Back to Journals » OncoTargets and Therapy » Volume 7
Development of anaplastic lymphoma kinase (ALK) inhibitors and molecular diagnosis in ALK rearrangement-positive lung cancer
Authors Iwama E, Okamoto I, Harada T, Takayama K, Nakanishi Y
Received 25 November 2013
Accepted for publication 29 December 2013
Published 5 March 2014 Volume 2014:7 Pages 375—385
DOI https://doi.org/10.2147/OTT.S38868
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 4
Eiji Iwama,1,2 Isamu Okamoto,3 Taishi Harada,2 Koichi Takayama,2 Yoichi Nakanishi2,3
1Department of Comprehensive Clinical Oncology, Faculty of Medical Sciences, Kyushu University, 2Research Institute for Diseases of the Chest, Graduate School of Medical Sciences, Kyushu University, 3Center for Clinical and Translational Research, Kyushu University Hospital, Fukuoka, Japan
Abstract: The fusion of echinoderm microtubule-associated protein-like 4 with anaplastic lymphoma kinase (ALK) was identified as a transforming gene for lung cancer in 2007. This genetic rearrangement accounts for 2%–5% of non-small-cell lung cancer (NSCLC) cases, occurring predominantly in younger individuals with adenocarcinoma who are never- or light smokers. A small-molecule tyrosine-kinase inhibitor of ALK, crizotinib, was rapidly approved by the US Food and Drug Administration on the basis of its pronounced clinical activity in patients with ALK rearrangement-positive NSCLC. Next-generation ALK inhibitors, such as alectinib, LDK378, and AP26113, are also being developed in ongoing clinical trials. In addition, the improvement and validation of methods for the detection of ALK rearrangement in NSCLC patients will be key to the optimal clinical use of ALK inhibitors. We here summarize recent progress in the development of new ALK inhibitors and in the molecular diagnosis of ALK rearrangement-positive NSCLC.
Keywords: ALK, rearrangement, NSCLC, ALK inhibitor, targeted therapy, diagnosis
Background
Lung cancer is the leading cause of cancer deaths worldwide. Non-small-cell lung cancer (NSCLC) accounts for 85% of lung cancer cases, and has usually achieved an advanced stage by the time of diagnosis.1 Cytotoxic chemotherapy has been the mainstay of treatment for metastatic NSCLC, but its efficacy has plateaued in recent years. Further improvement in the clinical outcome of individuals with NSCLC will thus depend on the development of new treatment strategies, such as molecularly targeted therapies. In 2004, the identification of activating mutations of the epidermal growth-factor receptor (EGFR) gene in a subset of NSCLC patients led to a change in treatment of the disease.2,3 Treatment of patients with NSCLC positive for EGFR mutations with such EGFR tyrosine-kinase inhibitors (TKIs) as gefitinib and erlotinib was found to have a high response rate and to result in both prolonged progression-free survival (PFS) and improved quality of life compared with cytotoxic chemotherapy.4,5 The discovery of EGFR mutations and the efficacy of EGFR TKIs in selected patients thus opened a new era of personalized treatment for NSCLC.
Anaplastic lymphoma kinase (ALK) is a receptor tyrosine kinase whose gene was initially identified in a subset of individuals with anaplastic large-cell lymphoma. A reciprocal translocation between chromosomes 2 and 5 apparent in such patients6 was found to result in the formation of a fusion gene comprising the 5′ portion of the nucleophosmin gene and the 3′ portion of ALK encoding the kinase domain.7 In 2007, a fusion gene formed by ALK and the echinoderm microtubule-associated protein-like 4 (EML4) gene was identified in the tumor of a 62-year-old Japanese man with adenocarcinoma of the lung, and was shown to possess pronounced oncogenic activity.8 This genetic rearrangement has since been found to occur in 2%–5% of NSCLC patients, predominantly in those with adenocarcinoma who are of younger age and never- or light smokers.9,10
The EML4–ALK fusion oncogene arises from a small inversion within the short arm of chromosome 2 that joins the 5′ region of EML4 (encoding the NH2-terminal portion of EML4, including its coiled-coil domain) to the 3′ region of ALK (encoding the COOH-terminal portion of ALK, including the tyrosine-kinase domain). It exists in multiple variants that encode the same intracellular tyrosine-kinase domain of ALK but different truncations of EML4.11,12 The most common variants are variant 1 (detected in 33% of patients), in which exon 13 of EML4 is fused to exon 20 of ALK (E13;A20), and variant 3a/b (detected in 29% of patients), in which exon 6 of EML4 is fused to exon 20 of ALK (E6a/b;A20).12 Two other rare fusion partners of ALK (tyrosine-kinase receptor-fused gene and kinesin family member 5B) in addition to EML4 have also been identified in individuals with NSCLC.
All of these ALK fusion proteins undergo ligand-independent dimerization mediated by the coiled-coil domain of the fusion partner, resulting in constitutive activation of the ALK tyrosine kinase.13,14 Such phosphorylation-mediated activation of the ALK fusion proteins results in activation of downstream signaling pathways – including the JAK–STAT, MEK–ERK, and PI3K–AKT pathways – that contribute to oncogenicity.15–17 TKIs that target the kinase activity of ALK (ALK TKIs) have been found to have pronounced antiproliferative and proapoptotic effects in EML4–ALK-positive lung cancer cells.14,18
Crizotinib
The first clinically available ALK TKI
The structure of crizotinib is shown in Figure 1. Crizotinib is an oral and potent small-molecule ALK TKI that was initially designed as an inhibitor of the tyrosine kinase MET. Crizotinib competes with adenosine triphosphate for binding to the tyrosine kinase pocket of ALK and thereby inhibits its tyrosine-kinase activity, leading to inhibition of downstream signaling and to anticancer effects. Crizotinib exerts proapoptotic activity, with a median effective concentration in the 5–25 nM range in vitro for cells with activated ALK or MET receptor tyrosine kinases.19,20
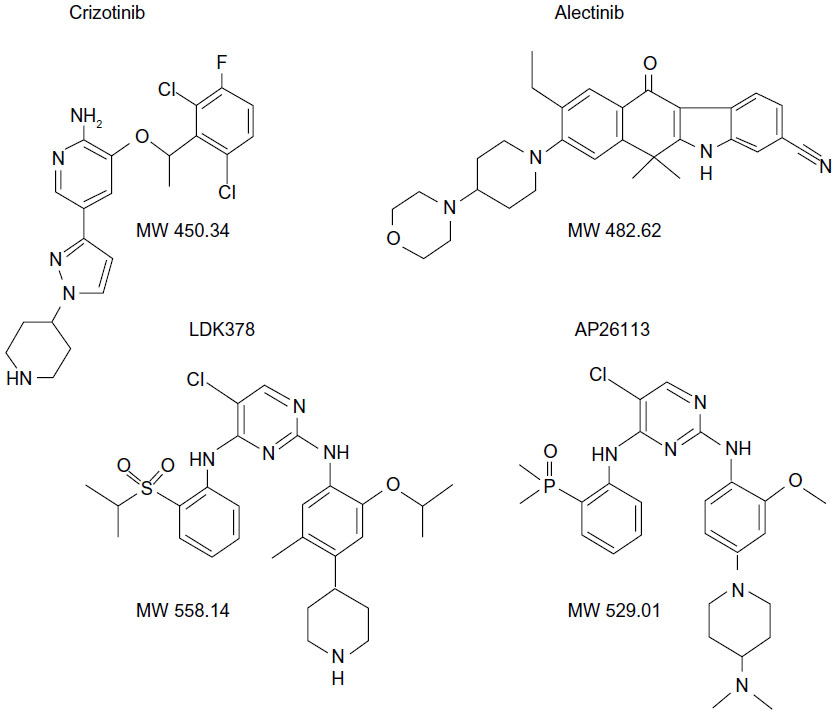 | Figure 1 Chemical structures and molecular weight (MW) of crizotinib, alectinib, LDK378, and AP26113. |
Crizotinib was the first ALK TKI introduced into clinical trials. A dose-escalation component of a Phase I trial (Profile 1001, NCT00585195) identified 250 mg twice daily (bid) as the recommended Phase II dose for crizotinib.21 Fatigue was the dose-limiting toxicity (DLT), occurring at grade 3 in two of the six patients treated with crizotinib at 300 mg bid. On the basis of promising results apparent in two patients with ALK rearrangement-positive NSCLC enrolled during the dose-escalation component, the protocol was amended to expand the cohort of such patients in the second part of this Phase I trial. A total of 149 ALK rearrangement-positive patients was thus enrolled, and 143 of these individuals were evaluated. The patients received crizotinib orally at 250 mg bid. The objective response rate (ORR) was 61%, independent of age, sex, performance status, or number of prior treatment regimens, and the median PFS was 9.7 months.22 On the basis of its pronounced clinical activity, crizotinib was approved by the US Food and Drug Administration (FDA) in August 2011.
In a subsequent randomized Phase III trial (Profile 1007, NCT00932893), 347 patients with ALK rearrangement-positive advanced NSCLC who had previously undergone platinum-based chemotherapy were randomly assigned to receive crizotinib (250 mg bid) or standard chemotherapy with either pemetrexed or docetaxel.23 Crizotinib treatment yielded a significantly better ORR (65% versus 20%, P<0.001) and longer PFS (hazard ratio 0.49, 95% confidence interval 0.37–0.64; P<0.001) compared with pemetrexed or docetaxel, whereas there was no significant difference in overall survival between the two treatment groups (hazard ratio 1.02, 95% confidence interval 0.68–1.54), as a result of crossover to the comparator treatment.23,24 Another randomized Phase III trial (Profile 1014, NCT01154140), designed to test the efficacy of crizotinib versus standard chemotherapy (pemetrexed–cisplatin or pemetrexed–carboplatin) as a first-line treatment for patients with ALK-rearranged NSCLC, is ongoing (Figure 2).25
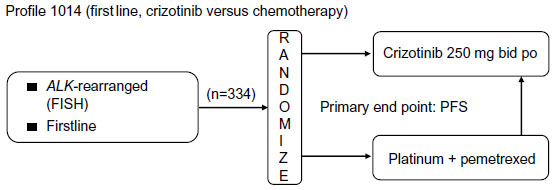 | Figure 2 Ongoing Phase III study (Profile 1014) of crizotinib for the treatment of ALK rearrangement-positive non-small-cell lung cancer. |
Most adverse events of crizotinib treatment appear to be mild (grade 1 or 2), with those that occur most frequently being visual effects, nausea, diarrhea, constipation, vomiting, and peripheral edema. Three warning adverse events – interstitial lung disease (ILD), hepatotoxicity, and prolongation of the QT interval – have been identified. Life-threatening or fatal treatment-related ILD was found to occur in 1.6% of patients.26 It remains unclear whether the risk factors for EGFR TKI-associated ILD, such as male sex, a history of smoking, and coincidence of interstitial pneumonia, also apply to crizotinib-associated ILD. It is thus important that patients treated with crizotinib be monitored for pulmonary symptoms and radiographic findings indicative of ILD, and the drug should be discontinued immediately on such a diagnosis. Elevated serum aminotransferase levels of grade 3 or 4 were detected in ~7% of crizotinib-treated patients, with such elevation usually being asymptomatic and reversible on discontinuation of crizotinib. Although crizotinib-induced hepatotoxicity with a fatal outcome has been reported in <1% of treated patients, routine evaluation of liver function, including measurement of aminotransferase and bilirubin levels, should be performed.
Mechanisms of crizotinib resistance
Although treatment with crizotinib has a pronounced clinical benefit for patients with ALK rearrangement-positive NSCLC, such individuals inevitably develop drug resistance. Several mechanisms of crizotinib resistance have been described to date, including secondary mutation or copy-number gain of ALK,27,28 inadequate drug delivery, and activation of alternative signaling pathways, such as those mediated by EGFR or KIT (Figure 3).29–31
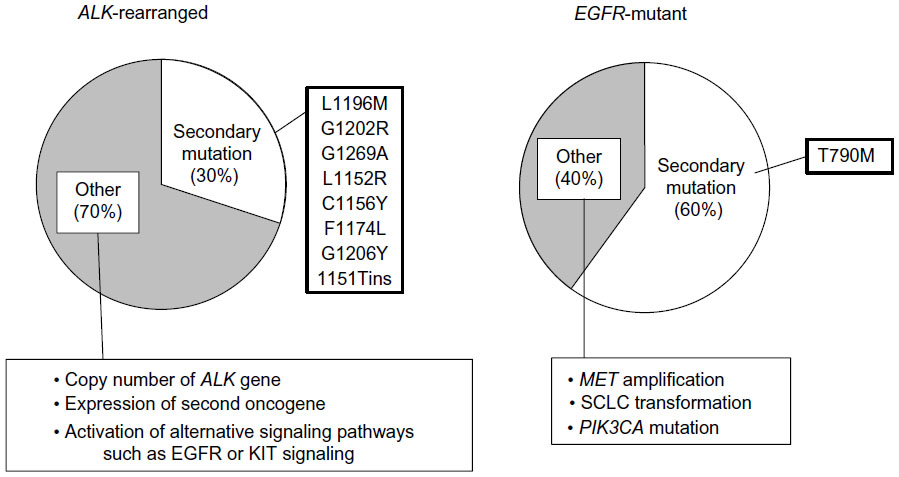 | Figure 3 Comparison of tyrosine-kinase inhibitor resistance mechanisms for ALK rearrangement-positive and EGFR mutation-positive non-small-cell lung cancer (NSCLC). Only 30% of cases of acquired crizotinib resistance in patients with ALK-rearranged NSCLC are attributable to various secondary mutations of ALK, with the remaining 70% of such cases being due to other mechanisms. In contrast, 60% of cases of acquired resistance to EGFR-tyrosine-kinase inhibitors in patients with EGFR mutation-positive NSCLC are caused by secondary mutation of EGFR, almost exclusively T790M, whereas only 40% of such cases are due to other resistance mechanisms. |
Two secondary mutations of ALK associated with crizotinib resistance – L1196M and C1156Y – were first detected in the same patient, who relapsed after achieving a partial response to the drug.32 The L1196M substitution occurs at the gatekeeper position of ALK (a position that controls the binding of nucleotides and TKIs), and corresponds to the T790M substitution in EGFR and the T315I substitution in the Bcr–Abl fusion protein, both of which confer resistance to corresponding TKIs. Multiple additional mutations in the ALK kinase domain have since been identified in patients who develop resistance to crizotinib.28,33 In contrast, T790M accounts for the vast majority of secondary mutations of EGFR that confer resistance to EGFR TKIs (Figure 3).34,35
Although the relative contributions of the different mechanisms to crizotinib resistance remain unclear because of the small numbers of patients examined, biopsy of tumors performed after the onset of acquired resistance has suggested that secondary mutations in the ALK kinase domain account for only ~30% of such cases of resistance.27,30 This situation also differs from that for EGFR mutation-positive NSCLC, for which the T790M substitution has been detected in up to 60% of tumors with acquired resistance to EGFR TKIs (Figure 3).36 Repeated biopsy and molecular analysis of relapsed tumors will be required in clinical trials of treatment strategies designed to overcome acquired resistance.
Clinical development of other ALK TKIs
Several new ALK TKIs are currently under development (Table 1).
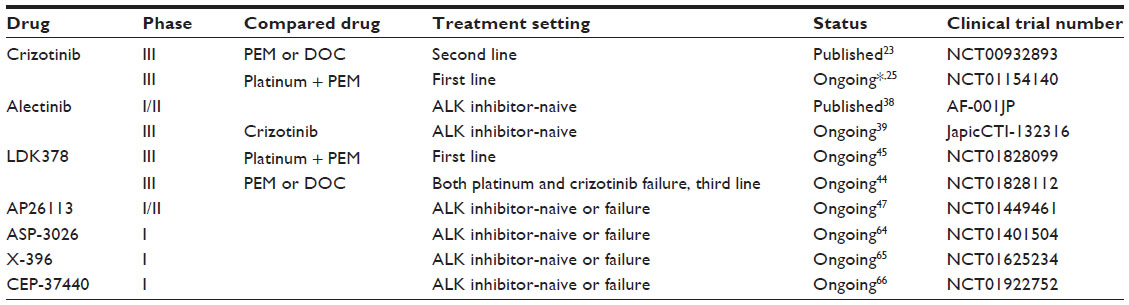 | Table 1 Selected clinical trials of anaplastic lymphoma kinase (ALK) inhibitors for the treatment of ALK rearrangement-positive non-small-cell lung cancer |
Alectinib (CH5424802)
Alectinib (Chugai Pharmaceutical, Tokyo, Japan) is a potent and selective ALK inhibitor with a median inhibitory concentration for ALK activity of 1.9 nM, and with little or no inhibitory activity for other protein kinases examined (Figure 1).37 The specific potency of alectinib for ALK inhibition appears to be related to its one-hinge hydrogen bond, whereas other ALK inhibitors, including crizotinib, form two- or three-hinge hydrogen bonds. In contrast to crizotinib, alectinib also shows substantial inhibitory activity against the L1196M mutant of ALK, apparently because it is able to maintain an efficient (CH/π) interaction with position 1196 even after the substitution of methionine for leucine.
A Phase I/II first-in-human study (AF-001JP) performed with previously treated and crizotinib-naive patients with ALK rearrangement-positive advanced NSCLC was performed in Japan.38 The participants were deemed to be ALK fusion gene-positive if a positive result was obtained either by reverse-transcription polymerase chain reaction (RT-PCR) analysis or by both immunohistochemistry (IHC) and fluorescence in situ hybridization (FISH). In the Phase I portion of the study, 24 patients received alectinib with a dose escalation from 20 to 300 mg bid, with the latter being determined as the highest planned dose on the basis of the available safety information for the additive formulation in Japan. Given that DLTs were not observed, the maximum tolerated dose (MTD) was not identified in this study. The highest planned dose (300 mg bid) was thus judged to be acceptable as the recommended dose for the 46 patients enrolled in the Phase II portion of the trial. Of these 46 patients, 43 individuals (93.5%) achieved an objective response, and 44 (95.7%) achieved disease control. The median PFS had not been determined by the time of publication. This excellent clinical activity was associated with mostly mild adverse events, with those of grade 3 being detected in only 17 (37.0%) patients and those of grade 4 or death in none. The most frequently reported treatment-related adverse events were dysgeusia and liver dysfunction, both of which were of grade 1 or 2 in almost all cases. The characteristic adverse events of crizotinib treatment, including visual effects and gastrointestinal disorders (diarrhea, vomiting, and nausea), occurred at a low rate in this study of alectinib. Application for approval of alectinib in Japan was submitted on October 7, 2013.
A Phase III clinical trial (JapicCTI-132316) comparing alectinib with crizotinib in terms of PFS for the treatment of patients with ALK rearrangement-positive NSCLC is ongoing in Japan.39 Major eligibility criteria include advanced or metastatic ALK-rearranged NSCLC (identified either by RT-PCR or by both IHC and FISH), no prior treatment with an ALK inhibitor, an Eastern Cooperative Oncology Group performance status of 0–2, and either no previous treatment or one line of prior treatment with chemotherapy (Figure 4).
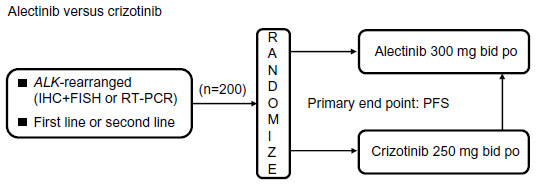 | Figure 4 Ongoing Phase III study (JapicCTI-132316) of alectinib for the treatment of ALK rearrangement-positive non-small-cell lung cancer.39 |
A dose-finding Phase I study (AF-002JG, NCT01588028) was also performed for alectinib in the US.40 Key eligibility criteria for this study included advanced NSCLC with ALK rearrangement confirmed by FISH, as well as failed crizotinib treatment. No treatment-related dose reductions were necessary up to a dose of 600 mg bid. Two of seven patients experienced DLTs (headache of grade 3, and neutropenia of grade 3 requiring dose-holding for 7 days) at the dose of 900 mg bid. On the basis of these results, 600 mg bid was determined as the recommended dose of alectinib for a Phase II study in the US. The ORR was 54.5% across all cohorts of the Phase I study, indicating that alectinib possesses significant clinical activity in ALK rearrangement-positive patients who are refractory to crizotinib. A global single-arm Phase II study of alectinib in patients with ALK-rearranged NSCLC resistant to crizotinib is ongoing (NCT01801111).41 The FDA granted breakthrough-therapy designation for alectinib on the basis of the NCT01588028 data, with early approval being expected.
LDK378
LDK378 (Novartis, Basel, Switzerland) is also a potent and selective small-molecule ALK inhibitor (Figure 1).42 In a Phase I study, 59 patients received LDK378 with dose escalation from 50 to 750 mg once daily (qd). DLTs were observed in two of the 14 patients who received the drug at 400 mg qd, in two of the nine patients at 600 mg qd, and in one of the nine patients at 750 mg qd. DLTs included diarrhea, vomiting, nausea, dehydration, and elevated serum aminotransferase levels. The MTD was thus defined as a dose of 750 mg qd. Among 88 evaluable ALK rearrangement-positive NSCLC patients who received LDK378 at 400–750 mg qd, the ORR was 70%. In the subset of 64 patients who had experienced crizotinib failure, the ORR was 73%.43 These results thus suggest that LDK378 may be effective for the treatment of patients with ALK-rearranged NSCLC who have developed acquired resistance to crizotinib.
A Phase III clinical trial (NCT01828112) comparing LDK378 with chemotherapy (pemetrexed at 500 mg/m2 or docetaxel at 75 mg/m2) for the treatment of ALK-rearranged NSCLC patients who have progressed after prior treatment with both crizotinib- and platinum-based chemotherapy is ongoing (Figure 5).44 In addition, a Phase III clinical trial (NCT01828099) comparing LDK378 with standard first-line chemotherapy (pemetrexed plus either cisplatin or carboplatin) in previously untreated ALK positive NSCLC patients assessed by IHC is also ongoing (Figure 5).45
 | Figure 5 Ongoing Phase III studies of LDK378 for the treatment of ALK rearrangement-positive non-small-cell lung cancer. |
AP26113
AP26113 (Ariad Pharmaceuticals, Inc., Cambridge, MA, USA) is another highly selective small-molecule ALK inhibitor that shows activity against the L1196M mutant (Figure 1).46 A Phase I/II study of AP26113 (NCT01449461) is ongoing.47 In the Phase I portion of the study, 44 patients received AP26113 with dose escalation from 30 to 300 mg qd. The most common adverse events were fatigue, nausea, and diarrhea, most of which were of grade 1 or 2. One DLT (increased serum alanine aminotransferase level of grade 3) was observed in one of nine patients treated at a dose of 240 mg, and one DLT (dyspnea of grade 4) was observed in one of two patients at a dose of 300 mg. Although the MTD has not been defined, a recommended Phase II dose was identified as 180 mg qd on the basis of safety, efficacy, and pharmacokinetic data. In the Phase I portion of the study, 24 patients with ALK-rearranged NSCLC were evaluable for response. Fifteen of these patients achieved an ORR of 63%, including 12 of the 16 individuals who had progressed after previous crizotinib therapy (ORR 75%). In addition, four of five patients showed objective responses for metastases in the central nervous system.
Other new ALK TKIs
Other new ALK TKIs, such as ASP-3026 (Astellas Pharma, Tokyo, Japan), NMS-E628 (Nerviano Medical Sciences, Milan, Italy), X-396 (Xcovery, West Palm Beach, FL, USA), CEP-37440 (Teva Pharmaceutical Industries Ltd, Petah Tikva, Israel), TSR-011 (Tesaro, Inc., Waltham, MA, USA), and PF-06463922 (Pfizer, New York, NY, USA), are currently introduced into clinical trials.
Molecular diagnosis of ALK rearrangement-positive NSCLC
FISH
Break-apart FISH analysis was applied for detection of ALK rearrangement in clinical trials with crizotinib. In this approach, the 5′ and 3′ portions of the ALK gene are separately labeled with red or green fluorescent probes (Figure 6). If the signals of the two probes overlap, resulting in yellow fluorescence, then there is no translocation. If a translocation is present, the two probes are spatially separated, and each is detected as an isolated signal (red or green). Tumors are deemed positive for ALK rearrangement if 15% or more of the tumor cells show isolated signals. Such analysis detects ALK rearrangement regardless of the ALK fusion partner or the specific EML4–ALK variant. The break-apart FISH assay is a unique diagnostic approach approved for screening for ALK rearrangement in NSCLC by the FDA. FISH has several disadvantages, however. First, it is an expensive and low-throughput method that requires technical expertise. Second, false-negative results sometimes occur because of difficulty in interpretation of separated signals. And third, the use of FISH alone (without IHC or RT-PCR) for screening may give rise to false-positive results. Indeed, in one study, the ORR for crizotinib was only 48% among patients screened with FISH alone, but increased up to 81% among those screened with FISH in combination with IHC or RT-PCR.48
 | Figure 6 Schematic illustration for break-apart fluorescence in situ hybridization for detecting ALK rearrangements. |
IHC
Given that ALK is not expressed in normal lung tissue or in lung cancer negative for ALK rearrangement, any level of ALK expression is considered to be abnormal and expected to be the result of ALK rearrangement. The abundance of ALK fusion proteins is relatively low, however, and initial attempts to detect such proteins by IHC were disappointing.49 The subsequent development of an intercalated antibody-enhanced polymer (iAEP) method for signal enhancement (which incorporates an intercalating antibody between the primary antibody to ALK and the dextran polymer-based detection reagents) resulted in a marked increase in the sensitivity of IHC for the detection of ALK fusion proteins.50 Several studies have since described the detection of ALK fusion proteins with high sensitivity and specificity by the application of IHC with improved detection methods (such as the iAEP method [Nichirei Biosciences] or EnVision™ FLEX+ [Dako, Glostrup, Denmark]) in combination with antibodies to ALK (ALK1, 5A4, and D5F3) (Table 2).51–59 The sensitivity of the improved IHC procedures is especially high with the D5F3 or 5A4 antibodies. Given that IHC is a routine methodology in most pathology laboratories, it may be suitable for screening of NSCLC patients for ALK rearrangement after appropriate clinical optimization and validation (Figure 7).
 | Table 2 Summary of the sensitivity and specificity of improved immunohistochemistry (IHC) procedures for the detection of anaplastic lymphoma kinase (ALK) in non-small-cell lung cancer specimens |
 | Figure 7 Proposed algorithm for testing for ALK rearrangement in patients with non-small-cell lung cancer. |
RT-PCR
RT-PCR is a highly sensitive and specific method for the identification of ALK rearrangement.60,61 In addition, unlike FISH or IHC, it can determine both the fusion partner of ALK (from among those previously identified) and the EML4–ALK variant.62 RT-PCR requires high-quality ribonucleic acid (RNA) extracted from nonfixed or freshly frozen specimens, however. It is generally difficult to extract suitable RNA from the paraffin-embedded specimens used in daily clinical practice.
Several new RT-PCR-based methods have recently been developed. MassARRAY is a nucleic acid-analysis platform for the detection of EML4–ALK that involves PCR amplification, single-base primer extension, and analysis by MALDI-TOF (matrix-assisted laser desorption ionization–time of flight) mass spectrometry.63 The region of EML4–ALK complementary deoxyribonucleic acid containing the fusion point is amplified by PCR, but given that the amplicons are relatively small (70–130 bp), the quality of RNA extracted from paraffin-embedded specimens is sufficient for the analysis. RT-PCR-based assays may thus come to be more convenient and a major tool for detection of ALK rearrangement if the use of paraffin-embedded tissue is validated.
Future perspectives
The identification of the EML4–ALK fusion gene has accelerated translational research and changed clinical practice for NSCLC, with crizotinib now being in clinical use as an ALK inhibitor for the treatment of patients with ALK rearrangement-positive NSCLC. Although crizotinib has an excellent initial therapeutic effect, all treated patients eventually develop resistance to this drug. The development of therapeutic strategies able to overcome crizotinib resistance, including those based on the administration of new ALK inhibitors, is thus warranted. In addition, given the existence of other drivers of NSCLC, such as EGFR mutations as well as reactive oxygen species 1 and RET fusion genes, it will be important to improve and validate methods for the detection of and screening for these various genetic changes, so that the appropriate drug can be prescribed.
Disclosure
The authors report no conflicts of interest in this work.
References
Ferlay J, Shin HR, Bray F, Forman D, Mathers C, Parkin DM. Estimates of worldwide burden of cancer in 2008: GLOBOCAN 2008. Int J Cancer. 2010;127(12):2893–2917. | |
Lynch TJ, Bell DW, Sordella R, et al. Activating mutations in the epidermal growth factor receptor underlying responsiveness of non-small-cell lung cancer to gefitinib. N Engl J Med. 2004;350(21):2129–2139. | |
Sordella R, Bell DW, Haber DA, Settleman J. Gefitinib-sensitizing EGFR mutations in lung cancer activate anti-apoptotic pathways. Science. 2004;305(5687):1163–1167. | |
Maemondo M, Inoue A, Kobayashi K, et al. Gefitinib or chemotherapy for non-small-cell lung cancer with mutated EGFR. N Engl J Med. 2010;362(25):2380–2388. | |
Mitsudomi T, Morita S, Yatabe Y, et al. Gefitinib versus cisplatin plus docetaxel in patients with non-small-cell lung cancer harbouring mutations of the epidermal growth factor receptor (WJTOG3405): an open label, randomised phase 3 trial. Lancet Oncol. 2010;11(2):121–128. | |
Le Beau MM, Bitter MA, Larson RA, et al. The t(2;5)(p23;q35): a recurring chromosomal abnormality in ki-1-positive anaplastic large cell lymphoma. Leukemia. Dec 1989;3(12):866–870. | |
Morris SW, Kirstein MN, Valentine MB, et al. Fusion of a kinase gene, ALK, to a nucleolar protein gene, NPM, in non-Hodgkin’s lymphoma. Science. 1994;263(5151):1281–1284. | |
Soda M, Choi YL, Enomoto M, et al. Identification of the transforming EML4-ALK fusion gene in non-small-cell lung cancer. Nature. 2007; 448(7153):561–566. | |
Shaw AT, Yeap BY, Mino-Kenudson M, et al. Clinical features and outcome of patients with non-small-cell lung cancer who harbor EML4-ALK. J Clin Oncol. 2009;27(26):4247–4253. | |
Camidge DR, Kono SA, Flacco A, et al. Optimizing the detection of lung cancer patients harboring anaplastic lymphoma kinase (ALK) gene rearrangements potentially suitable for ALK inhibitor treatment. Clin Cancer Res. 2010;16(22):5581–5590. | |
Choi YL, Takeuchi K, Soda M, et al. Identification of novel isoforms of the EML4-ALK transforming gene in non-small cell lung cancer. Cancer Res. 2008;68(13):4971–4976. | |
Sasaki T, Rodig SJ, Chirieac LR, Janne PA. The biology and treatment of EML4-ALK non-small cell lung cancer. Eur J Cancer. 2010;46(10):1773–1780. | |
Mano H. Non-solid oncogenes in solid tumors: EML4-ALK fusion genes in lung cancer. Cancer Sci. 2008;99(12):2349–2355. | |
Soda M, Takada S, Takeuchi K, et al. A mouse model for EML4-ALK-positive lung cancer. Proc Natl Acad Sci U S A. 2008;105(50):19893–19897. | |
McDermott U, Iafrate AJ, Gray NS, et al. Genomic alterations of anaplastic lymphoma kinase may sensitize tumors to anaplastic lymphoma kinase inhibitors. Cancer Res. 2008;68(9):3389–3395. | |
Takezawa K, Okamoto I, Nishio K, Janne PA, Nakagawa K. Role of ERK-BIM and STAT3-survivin signaling pathways in ALK inhibitor-induced apoptosis in EML4-ALK-positive lung cancer. Clin Cancer Res. 2011;17(8):2140–2148. | |
Okamoto I, Nakagawa K. Echinoderm microtubule-associated protein-like 4-anaplastic lymphoma kinase-targeted therapy for advanced non-small cell lung cancer: molecular and clinical aspects. Cancer Sci. 2012;103(8):1391–1396. | |
Koivunen JP, Mermel C, Zejnullahu K, et al. EML4-ALK fusion gene and efficacy of an ALK kinase inhibitor in lung cancer. Clin Cancer Res. 2008;14(13):4275–4283. | |
Christensen JG, Zou HY, Arango ME, et al. Cytoreductive antitumor activity of PF-2341066, a novel inhibitor of anaplastic lymphoma kinase and c-met, in experimental models of anaplastic large-cell lymphoma. Mol Cancer Ther. 2007;6(12 Pt 1):3314–3322. | |
Zou HY, Li Q, Lee JH, et al. An orally available small-molecule inhibitor of c-met, PF-2341066, exhibits cytoreductive antitumor efficacy through antiproliferative and antiangiogenic mechanisms. Cancer Res. 2007;67(9):4408–4417. | |
Kwak EL, Bang YJ, Camidge DR, et al. Anaplastic lymphoma kinase inhibition in non-small-cell lung cancer. N Engl J Med. 2010;363(18):1693–1703. | |
Camidge DR, Bang YJ, Kwak EL, et al. Activity and safety of crizotinib in patients with ALK-positive non-small-cell lung cancer: updated results from a phase 1 study. Lancet Oncol. 2012;13(10):1011–1019. | |
Shaw AT, Kim DW, Nakagawa K, et al. Crizotinib versus chemotherapy in advanced ALK-positive lung cancer. N Engl J Med. 2013;368(25):2385–2394. | |
Takeda M, Okamoto I, Sakai K, Kawakami H, Nishio K, Nakagawa K. Clinical outcome for EML4-ALK-positive patients with advanced non-small-cell lung cancer treated with first-line platinum-based chemotherapy. Ann Oncol. 2012;23(11):2931–2936. | |
Pfizer. A Clinical Trial Testing The Efficacy Of Crizotinib Versus Standard Chemotherapy Pemetrexed Plus Cisplatin Or Carboplatin In Patients With ALK Positive Non Squamous Cancer Of The Lung (PROFILE 1014). Available from: http://clinicaltrials.gov/show/NCT01154140. NLM identifier: NCT01154140. Accessed January 21, 2014. | |
Tamiya A, Okamoto I, Miyazaki M, Shimizu S, Kitaichi M, Nakagawa K. Severe acute interstitial lung disease after crizotinib therapy in a patient with EML4-ALK-positive non-small-cell lung cancer. J Clin Oncol. 2013;31(1):e15–e17. | |
Doebele RC, Pilling AB, Aisner DL, et al. Mechanisms of resistance to crizotinib in patients with ALK gene rearranged non-small cell lung cancer. Clin Cancer Res. 2012;18(5):1472–1482. | |
Zhang S, Wang F, Keats J, et al. Crizotinib-resistant mutants of EML4-ALK identified through an accelerated mutagenesis screen. Chem Biol Drug Des. 2011;78(6):999–1005. | |
Sasaki T, Koivunen J, Ogino A, et al. A novel ALK secondary mutation and EGFR signaling cause resistance to ALK kinase inhibitors. Cancer Res. 2011;71(18):6051–6060. | |
Katayama R, Shaw AT, Khan TM, et al. Mechanisms of acquired crizotinib resistance in ALK-rearranged lung cancers. Sci Transl Med. 2012;4(120):120ra17. | |
Tanizaki J, Okamoto I, Okabe T, et al. Activation of HER family signaling as a mechanism of acquired resistance to ALK inhibitors in EML4-ALK-positive non-small cell lung cancer. Clin Cancer Res. 2012;18(22):6219–6226. | |
Choi YL, Soda M, Yamashita Y, et al. EML4-ALK mutations in lung cancer that confer resistance to ALK inhibitors. N Engl J Med. 2010;363(18):1734–1739. | |
Heuckmann JM, Holzel M, Sos ML, et al. ALK mutations conferring differential resistance to structurally diverse ALK inhibitors. Clin Cancer Res. 2011;17(23):7394–7401. | |
Sequist LV, Waltman BA, Dias-Santagata D, et al. Genotypic and histological evolution of lung cancers acquiring resistance to EGFR inhibitors. Sci Transl Med. 2011;3(75):75ra26. | |
Balak MN, Gong Y, Riely GJ, et al. Novel D761Y and common secondary T790M mutations in epidermal growth factor receptor-mutant lung adenocarcinomas with acquired resistance to kinase inhibitors. Clin Cancer Res. 2006;12(21):6494–6501. | |
Yu HA, Arcila ME, Rekhtman N, etal. Analysis of tumor specimens at the time of acquired resistance to EGFR-TKI therapy in 155 patients with EGFR-mutant lung cancers. Clin Cancer Res. 2013;19(8):2240–2247. | |
Sakamoto H, Tsukaguchi T, Hiroshima S, et al. CH5424802, a selective ALK inhibitor capable of blocking the resistant gatekeeper mutant. Cancer Cell. 2011;19(5):679–690. | |
Seto T, Kiura K, Nishio M, et al. CH5424802 (RO5424802) for patients with ALK-rearranged advanced non-small-cell lung cancer (AF-001JP study): a single-arm, open-label, phase 1–2 study. Lancet Oncol. 2013; 14(7):590–598. | |
Chugai Pharmaceutical Co., Ltd. Open-label randomized PhaseIII Study of the Efficacy and Safety of CH5424802(AF802) in ALK-Positive Advanced or Recurrecnt Non-Small Cell Lung Cancer with Crizotinib control. Available from: http://www.clinicaltrials.jp/user/showCteDetailE.jsp?japicId=JapicCTI-132316. Identifier: JapicCTI-132316. Accessed January 21, 2014. | |
Ou S, Gadgeel S, Chiappori A, et al. Late breaking abstract: Safety and efficacy analysis of RO5424802/CH5424802 in anaplastic lymphoma kinase (ALK)-positive non-small cell lung cancer (NSCLC) patients who have failed crizotinib in a dose-finding Phase I study. Presented at: ECCO/ESMO September 30, 2013; Amsterdam. | |
Hoffmann-La Roche. A Study of RO5424802 in Patients With Non-Small Cell Lung Cancer Who Have ALK Mutation and Failed Crizotinib Treatment. Available from: http://clinicaltrials.gov/show/NCT01801111. NLM identifier: NCT01801111. Accessed January 21, 2014. | |
Marsilje TH, Pei W, Chen B, et al. Synthesis, structure-activity relationships, and in vivo efficacy of the novel potent and selective anaplastic lymphoma kinase (ALK) inhibitor 5-chloro-N2-(2-isopropoxy-5-methyl-4-(piperidin-4-yl)phenyl)-N4-(2-(isopropylsulf onyl)phenyl)pyrimidine-2,4-diamine (LDK378) currently in phase 1 and phase 2 clinical trials. J Med Chem. Epub June 26, 2013. | |
Shaw AT, Mehra R, Kim DW, et al. Clinical activity of the ALK inhibitor LDK378 in advanced, ALK positive NSCLC. J Clin Oncol. 2013;31 Suppl:8010. | |
Novartis Pharmaceuticals. LDK378 Versus Chemotherapy in ALK Rearranged (ALK Positive) Patients Previously Treated With Chemotherapy (Platinum Doublet) and Crizotinib. Available from: http://clinicaltrials.gov/show/NCT01828112. NLM identifier: NCT01828112. Accessed January 21, 2014. | |
Novartis Pharmaceuticals. LDK378 Versus Chemotherapy in Previously Untreated Patients With ALK Rearranged Non-small Cell Lung Cancer. Available from: http://clinicaltrials.gov/show/NCT01828099. NLM identifier: NCT01828099. Accessed January 21, 2014. | |
Katayama R, Khan TM, Benes C, et al. Therapeutic strategies to overcome crizotinib resistance in non-small cell lung cancers harboring the fusion oncogene EML4-ALK. Proc Natl Acad Sci U S A. 2011;108(18):7535–7540. | |
Camidge DR, Bazhenova L, Salgia R, et al. First-in-human dose-finding study of the ALK/EGFR inhibitor AP26113 in patients with advanced malignancies: updated results. J Clin Oncol. 2013;31 Suppl: 8031. | |
Chihara D, Suzuki R. More on crizotinib. N Engl J Med. 2011;364(8):776–777; author reply 778. | |
Martelli MP, Sozzi G, Hernandez L, et al. EML4-ALK rearrangement in non-small cell lung cancer and non-tumor lung tissues. Am J Pathol. 2009;174(2):661–670. | |
Takeuchi K, Choi YL, Togashi Y, et al. KIF5B-ALK, a novel fusion oncokinase identified by an immunohistochemistry-based diagnostic system for ALK-positive lung cancer. Clin Cancer Res. 2009;15(9):3143–3149. | |
Han XH, Zhang NN, Ma L, et al. Immunohistochemistry reliably detects ALK rearrangements in patients with advanced non-small-cell lung cancer. Virchows Arch. 2013;463(4):583–591. | |
Li Y, Pan Y, Wang R, et al. ALK-rearranged lung cancer in Chinese: a comprehensive assessment of clinicopathology, IHC, FISH and RT-PCR. PLoS One. 2013;8(7):e69016. | |
Ying J, Guo L, Qiu T, et al. Diagnostic value of a novel fully automated immunochemistry assay for detection of ALK rearrangement in primary lung adenocarcinoma. Ann Oncol. 2013;24(10):2589–2593. | |
Selinger CI, Rogers TM, Russell PA, et al. Testing for ALK rearrangement in lung adenocarcinoma: a multicenter comparison of immunohistochemistry and fluorescent in situ hybridization. Mod Pathol. 2013;26(12):1545–1553. | |
To KF, Tong JH, Yeung KS, et al. Detection of ALK rearrangement by immunohistochemistry in lung adenocarcinoma and the identification of a novel EML4-ALK variant. J Thorac Oncol. 2013;8(7):883–891. | |
Sholl LM, Weremowicz S, Gray SW, et al. Combined use of ALK immunohistochemistry and FISH for optimal detection of ALK-rearranged lung adenocarcinomas. J Thorac Oncol. 2013;8(3):322–328. | |
Park HS, Lee JK, Kim DW, et al. Immunohistochemical screening for anaplastic lymphoma kinase (ALK) rearrangement in advanced non-small cell lung cancer patients. Lung Cancer. 2012;77(2):288–292. | |
Yi ES, Boland JM, Maleszewski JJ, et al. Correlation of IHC and FISH for ALK gene rearrangement in non-small cell lung carcinoma: IHC score algorithm for FISH. J Thorac Oncol. 2011;6(3):459–465. | |
Mino-Kenudson M, Chirieac LR, Law K, et al. A novel, highly sensitive antibody allows for the routine detection of ALK-rearranged lung adenocarcinomas by standard immunohistochemistry. Clin Cancer Res. 2010;16(5):1561–1571. | |
Wallander ML, Geiersbach KB, Tripp SR, Layfield LJ. Comparison of reverse transcription-polymerase chain reaction, immunohistochemistry, and fluorescence in situ hybridization methodologies for detection of echinoderm microtubule-associated proteinlike 4-anaplastic lymphoma kinase fusion-positive non-small cell lung carcinoma: implications for optimal clinical testing. Arch Pathol Lab Med. 2012;136(7):796–803. | |
Wu YC, Chang IC, Wang CL, et al. Comparison of IHC, FISH and RT-PCR methods for detection of ALK rearrangements in 312 non-small cell lung cancer patients in Taiwan. PLoS One. 2013;8(8):e70839. | |
Takeuchi K, Choi YL, Soda M, et al. Multiplex reverse transcription-PCR screening for EML4-ALK fusion transcripts. Clin Cancer Res. 2008;14(20):6618–6624. | |
Sakai K, Okamoto I, Takezawa K, et al. A novel mass spectrometry-based assay for diagnosis of EML4-ALK-positive non-small cell lung cancer. J Thorac Oncol. 2012;7(5):913–918. | |
Astellas Pharma Inc. Study of an Investigational Drug, ASP3026, in Patients With Solid Tumors. Available from: http://clinicaltrials.gov/show/NCT01401504. NLM identifier: NCT01401504. Accessed January 21, 2014. | |
Xcovery Holding Company, LLC. Phase 1 Safety Study of X-396, an Oral ALK Inhibitor, in Patients With Advanced Solid Tumors. Available from: http://clinicaltrials.gov/show/NCT01625234. NLM identifier: NCT01625234. Accessed January 21, 2014. | |
Teva Branded Pharmaceutical Products, R&D Inc. To Determine the Maximum Tolerated Dose of Oral CEP-37440 in Patients With Advanced or Metastatic Solid Tumors. Available from: http://clinicaltrials.gov/show/NCT01922752. NLM identifier: NCT01922752. Accessed January 21, 2014. |
 © 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.