire de Biophysique Moléculaire aux Interfaces, Gembloux Agro-Bio Tech, Université de Liège, Gembloux, 3Centre for Microscopy and Molecular Imaging (CMMI), Charleroi, Belgium
Abstract: The aim of the present study was to develop a ghrelin-containing formulation based on liposomes coated with chitosan intended for nose–brain delivery for the treatment of cachexia. Among the three types of liposomes developed, anionic liposomes provided the best results in terms of encapsulation efficiency (56%) and enzymatic protection against trypsin (20.6% vs 0% for ghrelin alone) and carboxylesterase (81.6% vs 17.2% for ghrelin alone). Ghrelin presented both electrostatic and hydrophobic interactions with the anionic lipid bilayer, as demonstrated by isothermal titration calorimetry. Then, anionic liposomes were coated with N-(2-hydroxy)propyl-3-trimethyl ammonium chitosan chloride. The coating involved a size increment from 146.9±2.7 to 194±6.1 nm, for uncoated and coated liposomes, respectively. The ζ-potential was similarly increased from -0.3±1.2 mV to 6±0.4 mV before and after coating, respectively. Chitosan provided mucoadhesion, with an increase in mucin adsorption of 22.9%. Enhancement of permeation through the Calu3 epithelial monolayer was also observed with 10.8% of ghrelin recovered in the basal compartment in comparison to 0% for ghrelin alone. Finally, aerosols generated from two nasal devices (VP3 and SP270) intended for aqueous dispersion were characterized with either coated or uncoated liposomes. Contrarily to the SP270 device, VP3 device showed minor changes between coated and uncoated liposome aerosols, as shown by their median volume diameters of 38.4±5.76 and 37.6±5.74 µm, respectively. Overall, the results obtained in this study show that the developed formulation delivered by the VP3 device can be considered as a potential candidate for nose–brain delivery of ghrelin.
Keywords: nasal delivery, peptide, liposome, cachexia, brain targeting, enzyme
Introduction
Cachexia is defined as “weight loss, wasting of muscle, loss of appetite, and general debility that can occur during a chronic disease”.1 This pathologic syndrome is frequently associated with such diseases as cancers, heart failure, or chronic renal failure.2 For several years, much attention has been paid to ghrelin (Ghrl)3,4 for the management of cachexia. Ghrl is a naturally occurring orexigenic peptide hormone that is secreted mainly by the stomach and intestine into the blood.5 Ghrl is characterized by a cationic charge and an octanoyl group positioned on Ser3 residue.6 This fatty-acid chain confers relative hydrophobicity to the peptide and allows it to bind specific receptors located into the hypothalamus, known as growth-hormone secretagogue receptors (GHSR).7 Ghrl stimulates food intake via GHSR1As, thus inducing a release of orexigenic neuropeptides from hypothalamic neurons.8 Ghrl also causes a reduction in inflammatory cytokine levels involved in cachexia.9
In the body, this peptide hormone is subject to very rapid degradation, due to hepatic first-pass, plasmatic enzyme digestion (mainly by plasmatic butyrylcholinesterase),10 rapid renal elimination and pH-sensitive degradation (pH above physiological pH). These lead to a very short plasmatic half-life (t½ of acylated Ghrl in human plasma is about 9–13 minutes).11
In this context, the intranasal delivery of Ghrl through the nose–brain pathway could be promising. This pathway is characterized by direct transfer from the nose to the brain via intra or perineural routes through olfactory and/or trigeminal neurons.12 Nose–brain could be advantageously used for chronic brain administration of large and sensitive compounds, such as biotherapeutics.
However, nose–brain delivery suffers from major limitations. These include mucociliary clearance, enzymatic degradation, and limited permeation of large molecules.13 Therefore, a formulation able to protect Ghrl from nasal enzymes, increase its residence in the nasal cavity, and improve its transfer through the olfactory epithelium could be promising in the management of cachexia. To our knowledge, only a limited number of studies have focused on the development of formulations containing Ghrl, and these were intended for parenteral administration.14 The intravenous route, which is rather cumbersome for chronic administration,15 has been the most exploited for Ghrl administration,16 while intranasal delivery has been considered very rarely. A recently published study was focused on the nasal administration of a vaccine containing Ghrl-antigen for the management of obesity, but this was not intended for nose–brain delivery.17 Other studies have focused on chemical modifications of the peptide chain or on the use of GHSR agonists (eg, anamorelin) to gain better bioavailability and/or stability compared to physiological Ghrl.18,19 As such, the development of such a nose–brain treatment containing Ghrl has never been considered, and would provide better compliance (thanks to the noninvasiveness) associated with higher efficacy (eg, by avoiding plasmatic enzyme degradation). This could very clearly improve the prognosis of major pathologies (eg, cancer or heart failure) associated with this syndrome.
In this work, the formulation strategy involved liposomes, due to their potential application for nose–brain delivery20,21 and potential protection against enzymatic degradation. Chitosan, which has been well studied in the context of nasal delivery,22–24 was the second excipient selected, due to its permeation-enhancing effect and mucoadhesive properties. In this first study, the formulation was developed and optimized on the basis of the strategy described and characterized. The main objective was to study the formulation deeply before going further on its characterization in animals.
Materials and methods
Materials
Synthetic human octanoylated (1–28 amino-acid sequence, purity >98%) Ghrl was purchased from Scipeptide (Shanghai, China). Acetonitrile, methanol, trifluoroacetic acid (TFA), dichloromethane (all solvents high-performance liquid chromatography [HPLC] grade), dihexadecyl phosphate (DHDP; purity ≥98%), human isoform B carboxylesterase 1 (Ces1; activity ≥500 units/mg protein), cholesterol (purity ≥99%) and mucin type 1S were obtained from Sigma-Aldrich (St Louis, MO, USA). Minimal essential medium nonessential amino acids (MEM NEAAs), gentamicin 50 mg/mL, sodium pyruvate 100 mM, heat-inactivated FBS, L-glutamine 200 mM, 0.25% trypsin (Tryp)–EDTA phenol red, Hank’s balanced salt solution (HBSS) and penicillin–streptomycin were obtained from Thermo Fisher Scientific (Waltham, MA, USA). Soybean phosphatidylcholine (Lipoid S100) and 1,2-dioleoyl-3-trimethylammonium-propane (DOTAP; chloride salt) were purchased from Lipoid GmbH (Ludwigshafen, Germany). N-([2-hydroxy-3-trimethylammonium]propyl) chitosan chloride (HTCC) with 92 kDa molecular weight, deacetylation of 80%, and substitution of 33% was purchased from KitoZyme (Herstal, Belgium).
Ghrl-stability evaluation
Ghrl (1 mg/mL) solutions in Milli-Q water (PureLab system; Elga LabWater, High Wycombe, UK), phosphate buffered saline (PBS) pH 7.4, and HBSS pH 7.4 were exposed to various temperatures at 4°C, 25°C, 37°C, and 60°C and different pH levels of 2–10.4 (each buffer was exposed to all temperatures). The buffers were selected according to the requirements of the European Pharmacopoeia eighth edition (PBS pH 2, acetate buffer pH 4.4, sodium dihydrogen phosphate buffer pH 6, PBS pH 7.4, boric acid buffer pH 9, and borate buffer pH 10.4). Then, 500 μL samples were drawn at predefined times (0.5, 1, 2, 3, 4, 5, 6, 24, and 48 hours) to evaluate the potential kinetics of Ghrl degradation using a suitable HPLC/ultraviolet (UV) method.25
Preparation of liposomes and HTCC-coated liposomes
The lipid film-rehydration protocol was selected for liposome preparation.26 Briefly, 100 mg lipid mixture was introduced into a 50 mL round-bottomed flask. Neutral liposomes (NLs) were obtained using a mixture of neutral lipids composed of cholesterol and Lipoid S100 50%:50% (w:w). Replacing 10% w:w of neutral lipids with DHDP or DOTAP allowed the formation of anionic liposomes (ALs) or cationic liposomes (CLs), respectively (Table 1). Then, 10 mL of an organic solvent mixture (dichloromethane:methanol 50%:50% v:v) was added to obtain a final lipid concentration of 10 mg/mL. The organic phase was evaporated using a Rotavapor R-205 (Büchi Labortechnik, Flawil, Switzerland) at 60°C. Pressure was set at 250 mmHg for 10 minutes and 150 mmHg for the next 10 minutes. The lipid film was then rehydrated at 30°C for 1 hour with 10 mL PBS pH 7.4, which contained Ghrl (1 mg/mL). Before extrusion, large multilamellar vesicles were briefly sonicated by means of a VCX 500 probe sonicator (Vibra-Cell; Sonics and Materials, Newton, CT, USA) for 30 seconds with 30% amplitude. The large multilamellar vesicles were then extruded through a LiposoFast LF-50 extruder (Avestin, Ottawa, ON, Canada) to produce large unilamellar vesicles. The extrusion process was carried out through polycarbonate membranes characterized by porosities of 1, 0.4, and 0.1 μm (EMD Millipore, Billerica, MA, USA).
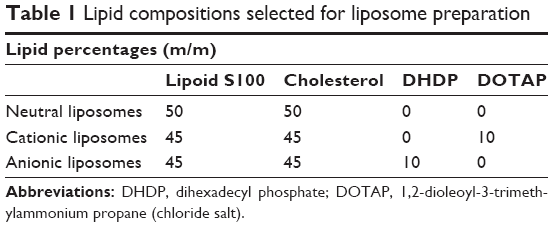 | Table 1 Lipid compositions selected for liposome preparation |
For preparation of HTCC-coated ALs (HTCC-ALs), a suitable amount of HTCC was weighed and dispersed under magnetic stirring in PBS pH 7.4 overnight to obtain a final solution (10 mg/mL) that was translucent and homogenous. Then, 9 mL ALs (10 mg/mL) were coated with 1 mL HTCC (10 mg/mL), which was added dropwise under magnetic stirring at 3,000 rpm to obtain a tenfold dilution of the initial HTCC solution (1 mg/mL). The dispersion of coated-liposomes was left for 1 hour under magnetic stirring before being left overnight at 4°C.
Evaluation of size, ζ-potential, and encapsulation efficiency
Z-average and ζ-potential of ALs, NLs, CLs, and HTCC-ALs containing or not containing Ghrl (1 mg/mL) were evaluated in triplicate at 25°C, with 1 minute of equilibration time and without dilution. Evaluations were done using dynamic light scattering and electrophoretic mobility, respectively (Zetasizer Nano ZS; Malvern Instruments, Malvern, UK). Size measurements were performed with “semimicro” disposable cuvettes in polystyrene (Brand GmbH, Wertheim, Germany), and results expressed in terms of Z-average (means ± SD) and polydispersity index (PDI). Disposable folded capillary cells were used for ζ-potential evaluation, and results expressed in terms of means ± SD.
To determine the Ghrl loading of liposomes, suspensions were centrifuged at 13,500 rpm through Amicon Ultra-15 centrifugal tubes with a 100 kDa cutoff (Merck KGaA, Darmstadt, Germany) for 30 minutes at 25°C. Additionally, a PBS pH 7.4 solution of unloaded Ghrl was centrifuged to confirm the nonadsorption of the peptide on the filter. The total amount of Ghrl (Ghrltotal) that was contained in the initial liposome suspensions was determined by HPLC. Following the solubilization of liposomes (confirmed by dynamic light scattering) in the mobile phase, Ghrl was released for quantification. The encapsulation efficiency (EE) of Ghrl in ALs, NLs, and CLs was determined indirectly by quantifying Ghrl in the filtrate and using the equation:
|
|
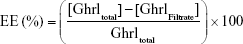
Tests were performed in triplicate, and percentages expressed as means ± SD.
HPLC/UV method for Ghrl quantification
The HPLC method adapted from the literature25 used a TSKgel ODS-120T 150×4.6 mm, 5 μm particle column coupled with a TSKgel ODS-120T 3.2×1.5 cm (Tosoh Bioscience, Tokyo, Japan) guard gel. The column was kept at 37°C. The mobile phase was a mixture of Milli-Q water–TFA 0.1% v:v (phase A) and acetonitrile–TFA 0.1% v:v (phase B). The mobile-phase gradient used was linear, from 12% (0 minutes) to 52% (32 minutes) in phase B. The wavelength was fixed at 210 nm, and the flow rate was set at 1 mL/min. The lower limit of quantification was 5 μg/mL. The HPLC system used was a 1100 series from Agilent (Santa Clara, CA, USA).
Enzymatic protection after trypsin and carboxylesterase-1 exposure
Both Ghrl in solution and Ghrl-loaded (1 mg/mL) ALs, NLs, and CLs (10 mg/mL) were exposed to Tryp solution (140 IU/mL) for 15 minutes at 37°C after dispersion in a vortex mixer (Vortex Genius 3; IKA, Staufen, Germany). Enzymatic digestion was stopped by the addition of 100 μL TFA (10% v:v). Ghrltotal was determined in the initial liposome suspensions using HPLC. The mobile phase containing acetonitrile allowed the destruction of liposomes and the release of Ghrl for the quantification of Ghrltotal. The remaining amount of nondegraded Ghrl was determined by withdrawing 500 μL solution for HPLC quantification. The percentage of Ghrl protected by liposomes was determined thus:
|
|
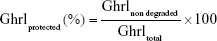
To get a better idea of the Ghrl position with regard to ALs, the lipid-film rehydration protocol was used and Ghrl (1 mg/mL) added during rehydration or after the rehydration step (postformation). If rehydration allowed Ghrl to access the AL internal aqueous compartments, it would be less impacted by enzymatic digestion than ALs produced by postformation. Indeed, Ghrl had more difficultly accessing the AL internal aqueous compartment and was only adsorbed at the exterior of liposomes when added after AL formation. Both formulations were then exposed to Tryp degradation for 15 minutes at 37°C. The remaining Ghrl was determined using HPLC/UV.
After this, ALs were exposed to a second enzyme: Ces1 (124 IU/mL, PBS pH 7.4, for 15 minutes at 37°C). The effective protection of loaded Ghrl was determined using the same protocol as for Tryp. The remaining Ghrl was determined by withdrawing 500 μL suspension for HPLC/UV analysis and using Equation 2. All experiments were repeated in triplicate, and are expressed as means ± SD.
Isothermal titration calorimetry
In order to study the interaction between Ghrl and ALs, isothermal titration calorimetry (ITC) experiments were performed as previously described.27 All measurements were performed at 25°C using a MicroCal VP-ITC (Malvern Instruments) with a sample cell volume of 1.4565 mL. Solutions were degassed in a sonic bath for 10 minutes prior to use. A 300 μL syringe was filled with a suspension of AL-free (anionic liposome without Ghrl) PBS 10 mg/mL, pH 7.4. The reference cell was loaded with Milli-Q water. For the blank and the sample, the measuring cell was loaded with PBS pH 7.4 and a 1 mg/mL Ghrl solution in PBS pH 7.4, respectively. The measuring cell was magnetically stirred at 300 rpm during the experiments. Titration experiments were performed by consecutive injections of 10 μL AL-free (10 mg/mL) into Ghrl solution (1 mg/mL). Each injection lasted 14.5 seconds. A delay of 600 seconds between each injection was applied until a steady state was reached. The resulting heat flows were recorded and raw data processed using the software provided by the manufacturer (Origin version 7; OriginLab, Northampton, MA, USA).
Raw ITC data were processed according to the cumulative model previously described28 and applied to molecules other than Ghrl27,29 to obtain the thermodynamic parameters characteristic of the interaction of Ghrl–AL-free. Thermodynamic parameters considered were binding coefficient (K), variation in Gibbs free energy (ΔG), enthalpy (ΔH), and entropy (TΔS). If the Ghrl–AL-free interaction is spontaneous, ΔG should be negative. The titration was repeated twice and expressed as the mean value.
Transmission electron microscopy
Here, 20 μL ALs and HTCC-ALs were deposited on Formvar carbon-coated electron-microscopy grids. Then, 1–2 μL glutaraldehyde 25% v:v was added to fix the liposomes. The preparation was left overnight at 4°C. Grids were transferred onto a drop of distilled water for washing and left for 2 minutes (three times). For contrasting and embedding, grids were placed onto a drop of methylcellulose–uranyl acetate (ratio 9:1 m/m) mixture for 10 minutes in an ice bath. The grids were removed and the excess fluid blotted by gently pushing the loop sideways on filter paper. A thin film was left over the section side of the grids. Observations were performed using a Tecnai 10 electron microscope (FEI, Hillsboro, OR, USA) operating at an accelerating voltage of 100 kV. Images were analyzed and processed using transmission electron microscopy (TEM) analysis (Olympus Soft Imaging Solutions, Münster, Germany).
Evaluation of osmolality and determination of mucoadhesion
Osmolality was determined in triplicate with an Osmomat 3000 (Gonotec, Berlin, Germany). The instrument used the freezing point to evaluate the osmolality. A volume of 50 μL ALs or HTCC-ALs was used to determine the value. For assessing mucoadhesion, a colorimetric method was used to determine the residual amount of free mucins that were not precipitated due to complexation with the formulation.30 A defined volume of 2 mL HTCC-ALs or ALs was added to 6 mL mucin solutions (0.5 mg/mL). The preparation was left for 20 minutes (similar to mucociliary clearance time)31 at 37°C and then centrifuged at 3,000 g for 4 minutes. Then, 2 mL supernatant was withdrawn to determine the amount of free mucin. The percentage of mucins fixed by the formulation was determined indirectly by subtracting the quantity of unfixed mucins in the supernatant from the initial quantity of mucin (0.5 mg/mL), and expressed as a percentage:
|
|
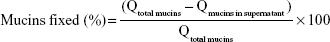
The test was performed in triplicate, and results are expressed as means ± SD.
Permeation through Calu3 monolayer
Frozen Calu3 cells obtained from ATCC (Manassas, VA, USA) were used with passages 15–20. The culture medium used was MEM NEAAs with 1 mL gentamicin (50 mg/mL), 5 mL sodium pyruvate (100 mM), 50 mL heat-inactivated FBS, 5 mL L-glutamine (200 mM), and 5 mL penicillin–streptomycin. The medium was renewed every couple of days. Cells were subcultured with a dilution factor of 1:3 from flask to flask. Cells were maintained at 37°C and 95% O2/5% CO2.
Inserts used to establish the air–liquid interface culture were composed of HA-mixed ester cellulose membranes with 0.45 μm porosity and a surface of 4.2 cm2 adapted for six-well plates (Merck). For inoculation in inserts, Calu3 cells from confluent T75 flasks were detached by means of a Tryp 0.25% w:v solution and suspended in 12 mL thawed culture medium. To the apical side of each insert, 2 mL cell suspension was added to get a fully filled six-well plate of inserts (5×105 cells/cm2). The basal side was then filled with 2 mL thawed MEM NEAAs. The apical compartment was emptied 24 hours after inoculation and the basal side medium renewed to allow the growth and polarization of the cells under the air–liquid interface.
Transepithelial electrical resistance (TEER) was monitored during the cell experiments by means of an epithelial volt/ohm meter (EVOM2; World Precision Instruments, Sarasota, FL, USA). TEER measurements reflect cell-monolayer integrity. Values are expressed after subtracting the value of the blank insert and normalizing for surface area (4.2 cm2). For routine TEER evaluations, 2 mL fresh medium was added to both the apical and the basal sides. Cells were allowed to equilibrate for 30 minutes prior to taking measurements. A polarized monolayer was obtained approximately 10 days after inoculation.
Before adding solutions or formulations of interest to the apical compartment, the monolayer was washed twice with HBSS pH 7.4. Then, 2 mL HBSS pH 7.4 were added to both the apical and the basal side. Cells were allowed to equilibrate for 30 minutes. TEER was then measured, and HBSS pH 7.4 from the apical side was replaced with the formulation or solution. Each formulation or solution was introduced in three different inserts to perform the test in triplicate. The compounds of interest were allowed to diffuse for 3 hours, and TEER was assessed at specific times during the experiments (ie, 30, 90, 150, and 180 minutes) and 24 hours after the end of the test to assess the recovery of the cells. After the 3 hours’ diffusion, 500 μL apical and basal medium each was sampled and quantified.
For the first permeation study, caffeine (Caf; 1 mg/mL), salmon calcitonin (Cal; 1 mg/mL), and Ghrl (1 mg/mL) solutions were dropped onto the apical side with or without solubilized HTCC (1 mg/mL). Caf,32 Cal,33 and Ghrl25 were then quantified using HPLC/UV. For the second part of the Calu3 experiments, both AL and HTCC-AL permeations were studied and compared.
Droplet-size distribution
The size distribution of the droplets generated from nasal devices was evaluated by laser diffraction using a Spraytec apparatus (Malvern Instruments). Liquid multidose spray VP3 pumps were kindly provided by Aptar Pharma (Le Vaudreuil, France). The VP3 device is presented as offering a high dose accuracy (volume delivered 93 μL per spray), even with viscosity changes that can be encountered with HTCC. The device is also suitable for suspensions and viscous formulations. The results generated with this device were compared to those collected from another standard device, the SP270 (Rexam Pharma, La Verpillière, France). Before each evaluation, nasal devices were manually shaken and the first five doses generated discarded in order to initiate the device. Spraytec parameters were fixed, with test duration of 3,000 milliseconds, data-acquisition rate of 1,000 Hz, and transmittance of 96%. Measurements were performed at room temperature and expressed in terms of median volume diameter (Dv50), mean Dv (Dv4,3) and percentage of volume distribution <10 μm.
Results and discussion
Evaluation of size, ζ-potential, and encapsulation efficiency
Before any formulation or development step, Ghrl was subjected to both temperature and pH variations to assess its stability for further experiments. Stability was optimal when both pH and temperature were decreased, as previously confirmed by other studies.25,34 At physiological pH, it was found that experimentation involving prolonged heating (eg, cell-culture or lipid-film rehydration) could be performed without any degradation.
Size measurements were performed, and all free liposomes were characterized by Z-average increase after addition of Ghrl. AL, NL, and CL formulations were 10–15 nm larger after loading (Table 2). This slight increase in size still reflected an interaction with Ghrl, most probably driven by hydrophobic interaction between the octanoyl chain of acylated Ghrl and the lipid bilayer. All formulations were characterized by low PDI, corresponding to a narrow and monomodal size distribution.
 | Table 2 Characterization of liposomes formulations with Ghrl and lipid concentrations of 1 mg/mL and 10 mg/mL, respectively |
In addition to the increase in Z-average, ζ-potential also increased (or was neutralized in the case of ALs) when Ghrl was incorporated in all liposomes. At pH 7.4, Ghrl is positively charged, as its isoelectric point is at 11.5.35 This could explain why the most radical change was observed with ALs (13.7 mV). Indeed, a lower increase in ζ-potential was observed with the other formulations, probably due to the absence of electrostatic attraction between cationic Ghrl and NLs or CLs. Study of the ζ-potential showed a neutral charge (0.5±0.4 mV) for NL-free, whereas negative (−14.0±0.7 mV) and positive (13.5±1.5 mV) charges were obtained for AL-free and CL-free (Table 2). The charge modulation was made possible by replacing 10% w:w of neutral lipids (cholesterol and Lipoid S100) with DHDP or DOTAP (Table 1). The negative charge of DHDP was conferred by the phosphate group and used to produce ALs, while the ammonium residue afforded a positive charge to DOTAP and was incorporated into the CLs.
As is often the case with interactions involving charged peptides and liposomes, both hydrophobic and electrostatic interactions are involved.36,37 However, when looking at EE, it can be seen that electrostatic interactions have a clear impact in the case of Ghrl.38 By providing a negative charge to ALs, the percentage of Ghrl loaded in ALs was fivefold higher compared to CLs (Table 2). This resulted in EE of 56.1%±7.8%, 21.3%±4.1%, and 9.8%±3.7% for ALs, NLs and CLs, respectively. The general trend followed by the Z-average, ζ-potential, and EE suggested that the theoretical Ghrl–AL interaction would be more pronounced than for other formulations.
Enzymatic protection after trypsin exposure
All liposome formulations (ALs, NLs and CLs) were exposed to Tryp, an endoprotease that cleaves peptide bonds between basic AAs, such as lysine or arginine, and other residues. As Ghrl contains seven lysine/arginine residues in its primary structure, sensitivity of the peptide upon contact with Tryp was compared between Ghrl in solution and Ghrl-loaded liposomes. Ghrl solution was used as a degradation reference, due to its rapid and complete digestion by Tryp within 15 minutes of incubation.
The protection afforded by all liposomes to Ghrl showed a logical evolution: where enzymatic protection increase, the ionic charge of liposomes became anionic (ALs) (Table 3). This trend may be explained by higher EE (Table 2) obtained with ALs in comparison with CLs and NLs. When EE is increased, less Ghrl is potentially exposed for enzymatic degradation. Protection reached 20.6%±4.2% for ALs compared to 10.2%±2.9% for NLs and 5.6%±1.4% for CLs. As can be seen, Tryp led to a consequent degradation of Ghrl, even when it was loaded into the liposomes. This can be explained by the fact that most Ghrl had its hydrophilic body outside the liposome and this body contains the sites of Tryp cleavage. These results support the fact that electrostatic attractions between ALs and Ghrl resulted in an increase in both loading and protection, while electrostatic repulsions with CLs decreased them.
 | Table 3 Enzymatic protection for Ghrl in the presence of trypsin (140 IU/mL, 15 minutes, 37°C, n=3) for Ghrl (1 mg/mL)-loaded liposomes |
In the nasal cavity, numerous enzymes, such as the CYP450 family, flavin-containing monooxygenase, aldehyde dehydrogenases, epoxide hydrolase, carboxyl esterase, and phase 2 enzymes, can be found.39 Moreover, the olfactory epithelium appears to have intense metabolic activity,40 and CYP450 activity can be even higher than in the liver.41 For instance, peptide hormones, such as insulin, can be rapidly metabolized after nasal delivery.31 These observations usually justify the use of enzyme inhibitors in formulations used for nasal drug delivery, and more particularly when the olfactory region is targeted. Indeed, Cal quantity after permeation through rat nasal mucosa has been found to be higher when Tryp inhibitors were added to the formulation.42 Therefore, it is relevant to develop a formulation that can both enhance nasal permeation and provide effective protection against enzymatic degradation, especially for biotherapeutics.
The next step was evaluation of Ghrl quantity effectively inserted inside liposomes instead of being adsorbed at their surface. The degradation of Ghrl was thus evaluated using the conventional protocol of rehydration of lipid film, where Ghrl was added during the formation of ALs in PBS pH 7.4, but also using a second method where Ghrl was allowed to diffuse in already-formed ALs (ie, the postformation method). Both formulations were exposed to Tryp degradation for 15 minutes at 37°C. With the postformation method, Ghrl could only be fixed onto the outer side of the liposome, with the octanoyl group embedded in the lipid bilayer and the peptide body easily accessible outside the liposomes. With this method, intact Ghrl remaining after digestion should be lower than with the rehydration method. With the rehydration method, the Ghrl peptide body should access the internal aqueous cavity of the lipid structure, resulting in higher protection. It was observed that the protection obtained with rehydration (24%±3.7%) was clearly higher than with postformation (0%) after Tryp exposure (Table 3).
This suggests that with the rehydration method, some of the encapsulated Ghrl could be incorporated inside the lipid structure. On the other hand, Ghrl added by postformation could not reach the internal aqueous cavity or the hydrophobic bilayer, and was thus more exposed to Tryp. This configuration, as well as the need of Tryp to access the main peptide body for digestion, would explain the complete degradation of Ghrl when the postformation method was used.
Enzymatic protection after carboxylesterase-1 exposure
Based on the positive results previously obtained with ALs, all further experiments were conducted with these liposomes from this point. It was decided to work with human Ces1 as an additional enzyme that targets another location of the peptide. As the nasal enzymatic activity of Ces is not exactly known,43 Ces1 concentration was considered fixed when raw Ghrl degradation was observable after 15 minutes (a period equivalent to mucociliary clearance,31 which could theoretically correspond to the time spent by Ghrl in the nasal cavity). Ces1 is strongly present in the nasal cavity on both ciliated and unciliated respiratory epithelia, but also in the olfactory area in Bowman’s gland of the lamina propria and the sustentacular cells of the epithelial barrier.44 Ces1 types are responsible for the hydrolysis of both ester and amide bond-containing drugs. They also hydrolyze long-chain fatty-acid esters and thioesters, such as the octanoyl group of Ghrl. Since the presence of the octanoyl chain was shown to be essential to preserving the physiological activity of Ghrl, it seemed necessary to demonstrate the ability of ALs to preserve the integrity of the octanoyl group of Ghrl from the activity of Ces1.
The protection obtained with ALs was 4.7-fold higher (81.6%) than with Ghrl in solution (17.2%) (Table 4). Globally, the degradation obtained with ALs in Ces1 was less than with Tryp (79.4% with Tryp and 18.4% with Ces1). It is known that Tryp preferentially cleaves lysine and arginine45 residues at the C-terminal side of the peptide sequence. The Ghrl structure contains three arginine and four lysine residues that are potential sites of Tryp cleavage. For its part, Ces1 selectively hydrolyzes the ester bond between the main peptide body and the octanoyl chain.46 This is most probably inserted inside the bilayer of the liposomes, as observed previously with the comparison between the postformation and rehydration methods after Tryp digestion. This configuration made the ester bond less accessible to Ces1 due to steric hindrance, while Tryp attacked the hydrophilic body more easily and thus induced higher degradation.
 | Table 4 Enzymatic protection of Ghrl in solution and loaded in ALs after Ces1 exposure |
Regarding overall enzymatic degradation, ALs showed very interesting results, which could make the administration of this type of unstable peptide possible and compatible with the conditions (presence of various enzymes, eg, aldehyde dehydrogenase, CYP450-dependent monooxygenase, Ces)40 encountered in the human nasal cavity.
Isothermal titration calorimetry
As potential Ghrl–AL interaction was suggested by size and ζ-potential data, ITC was selected to collect additional information on the binding of Ghrl to AL-free. The binding coefficients as well as the heat flow collected should be useful for a better understanding of Ghrl–AL interaction. The decrease in exothermic signal in the ITC profile after each injection of ALs (Figure 1) showed that the quantity of Ghrl available for binding to AL-free decreased after each addition of liposomes. This confirmed that Ghrl bound progressively onto AL-free.
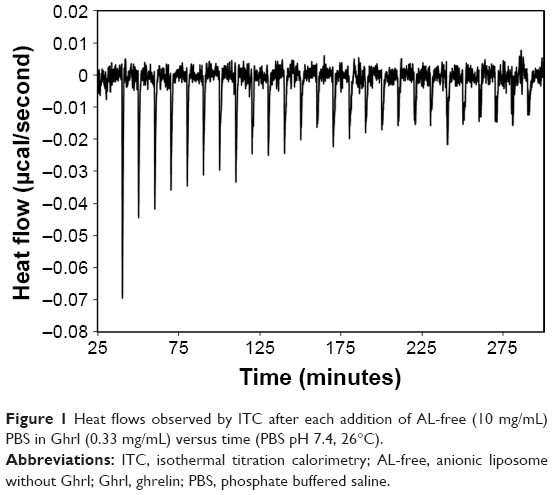 | Figure 1 Heat flows observed by ITC after each addition of AL-free (10 mg/mL) PBS in Ghrl (0.33 mg/mL) versus time (PBS pH 7.4, 26°C). |
The negative value (Table 5) observed for ΔG with Ghrl–AL-free interaction revealed the favorable and spontaneous nature of the interaction. Moreover, TΔS was much higher than ΔH, which suggested that the interactions involved were mainly driven by hydrophobic affinities.47 However, as previously observed, the charge modulation clearly impacted EE, which suggests a strong influence of the charge on the Ghrl–AL-free interaction.
 | Table 5 Binding coefficient (K), Gibbs free energy (ΔG), enthalpy (ΔH), and entropy (TΔS) from ITC analysis of AL-free PBS (10 mg/mL) and Ghrl (1 mg/mL) PBS |
The interaction between Ghrl and AL-free could take place in two ways. A first hypothetical Ghrl localization could include the body of the peptide inside the liposomes’ aqueous cavity, while the octanoyl chain would be inserted into the hydrophobic liposome bilayer. For this, Ghrl should pass through the liposome bilayer in order to reach the aqueous cavity. This passage would probably not be energetically favorable and thus not in accordance with ITC results. The second localization would be quite similar, except that the body of Ghrl would be present at the external part of the liposomal structure. In both theoretical localizations, the octanoyl fatty-acid group is inserted in the lipid structure. This interaction has already been described in other studies,37,38 and shown to provide significant protection to the octanoyl group, which is essential for preserving the physiological activity of Ghrl.34
HTCC-coated liposomes
It was shown that Ghrl interacted with ALs and that ALs improved the protection of the peptide. However, there are other areas that need to be optimized, which are residence time and absorption in the nasal cavity. Chitosans have been well studied for various administration routes, including intranasal administration. They offer several advantages, among which are mucoadhesion and tight junction (TJ) opening.48 To combine the beneficial effects of chitosans and liposomes, it was decided to coat ALs with HTCC. This derivative was selected as it is soluble at physiological pH (pH of the nasal cavity: 6.3). The nasal administration of peptide with such formulation strategies has already shown interesting properties (also with other administration routes).49,50
The HTCC-concentration range used after dilution in the AL suspensions for the coating was fixed between 1 mg/mL and 5 mg/mL.51 Higher HTCC concentrations (>1 mg/mL) did not provide satisfactory results with a PDI larger than 0.198 (data not shown). It is usually considered that a PDI larger than 0.2 reflects a nonmonomodal size distribution. Coating of ALs with 1 mg/mL HTCC was then performed, and showed a large increase in Z-average (48 nm) in comparison with ALs (Table 6). The ζ-potential of cationic Ghrl involved a switch from −14 mV to −0.6 mV before and after the addition of Ghrl to ALs, respectively. As the resultant charge remained slightly anionic (−0.6 mV), the coating with cationic HTCC was possible thanks to electrostatic attraction. Once ALs had been coated with HTCC, the resultant charge was positive (6 mV). The HTCC concentration of 1 mg/mL was selected for further characterization, since the Z-average and PDI were satisfactory. To visualize the morphology and confirm the size of both ALs and HTCC-ALs, TEM analyses were performed.
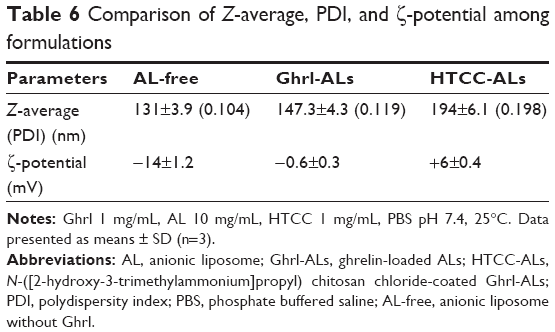 | Table 6 Comparison of Z-average, PDI, and ζ-potential among formulations |
TEM (Figure 2) confirmed the “large unilamellar vesicle” structure of liposomes, with a Z-average larger than 100 nm and a single lipid bilayer. Once the liposomes had been coated with HTCC, they became black, due to the polymer coating. The sizes observed by TEM for both ALs and HTCC-ALs were comparable with the Z-averages obtained by dynamic light scattering, which were 147.3±4.3 and 194±6.1 nm, respectively.
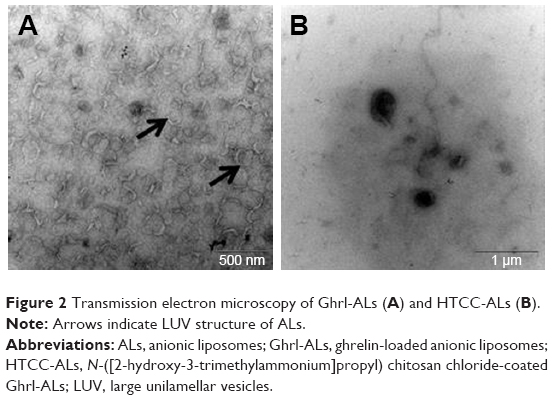 | Figure 2 Transmission electron microscopy of Ghrl-ALs (A) and HTCC-ALs (B). |
Permeation studies, mucoadhesion, and osmolality
Chitosan is known to provide mucoadhesion, but this compound and its derivatives are also known to open TJs. This action on TJs was assessed using the Calu3 cell line, epithelial cells isolated from lung adenocarcinoma. These cells may be used to evaluate both the permeability and the potential toxicity of nasal drug-delivery sytems.23,52 The use of lung cells to evaluate the response of nasal epithelial cells to a drug is explained by the multiple similarities between Calu3 and nasal mucosa. Indeed, Calu3 are characterized by mucus production, the presence of TJs, and cilia. These characteristics justify the cells’ use for permeation studies of formulations intended for nasal delivery.53,54 The particular cell-culture conditions under an air–liquid interface induce polarization of the cells, similarly to physiological conditions found in the respiratory tract.
The cellular transport of Ghrl through the blood–brain barrier has already been studied,7 but no data are available regarding its nose–brain transfer. As the transport of Ghrl is assumed to be paracellular, like most peptides,55 the addition of HTCC to the formulation could be justified by its ability to open TJs.48 Based on this theory, Calu3-transport studies were performed.
Ghrl permeation was compared with that of Cal. As it is known that paracellular transport is limited by molecular weight, Cal was selected, as it is a peptide hormone that uses paracellular transport and has a molecular weight close to that of Ghrl (3,454.9 and 3,370.9 Da for Cal and Ghrl, respectively). Cal was thus used as a positive control for paracellular transport.56 Caf permeation was also assessed as a transcellular positive control with low molecular weight (194.19 Da).57 Both compounds are physiologically active in the brain.58,59 Regardless of the molecule, the passage was followed with and without addition of HTCC (1 mg/mL). The impact of HTCC on permeation was assessed because of its enhancing effect on the permeation of peptides through nasal mucosal layers.22,60 The solutions evaluated for this experiment were PBS buffer pH 7.4 containing Cal, Caf, or Ghrl solubilized with or without HTCC. The degree of deacetylation of the HTCC was 33% to minimize cytotoxicity, which correlates with the degree of deacetylation.61 If Ghrl crosses a Calu3 monolayer using paracellular transport, the diffusion should be higher when HTCC is added (similarly to Cal).
HTCC addition allowed both Cal and Ghrl permeation (2.9 and 8.2% for Cal and Ghrl, respectively) to be increased (Table 7). The higher permeation for Ghrl could be explained by its amphiphilic property, which could make its transfer easier than Cal. It thus seems that like Cal, Ghrl is characterized by paracellular transport. This observation can be supported by the TEER values (Figure 3). These reduced when HTCC was used, confirming the TJs’ opening. The reversibility of this phenomenon was assessed and confirmed 24 hours after the test by TEER measurements (data not shown). In contrast, Caf showed overall higher permeation through the Calu3 monolayer, due to its low molecular weight and transcellular transport. However, as Caf is characterized by transcellular transport, permeation was not impacted by the addition of HTCC (Table 7). After paracellular transport of Ghrl had been confirmed, it was decided to study the complete formulation with HTCC-ALs further.
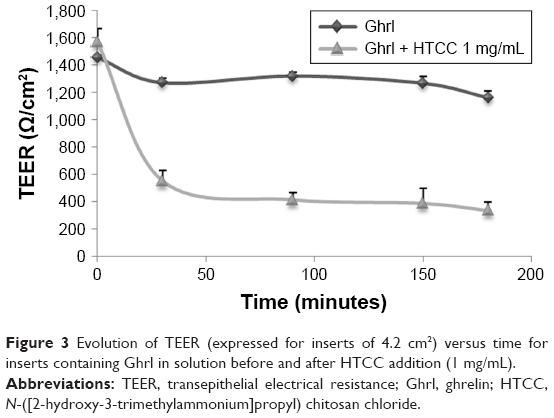 | Figure 3 Evolution of TEER (expressed for inserts of 4.2 cm2) versus time for inserts containing Ghrl in solution before and after HTCC addition (1 mg/mL). |
 | Table 7 Distribution (%) of Ghrl, Cal, and Caf solutions at a concentration of 1 mg/mL with or without HTCC (1 mg/mL) after 3 hours |
In order to show the effect of the HTCC-coating with regard to mucoadhesion, adhesion to mucins was determined for HTCC-ALs and compared to ALs. Mucins are the most represented glycoproteins in the nasal mucus (2%–5%).62 They contain sulfate and sialic acids, which confer a negative resultant charge on the mucus surface.63 Adhesion of formulations to mucins is essential to avoid rapid mucus clearance of the liposomes in the nasal cavity and to prolong diffusion to the brain.64 Both formulations showed the ability to complex mucins, with 39.8% and 62.7% adhesion for ALs and HTCC-ALs, respectively (Table 8). The coating with HTCC and the positive charge of the amino groups allowed electrostatic interactions with negative sialic acid of mucins. This led to a 22.9% increase in bioadhesion. This interaction with mucins could extend the residence of the formulation in the nasal cavity, and would thus optimize its transfer to the brain.
 | Table 8 Osmolality, mucoadhesion, and amounts in the basal side after permeation through Calu3 cells |
Before proceeding to permeation tests on Calu3 cells, the osmolality of both coated and uncoated formulations was assessed. It is known that a liquid formulation intended for nasal delivery should be close to or even slightly higher than 290 mOsm/kg and less than 500 mOsm/kg.65 Hypertonic formulations can be used occasionally, while isotonic solutions are suited for chronic use. Hypotonic formulations should be avoided. For ALs and HTCC-ALs, the values obtained were quite close, at 405 and 409 mOsm/Kg, respectively (Table 8). These values reflect the physiological conditions of the nasal cavity.
After this, the permeation of Ghrl (1 mg/mL) in ALs and HTCC-ALs was assessed. HTCC-ALs (10.8%) provided very limited improvement in permeation compared to HTCC in solution (8.2%, Table 7). HTCC coating of ALs loaded with Ghrl seemed to enhance the transport of Ghrl compared to the simple solution of HTCC and Ghrl. HTCC-ALs showed enhanced permeation compared to ALs (10.8±0.71 versus 3.6%±0.25% for HTCC-ALs and ALs, respectively). It appears that the coating of ALs with HTCC had a positive effect on their permeation in contrast with ALs without coating. ALs offered only a slight permeation increase in comparison with Ghrl solution (3.6%±0.25% versus 0 for ALs and Ghrl in solution, respectively), which suggests that their major function lies rather in enzymatic protection. Finally, Ghrl permeation of 10.8%±0.71% was obtained with HTCC-ALs in comparison with no permeation for the Ghrl solution. This formulation thus offers the advantage of combining the protective effects of ALs with an increase in permeability provided by HTCC.
Droplet-size distribution
The size of the droplets generated from a device is a predominant factor in the deposition site in the respiratory tract.66 As specified by US Food and Drug Administration and European Medicines Agency guidelines, the percentage of droplets <10 μm must be minimized to reduce their potential deposition in the lungs. Another phenomenon depending on the size distribution of the droplets is determination of the deposition site in the nasal cavity. For instance, it is well known that droplets or particles >10 μm are deposited in the anterior part of the nose.67 However, large particles are usually cleared more quickly from the nasal cavity, thanks to mucociliary clearance.68
The VP3 device coupled with the Aptar 144GI actuator generated only 3.2% of droplets <10 μm for ALs (Table 9), which could be potentially deposited in the lower airways. This percentage represents only a small fraction of the entire droplet population and suggests negligible loss in the lower airways. The Dv50 was 37.6±8.7 μm and Dv4,3 40.8±8.9 μm. When the HTCC-AL dispersion was introduced in the device, the Dv50 of the droplets generated was similar to that obtained with ALs (37.6±8.7 and 38.5±5.8 μm, respectively).
 | Table 9 Droplet-size data collected by laser diffraction (Spraytec) from aerosols |
Droplet-size distribution was then studied with the SP270 device. The Dv50 obtained with ALs in the conventional device was very close to that obtained with the VP3 device. However, a significant increase in Dv50 and Dv4,3 was observed for HTCC-ALs from the conventional device, showing droplets more than twice as large (Dv50 of 40.41±2.78 and 95.36±9.57 μm with ALs and HTCC-ALs, respectively). The potential viscosity afforded by HTCC could be responsible for this evolution, as has been already observed in another study.69 The SP270 device is clearly more impacted by viscosity variations than the VP3 device.
Conclusion
By changing the liposome compositions, it was possible to modulate the ionic charge and thus increase both EE and enzymatic protection (Tryp and Ces1). Electrostatic and hydrophobic interactions between Ghrl and ALs were demonstrated, and ALs appeared to be an interesting choice of formulation for Ghrl nasal delivery with brain targeting. The coating of ALs with HTCC was confirmed by both size and charge augmentations, but also by morphological assessments. HTCC-ALs showed stronger adhesion to mucins than ALs, and osmolality values were consistent with nasal administration.
Calu3 experimentation showed that Ghrl uses paracellular transport and that HTCC had an enhancing effect on Ghrl permeation. It was also underscored that ALs need to be coated with HTCC to obtain improved Ghrl permeation. By combining the beneficial effects of both ALs and HTCC, it was possible to protect Ghrl, increase bioadhesion, and optimize its transfer through Calu3 cells. The aerosol properties after actuation of the device were satisfactory with suitable size distributions.
Overall, the formulation developed could closely match the criteria required for efficient nose–brain delivery. The development of a treatment associated with this route of administration could offer a great opportunity to provide effective and high-compliance treatment to patients suffering from cachexia. However, the nasal administration of the formulation to mice should be the next step, in order to confirm the theoretical transfer from the nose to the brain by fluorescent labeling and confocal microscopy, for example.
Disclosure
The authors report no conflicts of interest in this work.
References
Editors of the American Heritage Dictionaries. The American Heritage Medical Dictionary. Boston: Houghton Mifflin; 2007. | ||
DeBoer MD. Emergence of ghrelin as a treatment for cachexia syndromes. Nutrition. 2008;24(9):806–814. | ||
Argilés JM, Stemmler B. The potential of ghrelin in the treatment of cancer cachexia. Expert Opin Biol Ther. 2013;13(1):67–76. | ||
Garcia J, Yan Y, Manning-Duus E, Friend J. Effects of the ghrelin receptor agonist anamorelin on lean body mass in cancer patients with cachexia; results from a Phase II randomized, double blind, multicenter study. Cancer Metab. 2014;2 Suppl 1:P19. | ||
Horvath TL, Diano S, Sotonyi P, Heiman M, Tschöp M. Minireview: Ghrelin and the regulation of energy balance – a hypothalamic perspective. Endocrinology. 2001;142(10):4163–4169. | ||
Kojima M, Hosoda H, Date Y, Nakazato M, Matsuo H, Kangawa K. Ghrelin is a growth-hormone-releasing acylated peptide from stomach. Nature. 1999;402(6762):656–660. | ||
Banks WA, Tschöp M, Robinson SM, Heiman ML. Extent and direction of ghrelin transport across the blood-brain barrier is determined by its unique primary structure. J Pharmacol Exp Ther. 2002;302(2):822–827. | ||
Barsh GS, Schwartz MW. Genetic approaches to studying energy balance: perception and integration. Nat Rev Genet. 2002;3(8):589–600. | ||
Akamizu T, Kangawa K. Ghrelin for cachexia. J Cachexia Sarcopenia Muscle. 2010;1(2):169–176. | ||
Brimijoin S, Chen VP, Pang YP, Geng L, Gao Y. Physiological roles for butyrylcholinesterase: a BChE-ghrelin axis. Chem Biol Interact. 2016;259(Pt B):271–275. | ||
Akamizu T, Takaya K, Irako T, et al. Pharmacokinetics, safety, and endocrine and appetite effects of ghrelin administration in young healthy subjects. Eur J Endocrinol. 2004;150(4):447–455. | ||
Thorne RG, Emory CR, Ala TA, Frey WH. Quantitative analysis of the olfactory pathway for drug delivery to the brain. Brain Res. 1995;692(1–2):278–282. | ||
Schipper NM, Verhoef JC, Merkus FH. The nasal mucociliary clearance: relevance to nasal drug delivery. Pharm Res. 1991;8(7):807–814. | ||
Moeller EH, Holst B, Nielsen LH, Pedersen PS, Østergaard J. Stability, liposome interaction, and in vivo pharmacology of ghrelin in liposomal suspensions. Int J Pharm. 2010;390(1):13–18. | ||
Graf S, Garcia J. Anamorelin hydrochloride in the treatment of cancer anorexia-cachexia syndrome: design, development, and potential place in therapy. Drug Des Devel Ther. 2017;11:2325–2331. | ||
Garin MC, Burns CM, Kaul S, Cappola AR. The human experience with ghrelin administration. J Clin Endocrinol Metab. 2013;98(5):1826–1837. | ||
Azegami T, Yuki Y, Sawada S, et al. Nanogel-based nasal ghrelin vaccine prevents obesity. Mucosal Immunol. 2017;10(5):1351–1360. | ||
Zollers B, Rhodes L, Heinen E. Capromorelin oral solution (ENTYCE) increases food consumption and body weight when administered for 4 consecutive days to healthy adult beagle dogs in a randomized, masked, placebo controlled study. BMC Vet Res. 2017;13(1):10. | ||
Temel JS, Abernethy AP, Currow DC, et al. Anamorelin in patients with non-small-cell lung cancer and cachexia (ROMANA 1 and ROMANA 2): results from two randomised, double-blind, phase 3 trials. Lancet Oncol. 2016;17(4):519–531. | ||
Migliore MM, Vyas TK, Campbell RB, Amiji MM, Waszczak BL. Brain delivery of proteins by the intranasal route of administration: a comparison of cationic liposomes versus aqueous solution formulations. J Pharm Sci. 2010;99(4):1745–1761. | ||
Salama HA, Mahmoud AA, Kamel AO, Hady MA, Awad GA. Phospholipid based colloidal poloxamer-nanocubic vesicles for brain targeting via the nasal route. Colloids Surfaces B Biointerfaces. 2012;100:146–154. | ||
Illum L, Farraj NF, Davis SS. Chitosan as a novel nasal delivery system for peptide drugs. Pharm Res. 1994;11(8):1186–1189. | ||
Van Woensel M, Wauthoz N, Rosière R, et al. Development of siRNA-loaded chitosan nanoparticles targeting galectin-1 for the treatment of glioblastoma multiforme via intranasal administration. J Control Release. 2016;227:71–81. | ||
Filipović-Grcić J, Skalko-Basnet N, Jalsenjak I. Mucoadhesive chitosan-coated liposomes: characteristics and stability. J Microencapsul. 2008;18(1):3–12. | ||
Staes E, Rozet E, Učakar B, Hubert P, Préat V. Validation of a method for the quantitation of ghrelin and unacylated ghrelin by HPLC. J Pharm Biomed Anal. 2010;51(3):633–639. | ||
Bangham AD. Properties and uses of lipid vesicles: an overview. Ann N Y Acad Sci. 1978;308:2–7. | ||
Zakanda FN, Lins L, Nott K, Paquot M, Lelo GM, Deleu M. Interaction of hexadecylbetainate chloride with biological relevant lipids. Langmuir. 2012;28(7):3524–3533. | ||
Heerklotz H, Seelig J. Titration calorimetry of surfactant-membrane partitioning and membrane solubilization. Biochim Biophys Acta. 2000;1508(1–2):69–85. | ||
Razafindralambo H, Dufour S, Paquot M, Deleu M. Thermodynamic studies of the binding interactions of surfactin analogues to lipid vesicles. J Therm Anal Calorim. 2009;95(3):817–821. | ||
Mantle M, Allen A. A colorimetric assay for glycoproteins based on the periodic acid/Schiff stain. Biochem Soc Trans. 1978;6(3):607–609. | ||
Tamilvanan S. Pharmaceutical Manufacturing Handbook: Production and Processes. Hoboken (NJ): Wiley; 2008. | ||
Erickson J. Determination of the concentration of caffeine, theobromine, and gallic acid in commercial tea samples. Concordia Coll J Anal Chem. 2011;2:31–35. | ||
Shah RB, Siddiqui A, Shah G, Khan MA. A validated HPLC assay for simultaneous analysis of salmon calcitonin and duck ovomucoid. Pharmazie. 2003;58(9):620–622. | ||
Hosoda H, Doi K, Nagaya N, et al. Optimum collection and storage conditions for ghrelin measurements: octanoyl modification of ghrelin is rapidly hydrolyzed to desacyl ghrelin in blood samples. Clin Chem. 2004;50(6):1077–1080. | ||
Min C, Ohta K, Kajiya M, et al. The antimicrobial activity of the appetite peptide hormone ghrelin. Peptides. 2012;36(2):151–156. | ||
Seelig J. Thermodynamics of lipid-peptide interactions. Biochim Biophys Acta. 2004;1666(1–2):40–50. | ||
Staes E, Absil PA, Lins L, et al. Acylated and unacylated ghrelin binding to membranes and to ghrelin receptor: towards a better understanding of the underlying mechanisms. Biochim Biophys Acta. 2010;1798(11):2102–2113. | ||
Østergaard J, Moeller EH. Ghrelin-liposome interactions: characterization of liposomal formulations of an acylated 28-amino acid peptide using CE. Electrophoresis. 2010;31(2):339–345. | ||
Margolis FL and Getchell TV. Molecular Neurobiology of the Olfactory System: Molecular, Membranous, and Cytological Studies. Heidelberg: Springer; 2013. | ||
Dahl AR, Hadley WM. Nasal cavity enzymes involved in xenobiotic metabolism: effects on the toxicity of inhalants. Crit Rev Toxicol. 1991;21(5):345–372. | ||
Sarkar MA. Drug metabolism in the nasal mucosa. Pharm Res. 1992;9(1):1–9. | ||
Morimoto K, Miyazaki M, Kakemi M. Effects of proteolytic enzyme inhibitors on nasal absorption of salmon calcitonin in rats. Int J Pharm. 1995;113(1):1–8. | ||
Lewis JL, Nikula KJ, Novak R, Dahl AR. Comparative localization of carboxylesterase in F344 rat, beagle dog, and human nasal tissue. Anat Rec. 1994;239(1):55–64. | ||
Bogdanffy M, Taylor M. Kinetics of nasal carboxylesterase-mediated metabolism of vinyl acetate. Drug Metab Dispos. 1993;21(6):1107–1111. | ||
Olsen JV, Ong SE, Mann M. Trypsin cleaves exclusively C-terminal to arginine and lysine residues. Mol Cell Proteomics. 2004;3(6):608–614. | ||
Taylor MS, Hwang Y, Hsiao PY, Boeke JD, Cole PA. Ghrelin O-acyltransferase assays and inhibition. Methods Enzymol. 2012;514:205–228. | ||
Chodera JD, Mobley DL. Entropy-enthalpy compensation: role and ramifications in biomolecular ligand recognition and design. Annu Rev Biophys. 2013;42:121–142. | ||
Casettari L, Illum L. Chitosan in nasal delivery systems for therapeutic drugs. J Control Release. 2014;190:189–200. | ||
Channarong S, Chaicumpa W, Sinchaipanid N, Mitrevej A. Development and evaluation of chitosan-coated liposomes for oral DNA vaccine: the improvement of Peyer’s patch targeting using a polyplex-loaded liposomes. AAPS Pharm Sci Tech. 2011;12(1):192–200. | ||
Mady MM, Darwish MM. Effect of chitosan coating on the characteristics of DPPC liposomes. J Adv Res. 2010;1(3):187–191. | ||
Guo J, Ping Q, Jiang G, Huang L, Tong Y. Chitosan-coated liposomes: characterization and interaction with leuprolide. Int J Pharm. 2003;260(2):167–173. | ||
Wen Z, Yan Z, He R, et al. Brain targeting and toxicity study of odorranalectin-conjugated nanoparticles following intranasal administration. Drug Deliv. 2011;18(8):555–561. | ||
Amoako-Tuffour M, Yeung P, Agu R. Permeation of losartan across human respiratory epithelium: an in vitro study with Calu-3 cells. Acta Pharm. 2009;59(4):395–405. | ||
Yang T, Hussain A, Paulson J, Abbruscato TJ, Ahsan F. Cyclodextrins in nasal delivery of low-molecular-weight heparins: in vivo and in vitro studies. Pharm Res. 2004;21(7):1127–1136. | ||
Ungell AL, Andreasson A, Lundin K, Utter L. Effects of enzymatic inhibition and increased paracellular shunting on transport of vasopressin analogues in the rat. J Pharm Sci. 1992;81(7):640–645. | ||
Torres-Lugo M, García M, Record R, Peppas NA. pH-sensitive hydrogels as gastrointestinal tract absorption enhancers: transport mechanisms of salmon calcitonin and other model molecules using the Caco-2 cell model. Biotechnol Prog. 2002;18(3):612–616. | ||
Prueksaritanont T, DeLuna P, Gorham LM, et al. In vitro and in vivo evaluations of intestinal barriers for the zwitterion L-767,679 and its carboxyl ester prodrug L-775,318: roles of efflux and metabolism. Drug Metab Dispos. 1998;26(6):520–527. | ||
Ribeiro JA, Sebastião AM. Caffeine and adenosine. J Alzheimers Dis. 2010;20 Suppl 1:S3–S15. | ||
Becskei C, Riediger T, Zünd D, Wookey P, Lutz TA. Immunohistochemical mapping of calcitonin receptors in the adult rat brain. Brain Res. 2004;1030(2):221–233. | ||
Prego C, Torres D, Alonso MJ. Chitosan nanocapsules: a new carrier for nasal peptide delivery. J Drug Deliv Sci Technol. 2006;16(5):331–337. | ||
Huang M, Khor E, Lim LY. Uptake and cytotoxicity of chitosan molecules and nanoparticles: effects of molecular weight and degree of deacetylation. Pharm Res. 2004;21(2):344–353. | ||
Carlstedt I, Lindgren H, Sheehan JK, Ulmsten U, Wingerup L. Isolation and characterization of human cervical-mucus glycoproteins. Biochem J. 1983;211(1):13–22. | ||
Khutoryanskiy VV. Advances in mucoadhesion and mucoadhesive polymers. Macromol Biosci. 2011;11(6):748–764. | ||
Naik A, Nair H. Formulation and evaluation of thermosensitive biogels for nose to brain delivery of doxepin. Biomed Res Int. 2014;2014:847547. | ||
Surber C, Elsner P, Farage MA. Topical Applications and the Mucosa. Basel, Switzerland: Karger; 2011. | ||
Se CM, Inthavong K, Tu JY. Unsteady particle deposition in a human nasal cavity. 2009. Available from: http://www.cfd.com.au/cfd_conf09/PDFs/016SE.pdf. Accessed March 27, 2017. | ||
Frank DO, Kimbell JS, Pawar S, Rhee JS. Effects of anatomy and particle size on nasal sprays and nebulizers. Otolaryngol Head Neck Surg. 2012;146(2):313–319. | ||
Harris AS, Svensson E, Wagner ZG, Lethagen S, Nilsson IM. Effect of viscosity on particle size, deposition, and clearance of nasal delivery systems containing desmopressin. J Pharm Sci. 1988;77(5):405–408. | ||
Pennington J, Pandey P, Tat H, Willson J, Donovan B. Spray pattern and droplet size analyses for high-shear viscosity determination of aqueous suspension corticosteroid nasal sprays. Drug Dev Ind Pharm. 2008;34(9):923–929. |
 © 2017 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2017 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.