Back to Journals » Clinical, Cosmetic and Investigational Dermatology » Volume 13
Development and Validation of an Immune-Related Gene Pair Signature in Skin Cutaneous Melanoma
Authors Xie R, Dong S ![]() , Jiang J, Yang C, Li L, Zhao S, Li Y, Wang C, Li S, Xiao Y, Chen L
, Jiang J, Yang C, Li L, Zhao S, Li Y, Wang C, Li S, Xiao Y, Chen L ![]()
Received 23 September 2020
Accepted for publication 26 November 2020
Published 15 December 2020 Volume 2020:13 Pages 973—986
DOI https://doi.org/10.2147/CCID.S281364
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Jeffrey Weinberg
Ran Xie,1,* Suwei Dong,2,* Jie Jiang,3 Conghui Yang,1 Lanjiang Li,4 Sheng Zhao,1 Yunlei Li,3 Chun Wang,1 Shujuan Li,1 Yanbin Xiao,2 Long Chen1
1PET/CT Center, Yunnan Cancer Hospital, The Third Affiliated Hospital of Kunming Medical University, Cancer Center of Yunnan Province, Kunming, Yunnan, People’s Republic of China; 2Second Orthopedic Ward, Yunnan Cancer Hospital, The Third Affiliated Hospital of Kunming Medical University, Cancer Center of Yunnan Province, Kunming, Yunnan, People’s Republic of China; 3Graduate School, Kunming Medical University, Kunming, Yunnan, People’s Republic of China; 4Department of Forensic Medicine, Kunming Medical University, Kunming, Yunnan, People’s Republic of China
*These authors contributed equally to this work
Correspondence: Yanbin Xiao
Second Orthopedic Ward, Yunnan Cancer Hospital, The Third Affiliated Hospital of Kunming Medical University, Cancer Center of Yunnan Province, Kunming 650118, Yunnan, People’s Republic of China
Email [email protected]
Long Chen
PET/CT Center, Yunnan Cancer Hospital, The Third Affiliated Hospital of Kunming Medical University, Cancer Center of Yunnan Province, Kunming 650118, Yunnan, People’s Republic of China
Email [email protected]
Introduction: Skin cutaneous melanoma (SKCM) is a common skin malignancy worldwide, and its metastasis and mortality rates are high. The molecular characteristics exhibited by tumor–immune interactions have drawn the attention from researchers. Therefore, increased knowledge and new strategies to identify effective immune-related biomarkers may improve the clinical management of SKCM by providing more accurate prognostic information.
Patients and Methods: In this study, we established a prognostic immune-related gene pair (IRGP) signature for predicting the survival of SKCM patients. The Cancer Genome Atlas (TCGA) and Gene Expression Omnibus (GEO) databases provided gene expression profiles together with clinical information, and the samples were randomly divided into three groups including the training, testing, and validation datasets. The regression model of least absolute shrinkage and selection operator (LASSO) helped to identify a 13-IRGP signature with a significant relation to the survival of SKCM patients.
Results: The training, TCGA, and independent sets have an average value of area under the curve of 0.79, 0.76, and 0.82, respectively. In addition, this 13-IRGP signature can noticeably divide SKCM patients into high-risk group and low-risk group with significantly different prognoses. Many biological activities such as gene family were enriched among the genes in our IRGP signature. While analyzing the risk signature and clinical characteristics, there was a large difference in the risk score between T stage and tumor stage grouping. Finally, we constructed a nomogram and forest plots of the risk score and clinical features.
Conclusion: In summary, we developed a robust 13-IRGP prognostic signature in SKCM, which can identify and provide new insights into immunological biomarkers.
Keywords: skin cutaneous melanoma, TCGA, immune-related gene pair, signature, bioinformatic
Introduction
Skin cutaneous melanoma (SKCM) acts as a common skin malignancy around the world.1 It was estimated that 287,723 new cases and 60,712 cancer-related deaths occurred in 2018.2 Thus, SKCM is a complex malignant tumor with high metastasis and mortality rates.3 SCKM accounts for 1% of all skin neoplasms while it is responsible for most of the skin cancer death.4 Significant progress has been made in understanding SKCM biology, genetics, and treatment methods; however, the prognosis is still poor due to the high probability of invasion and metastasis. The diagnosis of melanoma may indicate more aggressive treatment, lifelong monitoring, and a worse prognosis.5 Therefore, increased knowledge and new strategies to identify effective biomarkers may improve the clinical management of SKCM by providing more accurate prognostic information.
Recently, the number of approved cancer immunotherapies has become unprecedented, including immune checkpoint inhibitor monoclonal antibodies and adoptive cell therapy with chimeric antigen receptor (CAR)-T cell therapy.6 Cancer immunotherapy has become one of the most exciting and rapidly expanding fields. It is now considered to be one of the pillars of treatment alongside surgery, radiotherapy, and chemotherapy.7 In SKCM, most patients remain unresponsive to checkpoint inhibitors and resistance to anti-PD1-based immune checkpoint blockade remains a problem for the treatment of metastatic melanoma, while recent researches revealed that targeting the CD155 pathway might improve response to anti-PD1 therapy for patients with metastatic melanoma.8 Therefore, trials of different combinations of immunotherapies are being evaluated.9 Surgical treatment is still the most suitable treatment for metastatic SKCM currently, but targeted molecular therapy and immunotherapy can effectively assist in the development of melanoma therapies.10 At the same time, immunotherapy has shifted from therapy based on cytokine to the blockade of immune checkpoints of cytotoxic T-lymphocyte-associated antigen-4 (CTLA-4) and programmed cell-death protein 1 (PD-1) mediated by antibody.11 Targeted therapy with inhibitors of BRAF and MEK is currently an important cornerstone for the treatment of metastatic SKCM.12 Moreover, the role of monocyte-derived dendritic cells in melanoma has been redefined and possibly provides a novel strategy to increase the efficacy of T-cell-based immunotherapies in nonresponding individuals.13 Nevertheless, the molecular characteristics of tumor–immune interactions remain to be extensively studied in future studies.
With the development of genomics and transcriptomics, studies about immune-related genes (IRGs) or prognostic signatures in cancers are increasingly being reported. For example, Li et al,14 used transcriptomic data and clinicopathological information from the TCGA database to identify an immune-related signature specific to uveal melanoma prognosis. In another study of breast cancer,15 many tumor immune-related genes (IRGs) were identified for predicting patients’ prognosis. In SKCM, a novel IRG prognostic biomarker associated with the tumor microenvironment based on the TCGA and GEO databases was established.16 In addition, some other biological markers have been identified and discovered as prognostic biomarkers for SKCM. For example, alternative splicing events could be significant indicators for SKCM prognosis.17 Another study found a five-miRNA (miR-25, miR-204, miR-211, miR-510, miR-513c) signature as a potential significant biomarker for SKCM prognosis based on the GEO and TCGA databases.18
Recently, more and more studies have paid attention to immune-related gene pairs in human cancers. To be specific, a signature of 33 immune-related gene pairs that can predict clinical outcomes of hepatocellular carcinoma was established by Sun et al.19 In this study, they used the gene expression levels of IRGs from the TCGA database to construct IRGPs. Based on LASSO penalized Cox proportional hazards regression, the frequency of the 33-IRGP model was the highest. For colorectal cancer, Wu et al,20 also identified a 19 IRGP signature with 36 unique genes. The above studies helped us to better understand how to identify cancer patients with a high mortality. However, there is no report about IRGPs in SKCM.
In the present study, based on the TCGA and GEO databases, we constructed and validated a 13-IRGP prognostic signature for SKCM. This study provides new insights for prognosis prediction in SKCM patients.
Patients and Methods
Sources of Immune-Related Genes
In this study, the gene list of IRGs was downloaded from the InnateDB database (https://www.innatedb.com/),21 which recorded the endogenous IRGs of multiple species under the support of the literature with manual correction. Here, the endogenous IRGs of humans were used, and a total of 1039 IRGs were identified (the genes with duplicate names were removed). The list of genes is provided in Supplementary Table 1.
Data Source and Download
We used the TCGA GDC API to download the latest RNA-seq mRNA expression data together with clinical follow-up information of SKCM (Supplementary Table 2) from the TCGA database (https://portal.gdc.cancer.gov/)22 until December 2018. This dataset included 472 SKCM patient samples, of which 471 were tumor tissues and one was a normal sample. Three microarray datasets (GSE54467,23 GSE22155,24 and GSE65904)25 were also downloaded from the GEO database (http://www.ncbi.nlm.nih.gov/geo)26 for validation of this signature. In brief, we used the gene expression as MINiML files from NCBI. The GSE54467 dataset contained 79 SKCM samples with clinical features (Supplementary Table 3), the GSE22155 dataset contained 79 samples with clinical features (all stage III and IV samples, Supplementary Table 4), and the GSE65904 dataset contained 214 samples (Supplementary Table 5).
Data Preprocessing
The RNA-seq data of 471 samples from the TCGA database were preprocessed in the following steps: (1) samples with no clinical information or with an overall survival (OS) of 0 days were removed; (2) data from normal samples were removed; 3) genes with an FPKM value of 0 in over half of all samples were removed; and (4) only IRGs’ expression profiles were retained. For the GEO datasets, the samples were preprocessed as follows: (1) data from normal tissues were removed and only the primary tumor data were kept; (2) the overall survival (OS) time of was converted from years or months into days; (3) the microarray probes were mapped to the human gene symbol using the bioconductor R package; and (4) only the expression profiles of IRGs were retained. After data preprocessing, the sample sizes of the GSE54467 and GSE22155 datasets were only 70 and 43, respectively. We combined the above two datasets as an independent validation set. The clinical information of the three datasets is shown in Table 1.
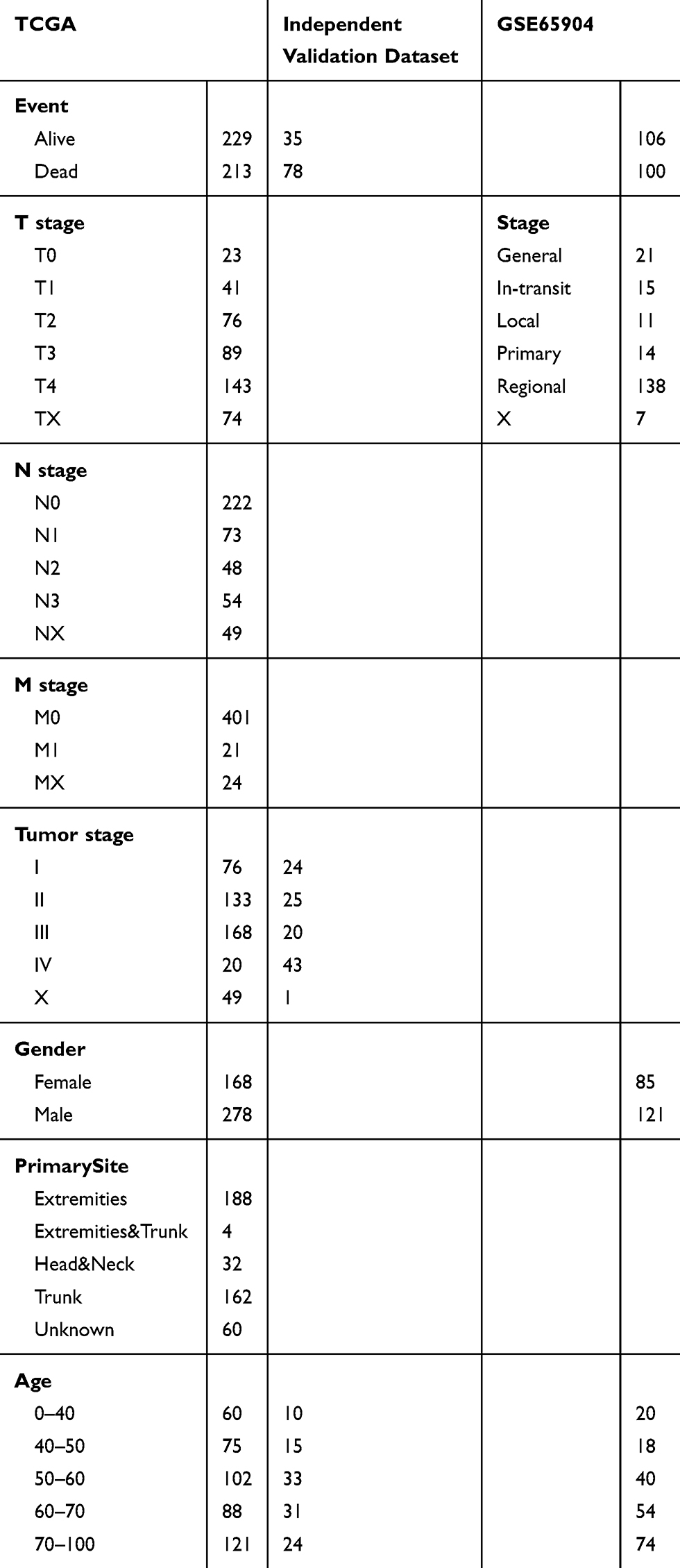 |
Table 1 The Clinical Information for Three Datasets |
Construction of IRGPs
The method to build the IRGPs was as described by Wu et al.20 In brief, we first constructed (1039*1038)/2 IRGPs on the basis of 1039 IRGs. IRGP values were calculated following rules below. If the first IRG in a particular IRGP held smaller gene expression level compared with the second IRG, their IRGP score = 1; otherwise, their IRGP score = 0. Next, we filtered IRGPs by computing the IRGPs for each dataset. If the IRGP was a unique value of 0 or 1 for all samples in that dataset, we removed the IRGPs (possibly due to platform factors).
Establishment and Validation of the Prognostic Signature Based on IRGPs
The TCGA dataset was used as the training set with a total of 220,718 IRGPs with 446 samples used for subsequent modeling and analysis. The IRGPs of the other two datasets were used as the validation sets to verify the signature. First, we divided 446 SKCM samples into two sets, the training set and the testing set. For avoiding the random allocation bias that could impact the stability exhibited by subsequent signature, all samples were resampled 100 times into random groups. Here, the group sampling was based on the training: testing set with a ratio of 0.5:0.5. We chose the most suitable training set and testing set considering two conditions: (1) the distribution of age, follow-up time, tumor stage, as well as death rate of patients in two groups were similar; and (2) the sample size of the two groups was similar after clustering by the gene expression profile. The final training set data are shown in Supplementary Table 6 and contained a total of 222 SKCM samples. The testing set data are shown in Supplementary Table 7 and contained a total of 224 samples.
The prognostic IRGPs (p-value < 0.05) in the training dataset were then selected via a univariate Cox proportional hazard regression model with a long-rank test. To further narrow the IRGP number while maintaining high accuracy, we performed LASSO regression for filtering the IRGPs in the risk model by using the glmnet R package.27 Here, we used the most stable gene pair for constructing the prognosis signature for SKCM. By calculating the risk score of each sample, we used the median risk score to divide samples in the training set into two groups, high-risk group (Risk-H) and low-risk group (Risk-L). The receiver operating characteristic (ROC) curves were plotted by virtue of the signature performance. The Kaplan–Meier curves of these samples were plotted with the KMsurv R package.
Functional Enrichment Analysis
First, we performed gene family enrichment analysis based on the IRGs in our signature with Fisher’s exact test. The clusterProfiler R package28 was then used for analyzing to the IRGPs for molecular function (MF), cellular component (CC), and biological process (BP) enrichment through studying the Gene Ontology (GO) terms, as well as the Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway enrichment.
The Nomogram and Forest Plots of the Risk Score and Clinical Features
The nomogram can visually and effectively show the results of the risk model and is convenient to use in the prediction of survival outcomes. It uses the length of the line to indicate the influence degree of different variables on the result as well as the influence of different values of variables on the result. Here, the nomogram was generated with the rms R package. Forest plots can simply and intuitively display the statistical summary results of different research factors. The general form of the forest plot is in a plane rectangular coordinate system, with an invalid line perpendicular to the X axis (usually the coordinate X = 1 or 0) as the center. Several line segments parallel to the X axis represent the magnitude of each study effect and its 95% confidence interval.
Statistical Analysis
We carried out all the statistical analyses in the R language (version 3.5.1) environment. The Log rank test assisted in evaluating the difference in survival between two groups. In the analysis of the risk signature and clinical characteristics, the Kruskal–Wallis test was used to determine differences between multiple groups. The survcomp R package helped to calculate the C-index. A p-value of less than 0.05 means the difference is statistically significant.
Results
Consistency Assessment of Datasets on Different Platforms
In this study, to evaluate the differences between RNA-seq data from the TCGA database and gene expression microarray data from the GEO database, we used IRG and IRGP data to cluster the correlation between SKCM samples. The results showed that IRG data can clearly separate the data from the two platforms (Supplementary Figure 1), while IRGP data can also separate the data from the two platforms (Supplementary Figure 2), but the difference between two platforms was significantly reduced, and the correlation was closer than that of IRGs (Supplementary Figure 3). This suggested that after IRGP construction from three datasets based on different platforms, the differences were eliminated.
Construction of the IRGP Signature in SKCM
We extracted the IRGPs shared by the three datasets. The TCGA dataset was used as the training set with a total of 220,718 IRGPs with 446 samples used for subsequent modeling and analysis. First, a total of 446 SKCM patients from the TCGA database were included in our study, which were split into a training dataset (n = 222) and a testing dataset (n = 224) in a random manner. The detailed clinical information is shown in Table 2. Based on the univariate Cox proportional hazards regression model, a total of 1614 significantly prognostic IRGPs were selected (Supplementary Table 8). As shown in Figure 1, we determined the relationships between the p-values of 1614 genes and the hazard ratios (HRs). From the results, the HRs corresponding to IRGPs with a significant p-value usually deviate from 1.
 |
Table 2 The Clinical Information of the Training and Testing Datasets |
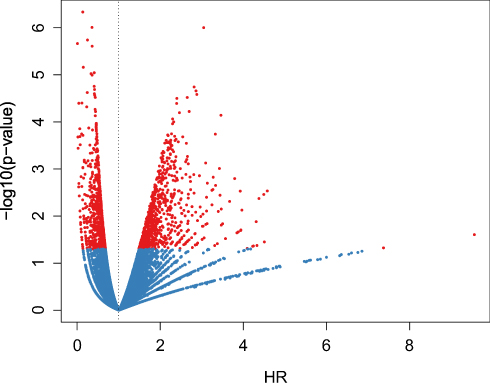 |
Figure 1 The relationships between p-values and HRs. The relationships between the p-values and HRs of 1614 significant prognostic IRGPs. Red indicates the IRGPs with p-value < 0.05. |
Next, aiming at further filtering above IRGPs for reducing the number in the risk model, we constructed a prognostic gene signature with the help of the LASSO regression model on the TCGA training dataset. We finally obtained 13 IRGPs to build this signature (Supplementary Table 9). Risk score = SIRPA_vs_IFI27*1.02 + IDO1_vs_APOBEC3A*-0.99 + ING4_vs_CNOT4*-1.49 + NLRP1_vs_EGFR*-0.67 + NFKBIE_vs_ITGB3*0.71 + IFIT5_vs_CASP6*-0.53 + RGMB_vs_ACHE*0.69 + CXCL14_vs_RSAD2*0.46 + CD36_vs_TLR8*0.55 + IFIT2_vs_FANCC*-0.25 + NFKBIE_vs_CXCL10*0.12 + TRIM45_vs_CD1D*0.12 + KCNJ8_vs_CTLA4*-0.02. Considering that the overall survival distribution of the samples was approximately 2 years (Supplementary Figure 4), we calculated the prediction effect on all datasets exhibited by the model for 1, 5, and 7 years. The average AUC of the training set was 0.79, of the TCGA dataset was 0.756, of the independent dataset was 0.818, and of the GSE65904 independent testing dataset was 0.675 (Figure 2A–D, left). The OS distribution of the samples in the Risk-H and Risk-L groups was also assessed (Figure 2A–D, middle). The Risk-H and Risk-L groups were not significantly different in terms of size in the 0- and 1-year periods. After the 5th year, the Risk-H group was gradually smaller than the Risk-L group, and the change became increasingly significant as the OS was extended (Figure 2A–D, right).
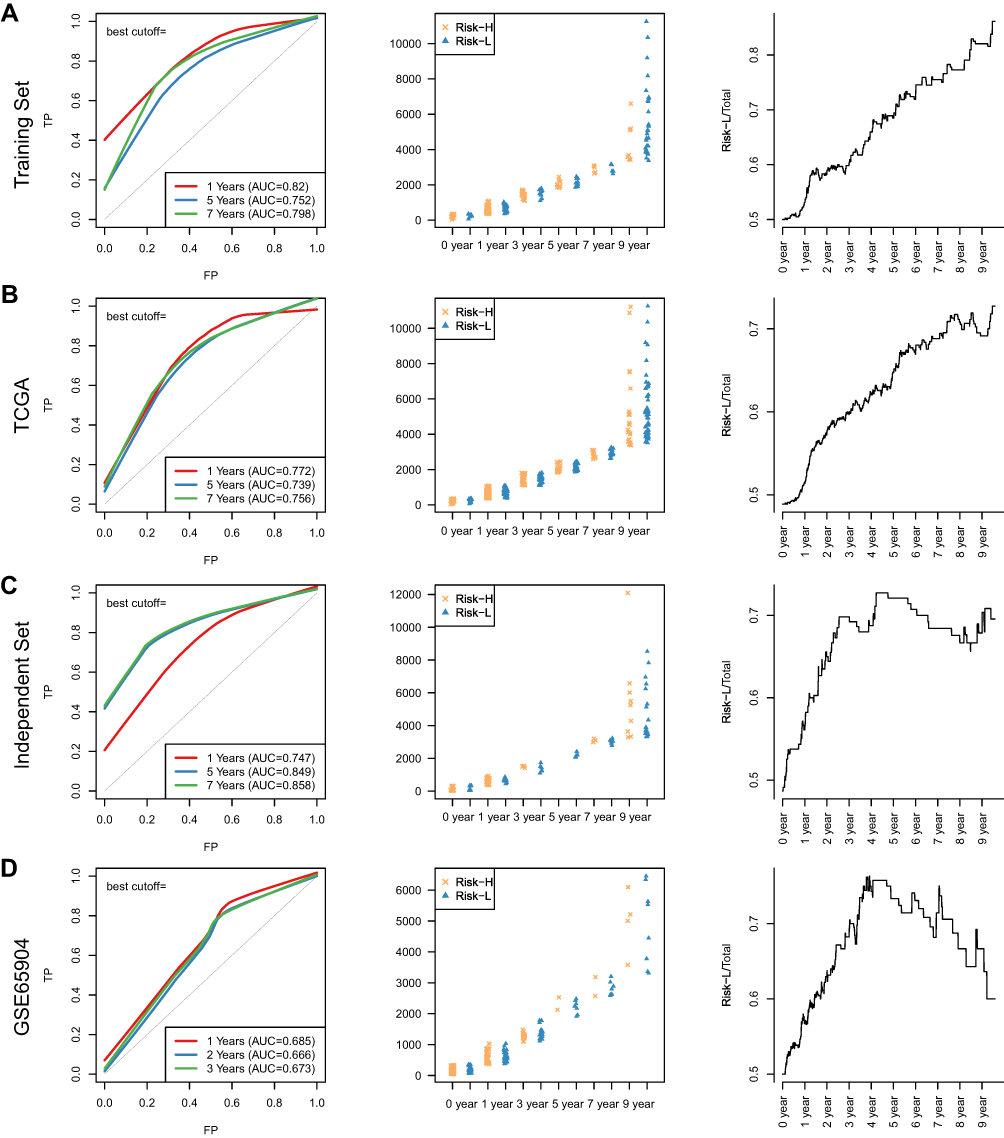 |
Figure 2 Construction of the IRGP signature in SKCM. (A) The risk model ROC curve of 13 IRGPs in the training set after LASSO regression. The statistics of Risk-H and Risk-L samples under different OS outcomes and the proportion of Risk-L samples in the total sample vary with OS. (B) The risk model ROC curve of 13 IRGPs in TCGA after LASSO regression. The statistics of Risk-H and Risk-L samples under different OS outcomes and the proportion of Risk-L samples in the total sample vary with OS. (C) The risk model ROC curve of 13 IRGPs in the independent set after LASSO regression. The statistics of Risk-H and Risk-L samples under different OS outcomes and the proportion of Risk-L samples in the total sample vary with OS. (D) The risk model ROC curves of 13 IRGPs in the GSE65904 set after LASSO regression. The statistics of Risk-H and Risk-L samples under different OS outcomes and the proportion of Risk-L samples in the total sample vary with OS. |
IRGP Signature Evaluation and Validation for Survival Prediction
The K-M survival curves of the Risk-H and Risk-L groups were predicted via the risk model based on the 13 IRGPs in the training set, TCGA dataset and two GEO datasets (Figure 3). As shown in Figure 3A, we found significant survival differences between the Risk-H and Risk-L groups (with Log rank test p-value less than 0.0001). Similar results were also validated in the TCGA, independent, and GSE65904 datasets (with Log rank test p-value less than 0.01, Figure 3B–D). Moreover, in the testing set of TCGA, Risk-H and Risk-L samples also had significant differences in prognosis (Supplementary Figure 5).
 |
Figure 3 IRGP signature evaluation and validation for survival prediction. (A) The K-M survival plot of the training set. (B) The K-M survival plot of the TCGA dataset. (C) The K-M survival plot of the independent dataset. (D) The K-M survival plot of the GSE65904 dataset. |
Biological Functions Related to the IRGP Signature
Based on the above IRGP signature, we next performed functional annotations of the IRGPs. As shown in Table 3, four gene families, namely, the repulsive guidance molecule family, Erb-b2 receptor tyrosine kinases, tetratricopeptide repeat domain containing, and integrin beta subunits, exhibited a significant enrichment (Fisher’s exact test p-value < 0.01). GO and KEGG pathway enrichment analyses were used for further confirming the biological activity of the 13 IRGPs (Figure 4, Supplementary Table 10). The enriched biological pathways are shown in Supplementary Table 11.
 |
Table 3 The Gene Family Enrichment Analysis of IRGPs Signature |
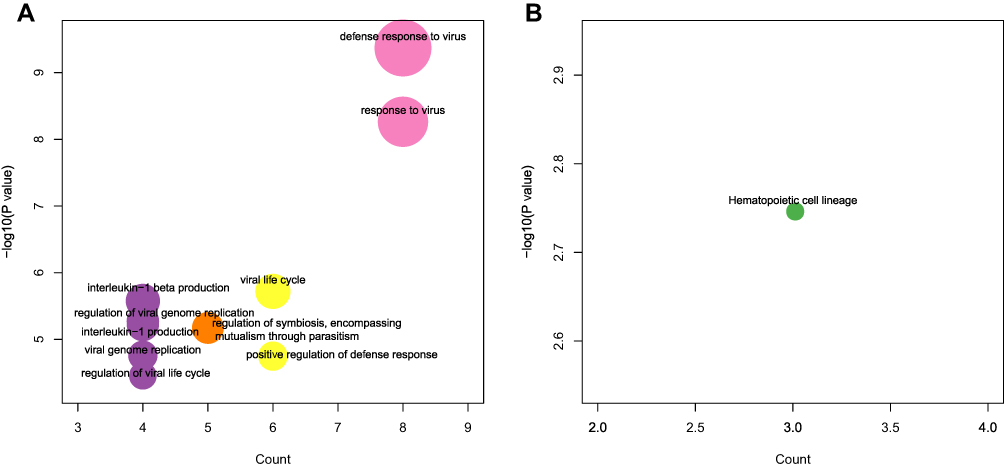 |
Figure 4 Biological functions related to the IRGP signature. (A) The results of GO enrichment of the 13-IRGPs. (B) The KEGG pathway enrichment results of the 13 IRGPs. |
Risk Signature and Clinical Characteristics Analysis
Furthermore, we analyzed the relationships between clinical features such as T stage, N stage, M stage, age and tumor stage and the risk score. The results are shown in Figure 5 (Supplementary Figure 6 for M stage) and revealed that there was a large difference in the risk score between the T stage and tumor stage groups (p-value < 0.05), indicating that our risk model was closely associated with the above clinical characteristics in SKCM. Because of the significant correlation between the clinical characteristics of T stage and tumor stage and the prognosis of SKCM, there were significant prognostic differences between the samples of the above clinical characteristic groupings (Supplementary Figure 7). TNM, age, and different pathological stage prognostic risk models were constructed. Then, we compared these models with our 13-IRGP risk signature. The C-index results of the models are shown in Figure 6. From the results, our 13-IRGP signature had the highest C-index, which demonstrated that the model exhibited a better performance compared with the other models. In addition, we constructed a T stage + tumor stage + risk score multifactor prognostic signature. The C-index of this model reached 0.76, indicating that the performance of the multifactor model was improved.
 |
Figure 5 The relationships between risk scores and clinical features. (A) The difference distribution of the risk score according to T stage. (B) The difference distribution of the risk score according to N stage. (C) The difference distribution of the risk score according to tumor stage. (D) The difference distribution of the risk score according to age. |
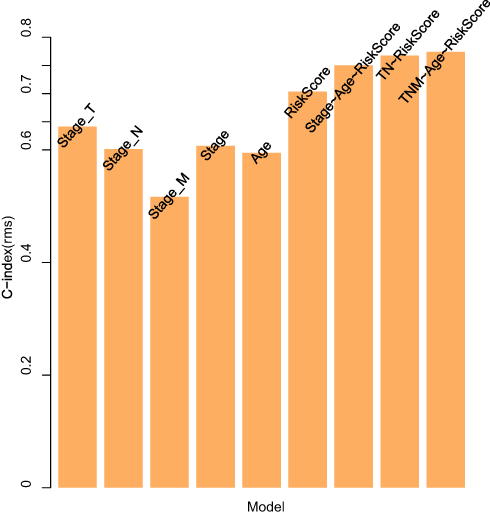 |
Figure 6 The C-index results of the prognostic risk models. |
The Nomogram and Forest Plots of the Risk Score and Clinical Features
Finally, we constructed a nomogram with clinical features including T stage, N stage, M stage, age, and risk score (Figure 7). Here, two nomograms, TNM stage + age + risk score and tumor stage + age + risk score, were constructed. From the results, the characteristics of the risk score the most significantly affected SKCM patient survival prediction, demonstrating the better prediction effect of the 13-IRGP risk model. Then, we described the clinical features, including T stage, N stage, M stage, and age, and the risk score by displaying forest plots (Figure 8). The TNM stage and tumor stage were used to construct forest plots with the risk score. From the results, the HR of the risk score was the largest in both models, approximately 1.9, with a p-value of <0.001 (Supplementary Table 12).
 |
Figure 7 The nomogram of risk score and clinical features. (A) The nomogram constructed based on clinical features including T stage, N stage, M stage, age and the risk score. (B) The nomogram constructed by clinical features including tumor stage, age and the risk score. |
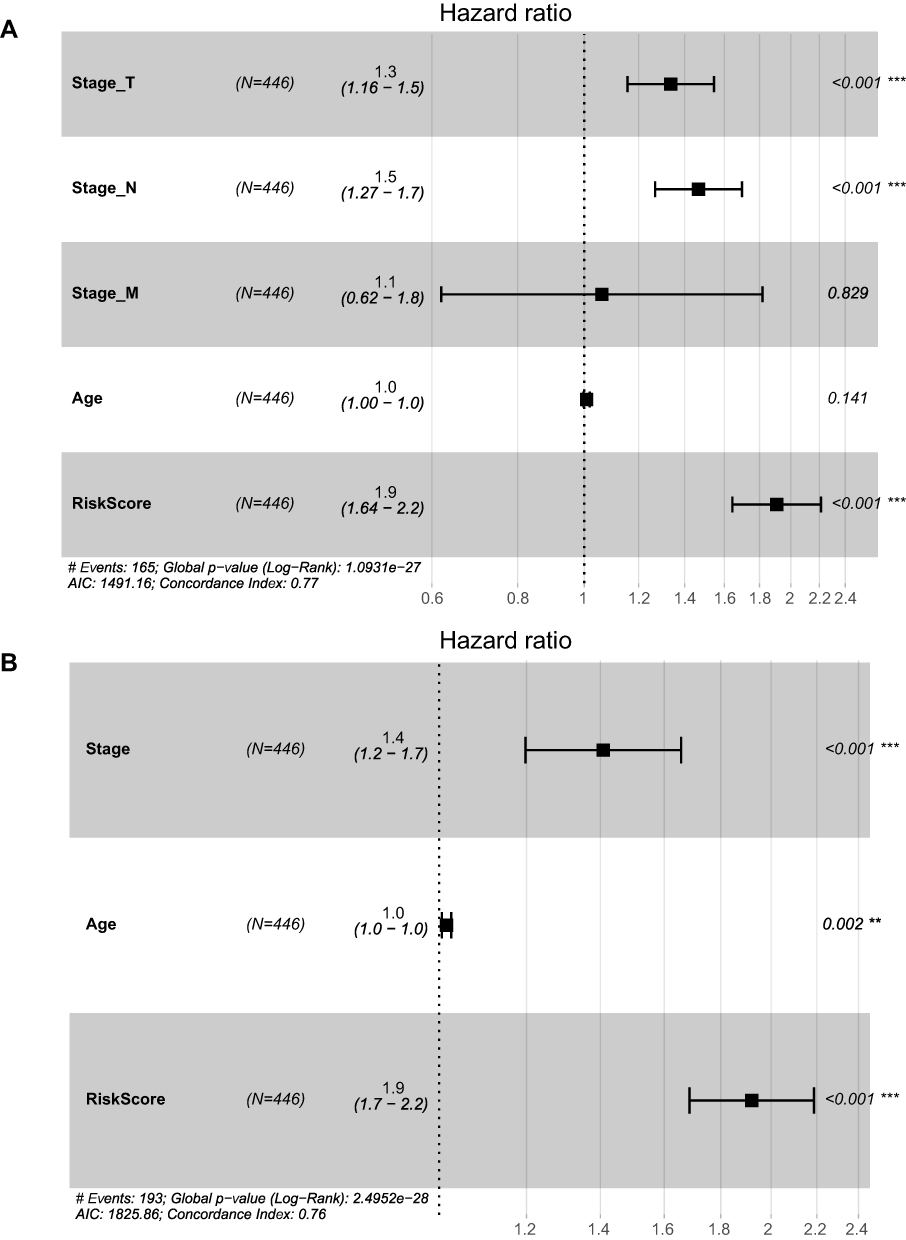 |
Figure 8 The forest plots of the risk score and clinical features. (A) The forest plots of the risk score and T stage, N stage, M stage and age. (B) The forest plots of the risk score, age and tumor stage. |
Discussion
In this study, a robust 13-IRGP signature was employed to estimate prognosis in SKCM, which can identify and provide new insights into immunological biomarkers.
The advent of immunotherapy has revolutionized the treatment of some cancers, including melanoma. Harnessing the immune system to improve tumor cell killing is now standard clinical practice and immunotherapy is the first line of defence for many cancers which were difficult to treat historically. However, although a considerable number of patients respond well to treatment, there are still some patients who do not respond, and some cancers cannot be treated with these therapies.29,30 Recently, with the development of genomics and transcriptomics, studies about IRGs or prognostic signatures in cancers, including melanoma, have been increasingly reported. In one study of melanoma, Huang et al,16 first found that 63 IRGs were associated with the OS of patients. Based on Cox regression and LASSO analysis, they identified an IRG signature including 8 IRGs, namely PSME1, CDC42, CMTM6, HLADQB1, HLA-C, CXCR6, CD8B, and TNFSF13, which could better predict the prognosis of patient compared with existing recorded data. For uveal melanoma, an immune-related signature with two genes of MANEAL and SLC44A3, was established.14 In addition, the researchers revealed a moderate association between the immune checkpoints that contained PD-1, CTLA-4, IDO, and TIGIT and such signature. Li et al,31 used RNA-seq data from 527 head and neck cancer patients from the TCGA database to establish a prognostic prediction model using multivariable Cox regression analysis. Finally, they identified a 10-IRG signature including SEMA3G, GNRH1, ZAP70, PLAU, SFTPA2, CCL26, DKK1, GAST, PDGFA and STC1. Other studies on IRG signatures in human cancers include breast cancer,15 colorectal cancer,32 hepatocellular cancer,33 lung cancer,34 and gliomas.35 Therefore, based on TCGA and microarray methods, IRG signatures for predicting the prognosis of patients have increasingly become the focus of research.
We constructed a 13-IRGP prognostic signature for SKCM in this study. There have been no reports about IRGP signatures for SKCM until now. However, in colorectal cancer,20 Wu et al used six public cohorts, including a training cohort (n = 565) together with five independent validation cohorts (n = 572, 290, 90,177, and 68). They established a 19-IRGP signature that contained 36 unique genes with an obvious relation to patients’ survival. When combined sex, stage, and other clinical factors, the IRGP signature and clinical factor combination showed a higher prognostic accuracy than the individual IRGP signature. In another study of hepatocellular carcinoma,19 researchers also identified a signature of 33 IRGPs that can predict clinical outcomes based on TCGA and GEO data. Compared with traditional studies about gene signatures, since pairwise comparison helped to generate our IRGP, the calculation of risk score was decided by the gene expression of the same patient. The obtained prognostic signature is capable of overcoming the batch effect of various platforms on the one hand and shows no need for scaling and normalization of data on the other hand.
In our study, LASSO penalized Cox regression was adopted for constructing a 13-IRGP prognostic signature of SKCM. We also validated this signature in testing and validation datasets. The training set, the TCGA dataset, and the independent dataset held an average AUC of 0.79, 0.756, and 0.818, respectively. The significant survival differences between the Risk-H and Risk-L groups were determined by K-M survival plots. Similar results were also validated in the TCGA, independent, and GSE65904 datasets. The above results proved the accuracy and robustness of our prognostic signature in SKCM patients.
Our 13-IRGP signature consists of a total of 25 IRGs in SKCM. Here, IFIT5_vs_CASP6 and NFKBIE_vs_CXCL10 were two IRGPs with the largest coefficients. IFIT5, as one of the human IFIT gene families, can affect different biological activities of cancers, such as antiviral immune response, host innate immunity, replication, PAMP recognition, as well as double-stranded RNA signaling.36 In bladder cancer, it was shown to be positively correlated with pathological characteristics and predicts poor prognosis in patients.37 Additionally, this gene is capable of inducing the epithelial–mesenchymal transition (EMT) as well as promoting the migration and invasion of cells.37 CXCL10, as a “key driver chemokine” as well as an effective target for treating autoimmune diseases,38 has been a target for novel cancer therapy in immune activation functions.39 In melanoma, CXCL10 was reported as a key candidate gene by integrated bioinformatics analysis.40 The above research results indicated that the IRGs in our signature may be involved in the occurrence and development of SKCM.
However, the study exhibits some limitations. First, adopting retrospective analysis, the study requires a prospective cohort for validating study results and this signature. Second, the molecular functions of the genes in the IRGP signature are still unknown. Finally, RNA-seq and microarray data were used to construct the signature in the study; thus, it is necessary to verify the model with IHC or Western blotting in a large sample size of clinical tissues.
To sum up, a 13-IRGP prognostic signature is developed here, which will serve as a suitable predictive method for identifying SKCM patients who may be better treated with immunotherapy.
Data Sharing Statement
All data generated or analyzed during this study are included in this published article and its supplementary tables and figures.
Acknowledgments
We gratefully thank American Journal Experts (https://www.aje.cn/) for editing the present paper (7876-83B2-73C2-1F71-2579).
Author Contributions
All authors made a significant contribution to the work reported, whether that is in the conception, study design, execution, acquisition of data, analysis and interpretation, or in all these areas; took part in drafting, revising, or critically reviewing the article; gave final approval of the version to be published; have agreed on the journal to which the article has been submitted; and agree to be accountable for all aspects of the work.
Funding
This study was supported by the Project funded by China Postdoctoral Science Foundation (No.2019M653501), the National Natural Science Foundation of China (No. 81960496, 81760495), the Funding Project of Oriented Postdoctoral Training in Yunnan Province, the 100 Young and Middle-aged Academic and Technical Backbone Incubation Projects of Kunming Medical University, Reserve candidates for Kunming’s young and middle-aged academic and technical leaders (17th), Special funding for the cultivation of high-level health technical personnel in Yunnan Province (H-2018006), Scientific Research Fund of Yunnan Provincial Department of Education(2019J1286).
Disclosure
The authors declare no conflicts of interest.
References
1. Lai V, Cranwell W. Epidemiology of skin cancer in the mature patient. Clin Dermatol. 2018;36(2):167–176. doi:10.1016/j.clindermatol.2017.10.008
2. Bray F, Ferlay J, Soerjomataram I, Siegel RL, Torre LA. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin. 2018;68(6):394–424.
3. Rastrelli M, Tropea S, Rossi CR. Melanoma: epidemiology, risk factors, pathogenesis, diagnosis and classification. Vivo. 2014;28(6):1005–1011.
4. Leonardi GC, Candido S, Falzone L, Spandidos DA. Cutaneous melanoma and the immunotherapy revolution (Review). Int J Oncol. 2020;57(3):609–618. doi:10.3892/ijo.2020.5088
5. Bsirini C. Histologic mimics of malignant melanoma. Singapore Med J. 2018;59(11):602–607. doi:10.11622/smedj.2018041
6. Koo SL, Wang WW. Cancer immunotherapy - the target is precisely on the cancer and also not. Ann Acad Med Singapore. 2018;47(9):381–387.
7. Bergman PJ. Cancer Immunotherapies. Vet Clin North Am Small Anim Pract. 2019;49(5):881–902. doi:10.1016/j.cvsm.2019.04.010
8. Lepletier A, Madore J, O’Donnell JS, et al. Tumor CD155 expression is associated with resistance to anti-pd1 immunotherapy in metastatic melanoma. Clin Cancer Res. 2020;26(14):3671–3681.
9. Simeone E, Grimaldi AM, Festino L, et al. Immunotherapy in metastatic melanoma: a novel scenario of new toxicities and their management. Melanoma Manag. 2019;6(4):MMT30. doi:10.2217/mmt-2019-0005
10. Pavri SN, Clune J, Ariyan S. Malignant melanoma: beyond the basics. Plast Reconstr Surg. 2016;138(2):330e–340e. doi:10.1097/PRS.0000000000002367
11. Luke JJ, Flaherty KT, Ribas A. Targeted agents and immunotherapies: optimizing outcomes in melanoma. Nat Rev Clin Oncol. 2017;14(8):463–482.
12. Albittar AA, Alhalabi O. Immunotherapy for melanoma. Adv Exp Med Biol. 2020;1244:
13. Santana-Magal N, Farhat-Younis L, Gutwillig A, et al. Melanoma-secreted lysosomes trigger monocyte-derived dendritic cell apoptosis and limit cancer immunotherapy. Cancer Res. 2020;80(10):1942–1956. doi:10.1158/0008-5472.CAN-19-2944
14. Li YZ, Huang Y, Deng XY. Identification of an immune-related signature for the prognosis of uveal melanoma. Int J Ophthalmol. 2020;13(3):458–465. doi:10.18240/ijo.2020.03.14
15. Yang H, Zhao K, Kang H, Wang M. Exploring immune-related genes with prognostic value in microenvironment of breast cancer from TCGA database. Medicine. 2020;99(14):e19561. doi:10.1097/MD.0000000000019561
16. Huang R, Mao M, Lu Y, Yu Q. A novel immune-related genes prognosis biomarker for melanoma: associated with tumor microenvironment. Aging. 2020;12.
17. Xue D, Cheng P, Jiang J, Ren Y, Wu D. Systemic analysis of the prognosis-related RNA alternative splicing signals in melanoma. Med Sci Monit. 2020;26:e921133.
18. Lu T, Chen S, Qu L, Wang Y, Chen HD. Identification of a five-miRNA signature predicting survival in cutaneous melanoma cancer patients. PeerJ. 2019;7:e7831. doi:10.7717/peerj.7831
19. Sun XY, Yu SZ, Zhang HP, Li J, Guo WZ. A signature of 33 immune-related gene pairs predicts clinical outcome in hepatocellular carcinoma. Cancer Med. 2020;9(8):2868–2878. doi:10.1002/cam4.2921
20. Wu J, Zhao Y, Zhang J, Wu Q. Development and validation of an immune-related gene pairs signature in colorectal cancer. Oncoimmunology. 2019;8(7):1596715. doi:10.1080/2162402X.2019.1596715
21. Breuer K, Foroushani AK, Laird MR, et al. InnateDB: systems biology of innate immunity and beyond–recent updates and continuing curation. Nucleic Acids Res. 2013;41(Database issue):D1228–1233. doi:10.1093/nar/gks1147
22. Weinstein JN, Collisson EA, Mills GB, Cancer Genome Atlas Research N. The cancer genome atlas pan-cancer analysis project. Nat Genet. 2013;45(10):1113–1120. doi:10.1038/ng.2764
23. Jayawardana K, Schramm SJ, Haydu L, et al. Determination of prognosis in metastatic melanoma through integration of clinico-pathologic, mutation, mRNA, microRNA, and protein information. Int J Cancer. 2015;136(4):863–874. doi:10.1002/ijc.29047
24. Jonsson G, Busch C, Knappskog S, et al. Gene expression profiling-based identification of molecular subtypes in stage IV melanomas with different clinical outcome. Clin Cancer Res. 2010;16(13):3356–3367. doi:10.1158/1078-0432.CCR-09-2509
25. Cirenajwis H, Ekedahl H, Lauss M, et al. Molecular stratification of metastatic melanoma using gene expression profiling: prediction of survival outcome and benefit from molecular targeted therapy. Oncotarget. 2015;6(14):12297–12309. doi:10.18632/oncotarget.3655
26. Edgar R, Domrachev M. Gene expression omnibus: NCBI gene expression and hybridization array data repository. Nucleic Acids Res. 2002;30(1):207–210. doi:10.1093/nar/30.1.207
27. Friedman J, Hastie T. Regularization paths for generalized linear models via coordinate descent. J Stat Softw. 2010;33(1):1–22. doi:10.18637/jss.v033.i01
28. Yu G, Wang LG, Han Y. clusterProfiler: an R package for comparing biological themes among gene clusters. OMICS. 2012;16(5):284–287. doi:10.1089/omi.2011.0118
29. Feeney OM, Gracia BDHS, Trevaskis NL, Cao E, Kaminskas LM. Lymph-directed immunotherapy - Harnessing endogenous lymphatic distribution pathways for enhanced therapeutic outcomes in cancer. Adv Drug Deliv Rev. 2020;160:115–135. doi:10.1016/j.addr.2020.10.002
30. Manspeaker MP, Thomas SN. Lymphatic immunomodulation using engineered drug delivery systems for cancer immunotherapy. Adv Drug Deliv Rev. 2020;160:19–35. doi:10.1016/j.addr.2020.10.004
31. Li L, Wang XL, Lei Q, Sun CZ, Xi Y, Chen R. Comprehensive immunogenomic landscape analysis of prognosis-related genes in head and neck cancer. Sci Rep. 2020;10(1):6395. doi:10.1038/s41598-020-63148-8
32. Lin K, Huang J, Luo H, et al. Development of a prognostic index and screening of potential biomarkers based on immunogenomic landscape analysis of colorectal cancer. Aging. 2020;12(7):5832–5857. doi:10.18632/aging.102979
33. Chen W, Ou M, Tang D, Dai Y. Identification and validation of immune-related gene prognostic signature for hepatocellular carcinoma. J Immunol Res. 2020;2020:5494858. doi:10.1155/2020/5494858
34. Qu Y, Cheng B, Shao N, Jia Y, Song Q, Tan B. Prognostic value of immune-related genes in the tumor microenvironment of lung adenocarcinoma and lung squamous cell carcinoma. Aging. 2020;12(6):4757–4777. doi:10.18632/aging.102871
35. Deng X, Lin D, Zhang X, et al. Profiles of immune-related genes and immune cell infiltration in the tumor microenvironment of diffuse lower-grade gliomas. J Cell Physiol. 2020;235:7321–7331. doi:10.1002/jcp.29633
36. Pidugu VK, Pidugu HB, Wu MM, Liu CJ. Emerging functions of human ifit proteins in cancer. Front Mol Biosci. 2019;6:148.
37. Huang J, Lo UG, Wu S, et al. The roles and mechanism of IFIT5 in bladder cancer epithelial-mesenchymal transition and progression. Cell Death Dis. 2019;10(6):437. doi:10.1038/s41419-019-1669-z
38. Karin N. Chemokines beyond chemo-attraction: CXCL10 and its significant role in cancer and autoimmunity. Cytokine. 2018;109:
39. Tokunaga R, Zhang W, Naseem M, et al. CXCL9, CXCL10, CXCL11/CXCR3 axis for immune activation – a target for novel cancer therapy. Cancer Treat Rev. 2018;63:
40. Huang L, Chen J, Zhao Y, et al. Key candidate genes of STAT1 and CXCL10 in melanoma identified by integrated bioinformatical analysis. IUBMB Life. 2019;71(10):1634–1644. doi:10.1002/iub.2103
 © 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.