")
Back to Journals » International Journal of Nanomedicine » Volume 14
Development of anti-HER2-targeted doxorubicin–core-shell chitosan nanoparticles for the treatment of human breast cancer
Authors Naruphontjirakul P , Viravaidya-Pasuwat K
Received 17 December 2018
Accepted for publication 1 April 2019
Published 4 June 2019 Volume 2019:14 Pages 4105—4121
DOI https://doi.org/10.2147/IJN.S198552
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Thomas Webster
Parichart Naruphontjirakul,1 Kwanchanok Viravaidya-Pasuwat1,2
1Biological Engineering Program, Faculty of Engineering, King Mongkut’s University of Technology Thonburi, Bangkok, Thailand; 2Department of Chemical Engineering, Faculty of Engineering, King Mongkut’s University of Technology Thonburi, Bangkok, Thailand
Purpose: Doxorubicin (DOX) encapsulated O-succinyl chitosan graft Pluronic® F127 (OCP) copolymer nanoparticles conjugated with an anti-HER2 monoclonal antibody were developed as targeted drug delivery vehicles for the treatment of HER2-overexpressing breast cancer.
Methods: Five percent and 10% (w/w) of O-succinyl chitosan was grafted onto Pluronic® F127 using 1-ethyl-3-(3-dimethylaminopropyl) carbodiimide hydrochloride (EDC) and N-hydroxysuccinimide (NHS) as mediated cross-linking agents. DOX was added to the copolymer solution to form DOX-nanoparticles before conjugation with anti-HER2 on the surface of the nanoparticles.
Results: DOX was encapsulated within the NP matrices at an encapsulation efficiency of 73.69 ± 0.53% to 74.65 ± 0.44% (the initial DOX concentration was 5 μg/mL). Anti-HER2 was successfully conjugated onto the surface of the nanoparticles at a moderately high conjugation efficiency of approximately 57.23 ± 0.38% to 61.20 ± 4.42%. In the in vitro DOX dissolution study, the nanoparticle formulations exhibited a biphasic drug release with an initial burst release followed by a sustained release profile at both pH 5.0 and pH 7.4. The drug was rapidly and completely released from the nanoparticles at pH 5.0. In the in vitro cytotoxicity, the anti-HER2 conjugated OCP copolymer nanoparticles showed the lowest IC50, which indicated an increase in the therapeutic efficacy of DOX to treat human breast cancer cells with the HER2 overexpression.
Conclusion: Our study shows that anti-HER2 conjugated OCP copolymer nanoparticles have the potential for the development of anticancer drug carriers.
Keywords: pluronic grafted chitosan, core-shell nanoparticles, doxorubicin, anti-HER2, MCF-7
Introduction
Drug delivery systems (DDSs), specifically nanoparticles (NPs), have recently emerged as an important advancement in cancer therapy.1–3 They have the potential to increase the efficacy of the chemotherapeutic agents by preferentially accumulating in tumor tissue due to the structurally irregular, leaky or disorganized tumor vasculature endothelium when compared to normal tissue.1,4–6 In addition, because of their nanometer size, the NPs can be assimilated by cells more efficiently than larger microparticles.7–9 As a result, undesirable adverse side effects of the anticancer drugs can be significantly reduced.
In recent decades, there has been a considerable interest in developing biodegradable, specifically polymer-based NPs as effective drug carriers to perform the sustained release of therapeutic agents to the specific target sites.10–12 One of the most widely investigated DDSs is prepared from amphiphilic block copolymers,13–16 which can spontaneously self-assemble into polymeric micelles consisting of a hydrophobic inner core and a hydrophilic outer shell in aqueous environments.13,17–19 A hydrophobic drug can be encapsulated in the hydrophobic core of the micelles,20,21 while the hydrophilic shell can significantly reduce phagocytosis and renal clearance, resulting in a prolonged circulation time.22,23 Among the amphiphilic block copolymers used in pharmaceutical research, Pluronic®, a tri-block copolymer composed of poly(ethylene oxide) (PEO) and poly(propylene oxide) (PPO), has shown great promise in delivery applications because of its low toxicity in the body and the ability to encapsulate any hydrophobic agents.16,24–28 However, the major problems with utilizing pluronic micelles are their instability and the fast drug release profile.29 To overcome these limitations, grafting pluronic with another biocompatible polymer, for example chitosan, is suggested.30–34 Furthermore, the functional group of the copolymer can be modified to attach targeting moieties to the surface of the NPs for an increase in the cellular uptake of the NPs.30,31,34
Active targeting techniques have been employed by applying ligands, such as antibodies, growth factors, transferrin, cytokines, folate and low-density lipoprotein,35,36 to NPs to be bound specifically to receptors on the cell surface.37,38 This ligand–receptor interaction induces the receptor-mediated endocytosis resulting in the internalization of the NPs and the drug release inside the target cells18. In breast cancer, human epidermal growth factor receptor 2 (HER2, or c-erbB-2) is one of the most attractive targeted receptors, as it plays an important role in the pathogenesis and progression of breast cancer.39–41 The expression of this receptor is markedly increased in breast cancer patients, while its expression is much less in normal adult tissue.42,43 At present, a monoclonal antibody against the HER2 receptor (anti-HER2) has been approved by the US Food and Drug Administration (FDA) for the treatment of HER2-positive breast cancer.44 Several clinical studies have shown that the combination of anti-HER2 and a conventional chemotherapeutic agent has led to an improved therapeutic outcome of the patients.41,45 Therefore, anti-HER2 would serve as a promising targeting ligand for a DDS.
In this study, we describe the synthesis and characterization of OCP NPs conjugated with anti-HER2 for the targeted doxorubicin (DOX) delivery to HER2-expressing breast cancer cells. O-succinyl chitosan, a water-soluble chitosan derivative, was chosen to be grafted with pluronic because of its water solubility and functional group, which permits conjugation with other molecules.46 The graft copolymer was then self-assembled to form core-shell structure NPs while encapsulating DOX at the particles’ core. The physical properties of these particles, such as CMC, encapsulation efficiency, conjugation efficiency, particle size and in vitro release profiles, were evaluated. The study of the in vitro anticancer effects of the NPs against the MCF-7 cells, the breast cancer cell model, was carried out to determine the targeting effectiveness of these particles.
Material and methods
Materials
Chitosan (molecular weight cut-off (MWCO) <50,000; degree of deacetylation, 90%) was purchased from Seafresh Chitosan Laboratory (Bangkok, Thailand). Pluronic® F127 (Poloxamer 407) was acquired from BASF (Lampertheim, Germany), and ethanol 99.7–100% was bought from BDH (East Yorkshire, UK). OX hydrochloride, 1-ethyl-3-(3-dimethylaminopropyl) carbodiimide (EDC), N-hydroxysuccinimide (NHS), pyridine, phthalic anhydride, acetone, sodium acetate, succinic anhydride, ethyl ether, and 1,4-Dioxane were procured from Sigma-Aldrich (St Louis, MO, USA). Sodium dodecyl sulfate was purchased from Promega Corporation (Fitchburg, WI, USA). DMEM, PBS, trypsin-EDTA, FBS, Trypan Blue, Vybant MTT cell proliferation kit, and anti-HER2, were obtained from Invitrogen (Waltham, MA, USA). Dimethyl sulfoxide (DMSO) was bought from Amresco (Cleveland, OH, USA). Triethylamine, dimethylformamide (DMF), and Hydrazine monohydrate were acquired from Carlo ERBA (Val-de-Reuil, France). Succinic anhydride was obtained from Fluka (Munich, Germany). Micro BCATM Protein Assay Kit was purchased from Thermo Scientific (Waltham, MA, USA).
Preparation of O-succinyl chitosan pluronic copolymer (OCP)
O-succinyl chitosan was synthesized following the procedure previously described by Zhang et al.46 Briefly, chitosan was added to phthalic anhydride in DMF to form phthalimide chitosan before adding succinic anhydride and pyridine to yield O-succinyl chitosan. O-succinyl chitosan then reacted with hydrazine monohydrate in distilled water. After the reaction, the suspension was filtered, and the precipitate was dried in a vacuum using a rotary evaporator. The residue was dissolved in water, dialyzed (MWCO 10,000) against distilled water for 4 days, and then lyophilized.
Pluronic F-127 was carboxylated with succinic anhydride to form monocarboxy pluronic.31 After that, monocarboxy pluronic (10.31 g) was coupled with 5% (0.57 g) and 10% (1.14 g) of O-succinyl chitosan using EDC (0.15 g)/NHS (0.10 g) at room temperature for 24 hrs. The solvent evaporation was carried out in a vacuum dryer overnight and in a desiccator for 7 days. Afterward, the product was dialyzed against distilled water using a membrane (molecular weight cut-off: 20,000) for 3 days and finally lyophilized.
Characterization of the graft copolymer
The functional groups of this copolymer were characterized using Fourier-transform infrared spectroscopy (FTIR) (Bruker, Vertex 70, Billerica, MA, USA) and 1H-NMR (Bruker, AVANCE 300, Billerica, MA, USA). The samples were milled, mixed with KBr, and then pressed into a thin layer before undertaking an FTIR analysis. For the 1H-NMR characterization, the samples were dissolved in DMSO-d6 and D2O.
Determination of the critical micelle concentrations (CMCs)
The CMCs of pluronic and the graft copolymer were determined using the dye solubilization method.47 The hydrophobic fluorescence probe, 1,6-diphenyl-1,3,5-hexatriene (DPH), was dissolved in 0.6 mM of methanol. Twenty microliters of the dye solution was mixed with 2.0 mL of pluronic and the copolymer in an aqueous solution to yield the dye concentrations ranging from 1×10–5 to 5% wt and gently stirred overnight at room temperature in the dark. The fluorescence of DPH in each sample was obtained by a microplate reader (TECAN Model InfiniteM200, Mannedorf, Switzerland), using an excitation wavelength of 360 nm and an emission wavelength of 460 nm. The CMCs of pluronic and the copolymer were obtained by plotting the first inflection point in the DPH fluorescence intensity versus the natural logarithm (ln) of the copolymer concentration.48
Preparation and characterization of the copolymer NPs and DOX-encapsulated nanoparticles (DOX-NPs)
To form the NPs, the solution containing 7% (w/v) of OCP copolymer in deionized water was stirred at 250 rpm for 12 hrs. The concentrations of the copolymer NPs used in this study were 7% (w/v), which was between its CMC and critical gel concentration. The formation of the DOX-NPs could be achieved by adding 5 to 25 µg/mL of the DOX concentrations to the copolymer solution while stirring at 250 rpm for 12 hrs in the dark. The NPs and DOX-NPs were separated by centrifugation at 25°C at 6,000 rpm for 2 hrs and then lyophilized. The particle sizes were examined by photon correlation spectroscopy (Nanosizer, Malvern Instruments, Malvern, UK).
Establishment of a calibration curve for DOX
A solution of DOX in PBS was diluted to concentrations between 0.01 and 50 μg/mL to generate a standard curve. The fluorescence intensities of different concentrations of DOX solutions were measured at an excitation wavelength of 485 nm and an emission wavelength of 590 nm. The calibration curve between DOX concentrations and fluorescence intensities was linear over the concentration range between 0.025 and 5 μg/mL with a correlation coefficient (R2) >0.99 (Figure S1). The limit of detection (LOD) and the limit of quantitation were found to be 0.03 μg/mL and 0.11 μg/mL, respectively, according to International Conference on Harmonization (ICH) of Technical Requirements for the Registration of Pharmaceuticals for Human Use, Validation of Analytical Procedures.
Drug encapsulation efficiency
The encapsulation efficiencies of DOX were calculated from the disappearance of DOX from its original solution. The remaining free DOX in the supernatant was measured using a fluorescence spectrophotometer (TECAN Model InfiniteM200) at an excitation wavelength of 485 nm and an emission wavelength of 590 nm. The concentration of DOX was calculated using the calibration curve in Figure S1. The DOX encapsulation efficiency was determined, as shown in Equation (1).
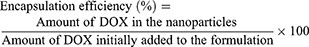
Preparation and characterization of the anti-HER2-conjugated nanoparticles (anti-HER2-NPs)
Twenty-five µg/mL of anti-HER2 and 0.5 mg/mL of EDC and NHS were added to 5 mg/mL of NPs in PBS at pH 7.4 and incubated for 20 mins at room temperature with gentle stirring. After the conjugation reaction, the anti-HER2-NPs were separated from the free anti-HER2 in the solution by centrifugation at 6,000 rpm for 2 hrs and then washed with PBS buffer and lyophilized. The conjugation efficiency on the surface of the NPs was evaluated using a Micro BCATM Protein Assay Kit. The protein concentration was determined by mixing supernatant with bicinchoninic acid at a ratio of 1:1 and incubated for 2 hrs at 37°C. Then, the protein concentration was measured using a UV/VIS spectrophotometer at 562 nm. The conjugation efficiency was calculated using Equation (2).
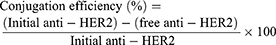
Evaluation of the in vitro DOX dissolution
An in vitro dissolution study was conducted at 37°C in the dark under a sink condition in which the amount of DOX was <10% of its solubility. 5 mg/mL of DOX-NPs and anti-HER2-conjugated nanoparticles containing DOX (anti-HER2-DOX-NPs) were dispersed in 1 mL PBS (receiving media) at pH 7.4 (using the initial DOX concentration of 5 µg/mL and anti-HER2 concentration of 25 µg/mL). At specific time points, the solutions containing DOX-NPs and anti-HER2-DOX-NPs were centrifuged to separate the NPs from the receiving media, and 0.1 mL of the supernatant was withdrawn. The receiving medium was replenished by adding 0.1 mL of fresh PBS to the test samples containing the NPs. The drug concentration was determined by a fluorescence spectrophotometer at an excitation wavelength of 485 nm and an emission wavelength of 590 nm. The cumulative DOX released was determined, as shown in Equation (3).
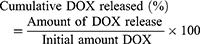
Cell culture
Human breast cancer cells (MCF-7) and an African green monkey kidney cell line (Vero) were obtained from the National Center for Genetic Engineering and Biotechnology (BIOTEC) (Bangkok, Thailand). The MCF-7 cells were used as a cancer cell model, while the Vero cells were used as a control cell line, which represented a normal healthy cell type. The MCF-7 and Vero cell lines were maintained in DMEM medium supplemented with 10% (v/v) of FBS, 100 units/mL of penicillin, and 100 µg/mL of streptomycin in a humidified atmosphere containing 5% of CO2 at 37°C.
Intracellular localization of the DOX-NP formulations
To observe the cellular localization of the NPs, the MCF-7 cells previously seeded on glass coverslips were incubated with various NP formulations. These included free DOX, DOX-NPs, and anti-HER2-DOX-NPs in DMEM supplemented with 10% of FBS to obtain the final equivalent DOX concentration of 0.5 µg/mL. This study was carried out at 4°C and 37°C. After the specific time points, the treated cells were washed twice with PBS to remove any remaining free DOX or NPs and fixed with 10% of paraformaldehyde. The accumulation of the free DOX or NP formations in the MCF-7 was detected using a laser scanning confocal microscope (Nikon Modular Confocal Microscope System C1, Tokyo, Japan). Fluorescence observation was carried out at 488 nm laser excitation.
In vitro cytotoxicity against breast cancer cell lines (MCF-7) and normal cell line (Vero)
The cytotoxicity of the free DOX, blank NPs, blank anti-HER2-NPs, DOX-NPs and anti-HER2-DOX-NPs against the MCF-7 cells and Vero cells was investigated using the MTT assay. Briefly, the cells were seeded in 96-well plates at a density of 5.0×103 cells/mL before being exposed to various concentrations of the free DOX, and nanoparticle samples were added. After 72 hrs of incubation, the spent medium was discarded and replaced with the MTT assay solution following the standard procedure (Molecular Probes, V-13,154, Eugene, OR, USA). The absorbance of formazan was measured at 570 nm (Infinite® 200 Tecan, Austria). The cell viability was presented as a percentage of the control cells not exposed to the NPs, as shown in Equation (4).

Statistical analysis
All experiments were performed at least in triplicate within two independent experiments. The data were reported as a mean ± SD. An unpaired t-test was used for data analyses in the encapsulation efficiency, conjugation efficiency, particle size, and cytotoxicity studies. A paired t-test was used for a statistical analysis of the in vitro DOX release data. A statistically significant difference was defined at a confidence level of 95%.
Results and discussion
Preparation of the OCP copolymer
Chitosan reacted with phthalic anhydride to form phthalimide chitosan, an intermediate of the reaction. The phthaloyl protection group was necessary since an amine group has a stronger nucleophilicity than a hydroxyl group. The O-succinylation of the chitosan was then carried out using succinic anhydride, and the protection group was later removed from the derivatives. Prior to the synthesis of the graft copolymer, pluronic was carboxylated with succinic anhydride to produce monocarboxy pluronic, followed by the reaction with EDC and NSH to form activated pluronic. Afterward, the activated pluronic was grafted onto the O-succinyl chitosan backbone through the reaction between the amino groups of the O-succinyl chitosan and the carboxyl groups of the pluronic.49 The synthesis scheme of the OCP copolymer is presented in Figure 1. The O-succinyl group of the chitosan was confirmed using 1H-NMR [refer to Figure S2], while the amide bond of the graft copolymer between the O-succinyl chitosan and pluronic was characterized by FTIR as previously reported49 and shown in Figure S3.
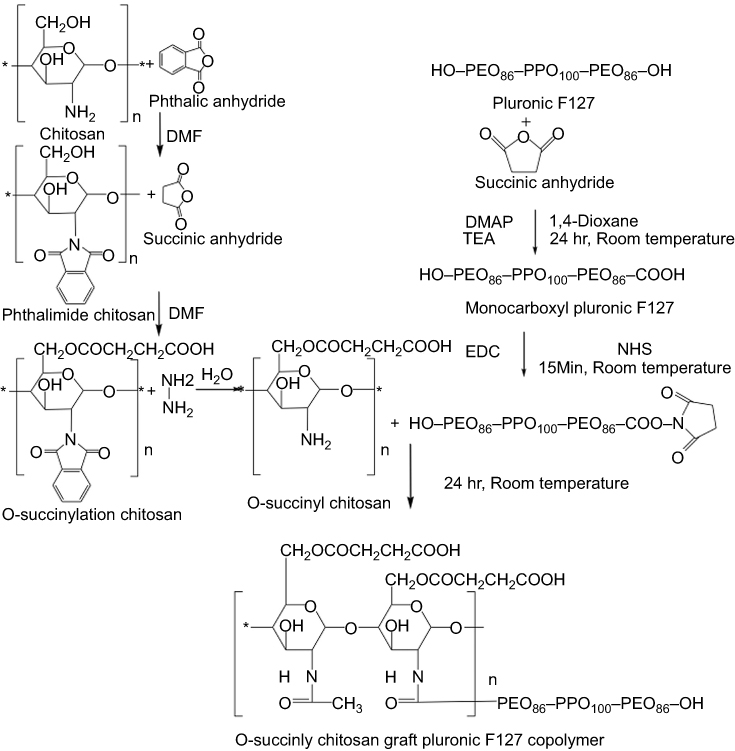 | Figure 1 Synthesis of the O-succinyl chitosan-pluronic copolymer using EDC/NHS.Abbreviations: EDC, 1-ethyl-3-(3-dimethylaminopropyl) carbodiimide hydrochloride; NHS, N-hydroxysuccinimide; DMF, dimethylformamide; DMAP, 4-dimethylaminopyridine; TEA, Triethylamine. |
CMC
To examine the formation of micelles with a core-shell NP structure, a fluorescence depolarization study was performed using DPH as a fluorescence probe. In an aqueous solution, DPH exhibits a very weak fluorescence while displaying a strong fluorescence in hydrophobic environments.50 The DPH probe normally becomes solubilized in the hydrophobic core of micelles giving rise to a drastic increase in its fluorescence signal.47,48
The CMCs of the pluronic, 5% of the OCP and 10% of the OCP at 25°C were found to be 0.18%, 0.39% and 0.69% (w/v), respectively (Figure 2). Clearly, grafting O-succinyl chitosan onto pluronic resulted in a higher CMC. More importantly, more O-succinyl chitosan content in the graft copolymer yielded a copolymer with a higher CMC. Previous studies have shown that micelle formation is driven by the hydrophobic part of amphiphiles. Polymers with a larger hydrophobic domain form micelles at lower concentrations leading to a lower CMC.47,51 In contrast, the micelle formation becomes more difficult with an increase in the hydrophilic molecules. The three polymers used in this study had the same hydrophobic segment (PPO), but varied hydrophilic segments (PEO and O-succinyl chitosan). Adding more O-succinyl chitosan to the graft copolymer resulted in an increase in the hydrophilic portion, thus lowering the ratio between the hydrophobic to hydrophilic portions. By decreasing the ratio between the hydrophobic and hydrophilic portions, the micelle formation was more difficult resulting in a higher CMC, as shown in Figure 2.
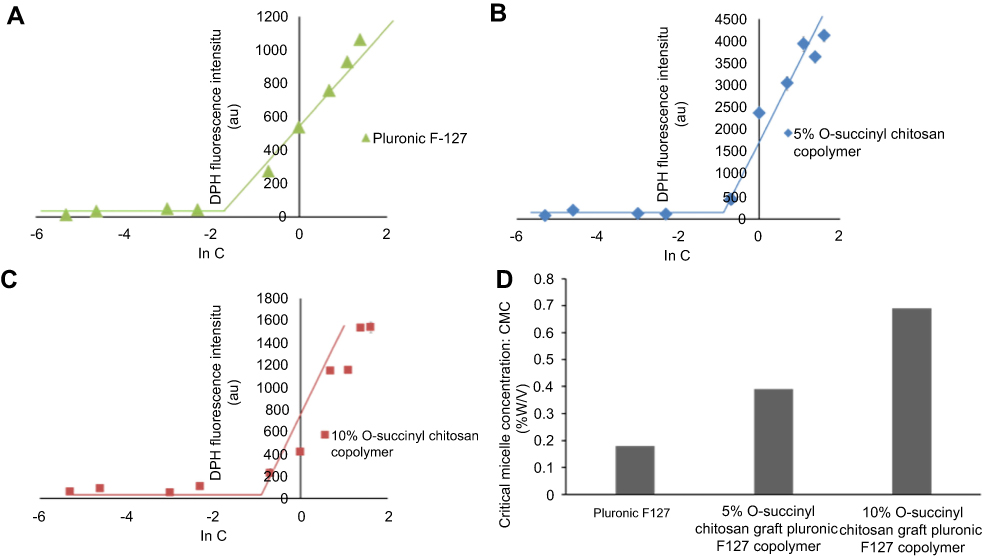 | Figure 2 Effect of the polymer concentrations on the fluorescence intensity of the DPH in the aqueous solutions of (A) pluronic, (B) 5% O-succinyl chitosan-pluronic and (C) 10% O-succinyl chitosan-pluronic, (D) the CMCs of pluronic, 5% O-succinyl chitosan-pluronic, and 10% O-succinyl chitosan-pluronic (n=3).Abbreviations: CMC, critical micelle concentrations; DPH, 1,6-diphenyl-1,3,5-hexatriene; C, concentration. |
Formation of the anti-HER2-conjugated OCP NPs containing DOX (anti-HER2-DOX-NPs)
In an aqueous solution, OCP copolymer self-assembled into core-shell NPs at the concentration above its CMC forming the hydrophobic inner core of PPO and the hydrophilic outer shell of PEO-O-succinyl chitosan. DOX, a hydrophobic anticancer drug, is, therefore, physically encapsulated into the hydrophobic core of the NPs during the self-assembly method.21,52 Afterward, the O-succinyl chitosan group on the outer shell forms the covalent complex through amidation with the primary amines of the anti-HER2 to yield the targeting ligand, which can specifically bind to the HER2 receptors on human breast cancer cells (refer to Scheme 1). According to our previous work, the concentration of the graft copolymer (5%, 7% and 10% w/v, respectively) had a minimal effect on the average size of the particle, the encapsulation efficiency, the DOX released and the in vitro cytotoxicity.49 Therefore, only the 5% and 10% OCP NPs at the concentration of 7% w/v were selected for further study.
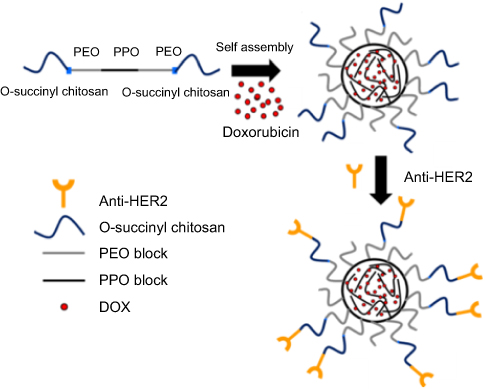 | Scheme 1 Preparation scheme of the anti-HER2-conjugated O-succinyl chitosan pluronic nanoparticles containing DOX (anti-HER2-DOX-NPs).Abbreviations: PEO, polyethylene oxide; PPO, polypropylene oxide; HER2, human epidermal growth factor receptor 2; DOX, doxorubicin. |
Encapsulation efficiency (%EE)
In this experiment, 5 to 25 µg of DOX was loaded into 5% and 10% OCP NPs. As shown in Table 1, the initial DOX concentration had an effect on the %EE, indicating that not all of the nominal DOX added was encapsulated into the particles. The %EE decreased as the initial DOX loading increased, which was consistent with the previous work.53,54 Interestingly, the amount of encapsulated DOX increased until the initial DOX concentration reached 20 µg/mL and then remained constant, indicating the saturation solubility point of DOX at the hydrophobic core of the NPs. In addition, the difference in the DOX encapsulation efficiencies between 5% and 10% OCP copolymer NPs was not statistically significant (P>0.05) because the hydrophobic parts of both formulations remained unchanged. Therefore, the NP ability to encapsulate a hydrophobic drug was not affected. Thus, 5 µg/mL of DOX was selected for further study because it showed the highest encapsulation efficiency and was, therefore, a more economical option.
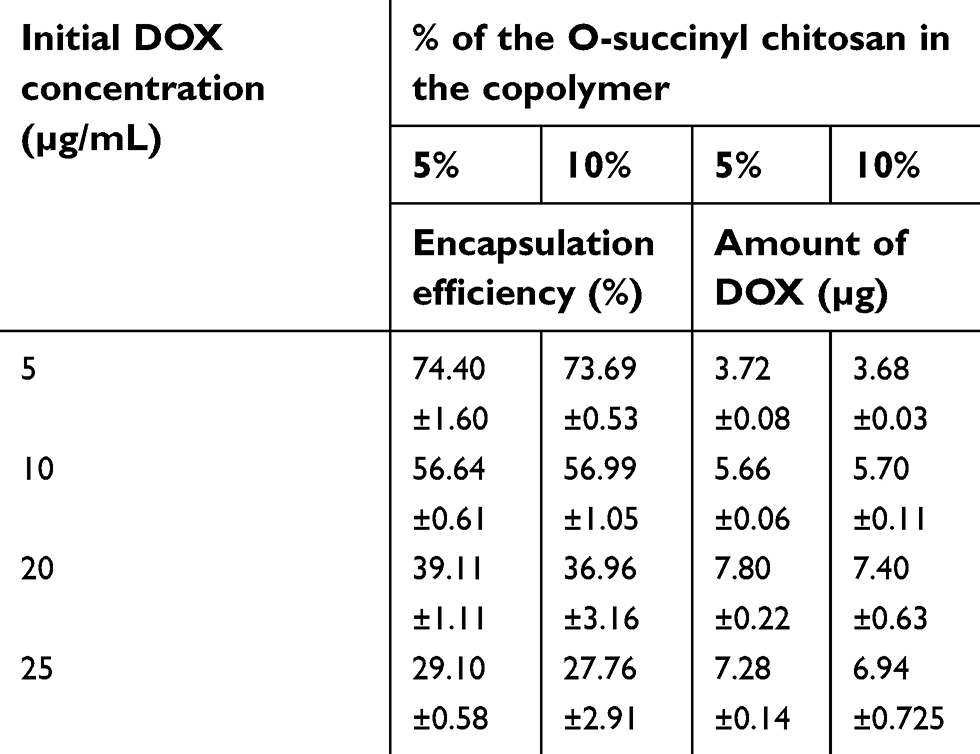 | Table 1 The encapsulation efficiencies of the O-succinyl chitosan pluronic NPs |
Preparation of the anti-HER2-conjugated OCP copolymer NPs
To functionalize NPs, after the formation and DOX encapsulation, the carboxyl groups of the OCP NP were preactivated by EDC and NHS and reacted with the terminal amine group (-NH2) of the anti-HER2 fragment (anti-HER2) to create a targeting moiety on the nanoparticles. As shown in Table 2, the conjugation efficiencies of anti-HER2 on the NP surfaces were relatively high, ranging from 57% to 61%. Interestingly, there was no statistically significant difference in the conjugation efficiencies between anti-HER2-DOX-NPs and anti-HER2-NPs (P>0.05), which indicated that the loaded DOX did not interfere with the conjugation process of the anti-HER2. In addition, the conjugation efficiency of the anti-HER2 to the 10% OCP NPs was slightly higher than that of the 5% OCP NPs (P<0.05) due to a higher number of available carboxyl groups (–COOH) of the O-succinyl chitosan to conjugate with the amine (-NH2) groups of anti-HER2.
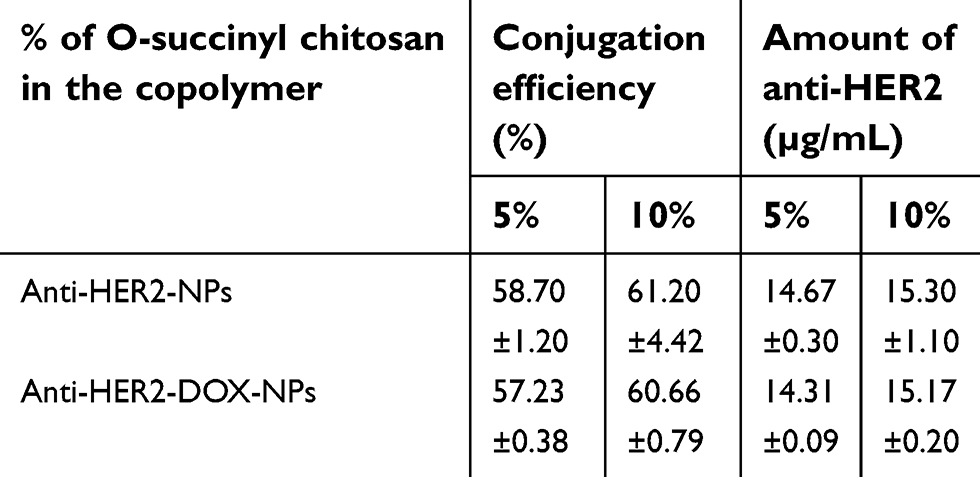 | Table 2 Conjugation efficiencies of the anti-HER2-conjugated O-succinyl chitosan pluronic NPs |
Average size of the OCP copolymer NPs
The average diameters of the 5% and 10% OCP NPs were smaller than those of the anti-HER2-NPs (P<0.05), as shown in Table 3. This demonstrated that the conjugation of anti-HER2 to the carboxyl groups of the copolymer caused an increase in the micelle sizes. Interestingly, the average size of the 10% OCP NPs was larger than that of the 5% OCP copolymer NPs. This may be a result of a higher O-succinyl chitosan content in the 10% OCP copolymer leading to a larger outer shell. The size distribution of the copolymer NPs displayed the NPs with an average size of <49 nm in diameter at 25°C. A narrow size distribution was observed in all cases, indicating uniform particle size distribution (refer to Figure S4).
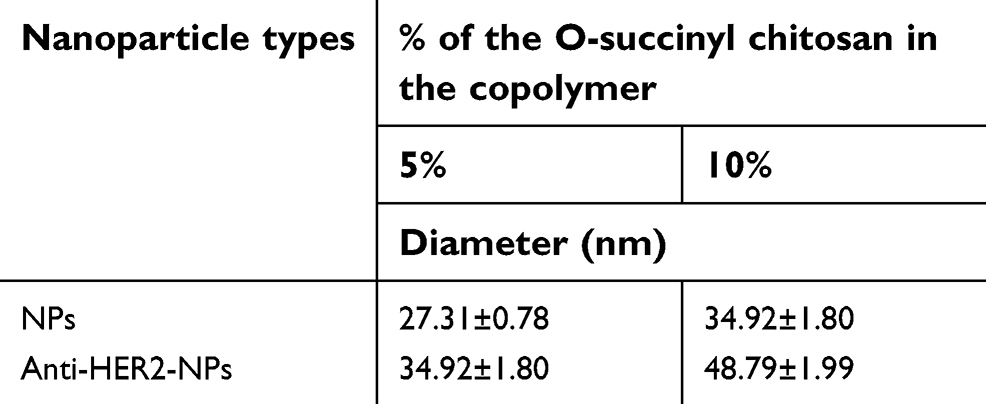 | Table 3 Average particle sizes of the NPs and anti-HER2-NPs |
Evaluation of the in vitro DOX dissolution
The study of the in vitro DOX releases from DOX-NPs and anti-HER2-DOX-NPs was carried out in a PBS solution at pH 7.4, which represented the actual physical environment to understand the mechanism governing the process of the drug release, and at pH 5.0, which represented the acidic environment of the cancer cells. The amount of DOX released was presented as the cumulative percentage release at 37°C over a period of 22 days (Figure 3). At pH 7.4 (Figure 3(b)), it was found that the DOX-NPs composed of 5% and 10% OCP copolymers exhibited similar release profiles with initial burst releases of up to 40% and 33%, respectively, in the first 24 hrs. This was followed by a sustained release of the encapsulated drug reaching 85% and 76% after 22 days, respectively. At the same condition, lower amounts of DOX were released from anti-HER2-DOX-NPs composed of 5% and 10% OCP copolymers (19.52% and 24.73%, respectively) in the first 24 hrs. As expected, the overall release of DOX was less than the anti-HER2-DOX-NPs. The conjugation of anti-HER2 on the OCP NP surface rendered slower drug release rates, which was possibly due to the steric hindrance of anti-HER2 on the NP structure. Fewer water molecules were able to reach the O-succinyl chitosan layer, resulting in slower degradation of chitosan and less DOX release from the NPs. At pH 5.0 (Figure 3(A)), the release profiles of different types of NPs showed similar trends with those at pH 7.4, which exhibited initial burst releases in the first 24 hrs followed by a sustained release of the encapsulated drug reaching 93% and 98% following a 22-day immersion. These particles tend to release more DOX in an acidic condition, which was likely to be beneficial for cancer treatment.
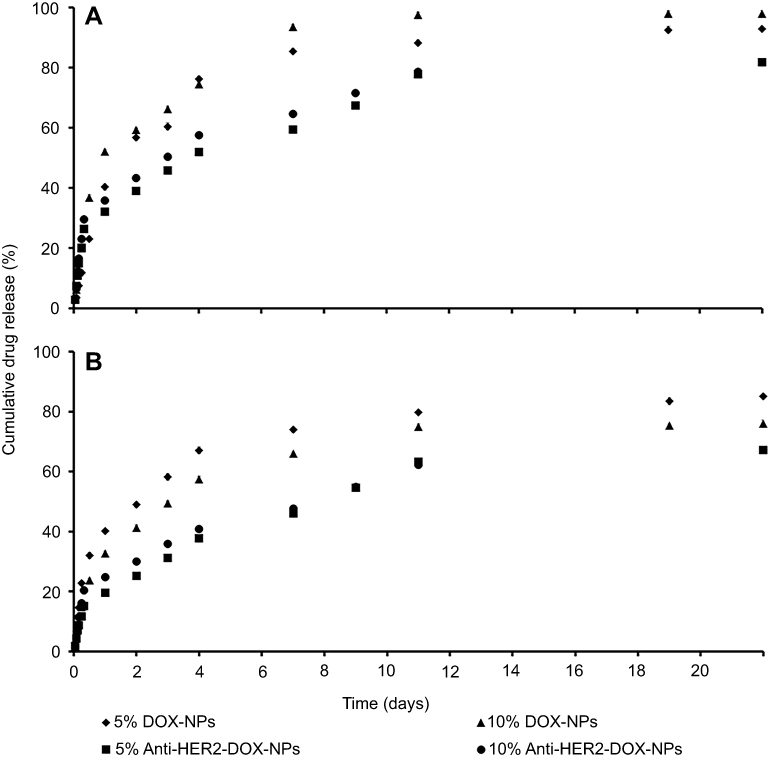 | Figure 3 DOX release profiles of DOX-NPs and anti-HER2-DOX-NPs in PBS at (A) pH 5.0 and (B) pH 7.4.Abbreviations: DOX, doxorubicin; NPs, nanoparticles; HER2, human epidermal growth factor receptor 2. |
Our result demonstrates that there were two phases in the DOX dissolution profile. First, an initial burst release of the DOX from the NPs took place in the first 24 hrs. This phenomenon may be attributable to DOX localized near the NP surface during the encapsulation process. In the next phase, a sustained release of the encapsulated DOX was shown after 24 hrs allowing for prolonged treatment. The sustained release was thought to be governed by two primary mechanisms, small-molecule diffusion and polymer degradation, which took place simultaneously.55,56
Intracellular localization of the DOX-NP formulations
The accumulation of DOX released from different NP formulations in human breast cancer, MCF-7, was investigated. This experiment was carried out at 37°C, which is equivalent to normal body temperature. The intracellular uptake of anti-HER2-DOX-NPs and DOX-NPs was compared to that of free DOX to provide insight into the mechanism of drug-encapsulated NP toxicity. Figure 4 shows the cellular distribution of each type of NP and free DOX in the MCF-7 cells at 1, 3, 6, and 24 hrs of incubation at 37°C. The red fluorescence of DOX was observed in the MCF-7 cells incubated with free DOX as early as 1 hr following the incubation. The fast uptake of the free drug was possibly due to the diffusion of small drug molecules through the cell membrane. After 3 hrs of incubation, more DOX, as shown in red fluorescence, from both DOX-NPs, and anti-HER2-DOX-NPs started to be localized in the cytoplasm and the nuclei of the cells. More intense red fluorescence was displayed from the MCF-7 cells exposed to all types of drug formulations after 6 hrs. Unlike free DOX, the uptake of the large particles like DOX-NPs and anti-HER2-DOX-NPs could not be accomplished through passive diffusion. Previous reports have shown that NPs could be localized into the cells mediated by endocytosis,57 which normally took longer. Endocytosis can be categorized as either nonspecific endocytosis or receptor-mediated endocytosis.58–60 It is believed that DOX-NPs were taken into the cells by an electrostatic interaction between the positively charged chitosan functional group and the negatively charged cell membrane. This mechanism was considered nonspecific. This result was consistent with the previous reports.61,62
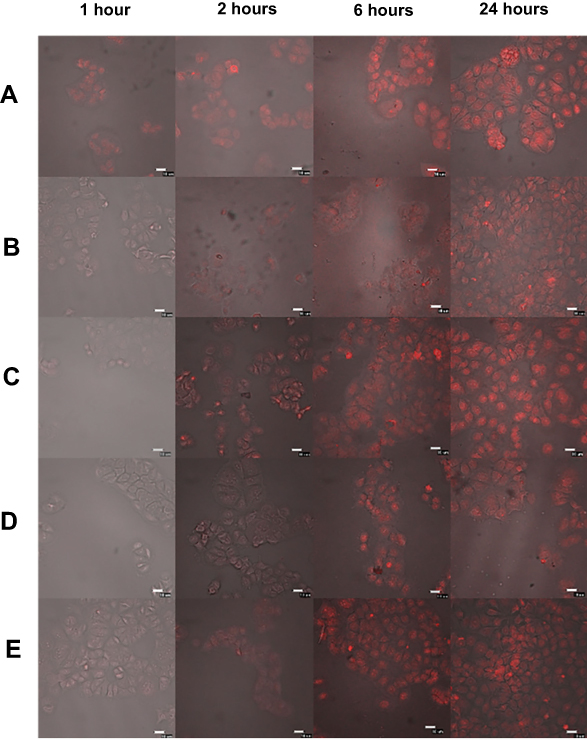 | Figure 4 Intracellular distribution of doxorubicin, doxorubicin-encapsulated nanoparticles, and anti-HER2-conjugated doxorubicin-encapsulated nanoparticles after the predetermined time of drug exposure in the MCF 7 cell line at 37°C (Merge image). The bar represents 10 μm. (A) Free DOX. (B) DOX-NPs: 5% O-succinyl chitosan copolymer. (C) Anti-HER2-DOX-NPs: 5% O-succinyl chitosan copolymer. (D) DOX-NPs: 10% O-succinyl chitosan copolymer. (E) Anti-HER2-DOX-NPs: 10% O-succinyl chitosan copolymer.Abbreviations: DOX, doxorubicin; NPs, nanoparticles; HER2, human epidermal growth factor receptor 2. |
On the other hand, anti-HER2-DOX-NPs, which had anti-HER2 as a targeting moiety on the particles’ outer shell, could be bound specifically to the HER2 receptors on the MCF-7 cells by means of a receptor-mediated endocytosis. As shown in Figure 4, more DOX was accumulated into the cells using the polymeric NPs with anti-HER2-targeting ligand, as compared to the NPs without the targeting ligand. The fluorescence intensities of the cells incubated with the DOX-NPs and anti-DOX-NPs were obviously different. This is possibly because the receptor-mediated endocytosis was shown to be more specific and efficient in delivering large molecules into the cells, resulting in a higher concentration of the drug at the target site.57,63
To further prove that the transport of the anti-HER2-DOX-NPs occurred by means of the receptor-mediated endocytosis, an additional bioaccumulation study was conducted at 4°C. In general, low temperature (0–4°C) treatment inhibits the functions of the cell’s receptors.64,65,66 If anti-HER2-DOX-NPs entered the cells by means of receptor-mediated endocytosis, a significant decrease in the uptake of the anti-HER2-DOX-NPs at 4°C would be observed. As shown in Figure 5, much less DOX was accumulated inside the MCF-7 cells when the cells were exposed to free DOX and DOX-NPs even after 24 hrs of incubation at 4°C. In addition, a longer incubation time was required to uptake these drug formulations. It is possible that the decrease of the uptake of free DOX, and the release of DOX from unconjugated NPs at a lower temperature was due to the increase in the level of the self-association of the DOX and aggregation at 4°C by the formation of the π-interaction.57 In addition, according to the Stokes–Einstein equation, the diffusion coefficient of a compound strongly depends on temperature.67 The diffusion coefficient of DOX at 4°C would be lower than that of 37°C, resulting in a lower passive diffusion through the cell membrane, hence, a lower DOX uptake.
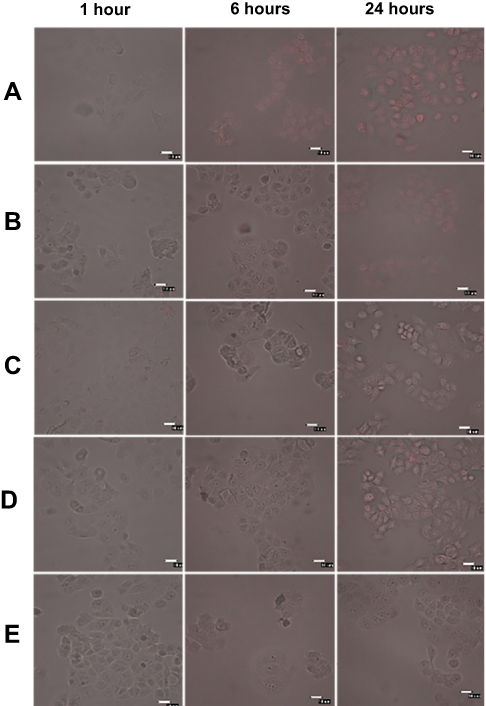 | Figure 5 Intracellular distribution of doxorubicin, doxorubicin-encapsulated nanoparticles, and anti-HER2-conjugated doxorubicin-encapsulated nanoparticles after the predetermined time of drug exposure in the MCF 7 cell line at 4°C (Merge image). The bar represents 10 μm. (A) Free DOX. (B) DOX-NPs: 5% O-succinyl chitosan copolymer. (C) Anti-HER2-DOX-NPs: 5% O-succinyl chitosan copolymer. (D) DOX-NPs: 10% O-succinyl chitosan copolymer. (E) Anti-HER2-DOX-NPs: 10% O-succinyl chitosan copolymer.Abbreviations: DOX, doxorubicin; NPs, nanoparticles; HER2, human epidermal growth factor receptor 2. |
Interestingly, no DOX was observed inside the cells exposed to the anti-HER2-DOX-NPs at 4°C. This indicated that the transport of DOX by means of HER2 receptor-mediated endocytosis was largely inhibited. The effect of the temperature on the binding of the ligand to the receptor as well as the lateral mobility of the ligand–receptor complex has been well documented.57 The inhibition of the anti-HER2-DOX-NP uptake might have been caused by the low binding between the targeting ligand on the NPs and HER2 receptors on the cells at 4 °C. In addition, incubation at a low temperature reduced the membrane’s fluidity, which could further restrict the movement of the ligand–receptor complex into the cells. Moreover, endocytosis, an energy-dependent mechanism, was inhibited at a low temperature.68 These observations confirmed that endocytosis was the major pathway for the anti-HER2-DOX-NP uptake.
In vitro cytotoxicity of the unconjugated and ligand-conjugated NPs
To ensure the safety of the NP formulations developed in this study for future therapeutic applications, the cytotoxicity of blank OCP NPs and the blank anti-HER2-NPs against the MCF-7 cells was assessed. Both NP formulations were shown to have minimal toxic effects and did not cause any cellular damage when used at a concentration <1 mg/mL (Figure 6). It was noted that the viability of the cells treated with blank anti-HER2-NPs had a statistically significant decrease (P<0.05) compared to the cells exposed to the unconjugated NPs at the same interval concentration (at a concentration >0.001 mg/mL) because anti-HER2 could inhibit cell growth by inducing diminished receptor signaling pathways.69 Anti-HER2 has been shown to block the shedding of the extracellular domain of the tyrosine kinase receptor of HER2 by inhibiting metalloproteinase activity resulting in possible cell damage.42,70
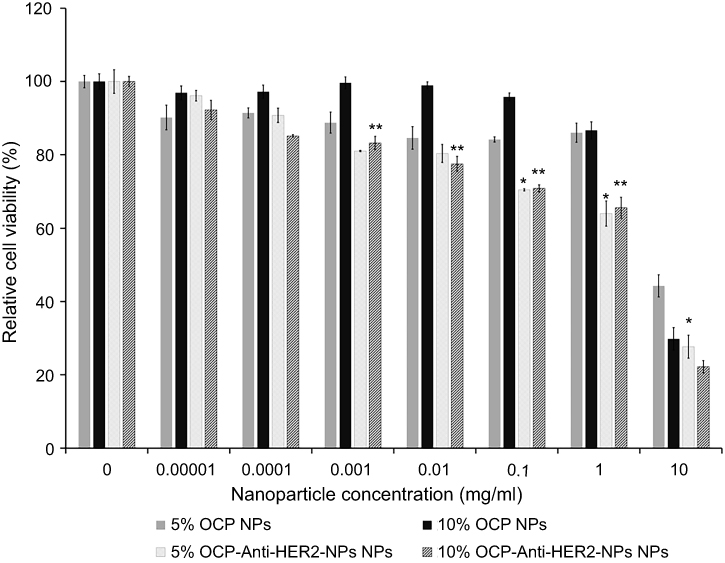 | Figure 6 The effect of blank 5% and 10% OCP nanoparticles and anti-HER2-conjugated nanoparticles on the cell viability of the MCF-7 cells based on the MTT assay (n=6 per group). The cells were treated with different concentrations of the particles (0.00001–10 mg/mL). *, ** indicates a statistical difference from the blank nanoparticles (*: 5% OCP and ** 10% OCP) at the same interval concentration, P<0.05.Abbreviations: DOX, doxorubicin; NPs, nanoparticles; HER2, human epidermal growth factor receptor 2; OCP, O-succinyl chitosan graft Pluronic® F127. |
When the cells were exposed to the NPs containing the DOX, the cell viability of the MCF-7 cells was clearly affected by the DOX content in the NP formulations (refer to Figure S5). The tumor cell numbers were markedly decreased as the equivalent drug concentrations increased. The differences in the numbers of the viable cells after treatment with unconjugated and conjugated NPs were more apparent at higher doses. When compared with all the NP formulations, the 10% OCP NPs conjugated with the anti-HER2 showed greater cytotoxicity toward the MCF-7 cells. This formulation could effectively reduce the number of viable cells, as compared to other formulations at the same DOX equivalent concentration. As shown in Figure 7A, the half maximal inhibitory concentration (IC50) of the 5% and 10% DOX-NPs against the MCF-7 cells was 405.73 and 205.90 ng/mL, respectively, which was about 1.66 and 3.26 times lower than that of free DOX (671.88 ng/mL). Interestingly, the IC50 values of the DOX-loaded 5% and 10% OCP NPs conjugated with the anti-HER2 were 186.01 and 14.74 ng/mL, respectively, which were approximately a 3.61-fold and 45.59-fold reduction of the IC50 of free DOX. Anti-HER2-DOX-NPs provided greater specificity to these cells than both the DOX-NPs and free DOX owing to the active targeting mechanism.71–73 In addition, it is believed that the combination of the anti-HER2 and DOX caused a synergistic effect in the cytotoxicity test. The 10% anti-HER2-DOX-NPs were more toxic against the MCF-7 cells than the 5% anti-HER2-DOX-NPs due to the higher anti-HER2 content, leading to more specificity and more toxicity to the MCF-7 cells.
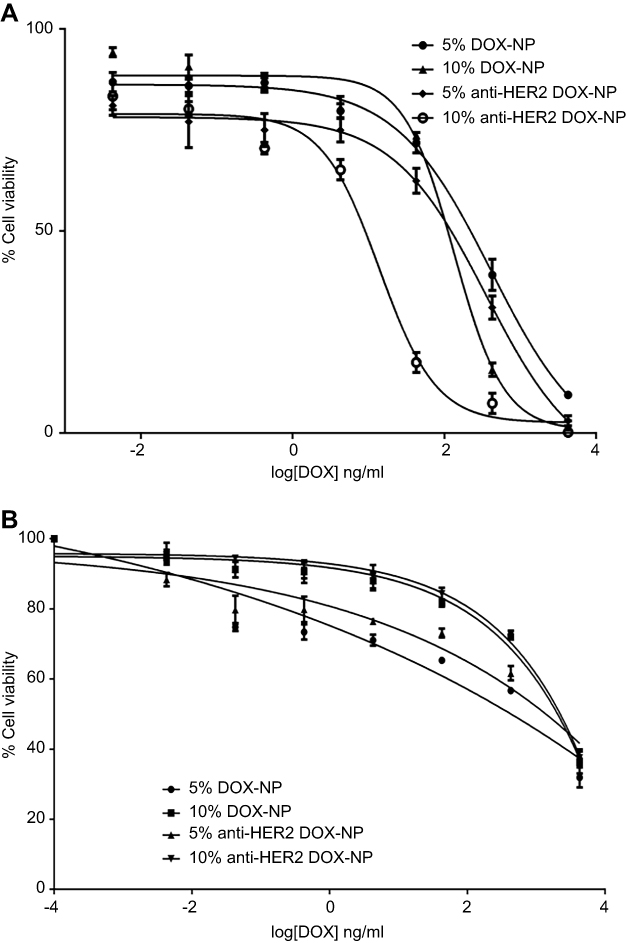 | Figure 7 Cytotoxicity profiles of DOX-encapsulated 5% and 10% OCP nanoparticles and anti-HER2-conjugated nanoparticles against (A) the MCF-7 cells and (b) Vero cells.Abbreviations: DOX, doxorubicin; NPs, nanoparticles; HER2, human epidermal growth factor receptor 2; OCP, O-succinyl chitosan graft Pluronic® F127. |
Furthermore, to evaluate the cytotoxic effect of these NP formulations toward healthy cells, the healthy cell model, Vero cell line, was exposed to the DOX-loaded OCP NPs with and without anti-HER2 conjugation. The IC50 concentrations of the DOX in various formulations against the Vero cells were significantly higher than those against the MCF-7 cells. This inferred that the healthy cells were less susceptible to the NP formulations containing DOX than the cancer cells (Figure 7B). Since the anti-HER2 receptors were not present on the Vero cells, the uptake of the NPs containing the DOX by means of the receptor-mediated endocytosis did not occur, inhibiting the internalization of the DOX NPs and the release of the anticancer drug. In addition, by encapsulating DOX inside the NPs, the polymeric NPs provided a protective layer to prevent the drug from freely diffusing into the cells. Hence, the IC50 values of all DOX NP formulations toward the healthy cells were higher than that of free DOX for healthy cells, thus indicating the lower toxicity of the anticancer drugs in the NP formulations for the normal cells.
To summarize, our results demonstrated that anti-HER2-DOX-NPs were shown to be beneficial in the treatment of cancer cells due to their ability to reduce the amount of DOX necessary for treatment. This is significantly important since DOX is cytotoxic to both cancer and normal cells when used in a free form; therefore, the reduction of the DOX doses helps lessen the damage to normal cells during the treatment process. Therefore, anti-HER2-DOX-NPs are promising as effective targeted drug carriers for cancer treatment because the carriers have been shown to selectively target cancer cells without damaging normal cells.
Conclusion
In this study, OCP copolymers were self-assembled to form NPs having core-shell structures while encapsulating DOX in its hydrophobic core at a relatively high encapsulation efficiency. Afterward, anti-HER2 was conjugated onto the OCP copolymer NPs as the targeting moiety. The in vitro dissolution profiles of DOX from the DOX-NPs and anti-HER2-DOX-NPs displayed initial burst releases followed by sustained releases for 22 days at pH 7.4. We have also demonstrated that the uptake of the anti-HER2-NPs occurred through receptor-mediated endocytosis, which was believed to be more specific and efficient. The in vitro cytotoxicity study indicated a higher therapeutic efficacy of the NP formulations. The targeted anti-HER2-DOX-NPs were found to provide higher anticancer activity toward the HER2-overexpressing cancer cells than both the nontargeted DOX-NPs and free DOX while showing much less toxicity toward the normal cells. Hence, the novel anti-HER2-conjugated DOX encapsulated OCP copolymer NPs are a promising alternative DOX carrier for effective breast cancer treatment.
Acknowledgments
This work was supported by the Research, Development and Engineering (RD&E) Fund through the National Nanotechnology Center (NANOTEC), the National Science and Technology Development Agency (NSTDA), Thailand (Project No. NN-B-22-EN6-e21-51-9), and the Higher Educational Research Promotion and National Research University Project of Thailand, the Office of the Higher Education Commission.
Disclosure
The authors report no conflicts of interest in this work.
References
1. Limeres MJ, Moretton MA, Bernabeu E, Chiappetta DA, Cuestas ML. Thinking small, doing big: current success and future trends in drug delivery systems for improving cancer therapy with special focus on liver cancer. Mater Sci Eng C. 2019;95:328 – 341. doi:10.1016/j.msec.2018.11.001
2. Aslan B, Ozpolat B, Sood AK, Lopez-Berestein G. Nanotechnology in cancer therapy. J Drug Target. 2013;21(10):904–913. doi:10.3109/1061186X.2013.837469
3. Qin S-Y, Zhang A-Q, Cheng S-X, Rong L, Zhang X-Z. Drug self-delivery systems for cancer therapy. Biomaterials. 2017;112:234–247. doi:10.1016/j.biomaterials.2016.10.016
4. Soga O, Van Nostrum CF, Fens M, et al. Thermosensitive and biodegradable polymeric micelles for paclitaxel delivery. J Control Release. 2005;103(2):341–353. doi:10.1016/j.jconrel.2004.12.009
5. Greish K. Enhanced permeability and retention (EPR) effect for anticancer nanomedicine drug targeting. Methods Mol Biol. 2010;624:25.
6. Markovsky E, Baabur-Cohen H, Satchi-Fainaro R. Anticancer polymeric nanomedicine bearing synergistic drug combination is superior to a mixture of individually-conjugated drugs. J Control Release. 2014;187:145–157. doi:10.1016/j.jconrel.2014.05.025
7. Kobiasi MA, Chua BY, Tonkin D, Jackson DC, Mainwaring DE. Control of size dispersity of chitosan biopolymer microparticles and nanoparticles to influence vaccine trafficking and cell uptake. J Biomed Mater Res A. 2012;100A(7):1859–1867. doi:10.1002/jbm.a.v100a.7
8. Otto DP, Otto A, de Villiers MM. Differences in physicochemical properties to consider in the design, evaluation and choice between microparticles and nanoparticles for drug delivery. Expert Opin Drug Deliv. 2015;12(5):763–777. doi:10.1517/17425247.2015.988135
9. Kohane DS. Microparticles and nanoparticles for drug delivery. Biotechnol Bioeng. 2006;96(2):203–209. doi:10.1002/bit.21301
10. Kumari A, Yadav SK, Yadav SC. Biodegradable polymeric nanoparticles based drug delivery systems. Colloids Surf B Biointerfaces. 2010;75(1):1–18. doi:10.1016/j.colsurfb.2009.09.001
11. Masood F. Polymeric nanoparticles for targeted drug delivery system for cancer therapy. Mater Sci Eng C. 2016;60:569–578. doi:10.1016/j.msec.2015.11.067
12. Crucho CIC, Barros MT. Polymeric nanoparticles: A study on the preparation variables and characterization methods. Mater Sci Eng C. 2017;80:771–784. doi:10.1016/j.msec.2017.06.004
13. Jones MC, Leroux JC. Polymeric micelles - a new generation of colloidal drug carriers. Eur J Pharm Biopharm. 1999;48:101–111.
14. Opsteen JA, Cornelissen JJLM, Hest J. Block copolymer vesicles. Pure Appl Chem. 2004;76:1309–1319. doi:10.1351/pac200476071309
15. Yang H, Zhao X, Zhang X, Ma L, Wang B, Wei H. Optimization of bioreducible micelles self-assembled from amphiphilic hyperbranched block copolymers for drug delivery. J Polym Sci A. 2018;56(13):1383–1394. doi:10.1002/pola.v56.13
16. Adams ML, Lavasanifar A, Kwon GS. Amphiphilic block copolymers for drug delivery. J Pharm Sci. 2003;92(7):1343. doi:10.1002/jps.10397
17. Kataoka K, Kwon G, Yokoyama M, Okano T, Sakurai Y. Block copolymer micelles as vehicles for drug delivery. J Control Release. 1992;24:119–132.
18. Kumari A, Yadav SK, Yadav SC. Biodegradable polymeric nanoparticles based drug delivery systems. Colloids Surf B. 2010;17:1–18. doi:10.1016/j.colsurfb.2009.09.001
19. Imran M, Shah MR, Shafiullah. Chapter 10 - Amphiphilic block copolymers–based micelles for drug delivery. In: Grumezescu AM, editor. Design and Development of New Nanocarriers. Norwich: William Andrew Publishing; 2018:365–400.
20. Gaucher G, Dufresne M-H, Sant VP, Kang N, Maysinger D, Leroux J-C. Block copolymer micelles: preparation, characterization and application in drug delivery. J Control Release. 2005;109:169–188. doi:10.1016/j.jconrel.2005.09.034
21. Missirlis D, Kawamura R, Tirelli N, Hubbell JA. Doxorubicin encapsulation and diffusional release from stable, polymeric, hydrogel nanoparticles. Eur J Pharm Sci. 2006;29:120–129. doi:10.1016/j.ejps.2006.06.003
22. Kataoka K, Harada A, Nagasaki Y. Block copolymer micelles for drug delivery: design, characterization and biological significance. Adv Drug Delivery Rev. 2001;47:113–131. doi:10.1016/S0169-409X(00)00124-1
23. Yoo HS, Park TG. Biodegradable polymeric micelles composed of doxorubicin conjugated PLGA–PEG block copolymer. J Control Release. 2001;70:63–70. doi:10.1016/S0168-3659(00)00340-0
24. Pillai SA, Patel VI, Ray D, Pal H, Aswal VK, Bahadur P. Solubilization and interaction of cinnamic acid and its analogues with Pluronic® micelles. Colloids Surf A Physicochem Eng Asp. 2018;559:314–324. doi:10.1016/j.colsurfa.2018.09.074
25. Managa M, Britton J, Prinsloo E, Nyokong T. Effects of Pluronic F127 micelles as delivering agents on the vitro dark toxicity and photodynamic therapy activity of carboxy and pyrene substituted porphyrins. Polyhedron. 2018;152:102–107. doi:10.1016/j.poly.2018.06.031
26. Zhao L-Y, Zhang W-M. Recent progress in drug delivery of pluronic P123: pharmaceutical perspectives. J Drug Target. 2017;25(6):471–484. doi:10.1080/1061186X.2017.1289538
27. Batrakova EV, Kabanov AV. Pluronic block copolymers: evolution of drug delivery concept from inert nanocarriers to biological response modifiers. J Control Release. 2008;130(2):98–106. doi:10.1016/j.jconrel.2008.04.013
28. Akash MSH, Rehman K. Recent progress in biomedical applications of Pluronic (PF127): pharmaceutical perspectives. J Control Release. 2015;209:120–138. doi:10.1016/j.jconrel.2015.04.032
29. Pruitt J, Husseini G, Rapoport NW. Stabilization of pluronic P-105 micelles with an interpenetrating network of N, N-diethylacrylamide. Macromolecules. 2000;33:9306–9309. doi:10.1021/ma0008544
30. Park KM, Bae JW, Joung YK, Shin JW, Park KD. Nanoaggregate of thermosensitive chitosan-Pluronic for sustained release of hydrophobic drug. Colloids Surf B Biointerfaces. 2008;63(1):1–6. doi:10.1016/j.colsurfb.2007.10.024
31. Chung HJ, Go DH, Bae JW, Jung IK, Lee JW, Park KD. Synthesis and characterization of Pluronic® grafted chitosan copolymer as a novel injectable biomaterial. Current Applied Physics. 2005;5(5):485–488. doi:10.1016/j.cap.2005.01.015
32. Hosseinzadeh H, Atyabi F, Dinarvand R, Ostad SN. Chitosan-Pluronic nanoparticles as oral delivery of anticancer gemcitabine: preparation and in vitro study. Int J Nanomedicine. 2012;7:1851–1863. doi:10.2147/IJN.S26365
33. Manaspon C, Viravaidya-Pasuwat K, Pimpha N. Preparation of folate-conjugated pluronic F127/chitosan core-shell nanoparticles encapsulating doxorubicin for breast cancer treatment. J Nanomater. 2012;2012:11. doi:10.1155/2012/593878
34. Domínguez-Delgado C, Fuentes-Prado E, Escobar-Chávez J, Vidal-Romero G, Rodríguez Cruz I, Díaz-Torres R. Chitosan and Pluronic® F-127: Pharmaceutical Applications. In: Mishra MK, editor. Encyclopedia of Biomedical Polymers and Polymeric Biomaterials. Florida: Taylor and Francis; 2016:1513 – 1535.
35. Li H, Qian ZM. Transferrin/transferrin receptor-mediated drug delivery. Med Res Rev. 2002;22:225–250.
36. You J, Li X, Cui FD, Du YZ, Yuan H, Hu FQ. Folate-conjugated polymer micelles for active targeting to cancer cells: preparation, in vitro evaluation of targeting ability and cytotoxicity. Nanotechnology. 2008;19(4):045102. doi:10.1088/0957-4484/19/04/045102
37. Byrne JD, Betancourt T, Brannon-Peppas L. Active targeting schemes for nanoparticle systems in cancer therapeutics. Adv Drug Deliv Rev. 2008;60:1615–1626. doi:10.1016/j.addr.2008.08.005
38. Kim BS, Taton TA. Multicomponent nanoparticles via self-assembly with cross-linked block copolymer surfactants. Langmuir. 2007;23:2198–2202. doi:10.1021/la062692w
39. van Rooijen JM, Qiu S-Q, Timmer-Bosscha H, et al. Androgen receptor expression inversely correlates with immune cell infiltration in human epidermal growth factor receptor 2–positive breast cancer. Eur J Cancer. 2018;103:52–60. doi:10.1016/j.ejca.2018.08.001
40. Yuan P, Gao S-L. Management of breast cancer brain metastases: focus on human epidermal growth factor receptor 2-positive breast cancer. Chronic Dis Transl Med. 2017;3(1):21–32. doi:10.1016/j.cdtm.2017.01.004
41. Loibl S, Gianni L. HER2-positive breast cancer. Lancet. 2017;389(10087):2415–2429. doi:10.1016/S0140-6736(16)32417-5
42. Diermeier S, Horvath G, Clarke RK, Hofstaedter F, SzfllIsi J, Brockhoff G. Epidermal growth factor receptor coexpression modulates susceptibility to Herceptin in HER2/neu overexpressing breast cancer cells via specific erbB-receptor interaction and activation. Exp Cell Res. 2005;304:604–619. doi:10.1016/j.yexcr.2004.12.008
43. Nielsen DL, Andersson M, Kamby C. HER2-targeted therapy in breast cancer. Monoclonal antibodies and tyrosine kinase inhibitors. Cancer Treat Rev. 2009;35:121–136. doi:10.1016/j.ctrv.2008.09.003
44. Parakh S, Gan HK, Parslow AC, Burvenich IJG, Burgess AW, Scott AM. Evolution of anti-HER2 therapies for cancer treatment. Cancer Treat Rev. 2017;59:1–21. doi:10.1016/j.ctrv.2017.06.005
45. Pondé N, Brandão M, El-Hachem G, Werbrouck E, Piccart M. Treatment of advanced HER2-positive breast cancer: 2018 and beyond. Cancer Treat Rev. 2018;67:10–20. doi:10.1016/j.ctrv.2018.04.016
46. Zhang C, Ping Q, Zhang H, Shen J. Synthesis and characterization of water-soluble O-succinyl-chitosan. Eur Polym J. 2003;39:1629–1634. doi:10.1016/S0014-3057(03)00068-5
47. Alexandridis P, Athanassiou V, Fukuda S, Hatton TA. Surface activity of poly(ethylene oxide)-block-poly(propylene oxide)-block-poly(ethylene oxide) copolymers. Langmuir. 1994;10:2604–2612. doi:10.1021/la00020a019
48. Pepic I, Grcic JF, Jalsenjak I. Bulk properties of nonionic surfactant and chitosan mixtures. Colloids Surf A. 2009;336:135–141. doi:10.1016/j.colsurfa.2008.11.034
49. Naruphontjirakul P, Viravaidya-Pasuwat K. Development of doxorubicin – core shell O-succinyl chitosan graft pluronic®127 copolymer nanoparticles to treat human cancer. Int J Biosci Biochem Bioinforma. 2011;1(2):131–136. doi:10.7763/IJBBB.2011.V1.24
50. Choo ESG, Yu B, Xue J. Synthesis of poly(acrylic acid) (PAA) modified pluronic P123 copolymers for pH-stimulated release of doxorubicin. J Colloid Interface Sci. 2011;358:462–470. doi:10.1016/j.jcis.2011.03.047
51. Kozlov MY, Melik-Nubarov NS, Batrakova EV, Kabanov AV. Relationship between pluronic block copolymer structure, critical micellization concentration and partitioning coefficients of low molecular mass solutes. Macromolecules. 2000;33:3305–3313. doi:10.1021/ma991634x
52. Chung YI, Kim JC, Kim YH, et al. The effect of surface functionalization of PLGA nanoparticles by heparin- or chitosan-conjugated pluronic on tumor targeting. J Control Release. 2010;43:374–382. doi:10.1016/j.jconrel.2010.01.017
53. Janes KA, Fresneau MP, Marazuela A, Fabra A, Alonso MJ. Chitosan nanoparticles as delivery systems for doxorubicin. J Control Release. 2001;73:255–267. doi:10.1016/S0168-3659(01)00294-2
54. Souto GD, Farhane Z, Casey A, Efeoglu E, McIntyre J, Byrne HJ. Evaluation of cytotoxicity profile and intracellular localisation of doxorubicin-loaded chitosan nanoparticles. Anal Bioanal Chem. 2016;408(20):5443–5455. doi:10.1007/s00216-016-9641-6
55. Colombo P, Bettini R, Santi P, Ascentiis AD, Peppas NA. Analysis of the swelling and release mechanisms from drug delivery systems with emphasis on drug solubility and water transport. J Control Release. 1996;39:231–237. doi:10.1016/0168-3659(95)00158-1
56. Heller J. Biodegradable polymers in controlled drug delivery. Crit Rev Ther Drug. 1984;1:39–90.
57. Stromhaug PE, Berg TO, Gjoen T, Seglen PO. Differences between fluid-phase endocytosis (pinocytosis) and receptor-mediated endocytosis in isolated rat hepatocytes. Eur J Cell Biol. 1997;73(1):28–39.
58. Albanese A, Tang PS, Chan WCW. The effect of nanoparticle size, shape, and surface chemistry on biological systems. Annu Rev Biomed Eng. 2012;14(1):1–16. doi:10.1146/annurev-bioeng-071811-150124
59. Wileman T, Harding C, Stahl P. Receptor-mediated endocytosis. Biochem J. 1985;232(1):1. doi:10.1042/bj2320001
60. Decuzzi P, Ferrari M. The role of specific and non-specific interactions in receptor-mediated endocytosis of nanoparticles. Biomaterials. 2007;28(18):2915–2922. doi:10.1016/j.biomaterials.2007.02.013
61. Cai H, Liang Z, Huang W, Wen L, Chen G. Engineering PLGA nano-based systems through understanding the influence of nanoparticle properties and cell-penetrating peptides for cochlear drug delivery. Int J Pharm. 2017;532(1):55–65. doi:10.1016/j.ijpharm.2017.08.084
62. Srinophakun P, Thanapimmetha A, Plangsri S, Vetchayakunchai S, Saisriyoot M. Application of modified chitosan membrane for microbial fuel cell: roles of proton carrier site and positive charge. J Clean Prod. 2017;142:1274–1282. doi:10.1016/j.jclepro.2016.06.153
63. Kanazaki K, Sano K, Makino A, et al. Development of anti-HER2 fragment antibody conjugated to iron oxide nanoparticles for in vivo HER2-targeted photoacoustic tumor imaging. Nanomedicine. 2015;11(8):2051–2060. doi:10.1016/j.nano.2015.07.007
64. AshaRani PV, Hande MP, Valiyaveettil S. Anti-proliferative activity of silver nanoparticles. BMC Cell Biol. 2009;10:1–14. doi:10.1186/1471-2121-10-1
65. Chithrani BD, Chan WCW. Elucidating the mechanism of cellular uptake and removal of protein-coated gold nanoparticles of different sizes and shapes. Nano Letter. 2007;7:1542–1550. doi:10.1021/nl070363y
66. Thurn KT, Arora H, Paunesku T, et al. Endocytosis of titanium dioxide nanoparticles in prostate cancer PC-3M cells. Nanomedicine. 2011;7:123–130. doi:10.1016/j.nano.2010.10.012
67. Miller CC. The Stokes-Einstein law for diffusion in solution. Proc R Soc London Ser A Containing Pap Math Phys Charact. 1924;106(740):724–749.
68. Yadav JS, Das PP, Reddy TL, et al. Sub-cellular internalization and organ specific oral elivery of PABA nanoparticles by side chain variation. J Nanobiotechnology. 2011;9(1):10. doi:10.1186/1477-3155-9-10
69. Le XF, McWatters A, Wiener J, Wu JY, Mills GB, Bast RC
70. Valabrega G, Montemurro F, Aglietta M. Trastuzumab: mechanism of action, resistance and future perspectives in HER2-overexpressing breast cancer. Ann Oncol. 2007;18:977–984. doi:10.1093/annonc/mdl475
71. Park JW, Kirpotin DB, Hong K, et al. Tumor targeting using anti-her2 immunoliposomes. J Control Release. 2001;74:95–113. doi:10.1016/S0168-3659(01)00315-7
72. Shi M, Ho K, Keating A, Shoichet MS. Doxorubicin-conjugated immuno-nanoparticles for intracellular anticancer drug delivery. Adv Funct Mater. 2009;19:1–8.
73. Yousefpour P, Atyabi F, Vasheghani-Farahani E, Movahedi AA, Dinarvand R. Targeted delivery of doxorubicin-utilizing chitosan nanoparticles surface-functionalized with anti-Her2 trastuzumab. Int J Nanomedicine. 2011;6:1977–1990. doi:10.2147/IJN.S21523
Supplementary materials
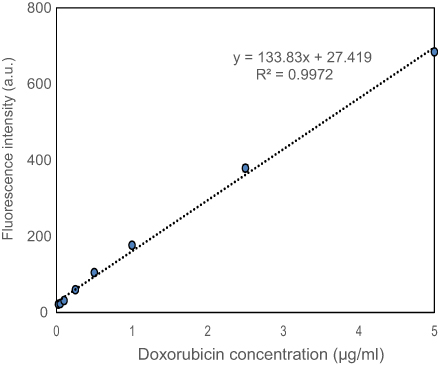 | Figure S1 Calibration curve of DOX in PBS.Abbrevations: DOX, doxorubicin; PBS, phosphate buffer saline. |
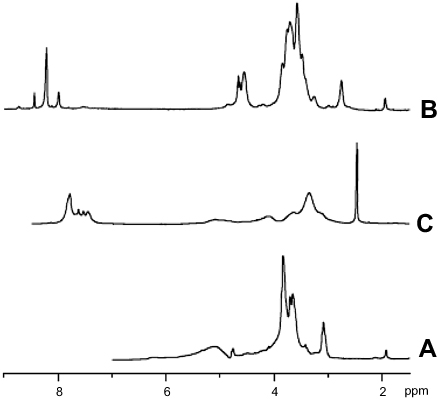 | Figure S2 1H-NMR spectra of (A) chitosan, (B) O-succinyl-chitosan and (C) phthalimide chitosan. |
 | Figure S3 Fourier-transform infrared spectroscopy spectra of (A) Pluronic® F127, (B) Monocarboxy pluronic and (C) O-succinyl chitosan graft pluronic copolymer. |
 | Figure S4 Intensity particle size distribution of 5% OCP copolymer (A) and 10% OCP copolymer nanoparticles (B) dispersed in distilled water (n=3). n: number of samples. Abbreviation: OCP, O-succinyl chitosan graft Pluronic® F127. |
 | Figure S5 The cell viability of MCF-7 and Vero cells after exposure to various concentrations of doxorubicin for 3 days. The calculated IC50 of doxorubicin for MCF-7 and Vero cells are 672 and 758 ng/mL, respectively. |
 © 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.