Back to Journals » OncoTargets and Therapy » Volume 13
Development and Validation of a Novel 8 Immune Gene Prognostic Signature Based on the Immune Expression Profile for Hepatocellular Carcinoma
Authors Xu D, Wang Y, Zhou K, Wu J, Zhang Z, Zhang J, Yu Z, Liu L, Liu X, Li B, Zheng J
Received 16 May 2020
Accepted for publication 29 July 2020
Published 14 August 2020 Volume 2020:13 Pages 8125—8140
DOI https://doi.org/10.2147/OTT.S263047
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Sanjeev K. Srivastava
Dafeng Xu,1 Yu Wang,2 Kailun Zhou,1 Jincai Wu,1 Zhensheng Zhang,1 Jiachao Zhang,1 Zhiwei Yu,1 Luzheng Liu,1 Xiangmei Liu,1 Bidan Li,1 Jinfang Zheng1
1Department of Hepatobiliary and Pancreatic Surgery, Hainan General Hospital, Hainan Affiliated Hospital of Hainan Medical University, Haikou, Hainan 570311, People’s Republic of China; 2Geriatrics Center, Hainan General Hospital, Hainan Affiliated Hospital of Hainan Medical University, Haikou, Hainan 570311, People’s Republic of China
Correspondence: Jinfang Zheng
Department of Hepatobiliary and Pancreatic Surgery, Hainan General Hospital, Hainan Affiliated Hospital of Hainan Medical University, Xiuhua Road 19, Xiuying District, Haikou City, Hainan Province 570311, People’s Republic of China
Email [email protected]
Background: The immune microenvironment plays a vital role in the development of hepatocellular carcinoma (HCC). This study explored novel immune-related biomarkers to predict the prognosis of patients with HCC.
Methods: RNA-Seq data were downloaded from The Cancer Genome Atlas (TCGA). Univariate Cox regression was used to identify prognosis-related genes; the Lasso method was used to construct the prognosis risk model. Validation was performed on the International Cancer Genome Consortium (ICGC) cohort, and the C-index was calculated to evaluate its overall predictive performance. Western blots were conducted to evaluate the expression of genes.
Results: There were 320 immune-related genes, 40 of which were significantly related to prognosis. Eight immune gene signatures (CKLF, IL12A, CCL20, PRELID1, GLMN, ACVR2A, CD7, and FYN) were established by Lasso Cox regression analysis. This immune signature performed well in different cohorts and can be an independent risk factor for prognosis. In addition, the overall predictive performance of this model was higher than the other models reported previously.
Conclusion: The predictive immune model will enable patients with HCC to be more accurately managed in immunotherapy.
Keywords: HCC, immune gene, prognostic markers, TCGA, ICGC
Introduction
Hepatocellular carcinoma (HCC) is the second leading cause of cancer-related mortality and the fifth most common malignancy worldwide.1 In 2012, it was estimated that approximately 700,000 people globally die of hepatocellular carcinoma each year.2 The incidence of HCC varies by geographic region, gender, age, and risk factors associated with cancer development.3 Despite advances in treatments such as liver transplantation, surgical resection, and radiofrequency ablation, the prognosis in patients with HCC is still very poor, with a 5-year survival rate of less than 30%.4 In addition, tumor heterogeneity and immune microenvironment play key roles in the progression, treatment, and prognosis of HCC. Therefore, an in-depth exploration of immune phenotypic changes and their impact on prognosis is necessary to improve patient survival.
HCC is mainly caused by chronic inflammation of the liver and is considered a typical immunogenic cancer.5 The immune response plays an important role in the development of HCC.6 There are many antigen-presenting cells in the liver. When presenting antigens to T cells in the liver, they usually express immunosuppressive surface molecules or secrete immunosuppressive cytokines, thereby mediating liver immune tolerance.7 These antigen-presenting cells play an important role in HCC and are involved in the immune escape process of HCC cells.8 In addition, in the microenvironment of HCC, infiltrating immune cells are impaired by the expression of inhibitory signal molecules such as PD-1 on the surface, leading to tumor immunosuppression. These inhibitory signal molecules are immune checkpoints.9 In recent years, immune checkpoint inhibitors have become an effective means of treating tumors.10,11 Therefore, it is of vital importance to further explore the clinical relevance of HCC immunophenotypes and their relationship with prognosis.
Several previous studies have explored the role of tumor microenvironment in HCC. One investigation detected the expression of 49 immune genes in 61 HCC tissues by immunohistochemistry and PCR, indicating that these genes were correlated with the prognosis of HCC.12 This research clarified the potential effect of the immune microenvironment on prognosis and initially confirmed that the expression level of immune system genes had great prognostic value in HCC. In addition, tumor-infiltrating lymphocytes (TILs), as an important part of the immune microenvironment, have been widely confirmed to be correlated with the prognosis of HCC.13,14 For example, the low expression of IL-36αwas associated with to the poor prognosis of HCC, the specific mechanism of which was related to T cells.15 Several studies have also confirmed the role of immune checkpoint-related genes in predicting prognosis. For example, studies have demonstrated that overexpression of PD-L1 and PD-L2 was associated with poor prognosis.16,17 However, as this research has thus far been limited, there is still no systematic understanding of the immunophenotype in the HCC microenvironment and its relationship to prognosis.
In this study, we integrated multiple gene expression cohorts to develop an HCC prognostic signature based on immune-related genes. The stability and reliability of the model are proven through the external independent cohort and laboratory experiments.
Materials and Methods
Inclusion Criteria
RNA-seq expression profiles were retrospectively collected from two public cohorts: The Cancer Genome Atlas hepatocellular carcinoma cohort (TCGA-LIHC), and the International Cancer Genome Consortium cohort (ICGC-LIRI-JP). All patients had undergone primary tumor removal. Only those patients with available follow-up time and gene expression profiles were included. The primary outcome was overall survival, and staging was evaluated according to the AJCC system.
For the TCGA-LIHC cohort, the GDC (https://portal.gdc.cancer.gov/) API was used to download the RNA-seq expression profile and clinical follow-up information. The cohort contained 423 samples, which were further converted into gene symbols by the hgu133plus2.db. R package. We downloaded the ICGC-LIRI-JP cohort as the external validation cohort, which has 212 samples.
Processing of Gene Expression Profiles
All data were mapped to Entrez id. If multiple probes corresponded to the same Entrez ID, the probe with the highest average signal was selected as the expression level of its corresponding gene. The details of sample quality control are described in Table 1. Finally, 583 cases of HCC samples including TCGA-LIHC and ICGC-LIRI-JP cohorts were included in this study.
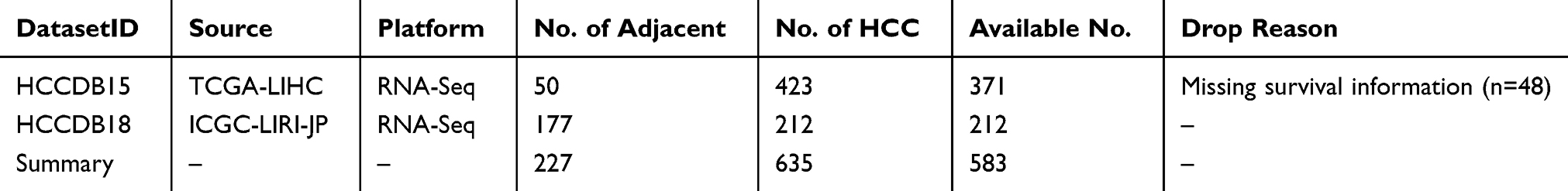 |
Table 1 Details of Sample Quality Control |
Identification of Immune-Related Genes
The immune-related genes were downloaded from the Molecular Signatures Database v4.0 (immune system process, M13664; immune response, M19817; immune effector process, M14818; and immune system development, M3457). Finally, 321 immune-related genes were included in the subsequent analysis.
Establishment of Immune Prognostic Signature of HCC (IPSHCC)
First, the TCGA training cohort was used to identify genes related to survival. There were a total of 321 genes in the immune-related gene set, and a total of 320 genes in this gene set were detected in the TCGA-LIHC training cohort. A Cox proportional hazards model was used to exclude genes with p-values > 0.05.
Because there might be interactions between genes, the Lasso method was used for further screening of genes. Lasso was more suitable for high-dimensional, strong correlation, and small sample survival data. The basic goal of using Lasso was to minimize the sum of squared residuals under the constraint that the sum of the absolute values of the regression coefficients was less than a constant, so that some regression coefficients were strictly equal to 0, thereby obtaining an interpretable model.
minβ∑i=1n(yi−∑j=1pβjxij)2, subject to∑j=1p||βj||≤λminβ∑i=1n(yi−∑j=1pβjxij)2, subject to ∑j=1p|βj|≤λ
As l increased, the terms ∑j = 1p||βi||∑j = 1p | βi | would decrease, and the coefficients of some independent variables would be gradually compressed to 0, to achieve dimensionality reduction of high-dimensional data. The dimension reduction of the Lasso method was achieved by penalizing the number of regression coefficients.
Cross-validation, such as k-fold cross-validation, was a standard method to estimate the adjustment parameter l. In the k-fold cross-validation, all data observations were roughly divided into k equal parts, and the data were fitted in turn with all possible k-1 parts as the training cohort, and the remaining one was used as the testing cohort. The ratio of the number of observations of the testing cohort to the training cohort was approximately 1: (k-1). A total of k calculations were performed to obtain k indicators (such as the error rate or other indicators) when fitting the testing cohort and then averaged. This performance was repeated for each model; the one with the smallest error rate was finally selected.
The R package glmnet was used to perform Lasso regression using 10-fold cross-validation. The coefficients of each category were further determined by multivariate Cox regression using the reduced-dimensional genes:
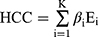
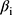 was the regression coefficient of the i-th gene, and
was the regression coefficient of the i-th gene, and 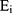 was the expression level of the i-th gene.
was the expression level of the i-th gene.
Validation of IPSHCC
We used the median risk score to divide patients into high-risk and low-risk groups. After the IPSHCC prognostic risk score was determined in the training cohort, it was calculated in the validation cohort.
Pathway Enrichment Analysis
To further understand the gene function in IPSHCC, single-sample GSEA analysis was performed using the GSVA. R package, based on the KEGG database, and the enrichment scores of each sample in each pathway were calculated. Pathways significantly related to IPSHCC were further screened based on the correlation between the enrichment score and IPSHCC.
Comparison with Previous Prognostic Signatures
To assess the survival classification and predictive performance of IPSHCC, five published prognostic signatures were retrospectively collected for comparison, ranging from 3 to 9 genes (Table 2).18–22 A continuous prognostic risk score was calculated for each signature. The p-value and the overall concordance index (C-index) of the univariate Cox model were compared among the other five signatures.
 |
Table 2 Review of 5 Liver Cancer Prognosis Studies |
Validation of Expression of the Immune Genes
The Human Protein Atlas (HPA) provides information on the tissue and cell distribution of 26,000 human proteins. It primarily uses specific antibodies to study protein expression in cell lines, normal tissues, and tumor tissues. We explored the expression of 8 genes (ACVR2A, CCL20, CD7, CKLF, FYN, GLMN, IL12A, PRELID1) in normal and tumor tissues. We explored the expression of 8 genes in HCC and normal tissues in GSE6764 and GSE14520, and draw boxplots on the expression of genes.
Genetic Alterations of the 8 Immune Genes
cBioportal integrates genomic data including somatic mutations, DNA copy number, alternations (CNAs), mRNA and microRNA (miRNA) expression, DNA methylation, protein enrichment, and phosphorylated protein enrichment. The mutation correlation analysis of the 8 predictive genes (OncoPrint and histogram display of gene mutation) was conducted on hepatocellular carcinomas (INSERM, Nat Genet 2015), liver hepatocellular carcinoma (AMC, Hepatology 2014), and liver hepatocellular carcinoma (TCGA, Firehose Legacy) data sets of cBioportal database.
Sample Collection
HCC and adjacent tissues were collected from 3 patients, immediately placed in liquid nitrogen, and preserved at −80°C. Patients and their families were fully informed, and informed consent was obtained from the participants. The Ethics Committee of Department of hepatobiliary and pancreatic surgery, Hainan General Hospital, approved this study.
Western Blotting
Western blotting was carried out according to standard protocols. We used primary antibodies raised against GAPDH (Santa-Cruz Biotechnology, CA, USA), ACVR2A, CCL20, CD7, CKLF, FYN (Cell Signaling Technology, MA, USA), and GLMN, IL12A, PRELID1 (Proteintech, China). Goat anti-mouse and anti-rabbit antibodies conjugated with horseradish peroxidase were used as secondary antibodies (Jackson Immuno Research, PA, USA), and we detected the blots using enhanced chemiluminescence (ECL) (Dura, Pierce, NJ, USA)
RNA Extraction and Real-Time Polymerase Chain Reaction (PCR) Assay
Total RNA was extracted using TRIzol Reagent (Invitrogen, CA, USA) following the manufacturer’s protocol and was reverse-transcribed into complementary DNA (cDNA) using a Superscript Reverse Transcriptase Kit (Transgene, France). Super SYBR Green Kit (Transgen, France) was used to carry out real-time PCR in the ABI7300 real-time PCR system (Applied Biosystems). The primers pairs were:
ACVR2A Forward GTTTGCCGTCTTTCTTATCTCCT,
ACVR2A Reverse GTCACCATAACACGGTTCAACA;
CCL20 Forward ACTGTTGCCTCTCGTACATACA,
CCL20 Reverse GAGGAGGTTCACAGCCCTTTT;
CD7 Forward GCCTGGGAGCTTACGATTTTG,
CD7 Reverse TAGTGCCCTGGTACTGGTCG;
CKLF Forward AACCTGAGTCATCCGAGGCA,
CKLF Reverse CAGTCTCCGGTTGTTTCAAGT;
FYN Forward CTCTGGGAGTACCTAGAACCC,
FYN Reverse AGCCTGGTAATCAAACAAAGCC;
GLMN Forward TCCTGTTGTTCGATGCCTTTT,
GLMN Reverse TCAATCAGTTCAAGCAAACCCAA;
IL12A Forward CCTTGCACTTCTGAAGAGATTGA,
IL12A Reverse ACAGGGCCATCATAAAAGAGGT;
PRELID1 Forward CTTGACGGAAGACATAGTACACC,
PRELID1 Reverse ACATTGGCAGGAAATAGTCGC.
Statistical Analysis
The Cox proportional hazard model was used to assess the relationship between clinical features and overall survival. Kaplan-Meier (KM) survival curves were plotted, and Log rank tests were used to detect differences among subgroups. The survival and survRM2. R packages were used to estimate the C-index.
Results
Immune Pathways Were Significantly Suppressed in HCC
We performed a single-sample Gene Set Enrichment Analysis (ssGSEA) analysis in TCGA-LIHC cohort and calculated the enrichment scores of each sample in the four immune pathways (Figure 1A). We observed that the scores in tumor samples were significantly lower (Figure 1B), indicating that immunity was significantly suppressed.
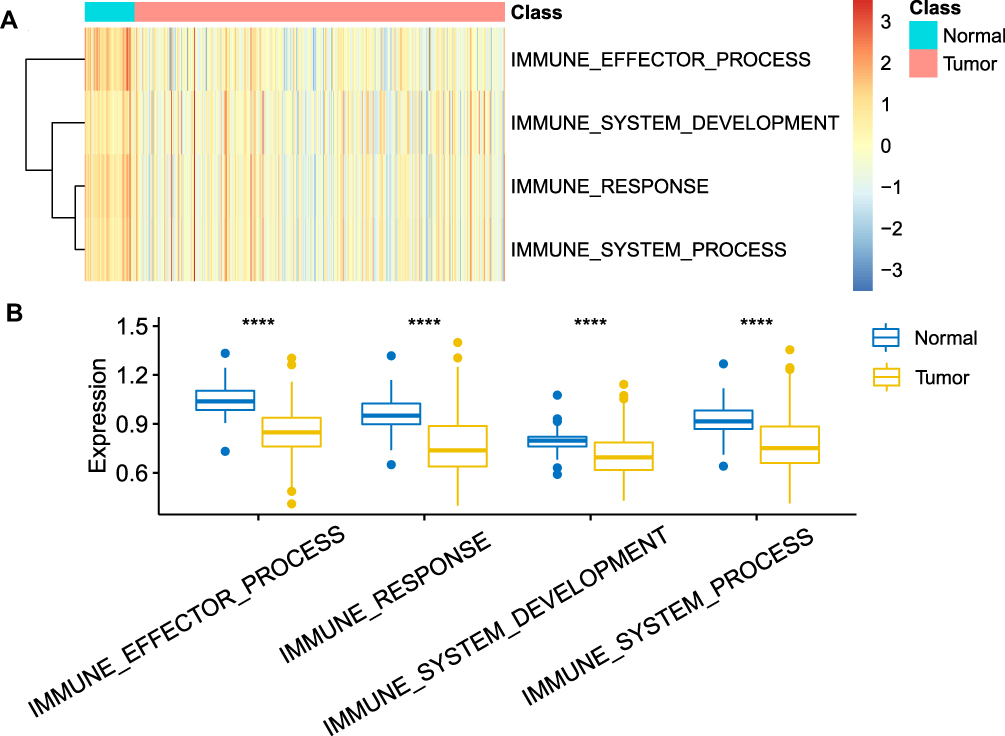 |
Figure 1 (A) Heat map of enrichment scores of four pathways in normal HCC samples and tumor samples; (B) Difference in enrichment scores of four pathways in normal hepatocellular carcinoma samples and tumor samples, using the Student’s t-test, ****indicates p < 0.0001. |
Development and Definition of 8- IPSHCC Signature
The TCGA cohort was randomized into the training cohort (N = 186) and the internal testing cohort (N = 185). In the training cohort, a univariable analysis was performed on each immune gene to identify a total of 40 prognostic genes. Lasso regression was used for dimensionality reduction analysis, and 10-fold cross-validation was selected. The minimum error rate when λ = 0.07388702 is shown in Figure 2A and B. The 8-IPSHCC signature was established using multivariable Cox regression analysis (Table 3).

 |
Table 3 Information of 8 Characteristic Genes |
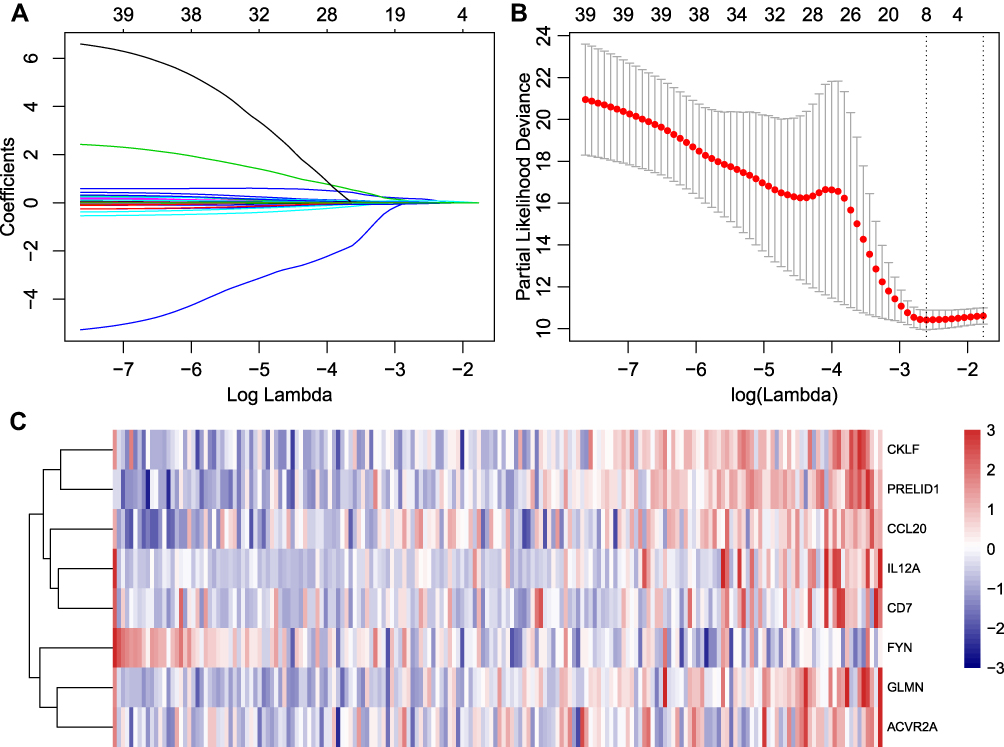 |
Figure 2 Lasso regression analysis results: (A) the trajectory of each independent variable, the horizontal axis represents the log value of the independent variable lambda, and the vertical axis represents the coefficient of the independent variable; (B) average error rate interval for each lambda; (C) the relationship between 8 individual immune genes and risk scores. |
The risk score of each sample was calculated, and samples were grouped according to the median risk score. The low expression of FYN was associated with high risk and was thus considered a protective factor. High expression of the other seven genes was related to high risk, and thus were risk factors (Figure 2C).
Predictive Performance of the Signature in Different Cohorts
The model was used to predict risk scores of the training cohort, testing cohort, and external validation cohort, and samples were classified according to their median risk score. The average area under the curve
(AUC) of the 8-IPSHCC signature in the training cohort was 0.76 (Figure 3A). The prognoses of the high-risk and the low-risk groups were significantly different (Hazard ratio, HR = 2.72, 95% CI: 2.07–3.57, Figure 3B). The average AUC in the testing cohort was 0.65 (Figure 3C). The prognosis of high-risk samples was significantly worse than low-risk samples (HR = 1.71, 95% CI: 1.26–2.32, Figure 3D), and the average AUC in the external validation cohort was 0.73 (Figure 3E). The overall survival of high-risk samples was significantly worse than the low-risk samples with p = 0.00016, which was consistent with the training cohort (HR = 73.75, 95% CI: 11.59–469.4, Figure 3F). In short, 8-IPSHCC signature provided effective prognostic classification in multiple cohorts.
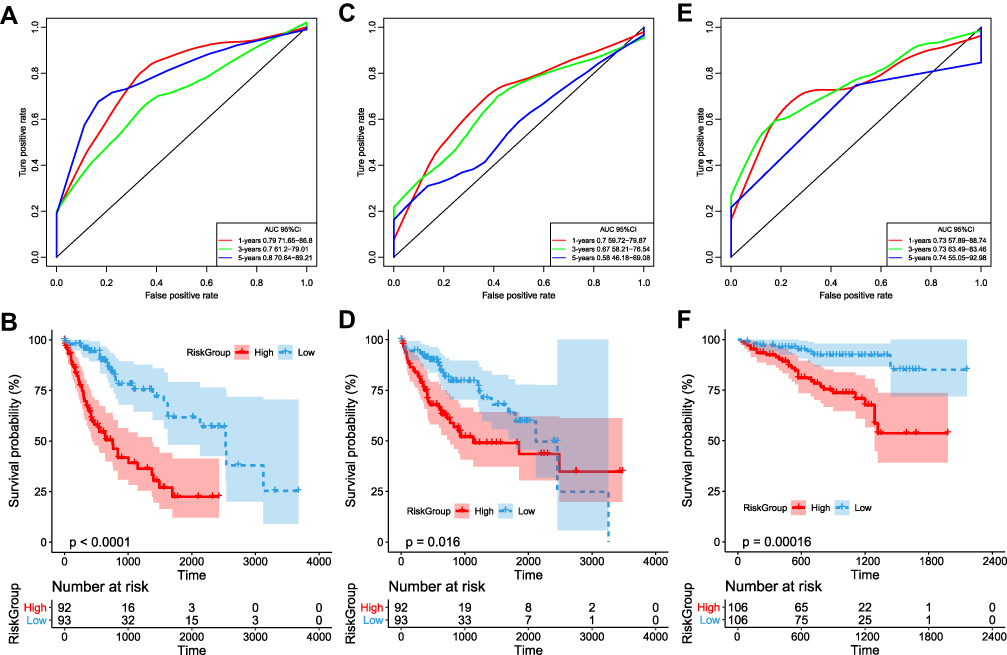 |
Figure 3 (A) Receiver operating characteristic (ROC) curve of TCGA training cohort; (B) Kaplan-Meier curve of prognosis difference between high and low-risk groups in TCGA training cohort; (C) ROC curve of TCGA testing cohort; (D) Kaplan-Meier curve of prognosis difference between high and low-risk groups in TCGA testing cohort; (E) ROC curve of ICGC external validation cohort; (F) Kaplan-Meier curve of prognostic difference between high- and low-risk groups in ICGC external validation. |
Univariate and Multivariate Analysis of IPSHCC
To identify the independence of the IPSHCC in clinical applications, we used Cox regression to analyze the clinical information, including age, stage, gender, and risk score (Table 4). It showed that the risk score was independent prognostic risk factors in multivariate Cox regression analysis (HR = 2.383, 95% CI = 1.7849–3.1825, p = 3.94E-09). We plotted survival curves for the age and stage (Figure 4A–D).
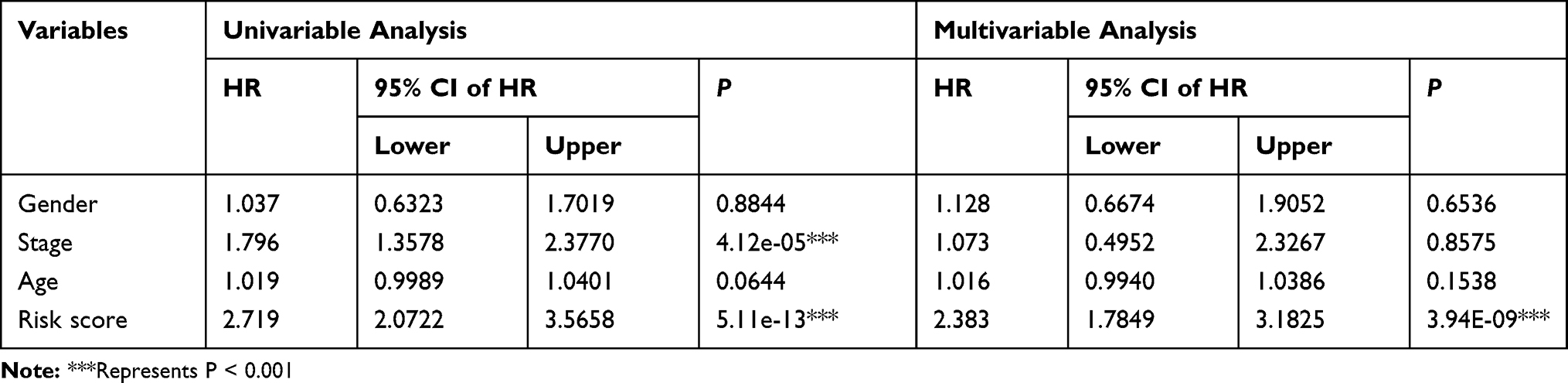 |
Table 4 Univariate and Multivariate Analyses of 8-Gene Signature |
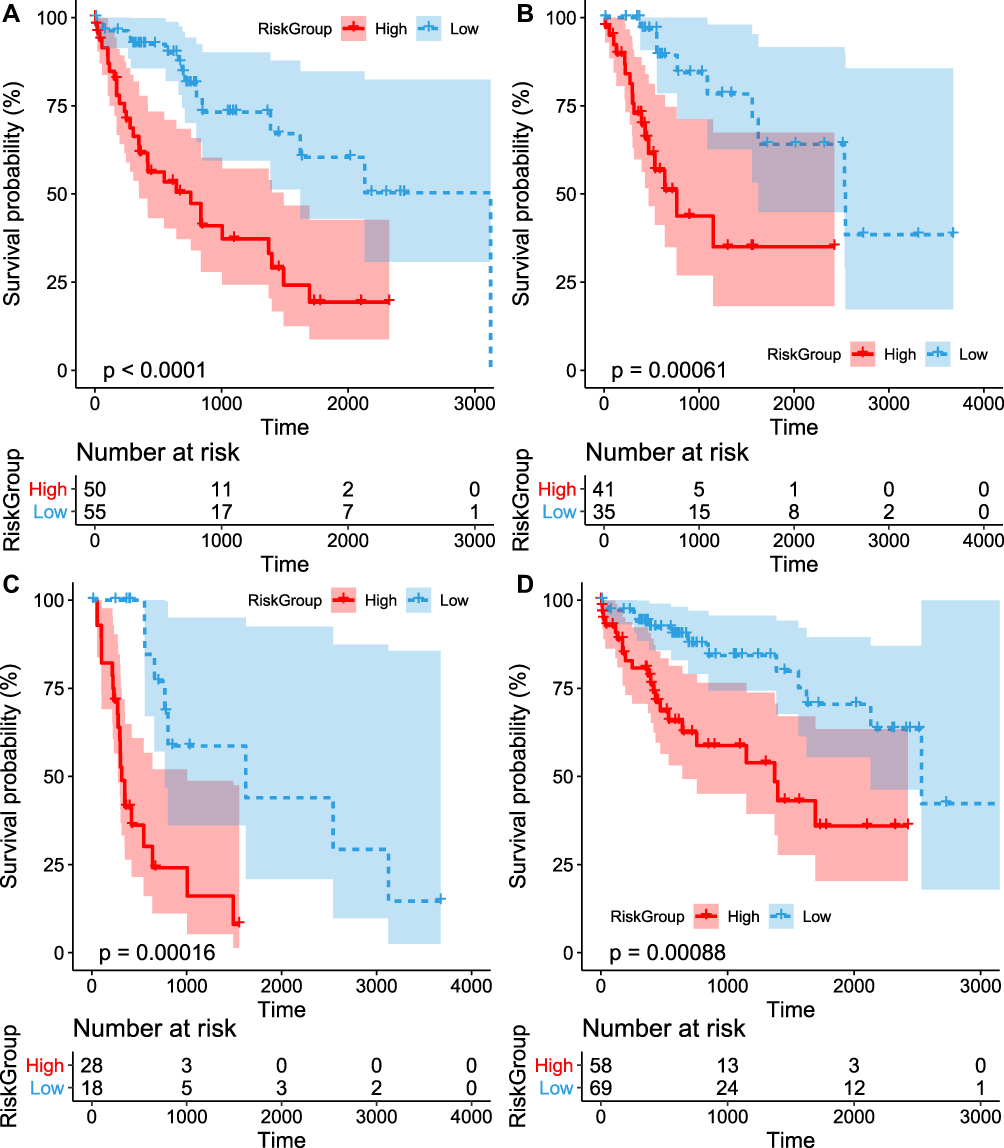 |
Figure 4 Subgroup and sensitivity analysis for HCC. (A) the difference in classification prognosis in the sample applied to samples older than 60; (B) the difference in classification prognosis in the sample applied to samples older than 60; (C) the difference in classification prognosis in the sample applied to Stage I + Stage II; (D) the differences in classification prognosis in the model applied to the samples of Stage III + Stage IV. |
Pathway Enrichment Analysis of IPSHCC
The risk score of each sample was calculated based on the immune gene signature in the training cohort, and Kyoto Encyclopedia of Genes and Genome (KEGG) pathway analysis was performed using ssGSEA. The Pearson correlation between the immune gene signature and sample pathway enrichment score was calculated. A total of 50 pathways had false discovery rate (FDR) <0.01. Among these pathways, there was a total of 12 significant positive correlations and 38 significant negative correlations (Figure 5A). There were pathways related to metabolism, cancer, the cell cycle, DNA repair, and other functions. Most of these pathways were significantly related to tumors: the six most relevant pathways included “FATTY ACID METABOLISM”, “BETA-ALANINE METABOLISM”, “SPLICEOSOME”, “CALCIUM SIGNALING PATHWAY”, “CELL CYCLE”, and the “ADIPOCYTOKINE SIGNALING PATHWAY”. These pathways had significant different enrichment scores between the high and low-risk groups (Figure 5B).
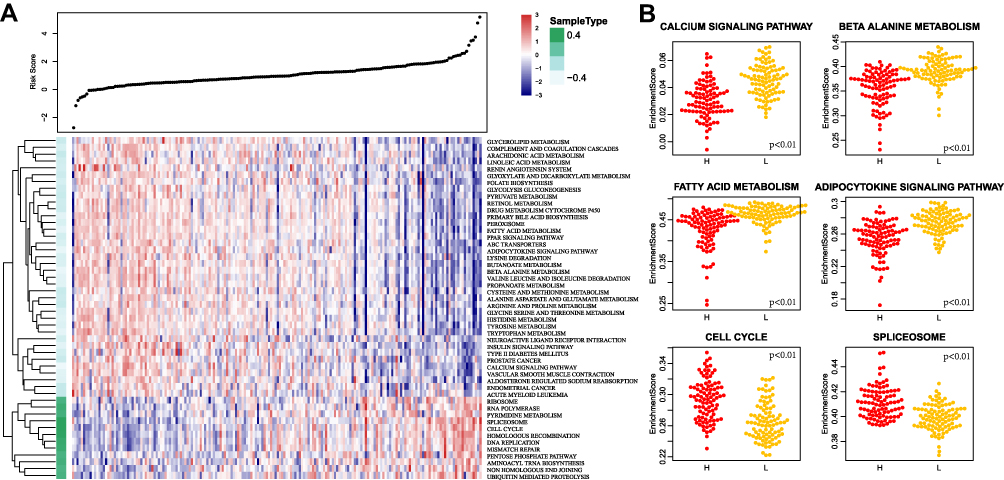 |
Figure 5 (A) Heat map of the enrichment scores of the 50 KEGG pathways that were significantly enriched. The risk scores of the samples increase from left to right; (B) The difference of the enrichment scores of the six most relevant KEGG pathways in the high- and low-risk groups. |
Comparison of Prognostic and Clinical Characteristics with Other Signatures
To assess the survival classification and predictive performance of IPSHCC, the accuracy of the IPSHCC model in predicting HCC prognosis was compared with 5 published prognostic signatures of HCC (Table 2). Continuous prognostic scores were calculated based on each feature, and cohorts were compared. The C-indices of prognostic signatures in the training cohort and the external validation cohort are shown in Figure 6A and B. Among these, IPSHCC had the highest (0.725) average C-index in both cohorts, demonstrating that IPSHCC had a good predictive performance.
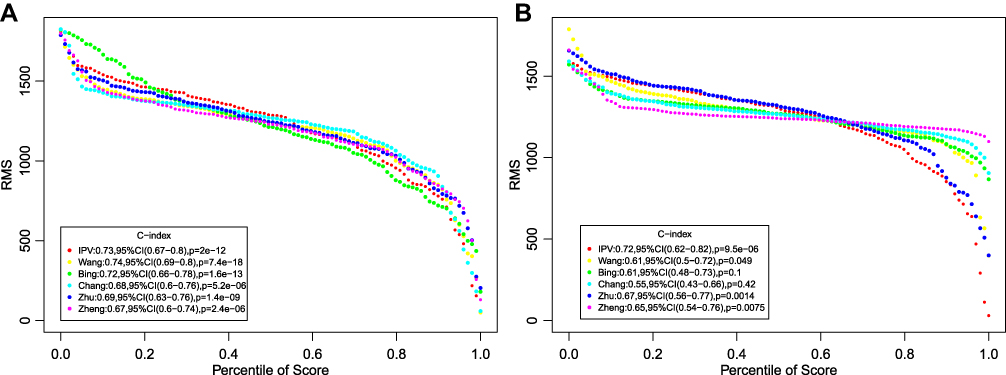 |
Figure 6 Comparison with other prognostic signature. (A) Training cohort. (B) External validation cohort. Restricted mean survival (RMS) curve for 8-IPSHCC signature and five other signatures. |
External Validation of the IPSHCC Expression
The protein expressions of the presumptive 8 predictive genes were analyzed using the HPA database. Among them, CCL20 and CKLF were not collected in HPA database, CD7 was negative in tumor and normal tissue. There were no significant differences in the expression of FYN and IL12A in tumor and normal tissue, and ACVR2A, GLMN, PRELID1 was relatively highly expressed in tumors (Figure 7). The differential expression of the 8 genes in GSE6764 and GSE14520 cohort was analyzed, among which expression of GLMN, CKLF, and CCL20 was high in tumors, whereas the expression of FYN, IL12A in tumors was low (Figure 8).
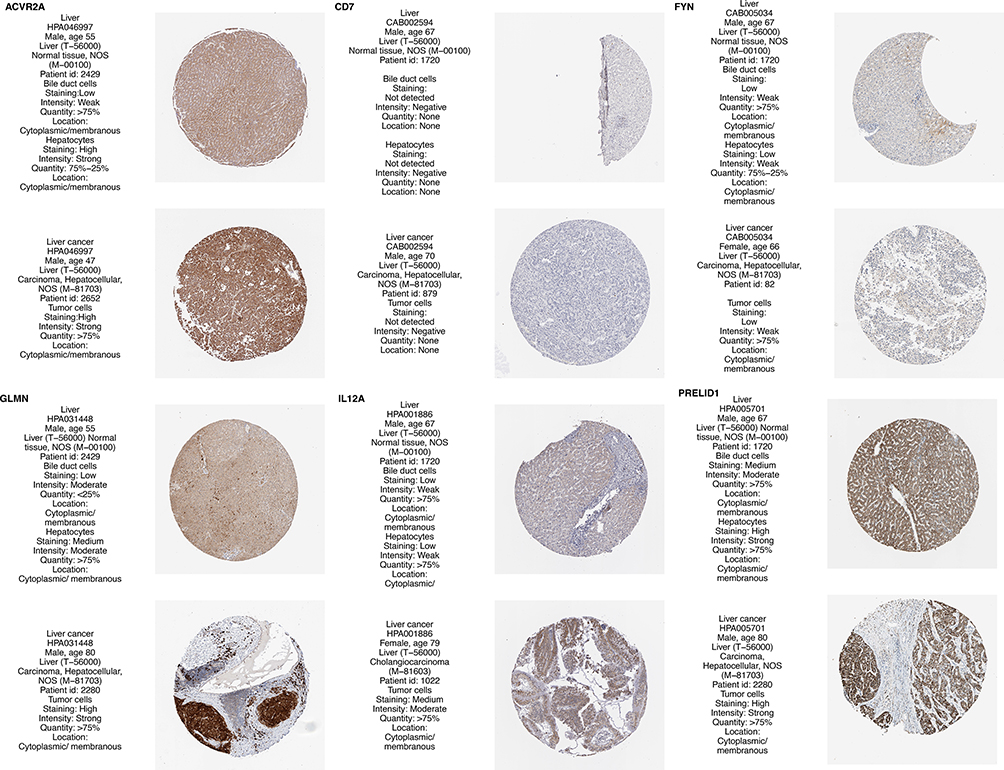 |
Figure 7 Analysis of protein expression of 8 genes in the HPA database. |
 |
Figure 8 Differential expression analysis of 8 genes in the GEO database. (A) Differential expression analysis of 8 genes in GSE6764; (B) Differential expression analysis of 8 genes in GSE14520.* represents P<0.05; ** represents P<0.01; *** represents P <0.001. |
Genetic Alterations of 8 Genes in IPSHCC
The mutations of the 8 genes were explored in the cBioportal database. Among them, the gene with the highest mutation rate was CD7, accounting for 4%, and the primary type of mutation was amplification. ACVR2A mutations accounted for 3%, and the mutation form is mainly deep deletion (Figure 9).
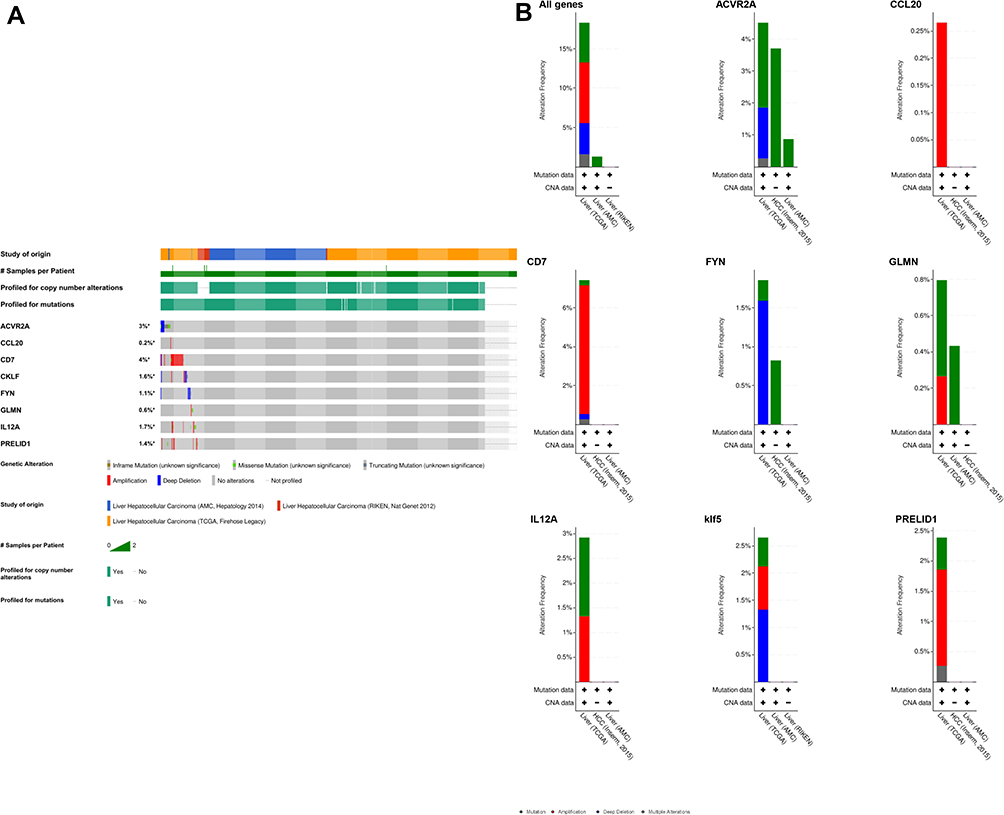 |
Figure 9 Analysis of mutations of 8 genes in the cBioportal liver cancer cohort. (A) Oncoprint display of gene mutation distribution. (B) Histogram display of gene mutation distribution. |
Clinical Validation of Protein and mRNA Levels of 8 Genes
We analyzed 3 pairs of HCC tissues and para-cancerous controls to validate the protein and mRNA levels of 8 genes. The results showed that the expressions of CD7, IL12A, and FYN in cancer and normal tissue was not significant. The expressions of GLMN, CKLF, CCL20, PRELID1, and ACVR2A was relatively highly expressed in tumors. The trend of our experimental results was almost consistent with data analysis (Figure 10).
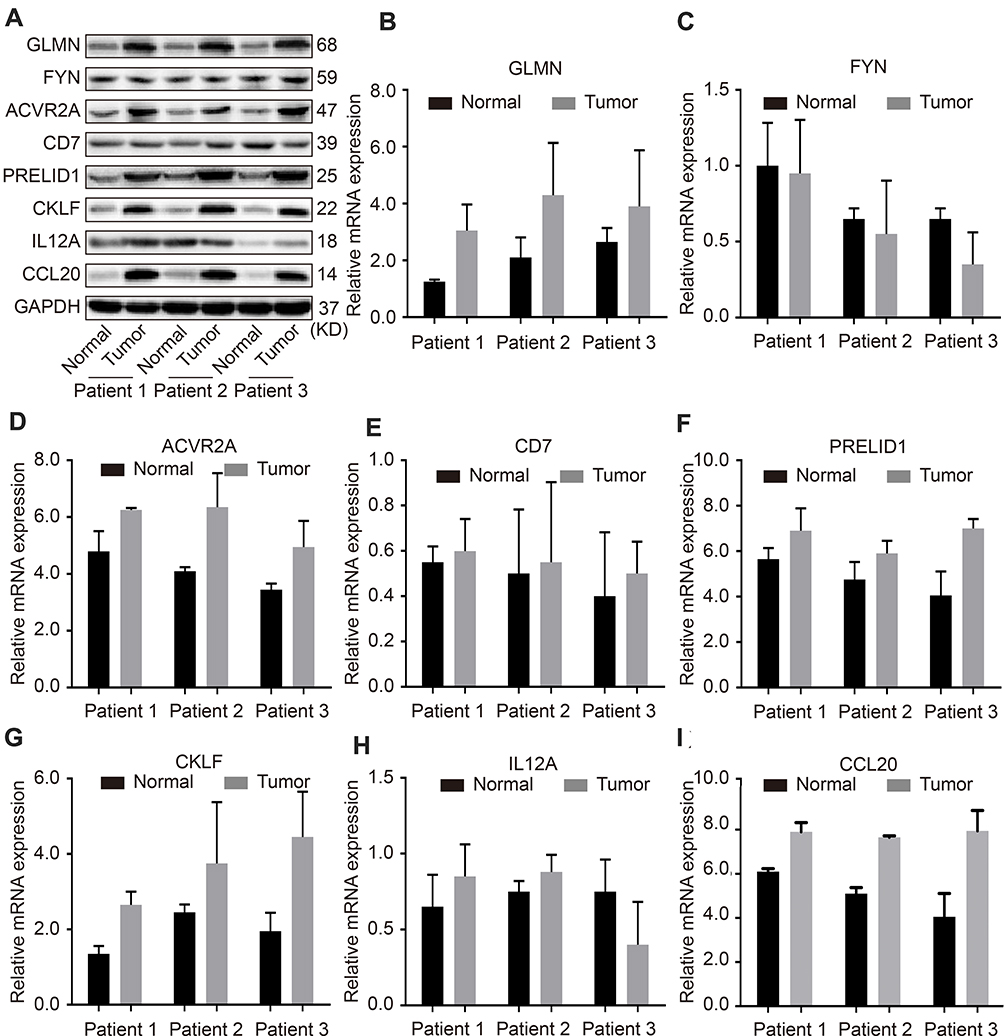 |
Figure 10 Validation of protein and mRNA levels of 8-IPSHCC signature. Notes: (A) The protein expression of 8 genes in 3 pairs of HCC tissues and para-cancerous tissues. (B) The mRNA expression of GLMN in 3 pairs of HCC tissues and para-cancerous tissues. (C)The mRNA expression of FYN in 3 pairs of HCC tissues and para-cancerous tissues. (D)The mRNA expression of ACVR2A in 3 pairs of HCC tissues and para-cancerous tissues. (E)The mRNA expression of CD7 in 3 pairs of HCC tissues and para-cancerous tissues. (F)The mRNA expression of PRELID1 in 3 pairs of HCC tissues and para-cancerous tissues. (G) The mRNA expression of CKLF in 3 pairs of HCC tissues and para-cancerous tissues. (H)The mRNA expression of IL12A in 3 pairs of HCC tissues and para-cancerous tissues. ( I) The mRNA expression of CCL20 in 3 pairs of HCC tissues and para-cancerous tissues. |
Discussion
HCC is a highly aggressive malignancy and is currently the second leading cause of tumor-related death.1 Despite plenty of advances in comprehensive treatment strategies for hepatocellular carcinoma, effective molecular targeted therapies are still lacking.23 The complex immune microenvironment promotes the development of HCC and mediates the poor prognosis of patients.24,25 Therefore, it is critical to integrate immune-related genes and conduct comprehensive research on HCC, to improve prognoses. In this study, we conducted the first ssGSEA analysis of HCC samples. The results showed that immunity was significantly suppressed, suggesting the important role of the immune microenvironment in the progression of HCC.
Furthermore, an 8-IPSHCC signature was constructed by Lasso and multivariable analysis, and it showed stable and consistent predictive performance in both the internal and external validation cohort. More importantly, it was an independent prognostic risk factor in HCC patients.
In IPSHCC signature, CKLF, IL12A, CCL20, PRELID1, GLMN, ACVR2A, and CD7, were risk factors, and FYN was protective. Previous studies have confirmed that CKLF is related to the prognosis of patients with colon cancer, and the specific mechanism was related to leukocyte infiltration.26 In addition, the gene polymorphism of IL12A was correlated with the risk and prognosis of various tumors, including lung cancer,27 HCC,28 cervical cancer,29 and gastric cancer.30 As a chemokine, CCL20 plays an important role in the progression of tumors and is closely related to the prognosis of patients with solid tumors such as colon cancer,31 lung cancer,32 and pancreatic cancer.33 PRELI was originally thought to be expressed on germinal center B lymphocytes.34 Recent studies have confirmed that it is associated with the prognosis of patients with cervical squamous cell carcinoma, pancreatic adenocarcinoma, and acute myeloid leukemia.35 ACVR2A has been shown to correlate with the prognosis of patients with colon cancer.36,37 CD7 is a common antigen in lymphoid hematopoietic tissues, often expressed in T cells and NK cells, and used in the diagnosis of T-cell lymphoma and leukemia.38,39 The primary type of mutation of gene CD7 was amplification, accounting for 4%. FYN is a non-receptor tyrosine kinase that belongs to the Src family of kinases.40 By regulating cell growth, apoptosis, morphogenetic transformation, and motor capacity, it promotes the development and progress of tumors and is closely related to the prognosis of patients with various tumors.41,42 GLMN is not associated with the prognosis of tumor patients, and it was first discovered in this study as a prognostic marker for HCC.
Finally, the results of GSEA enrichment analysis revealed potential regulatory pathways of IPSHCC. The liver is vital in lipid metabolism,43 and the occurrence and development of liver cancer are often accompanied by complex metabolic abnormalities.44,45 The 8-IPSHCC signature may promote the metabolic reprogramming of liver cancer cells by regulating the metabolic pathway, such as the “FATTY ACID METABOLISM,” “BETA-ALANINE METABOLISM,” and “ADIPOCYTOKINE SIGNALING PATHWAY”, thereby exerting a cancer-promoting effect. The “SPLICEOSOME” pathway catalyzes the process of alternative splicing, which is a post-transcriptional modification mechanism to ensure high transcript and protein diversity.46,47 We suggest that the IPSHCC may regulate fundamental processes in HCC cells by alternative splicing via the “SPLICEOSOME” pathway. Ca2+ signaling mediates fundamental cellular processes such as gene transcription, motility, cell proliferation, and apoptosis.48,49 The “CALCIUM SIGNALING PATHWAY” was enriched, suggesting that the IPSHCC may function via regulation of Ca2+ ion transporters and channels. In addition, an infinitely limited growth and cell cycle disorder is the main characteristic of tumors.50 Notably, the occurrence of the vast majority of HCC is related to abnormalities in the regulatory mechanism of the G1 phase of the cell cycle.51 So, the presence of the “CELL CYCLE” pathway in the GSEA results suggests the 8 immune gene signature may be related to the proliferation ability of HCC cells. Therefore, the enrichment analysis pathway in this study requires further research to elucidate the mechanism of HCC occurrence and progression.
Moreover, the results of GSEA enrichment analysis also showed that the IPSHCC signature was significantly related to pathways such as cell cycle, and DNA repair. Many previous studies have confirmed the significant effects of metabolic pathways and cell cycle changes on the biological behavior of HCC cells and the prognosis of clinical patients,52,53 which was consistent with our results. Therefore, the pathway of enrichment analysis in this study needed further research to deepen the understanding of the mechanism of pancreatic cancer occurrence and progression.
Many previous studies have attempted to screen and construct prognostic marker models for HCC, including one that utilized 6 genes based on the TCGA cohort,18 and another the utilized 3 genes (UPB1, SOCS2, and RTN3) from the same cohort.19 A prognostic signature based on the GEO and TCGA cohorts constructed for patients with HCC contained 4 genes (SPINK1, TXNRD1, LCAT, and PZP).20 A genetic marker combining hypoxia and lymphocyte infiltration was used to construct an 8-gene prognostic signature for patients with multiple solid tumors, including HCC.21 A 9-gene prognostic signature has also been constructed.22
To confirm the advantages of our signature based on immunophenotypes, we analyzed the above 5 genetic models simultaneously. The results showed that the 8-IPSHCC signature in our study was better than these models in predicting the prognosis. Further C-index analysis confirmed our model’s overall superior performance. Therefore, this model could help clinicians predict individual risk of patients and guide patient assessment and treatment decisions.
Finally, we used 3 pairs of HCC tissues and normal controls to verify the protein and mRNA levels of 8 genes. The results showed that the expressions of CD7, IL12A, and FYN in the tumor and control groups were relatively low. GLMN, CKLF, CCL20, PRELID1, and ACVR2A were highly expressed in tumor tissues. Our experimental results are generally consistent with the results from the HPA and GEO databases, which confirms our model’s reliability.
Conclusions
In summary, we integrated multiple gene expression cohorts of 583 samples to develop and validate an 8-IPSHCC signature. It performed well in different cohorts and could improve prediction accuracy of overall survival in HCC patients.
Abbreviations
HCC, hepatocellular carcinoma; TCGA, The Cancer Genome Atlas; ICGC, International Cancer Genome Consortium; IPSHCC, immune prognostic signature of HCC; AUC, area under the curve; FDR, false discovery rate; HR, hazard ratio; KEGG, Kyoto Encyclopedia of Genes and Genomes; KM, Kaplan Meier; OS, overall survival; ROC, receiver operating characteristic; ssGSEA, single-sample Gene Set Enrichment Analysis; C-index, concordance index; RMS, Restricted mean survival.
Data Sharing Statement
The data sets used and/or analyzed during the current study are available from the corresponding author on reasonable request.
Acknowledgment
We thank the numerous individuals who participated in this study.
Author Contributions
Dafeng Xu designed the current study. Dafeng Xu, Yu Wang, Kailun Zhou and Jincai Wu collected the data. Dafeng Xu, Zhensheng Zhang, Jiachao Zhang, Zhiwei Yu and Luzheng Liu analyzed and interpreted the data. Dafeng Xu wrote the manuscript. Xiangmei Liu, Bidan Li and Jinfang Zheng supervised the study. All authors contributed to data analysis, drafting or revising the article, have agreed on the journal to which the article will be submitted, gave final approval of the version to be published, and agree to be accountable for all aspects of the work.
Disclosure
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest and report no conflicts of interest for this work.
References
1. Li D, Sedano S, Allen R, Gong J, Cho M, Sharma S. Current treatment landscape for advanced hepatocellular carcinoma: patient outcomes and the impact on quality of life. Cancers (Basel). 2019;11(6):841. doi:10.3390/cancers11060841
2. Ferlay J, Soerjomataram I, Dikshit R, et al. Cancer incidence and mortality worldwide: sources, methods and major patterns in GLOBOCAN 2012. Int J Cancer. 2015;136(5):E359–386. doi:10.1002/ijc.29210
3. Mak L-Y, Ka-Ho Wong D, Pollicino T, Raimondo G, Hollinger FB, Yuen M-F. Occult hepatitis B infection and hepatocellular carcinoma: epidemiology, virology, hepatocarcinogenesis and clinical significance. J Hepatol. 2020. doi:10.1016/j.jhep.2020.05.042
4. Wang J, Ha J, Lopez A, Bhuket T, Liu B, Wong RJ. Medicaid and uninsured hepatocellular carcinoma patients have more advanced tumor stage and are less likely to receive treatment. J Clin Gastroenterol. 2018;52:437–443.
5. Ringelhan M, Pfister D, O’Connor T, Pikarsky E, Heikenwalder M. The immunology of hepatocellular carcinoma. Nat Immunol. 2018;19(3):222–232. doi:10.1038/s41590-018-0044-z
6. Giannone G, Ghisoni E, Genta S, et al. Immuno-metabolism and microenvironment in cancer: key players for immunotherapy. Int J Mol Sci. 2020;21(12):4414. doi:10.3390/ijms21124414
7. Kubes P, Jenne C. Immune responses in the liver. Annu Rev Immunol. 2018;36(1):247–277. doi:10.1146/annurev-immunol-051116-052415
8. Grunebaum E, Avitzur Y. Liver-associated immune abnormalities. Autoimmun Rev. 2019;18(1):15–20. doi:10.1016/j.autrev.2018.06.016
9. Greten TF, Sangro B. Targets for immunotherapy of liver cancer. J Hepatol. 2017.
10. Flynn MJ, Sayed AA, Sharma R, Siddique A, Pinato DJ. Challenges and opportunities in the clinical development of immune checkpoint inhibitors for hepatocellular carcinoma. Hepatology. 2019;69(5):2258–2270. doi:10.1002/hep.30337
11. Hui E. Immune checkpoint inhibitors. J Cell Biol. 2019;218(3):740–741. doi:10.1083/jcb.201810035
12. Chew V, Tow C, Teo M, et al. Inflammatory tumour microenvironment is associated with superior survival in hepatocellular carcinoma patients. J Hepatol. 2010;52(3):370–379. doi:10.1016/j.jhep.2009.07.013
13. Yao W, He J-C, Yang Y, et al. The prognostic value of tumor-infiltrating lymphocytes in hepatocellular carcinoma: a systematic review and meta-analysis. Sci Rep. 2017;7(1):7525. doi:10.1038/s41598-017-08128-1
14. Ding W, Xu X, Qian Y, et al. Prognostic value of tumor-infiltrating lymphocytes in hepatocellular carcinoma: a meta-analysis. Medicine. 2018;97(50):e13301. doi:10.1097/MD.0000000000013301
15. Pan Q-Z, Pan K, Zhao -J-J, et al. Decreased expression of interleukin-36α correlates with poor prognosis in hepatocellular carcinoma. Cancer Immunol Immunother. 2013;62(11):1675–1685. doi:10.1007/s00262-013-1471-1
16. Jung HI, Jeong D, Ji S, et al. Overexpression of PD-L1 and PD-L2 is associated with poor prognosis in patients with hepatocellular carcinoma. Cancer Res Treat. 2017;49(1):246–254. doi:10.4143/crt.2016.066
17. Ma L-J, Feng F-L, Dong L-Q, et al. Clinical significance of PD-1/PD-Ls gene amplification and overexpression in patients with hepatocellular carcinoma. Theranostics. 2018;8(20):5690–5702. doi:10.7150/thno.28742
18. Wang Z, Teng D, Li Y, Hu Z, Liu L, Zheng H. A six-gene-based prognostic signature for hepatocellular carcinoma overall survival prediction. Life Sci. 2018;203:83–91. doi:10.1016/j.lfs.2018.04.025
19. Li B, Feng W, Luo O, et al. Development and validation of a three-gene prognostic signature for patients with hepatocellular carcinoma. Sci Rep. 2017;7(1):5517. doi:10.1038/s41598-017-04811-5
20. Zheng Y, Liu Y, Zhao S, et al. Large-scale analysis reveals a novel risk score to predict overall survival in hepatocellular carcinoma. Cancer Manag Res. 2018;10:6079–6096. doi:10.2147/CMAR.S181396
21. Chang WH, Forde D, Lai AG. A novel signature derived from immunoregulatory and hypoxia genes predicts prognosis in liver and five other cancers. J Transl Med. 2019;17(14). doi:10.1186/s12967-019-1775-9
22. Zhu G-Q, Yang Y, Chen E-B, et al. Development and validation of a new tumor-based gene signature predicting prognosis of HBV/HCV-included resected hepatocellular carcinoma patients. J Transl Med. 2019;17(1):203. doi:10.1186/s12967-019-1946-8
23. Yang JD, Hainaut P, Gores GJ, Amadou A, Plymoth A, Roberts LR. A global view of hepatocellular carcinoma: trends, risk, prevention and management. Nat Rev Gastroenterol Hepatol. 2019;16(10):589–604. doi:10.1038/s41575-019-0186-y
24. Nishida N, Kudo M. Immunological microenvironment of hepatocellular carcinoma and its clinical implication. Oncology. 2017;92(Suppl 1):40–49. doi:10.1159/000451015
25. Yarchoan M, Xing D, Luan L, et al. Characterization of the immune microenvironment in hepatocellular carcinoma. Clin Cancer Res. 2017;23(23):7333–7339. doi:10.1158/1078-0432.CCR-17-0950
26. Dunne PD, O’Reilly PG, Coleman HG, et al. Stratified analysis reveals chemokine-like factor (CKLF) as a potential prognostic marker in the MSI-immune consensus molecular subtype CMS1 of colorectal cancer. Oncotarget. 2016;7(24):36632–36644. doi:10.18632/oncotarget.9126
27. Pérez-Ramírez C, Cañadas-Garre M, Alnatsha A, et al. Interleukins as new prognostic genetic biomarkers in non-small cell lung cancer. Surg Oncol. 2017;26(3):278–285. doi:10.1016/j.suronc.2017.05.004
28. Elsayed HM, Nabiel Y, Sheta T. IL12 gene polymorphism in association with hepatocellular carcinoma in hcv-infected egyptian patients. Immunol Invest. 2017;46(2):123–133. doi:10.1080/08820139.2016.1229789
29. Chen X, Han S, Wang S, et al. Interactions of IL-12A and IL-12B polymorphisms on the risk of cervical cancer in Chinese women. Clin Cancer Res. 2009;15(1):400–405. doi:10.1158/1078-0432.CCR-08-1829
30. Navaglia F, Basso D, Zambon CF, et al. Interleukin 12 gene polymorphisms enhance gastric cancer risk in H pylori infected individuals. J Med Genet. 2005;42(6):503–510. doi:10.1136/jmg.2004.022723
31. Wang D, Yuan W, Wang Y, et al. Serum CCL20 combined with IL-17A as early diagnostic and prognostic biomarkers for human colorectal cancer. J Transl Med. 2019;17(1):253. doi:10.1186/s12967-019-2008-y
32. Wang G-Z, Cheng X, Li X-C, et al. Tobacco smoke induces production of chemokine CCL20 to promote lung cancer. Cancer Lett. 2015;363(1):60–70. doi:10.1016/j.canlet.2015.04.005
33. Rubie C, Frick VO, Ghadjar P, et al. CCL20/CCR6 expression profile in pancreatic cancer. J Transl Med. 2010;8(1):45. doi:10.1186/1479-5876-8-45
34. Guzman-Rojas L, Sims JC, Rangel R, et al. PRELI, the human homologue of the avian px19, is expressed by germinal center B lymphocytes. Int Immunol. 2000;12(5):607–612. doi:10.1093/intimm/12.5.607
35. Gillen AE, Brechbuhl HM, Yamamoto TM, et al. Alternative polyadenylation of PRELID1 regulates mitochondrial ROS signaling and cancer outcomes. Mol Cancer Res. 2017;15(12):1741–1751. doi:10.1158/1541-7786.MCR-17-0010
36. Zhuo C, Hu D, Li J, et al. Downregulation of activin a receptor type 2A is associated with metastatic potential and poor prognosis of colon cancer. J Cancer. 2018;9(19):3626–3633. doi:10.7150/jca.26790
37. Ge W, Cai W, Bai R, et al. A novel 4-gene prognostic signature for hypermutated colorectal cancer. Cancer Manag Res. 2019;11:1985–1996. doi:10.2147/CMAR.S190963
38. Gomes-Silva D, Atilla E, Atilla PA, et al. CD7 CAR T cells for the therapy of acute myeloid leukemia. Mol Ther. 2019;27:272–280. doi:10.1016/j.ymthe.2018.10.001
39. Haftcheshmeh SM, Tajbakhsh A, Kazemi M, et al. The clinical importance of CD4 CD7 in human diseases. J Cell Physiol. 2019;234:1179–1189.
40. Elias D, Ditzel HJ. Fyn is an important molecule in cancer pathogenesis and drug resistance. Pharmacol Res. 2015;100:250–254. doi:10.1016/j.phrs.2015.08.010
41. Goel RK, Lukong KE. Understanding the cellular roles of Fyn-related kinase (FRK): implications in cancer biology. Cancer Metastasis Rev. 2016;35:179–199. doi:10.1007/s10555-016-9623-3
42. Saito YD, Jensen AR, Salgia R, Posadas EM. Fyn: a novel molecular target in cancer. Cancer. 2010;116:1629–1637.
43. Leussink S, Aranda-Pardos I, A-Gonzalez N. Lipid metabolism as a mechanism of immunomodulation in macrophages: the role of liver X receptors. Curr Opin Pharmacol. 2020;53:18–26. doi:10.1016/j.coph.2020.02.003
44. Streba LAM, Vere CC, Rogoveanu I, Streba CT. Nonalcoholic fatty liver disease, metabolic risk factors, and hepatocellular carcinoma: an open question. World J Gastroenterol. 2015;21:4103–4110.
45. Jinjuvadia R, Patel S, Liangpunsakul S. The association between metabolic syndrome and hepatocellular carcinoma: systemic review and meta-analysis. J Clin Gastroenterol. 2014;48(2):172–177. doi:10.1097/MCG.0b013e3182a030c4
46. Lee SE, Alcedo KP, Kim HJ, Snider NT. Alternative splicing in hepatocellular carcinoma. Cell Mol Gastroenterol Hepatol. 2020.
47. Eymin B. Targeting the spliceosome machinery: a new therapeutic axis in cancer? Biochem Pharmacol. 2020;114039.
48. Racioppi L, Means AR. Calcium/calmodulin-dependent protein kinase kinase 2: roles in signaling and pathophysiology. J Biol Chem. 2012;287(38):31658–31665. doi:10.1074/jbc.R112.356485
49. Mangla A, Guerra MT, Nathanson MH. Type 3 inositol 1,4,5-trisphosphate receptor: a calcium channel for all seasons. Cell Calcium. 2020;85:102132. doi:10.1016/j.ceca.2019.102132
50. Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011;144(5):646–674. doi:10.1016/j.cell.2011.02.013
51. Wu CX, Wang XQ, Chok SH, et al. Blocking CDK1/PDK1/β-Catenin signaling by CDK1 inhibitor RO3306 increased the efficacy of sorafenib treatment by targeting cancer stem cells in a preclinical model of hepatocellular carcinoma. Theranostics. 2018;8(14):3737–3750. doi:10.7150/thno.25487
52. De Matteis S, Ragusa A, Marisi G, et al. Aberrant metabolism in hepatocellular carcinoma provides diagnostic and therapeutic opportunities. Oxid Med Cell Longev. 2018;2018:7512159. doi:10.1155/2018/7512159
53. Khemlina G, Ikeda S, Kurzrock R. The biology of hepatocellular carcinoma: implications for genomic and immune therapies. Mol Cancer. 2017;16(149).
 © 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.