Back to Journals » Drug Design, Development and Therapy » Volume 14
Development of a Novel Controlled-Release Tablet of Pregabalin: Formulation Variation and Pharmacokinetics in Dogs and Humans
Authors Kim KH, Lim SH, Shim CR, Park J, Song WH, Kwon MC ![]() , Lee JH, Park JS, Choi HG
, Lee JH, Park JS, Choi HG
Received 24 July 2019
Accepted for publication 5 December 2019
Published 30 January 2020 Volume 2020:14 Pages 445—456
DOI https://doi.org/10.2147/DDDT.S222505
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Professor Yan Zhu
Kyung Hun Kim,1,2 Seo Hyun Lim,2 Cho Rok Shim,2 Junsung Park,2 Woo Heon Song,2 Min Chang Kwon,2 Jong Hyuk Lee,3 Jun Sang Park,2 Han-Gon Choi1
1College of Pharmacy and Institute of Pharmaceutical Science and Technology, Hanyang University, Ansan, Republic of Korea; 2GL PharmTech Corp, Seongnam, Republic of Korea; 3Department of Pharmaceutical Engineering, College of Life and Health Sciences, Hoseo University, Asan, Republic of Korea
Correspondence: Jun Sang Park
GL PharmTech Corp, #714, Jungang Induspia V, 137 #714, Jungang Induspia V, 137, Seongnam-Si, Gyeonggi-Do 13202, South Korea
Tel +82-31-739-5220
Fax +82-31-739-5224
Email [email protected]
Han-Gon Choi
College of Pharmacy and Institute of Pharmaceutical Science and Technology, Hanyang University, Ansan, Republic of Korea
Email [email protected]
Background: Novel three-layered (TL) tablet systems were compared with both monolithic matrix (MM) formulations and a commercial immediate-release (IR) capsule to develop once-a-day (OAD) pregabalin tablets.
Methods: The physical properties of the TL tablets, including dissolution and swelling rates, were compared with those of the MM tablets and the pharmacokinetic parameters of the TL tablet were compared with those of an IR capsule in beagles and humans.
Results: Our results indicated that the same amount of a hydrophilic polymer in the formulations had similar dissolution profiles at 12 h, regardless of the tablet geometry. However, the degree of tablet swelling differed, with larger amounts of polymer in the tablets showing a greater degree of swelling. In addition, TL tablets swelled more rapidly compared with MM tablets. For the pharmacokinetic study of the TL tablet, the beagles demonstrated absorption results similar to those of an IR capsule, whereas the humans demonstrated low total absorption compared with an IR capsule. The time of the peak plasma concentration at 6 h in the fed state of humans coincided with the results of the study on beagles.
Conclusion: The novel TL tablet system of pregabalin may prove to be helpful in developing improved formulations with better continuous drug absorption for OAD administration.
Keywords: controlled-release, three-layered tablet, polyethylene oxide, high swellable, once-a-day
Introduction
Pregabalin (S-[+]-3-isobutyl GABA or (S)-3-(aminomethyl)-5-methylhexanoic acid) is a ligand of the auxiliary α2δ protein subunit associated with voltage-gated calcium channels, which are widely distributed throughout the peripheral and central nervous systems. It has analgesic, anxiolytic, and anticonvulsant properties.1,2 Pregabalin is classified as a biopharmaceutics classification system (BCS) Class 1 compound (highly permeable and highly soluble) and its bioavailability is higher than or equal to 90% and is independent of dose.3 Maximum plasma concentration (Cmax) and area under the plasma concentration-time curve (AUC) of pregabalin increase linearly with dose increasing.4 However, food reduces the rate of pregabalin absorption, resulting in lower Cmax and delayed time to peak plasma concentration (Tmax).5,6 In addition, pregabalin absorption is limited to the ascending colon and is excreted unchanged in the urine.7,8
The commercial products of pregabalin, including Lyrica® capsules, are immediate-release (IR) formulations with available doses of 75, 150, and 300 mg, which are administered at 150–600 mg twice daily. Patients with chronic diseases have been reported to be more compliant with once-a-day (OAD) formulations than with medicines scheduled more frequently.9 As such, improving adherence may result in a subsequent decrease in related healthcare costs.10 Therefore, controlled-release (CR) tablets that improve patient compliance by reducing the frequency of dosing need to be developed.
CR technologies using hydrophilic polymers were developed originally in an attempt to improve patient adherence by reducing the inconvenience caused by the frequent dosing of conventional formulations.11 A matrix in which the drug is uniformly dissolved or dispersed is the most common system, with hydrophilic polymers typically incorporated. However, the hydrophilic matrix shows a high release rate during the initial stage because of the existence of the drug at the surface of the matrix (ie, the burst effect).12 This undesirable phenomenon may result in adverse effects. To overcome any undesirable effects due to the hydrophilic matrix, various geometric matrix tablets have been suggested to avoid the burst effect and to achieve a constant release rate. The multi-layered matrix tablet represents one of these techniques and is usually composed of a matrix core containing the drug and one or more layers acting as barriers. In the barrier layers, hydrophilic polymers, such as hydroxypropyl methylcellulose and polyethylene oxide (PEO), primarily perform swelling and erosion processes during contact with the aqueous medium. The barrier layers thus control the surface area and the diffusion rate of drug release from the core layer.13,14 Therefore, the physical properties of the barrier layers themselves are important in achieving successful new formulations.15
In our previous research, the novel geometric system using a three-layered (TL) tablet (GLARS®, Geometrically Long Absorption Regulated System), consisting of an inner IR layer and two extended release barrier layers with swellable hydrophilic polymers, was suggested for OAD formulations (Figure 1).16 In this TL tablet system, the aqueous medium rapidly penetrates into the mid-layer, which is composed of water-soluble excipients, and the two barrier layers, composed of hydrophilic polymers, swell and wrap around the lateral side of the mid-layer. After oral administration, tablet hydration occurs more quickly than conventional matrix tablets, and the fully hydrated tablet can reach the colon where water is rarely available. Accordingly, the hydrated state of the tablet was assumed to induce continuous drug release and absorption throughout the gastrointestinal (GI) tract, even in the colon, irrespective of the unfavorable environment for OAD administration.17
 |
Figure 1 Difference of swelling and gelation behaviors between (A) MM tablets and (B) TL tablets containing a water-penetrating layer. |
In this study, to develop a pregabalin CR dosage form for OAD administration, various physical properties of formulations using the novel TL tablet system were investigated and compared with monolithic matrix (MM) tablets in vitro. In addition, the pharmacokinetic parameters of the optimized formulation were further studied and compared with IR formulations in beagles and humans.
Materials and Methods
Pregabalin was purchased from MSN Pharmachem (India). PEO (Polyox WSR 303) was purchased from Dow Chemical. Microcrystalline cellulose (MCC, Vivapur® 12) was purchased from JRS Pharma (Germany), povidone (Kollidon® 90F) from BASF (Germany), L-tartaric acid from Caviro (Italy), and D-mannitol from Roquette. All other excipients used were of pharmaceutical grade. All reagents used in the analyses were of analytical grade. An IR formulation (Lyrica Capsules, Pfizer) was used as a reference drug for pharmacokinetic studies.
Preparation of Tablets
To prepare MM tablets, pregabalin was mixed with PEO and MCC, and then blended with magnesium stearate (Mg.S). The dimensions of the punch were as follows: the major axis was 14 mm and the minor axis was 8 mm. The mixture was then compressed directly on the rotary tableting machine (Erweka, Germany). To manufacture the TL tablets, the drug and additives of each layer were granulated separately using a high-speed mixer, dryer, and sieve. The three kinds of granules were then lubricated separately with Mg.S. The TL tablets were next compressed using a TL tableting machine (Flexi-Tab, Bosch, Germany) (Table 1). The manufactured tablets had a hardness of more than 10 kp and they showed no separation between layers. The tablets were confirmed to be stable for 6 months under accelerated storage conditions (data not shown).
 |
Table 1 Composition of CR Tablets of Pregabalin |
In vitro Drug Release
Drug release was studied using the USP dissolution Apparatus II method (paddle) (VK7000, Varian Inc., Edison, NJ, USA) at 100 rpm with 900 mL of simulated gastric fluid (pH 1.2) at 37 ± 0.5°C. Drug concentrations were analyzed by high-performance liquid chromatography (HPLC; Agilent Technologies, CA, USA) at a wavelength of 210 nm. The column consisted of a Capcell Pak® C18, 4.6 x 150 mm, 5 μm (Shiseido, Tokyo, Japan). A mixture of phosphate buffer (pH 6.5) and methanol at a volume ratio of 65:35 was used for the mobile phase, with a flow rate of 0.8 mL/min.
Tablet Evaluation: Degree of Swelling
For this procedure, one tablet was placed in a vessel with 900 mL of simulated gastric fluid (pH 1.2) at 37 ± 0.5°C, followed by stirring at 50 rpm using the USP dissolution method 2 (paddle). The size of the tablet was measured with a Vernier caliper each time to calculate the volume. The volume and the volume increase rate of the tablet were calculated based on the following equations:
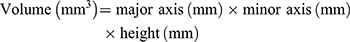

Pharmacokinetic Study in Beagles
Animals, Study Design, Treatment and Plasma Sampling
The protocols used for animal studies were consistent with the National Institutes of Health Policy and Animal Welfare Act and were approved by the Institutional Animal Care and Use Committee at Orient Bio, Inc. (Seongnam, Korea). Briefly, twelve male beagles (Covance Research Products Inc., USA) were fasted for 16 h before the experiment. They were divided into two groups and given 80 g of foodstuff 40 min before the oral administration of the drug formulation. An IR capsule of 75 mg (Lyrica capsules, Pfizer) was administered twice daily to group 1 for reference, while group 2 received a TL1 tablet of 150 mg once daily. Each drug of IR capsule and TL1 tablet was administered with 10 mL of water. Blood samples were then collected from the cephalic vein of each dog at the scheduled times. The IR capsule (pregabalin 75 mg) was given twice a day (0 and 10 h) and blood samples were collected at predetermined time points (0, 0.5, 1, 1.5, 2, 4, 6, 8, 10, 10.5, 11, 11.5, 12, 14, 16, 18, and 24). The TL1 tablet (pregabalin 150 mg) was given once a day and blood samples were withdrawn at predetermined time points (0, 0.5, 1, 2, 3, 4, 5, 6, 8, 10, 12, 14, 18, and 24 h). Samples were immediately centrifuged and stored at −70°C prior to analysis.
Analytic Method
Liquid chromatography–combined mass spectrometry (LC/MS/MS; API Qtrap 5500, Waters, USA) was used to determine the concentration of pregabalin in the plasma. Separation was performed on a Capcell Pak® MG C18, 2.0 x 50 mm, 3 µm column (Shiseido, Tokyo, Japan). The mobile phase consisted of solutions A (10 mM ammonium acetate buffer) and B (acetonitrile) at a volume ratio of 85:15 at a flow rate of 0.2 mL/min, and was maintained for 2 min. The peak detection method used multiple reaction monitoring and triple–quadrupole mass spectrometry; the ionization used a turbo ion-spray for electrospray ionization. The detection of pregabalin and IS (Losartan) using MRM mode was analyzed, each protonated molecular ion is m/z 160.1 → 142.3 and 424.3 → 207.3. The pregabalin stock solution was dissolved in 50% methanol to reach a concentration of 1 mg/mL. A dilute stock solution consisting of 50% methanol was prepared, along with calibration solutions prepared at pregabalin concentrations of 0.1, 0.5, 1, 5, 10, and 50 μg/mL. For an internal standard solution, losartan was dissolved in acetonitrile (1 mg/mL) to reach a 10-μg/mL final concentration. After thawing the samples at room temperature, each sample (20 μL) was pipetted into an Eppendorf tube. Then, 20 μL of the internal standard solution and 15 μL of H3PO4 were added together and vortexed. Cold acetonitrile (1.5 mL) was then added. After shaking for 10 min, 2 μL of the filtered solution was passed through a 0.22-μm filter and injected into the LC–MS/MS system.
Statistical Analysis
Non-compartmental analysis (WinNonlin® 5.3, Pharsight Co., CA, USA) was used to assess the area under the drug concentration–time curve from zero to infinity (AUC) and the time taken to reach the Tmax and the Cmax. Levels of statistical significance (p < 0.05) were determined using the analysis of variance (ANOVA) test. All data are denoted as the mean ± standard deviation (S.D.).
Pharmacokinetic Study in Humans
Subjects, Ethics Approval, Study Design, Treatment and Plasma Sampling
Healthy Korean male volunteers aged 20–45 years were eligible for inclusion in this study if they weighed ≥50 kg. Subjects were excluded from the study if they had any evidence or history of clinically significant diseases or an abnormal laboratory test result related to clinical biochemistry, hematology, or urinalysis. This study was conducted as a randomized, open-label, three-way crossover clinical trial to compare the pharmacokinetics of a TL3 tablet of pregabalin (150 mg) with an IR capsule of pregabalin (75 mg), and to assess the effects of a food on TL3 tablets. They were given high-fat meal (989 kcal) of foodstuff 30 min before the oral administration of the TL3 tablet. Each drug of IR capsule and TL3 tablet was administered with 240 mL of water.
Thirty subjects were randomized into six groups and treated in sequence of the clinical study design. A sufficient period of time for the excretion of pregabalin, considering the half-life (T1/2) of the drug, was established between each treatment. The IR capsule (pregabalin 75 mg) was given twice a day (0 and 12 h – fasted state) and blood samples were collected at predetermined time points (0, 0.33, 0.67, 1, 1.5, 2, 4, 6, 12, 12.33, 12.67, 13, 13.5, 14, 16, 24, and 36 h). The TL3 tablet (pregabalin 150 mg) was given OAD and blood samples were also collected at predetermined time points (0, 0.5, 1, 2, 3, 4, 5, 6, 8, 10, 12, 14, 24, and 36 h). Samples were immediately centrifuged and stored at −70°C prior to analysis. During the clinical trial, two subjects did not complete the study. In total, twenty-eight subjects completed the study. All subjects provided their written informed consent before being screened for the study. The study protocols were reviewed and approved by the Institutional Review Board of the Catholic University of Korea, Seoul St. Mary’s Hospital (ClinicalTrials.gov registry no: NCT01635751). The studies were conducted in accordance with the principles stipulated in the Declaration of Helsinki, as amended in 2008 (Seoul, Korea) and the Good Clinical Practice (GCP) guidelines of the International Council for Harmonization (ICH).
Analytic Methods
A HPLC system (Shiseido Nanospace SI-2, Shiseido, Japan), in conjunction with MS/MS (API 4000, AB SCIEX, USA), was used to determine the concentration of pregabalin in the plasma. Separation was performed on a Unison UK-C18 column (2.0 mm x 75 mm, 3 μm, Imtakt, Japan). The mobile phase consisted of 0.1% formic acid containing acetonitrile and 10 mM ammonium formate (50:50, v/v), with a flow rate of 0.2 mL/min. The detection of pregabalin and IS using MRM mode was analyzed, each protonated molecular ion is m/z 160.1 → 142.1 and 163.3 → 145.0. A stock solution of pregabalin at 0.1 mg/mL in methanol was prepared separately. Then, a working standard was diluted with a 50% acetonitrile solution. A calibration solution was then prepared at pregabalin concentrations of 0.05, 0.1, 0.5, 1, 5, 10, and 20 μg/mL. For sample preparation, each plasma sample (100 μL) was pipetted into a polypropylene tube and deproteinated with acetonitrile (1 mL) with the addition of the Internal standard solution (100 μg/mL). The mixture was then vortexed for 1 min and centrifuged at 13,000 rpm for 5 min. Each sample of 5 μL was then injected into the LC–MS/MS.
Statistical Analysis
Non-compartmental analysis (WinNonlin® 6.3, Pharsight Co., CA, USA) was used to assess the Cmax, Tmax, AUC, and T1/2. The results of bioequivalence were calculated as the ratio of Cmax and the AUC of the TL3 tablet compared with those of the IR capsule. Fraction-absorbed calculations employed the Wagner–Nelson Method. The percentages absorbed vs time were calculated using the following formula:
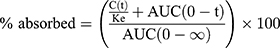
where C(t) is the plasma concentration at time t, Ke is the elimination rate constant, and AUC(0-t) and AUC(0-∞) are the area under the plasma concentration–time curve from zero to time t and to infinity of IR capsule, respectively.18 The relationships between the pharmacokinetic data in beagles and humans were examined by plotting the percentage of drug absorbed after 0.5, 1, 2, 3, 4, 5, 6, 8, 10, 12, and 14 h in each study. Levels of statistical significance (p < 0.05) were determined using the ANOVA test. All data are denoted as the mean ± S.D.
Result and Discussion
In vitro Release Profiles of MM and TL Formulations
The release profiles of MM tablets in pH 1.2 buffer are shown in Figure 2. Each formulation contained 100–300 mg of PEO as a release controller. The release profiles of MM1 and MM2 showed similar rates, but MM3 showed rates faster than other formulations. The amount of 200 mg PEO in MM2 appeared to be enough to control the release rate of pregabalin, which is soluble in water. When the matrix tablets contained more than 200 mg of PEO, which has the highest molecular weight and viscosity among the PEO polymers, the diffusion rate of pregabalin through the gelled polymer in the swollen matrix tablets predominated the release compared to the erosion rate of swollen PEO.19
 |
Figure 2 Influence of the amount of PEO on the dissolution profiles of MM tablets in pH 1.2 buffer. |
TL formulations were manufactured according to the compositions in Table 1. The release profiles of TL tablets are compared in Figure 3. Formulations of MM1 and TL1, which had the same amounts of PEO (300 mg in a tablet) showed similar release rates despite different geometries, such as conventional tablets and TL tablets. Our results also showed that the release rates of TL2 and MM2 were similar, although TL2 released pregabalin a little faster than MM2. The release rates of TL1 and TL3 were the same (data not shown). This may suggest that the amount of hydrophilic polymer to some extent primarily modulated the release rate of pregabalin, while the geometry of the tablet may not play a key role in controlling these formulations. However, the drug release profiles could be modified by the disposition ratio of the drug in each layer, because a sigmoid type of release pattern was observed when the drug was incorporated only in the middle layer, while the drug in the swellable outer layers was released in a typical hyperbolic pattern.20
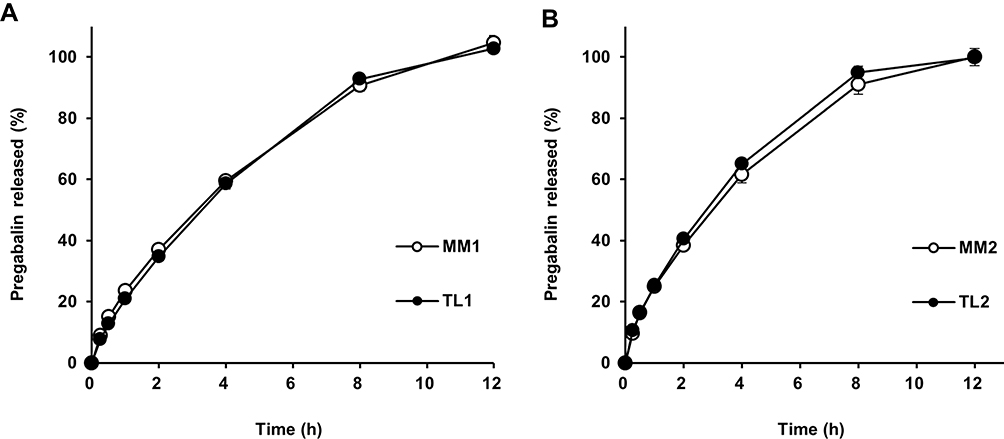 |
Figure 3 Drug release profiles comparing MM and TL tablets containing the same amount of PEO in a tablet in pH 1.2 buffer: (A) 300 mg; (B) 200 mg. |
The drug release mechanism from the TL formulation using the novel TL tablet can be described in three steps: (1) initial release from the uncovered lateral area of the middle layer; (2) the diffusion of the drug from the middle layer into the swollen outer layers and throughout the covered region surrounding the middle layer; and (3) the diffusion of the drug, with erosion of the swollen layers.21
Different Swelling Behavior Between MM and TL Tablets
The expanded volume and swelling behaviors of MM and TL during the swelling test are shown in Table 2 and Figure 4. All formulations swelled and expanded more than twice after 1 h compared with their initial volume. Larger amounts of PEO in the tablets showed greater degrees of swelling. The same amount of PEO in the formulation, regardless of the geometry of the tablets, showed very similar dissolution profiles. However, the swelling degrees of the tablets were all different, with TL formulations swelling more rapidly than MM formulations. The amount of PEO played an important role in the tablet size increase, while the different geometries of the tablets affected water absorption into the core of the tablet. The quickly absorbed medium through the middle layer appeared to move into the upper and lower layers, causing a rapid increase in the gelation and swelling of the TL tablets.
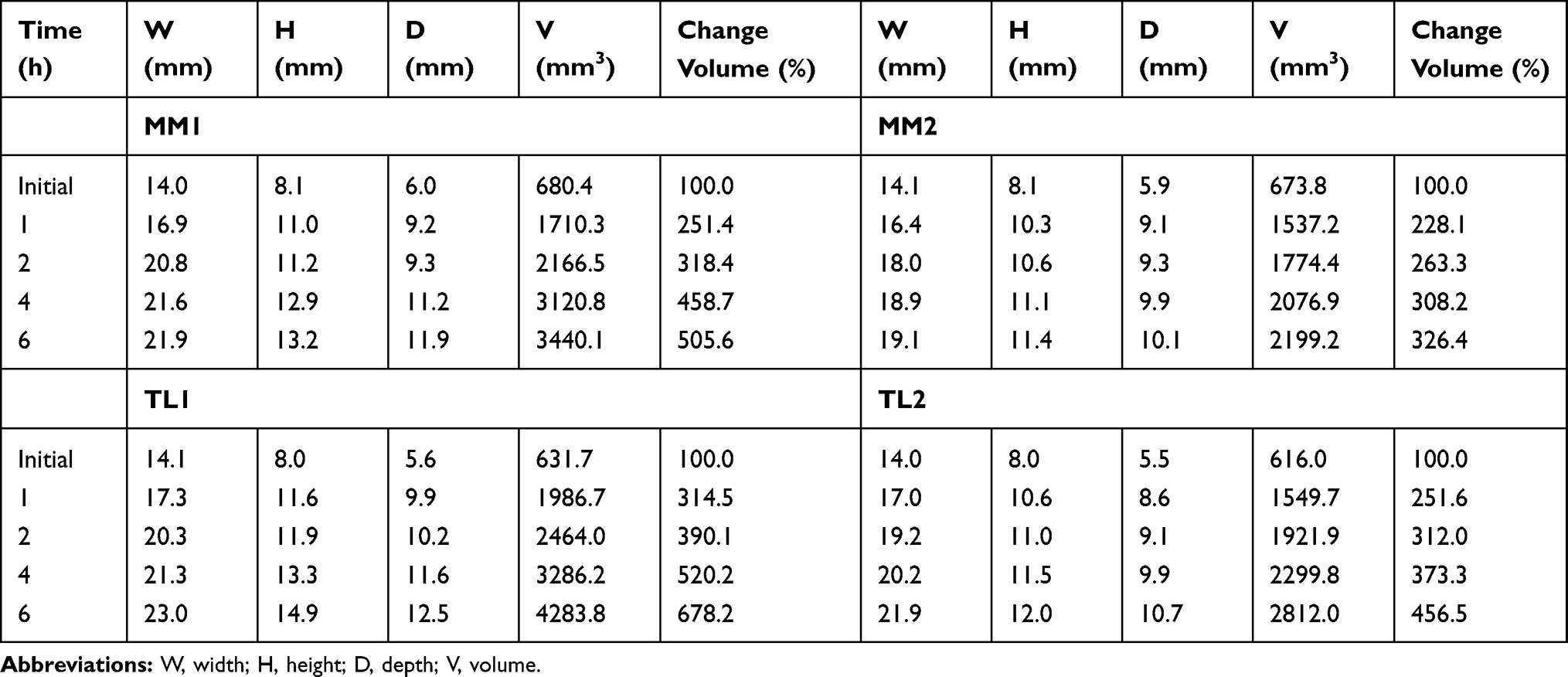 |
Table 2 Comparison of the Change in Tablet Size by Different Swelling Rates of CR Tablets at pH 1.2 |
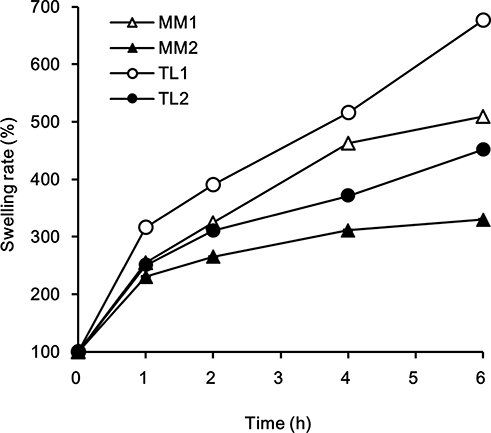 |
Figure 4 Comparison of swelling rates between MM and TL tablets in pH 1.2 buffer. |
Pregabalin, after oral administration, is absorbed at limited sites, with preference to the upper segment of the GI tract down to the ascending colon.22 Therefore, a gastro-retentive system would be the most preferable dosage form in order to obtain the maximum absorption of pregabalin from the GI tract for an extended period for OAD oral administration.23 TL tablets have been confirmed to swell rapidly and increase in size by swift hydration after oral administration. When the swollen tablet becomes bigger than the diameter of the pyloric sphincter, it allows the TL tablet to stay in the stomach longer, thus extending the GI transit time and providing a greater chance for absorption. In other words, TL formulations consisting of the novel TL tablet system may provide gastro-retentive properties with the rapid expanded tablet volume.
Pharmacokinetic Study in Beagles
A pharmacokinetic study in beagles was performed to compare the absorption properties of TL1 tablets as a once-daily administration compared with the pregabalin IR capsule. The TL1 formulation was designed to be released completely within 12 h in vitro for OAD administration, although many other OAD formulations have longer release periods. The mean plasma concentration–time profiles of pregabalin and the statistical summary are presented in Figure 5 and Table 3. The Tmax of the TL1 tablet and the reference were 5.86 and 0.85 h, respectively. This confirmed that the TL1 tablet controlled the absorption rate of pregabalin to show a peak plasma concentration at 6 h, while the drug from the reference was absorbed rapidly within 1 h after oral administration. The total drug exposure of the TL1 tablet with OAD administration, the AUC(0–24), was comparable to one pregabalin IR capsule with twice-daily administration because the T/R ratio for AUC(0–24), defined as the relative ratio of the corresponding value of the test divided by the equivalent value of the reference, was 1.05. The peak plasma concentration of the TL1 tablet was also comparable with that of the reference because the T/R ratio for the Cmax was 0.85. The T1/2 values of the TL1 tablet and the reference were 6.5 and 7.0 h, respectively, indicating that they had similar elimination phases irrespective of the different absorption rates. All pharmacokinetic parameters in the beagles suggested that the TL1 tablet may be appropriate for OAD administration, and further pharmacokinetic studies in humans were warranted.
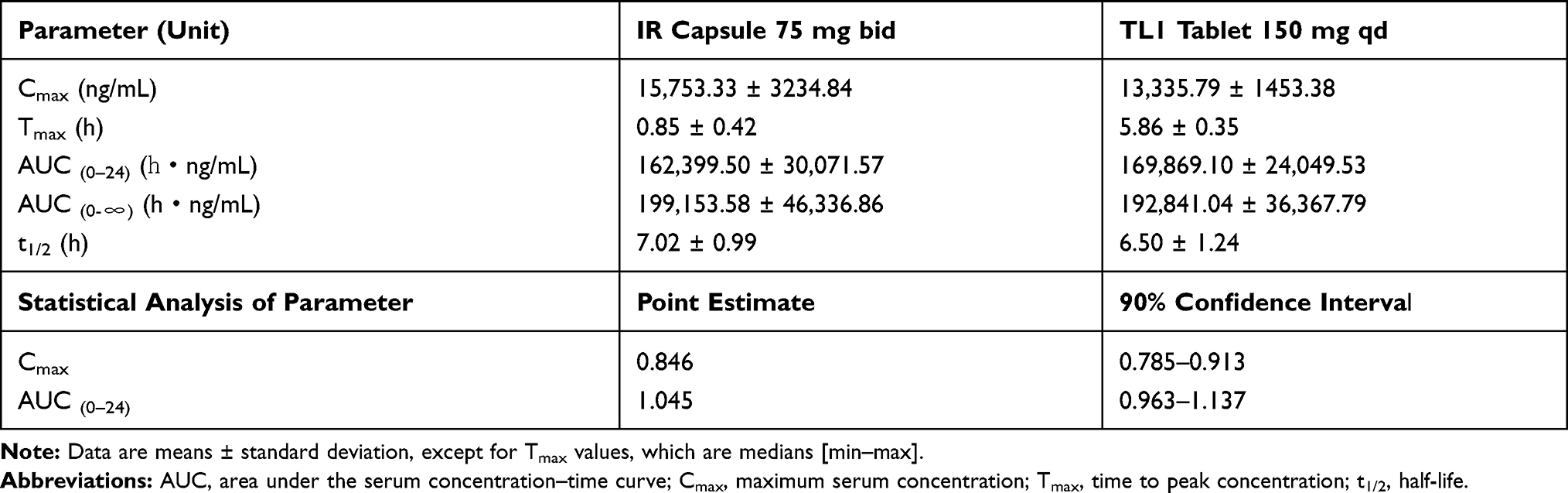 |
Table 3 Summary of Pregabalin Pharmacokinetic Parameters and Statistical Analysis Parameters in Beagles |
 |
Figure 5 Plasma concentration–time profiles of pregabalin after oral administration to beagles of the IR capsule (75 mg), twice a day, and the TL1 tablet (150 mg), once daily. Each value represents the mean ± S.D. |
Though some formulations were designed by gastric retentive system because pregabalin is absorbed poorly in the lower GI tract, they have needed to increase the dose amount by 10% to obtain an equivalent absorption for once-daily formulations compared with the twice-daily administration of the pregabalin IR capsule.4,24 On the other hand, Sako et al verified the importance of the rapid gelling rate of CR tablets for their continuous and more absorption in the colon. Although two CR formulations have been designed to show similar in vitro release patterns in several dissolution conditions, continuous drug release from the rapid-gelled formulation results in an increased AUC compared with the slow-gelled formulation.25,26 It was assumed that the swelling of TL tablets could make pregabalin absorbed longer for OAD formulation by gastric retentive property and the rapid gelation could improve the extent of absorption which did not need an extra dose amount of the drug to obtain absorption comparable to IR capsules.
Pharmacokinetic Study in Humans
In order to get tablets more processable for scale-up manufacturing, TL3 was optimized a little to get more cohesiveness between each layer during manufacturing based on TL1. There was no difference in dissolution and swelling behavior between TL1 and TL3. And then TL3 was taken in clinical human trials.
The human plasma concentration profiles of pregabalin formulations are shown in Figure 6, with the pharmacokinetic parameters summarized in Table 4. After oral administration, the TL3 tablets showed peak plasma concentrations at 6 h in the fed state, which coincided with the results of the in vivo study in beagles. In contrast, the drug in the IR capsule was absorbed rapidly, with a peak plasma concentration within 1 h, which is similar to previous reports.
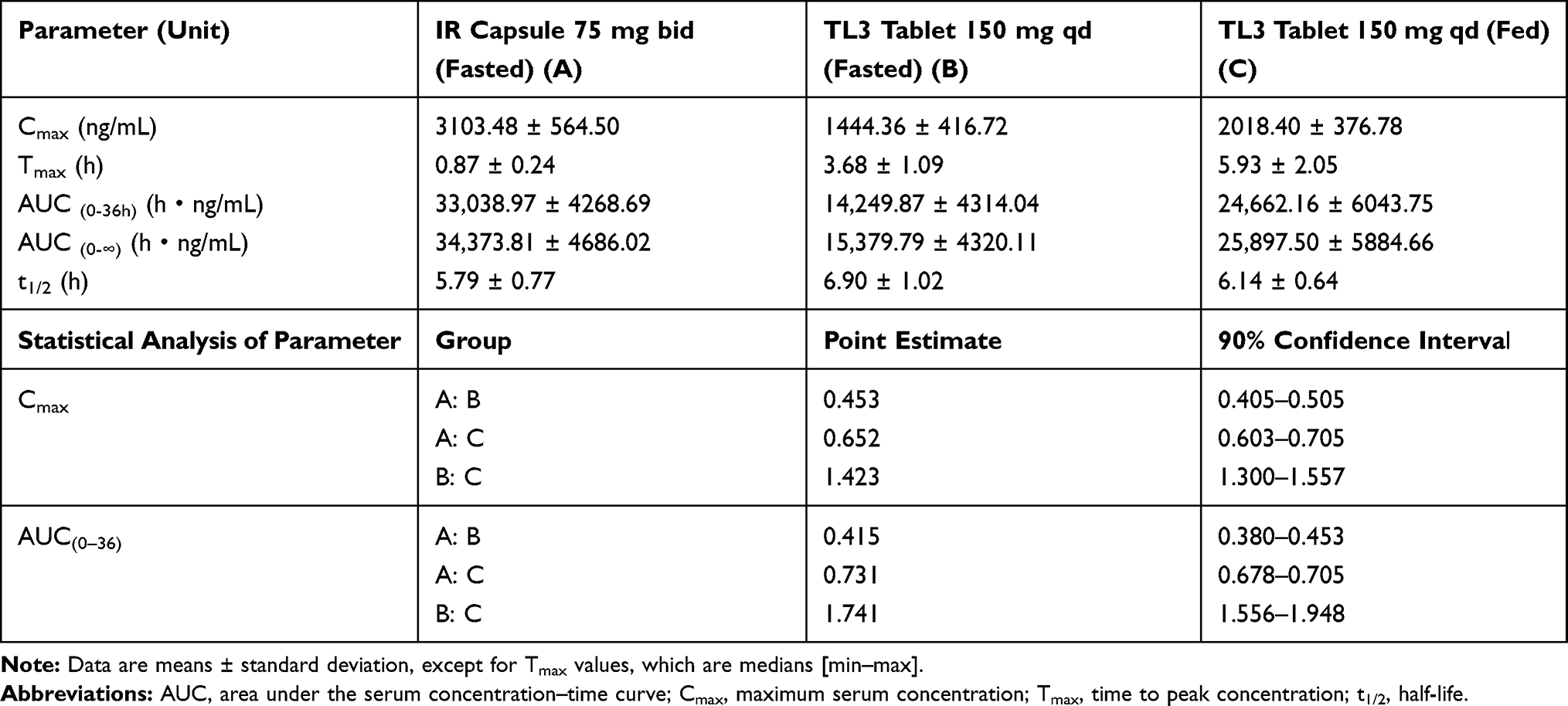 |
Table 4 Summary of Pregabalin Pharmacokinetic Parameters and Statistical Analysis Parameters in Humans |
 |
Figure 6 Plasma concentration–time profiles of pregabalin after oral administration to humans of the IR capsule (75 mg), twice a day, and the TL3 tablet (150 mg), once daily. Each value represents the mean ± S.D. |
IR capsules were taken twice a day, but the first dosing was administered in the fasted state and the second dosing in the fed state, which caused the Cmax of the first dosing to be significantly higher than the second dosing, which is similar to what has been reported previously.6
However, TL3 tablets showed lower total absorption amounts in the fasted state, which differed from the IR capsules. This is probably because of the limited absorption site of pregabalin, suggesting that, in the fasted state, the gastric emptying time is short and the tablet could not be swelled sufficiently to stay longer in the stomach; thus, it passed the absorption site quickly. However, in the fed state, the TL3 tablets showed increases in the Cmax and AUC by 42% and 74%, respectively, compared with the fasted state. This is probably because the gastric emptying time was longer; thus, the TL3 tablets were able to swell enough to stay longer in the stomach. Therefore, pregabalin in this tablet formulation had a greater chance for absorption.7
The fractional absorption of pregabalin in beagles and humans was calculated and is shown in Figure 7, with the correlation between them. Beagles appeared to absorb pregabalin slightly faster and in greater amounts than humans, with absorption terminated at 8 h. In the fed state, the absorption rate in the initial stage and the final absorbed fraction compared with the IR capsules in the beagles were higher than those in humans, which could be because dogs usually have longer gastric emptying times than humans.27 However, a good correlation of the absorption rate by time between the two in vivo results was observed, with a value of R2_0.989. In the human study, the effect of food was also observed, which could be explained by the gastric emptying time difference with the meal because of the limited absorption site of pregabalin.
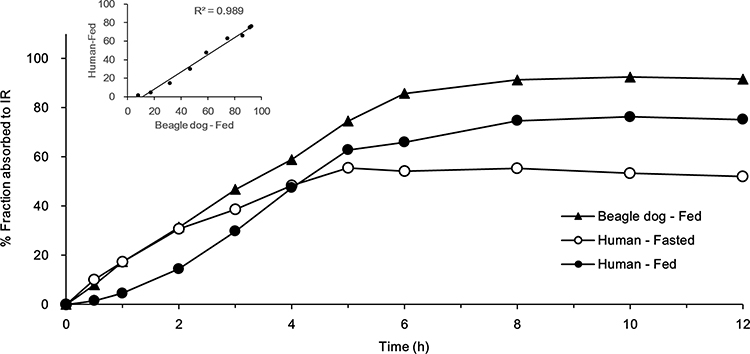 |
Figure 7 In vivo fraction-absorbed to IR capsule of TL1 and TL3 tablets and correlation between beagle and human fractions absorbed (Fed). |
Our attempt to perform a level A in vitro–in vivo correlation showed that the in vivo drug absorption rate of TL1 and TL3 tablets had a high correlation with the in vitro drug release rate of the formulation (Figure 8). The fed state (R2_0.980) showed a more linear relationship than the fasted state (R2_0.879) because of the decreased absorption of the fasted state in the late stage due to the short gastric emptying time. This result suggested that the in vitro dissolution profiles and release kinetics may be useful in predicting absorption kinetics, as well as the overall plasma drug concentration–time profiles for further studies.
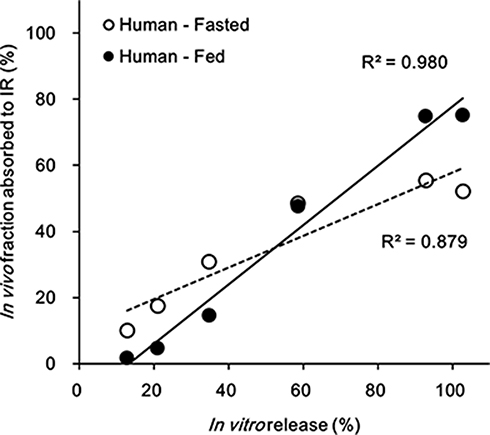 |
Figure 8 In vivo–in vitro correlation, level A, comparing the effect of food on the TL3 tablet in humans. |
Conclusion
Novel TL tablet formulations were evaluated and compared with MM formulations. The same amount of a hydrophilic polymer in the formulation showed very similar dissolution profiles for 12 h, regardless of the tablet geometry. This may suggest that the amount of the hydrophilic polymer mainly modulated the release rates of pregabalin, while the tablet geometry may not play a key role in controlling these formulations. However, the degree of swelling for the different tablets differed. Larger amounts of the polymer in the tablets resulted in a greater degree of swelling, with TL formulations swelling more rapidly than MM tablets. The different geometries of the tablets influenced water absorption into the core of the tablet, and the amount of the polymer played an important role in increasing the tablet size. Therefore, the novel TL tablet system may supply gastro-retentive properties given the rapid expanded volume of the tablets.
For the pharmacokinetic study after a meal in beagles, the mean ratio of the AUC and Cmax for the test formulation were comparable to the reference. In humans, lower absorption amounts for the test formulation in the fasted state were observed than in the fed state, which was different from the IR capsules. However, the time of peak plasma concentration at 6 h in the fed state of humans coincided with the results of the study on beagles. In this study, the novel TL tablet system of pregabalin, which has limited absorption sites in the GI tract, was formulated; these results may prove to be helpful in developing improved formulations with better continuous drug absorption for OAD administration.
Data Sharing Statement
The raw data of this study will not be shared because of confidentiality.
Disclosure
Kyung Hun Kim, Seo Hyun Lim, Cho Rok Shim, Junsung Park, Woo Heon Song, Min Chang Kwon, Jun Sang Park report grants from Ministry of Trade, Industry and Energy, Republic of Korea, during the conduct of the study. These authors have patents KR102039344B1 and KR102039345B1 registered to GL PharmTech. Jong Hyuk Lee and Han-Gon Choi has nothing to disclose. The authors report no other conflicts of interest in this work.
References
1. Bian F, Li Z, Offord J, et al. Calcium channel alpha2-delta type 1 subunit is the major binding protein for pregabalin in neocortex, hippocampus, amygdala, and spinal cord: an ex vivo autoradiographic study in alpha2-delta type 1 genetically modified mice. Brain Res. 2006;1075(1):68–80. doi:10.1016/j.brainres.2005.12.084
2. Gee NS, Brown JP, Dissanayake VU, Offord J, Thurlow R, Woodruff GN. The novel anticonvulsant drug, gabapentin (Neurontin), binds to the alpha2delta subunit of a calcium channel. J Biol Chem. 1996;271(10):5768–5776. doi:10.1074/jbc.271.10.5768
3. Jack C, William A, Yunhui HW. Application of the biopharmaceutical classification system in clinical drug development – an industrial view. AAPS J. 2008;10(2):306–310. doi:10.1208/s12248-008-9036-5
4. Chew ML, Alvey CW, Plotka A, et al. Pregabalin controlled-release pharmacokinetics in healthy volunteers. Clin Drug Investig. 2014;34(9):627–637. doi:10.1007/s40261-014-0221-2
5. Chew ML, Plotka A, Alvey CW, et al. Pharmacokinetics of pregabalin controlled-release in healthy volunteers: effect of food in five single-dose, randomized, clinical pharmacology studies. Clin Drug Investig. 2014;34(9):617–626. doi:10.1007/s40261-014-0211-4
6. Bockbrader HN, Radulovic LL, Posvar EL, et al. Clinical pharmacokinetics of pregabalin in healthy volunteers. J Clin Pharmacol. 2010;50(8):941–950. doi:10.1177/0091270009352087
7. Piyapolrungroj N, Li C, Bockbrader H, Liu G, Fleisher D. Mucosal uptake of gabapentin (neurontin) vs. pregabalin in the small intestine. Pharm Res. 2001;18(8):1126–1130. doi:10.1023/A:1010970809090
8. French JA, Kugler AR, Robbins JL, Knapp LE, Garofalo EA. Dose-response trial of pregabalin adjunctive therapy in patients with partial seizures. Neurology. 2003;60(10):1631–1637. doi:10.1212/01.WNL.0000068024.20285.65
9. Coleman CI, Limone B, Sobieraj DM, et al. Dosing frequency and medication adherence in chronic disease. J Manag Care Pharm. 2012;18(7):527–539. doi:10.18553/jmcp.2012.18.7.527
10. Srivastava K, Arora A, Kataria A, Cappelleri JC, Sadosky A, Peterson AM. Impact of reducing dosing frequency on adherence to oral therapies: a literature review and meta-analysis. Patient Prefer Adherence. 2013;7:419–434. doi:10.2147/PPA.S44646
11. Ma L, Deng L, Chen J. Applications of poly(ethylene oxide) in controlled release tablet systems: a review. Drug Dev Ind Pharm. 2014;40(7):845. doi:10.3109/03639045.2013.831438
12. Huang X, Brazel CS. On the importance and mechanisms of burst release in matrix-controlled drug delivery systems. J Control Release. 2001;73(2–3):121–136. doi:10.1016/S0168-3659(01)00248-6
13. Colombo P, Conte U, Gazzaniga A, et al. Drug release modulation by physical restriction of matrix swelling. Int J Pharm. 1990;63(1):43–48.
14. Conte U, Maggi L, Torre ML, Giunchedi P, La Manna A. Press-coated tablets for time-programmed release of drugs. Biomaterials. 1993;14(13):1017–1023. doi:10.1016/0142-9612(93)90195-8
15. Conte U, Maggi L, Colombo P, La Manna A. Multi-layered hydrophilic matrices as constant release devices (Geomatrix Systems). J Control Release. 1993;26(1):39–47.
16. Park JS, Shim JY, Park JS, Choi YW, Jeong SH. A novel three-layered tablet for extended release with various layer formulations and in vitro release profiles. Drug Dev Ind Pharm. 2011;37(6):664–672. doi:10.3109/03639045.2010.535211
17. Hodges LA, Sime KA, Creech LA, et al. Pharmacoscintigraphy confirms consistent tamsulosin release from a novel triple-layered tablet. Int J Pharm. 2013;454(1):41–46. doi:10.1016/j.ijpharm.2013.06.065
18. Wagner JG. The Wagner-Nelson method applied to a multicompartment model with zero order input. Biopharm Drug Dispos. 1983;4(4):359–373. doi:10.1002/bdd.2510040408
19. Ma L, Deng L, Chen J. Applications of poly (ethylene oxide) in controlled release tablet systems: a review. Drug Dev Ind Pharm. 2014;40(7):845–851. doi:10.3109/03639045.2013.831438
20. Park JS, Shim JY, Nguyen KV, et al. A pharma-robust design method to investigate the effect of PEG and PEO on matrix tablets. Int J Pharm. 2010;393(1–2):79–87. doi:10.1016/j.ijpharm.2010.04.009
21. Choi Du H, Kim KH, Park JS, Jeong SH, Park K. Evaluation of drug delivery profiles in geometric three-layered tablets with various mechanical properties, in vitro-in vivo drug release, and Raman imaging. J Control Release. 2013;172(3):763–772. doi:10.1016/j.jconrel.2013.08.301
22. Bockbrader HN, Wesche D, Miller R, Chapel S, Janiczek N, Burger P. A comparison of the pharmacokinetics and pharmacodynamics of pregabalin and gabapentin. Clin Pharmacokinet. 2010;49(10):661–669. doi:10.2165/11536200-000000000-00000
23. Kanwar N, Kumar R, Sarwal A, Sinha VR. Preparation and evaluation of floating tablets of pregabalin. Drug Dev Ind Pharm. 2016;42(4):654–660. doi:10.3109/03639045.2015.1062895
24. Bockbrader HN, Cho YH, Santiago SD, et al. (Warner-Lambert Co LLC., USA). Solid pharmaceutical compositions containing pregabalin. US Patent 9,144,559 B2. 2015 Sep 29.
25. Sako K, Nakashima H, Sawada T, Fukui M. Relationship between gelation rate of controlled-release acetaminophen tablets containing polyethylene oxide and colonic drug release in dogs. Pharm Res. 1996;13(4):594–598. doi:10.1023/A:1016006423601
26. Yogesh J, David M, Hossein O. Enhanced bioavailability via extended gastric retention. J Develop Drugs. 2013;2(1):105–109.
27. Sutton SC. Companion animal physiology and dosage form performance. Adv Drug Deliv Rev. 2004;56(10):1383–1398. doi:10.1016/j.addr.2004.02.013
 © 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.