Back to Journals » Drug Design, Development and Therapy » Volume 8
Development and use of sulodexide in vascular diseases: implications for treatment
Authors Coccheri S, Mannello F
Received 11 June 2013
Accepted for publication 13 August 2013
Published 24 December 2013 Volume 2014:8 Pages 49—65
DOI https://doi.org/10.2147/DDDT.S6762
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Sergio Coccheri,1 Ferdinando Mannello2
1Cardiovascular Medicine, University of Bologna, Bologna, 2Department of Biomolecular Sciences (Section Clinical Biochemistry and Cell Biology), University 'Carlo Bo', Urbino, Italy
Abstract: Sulodexide (SDX), a sulfated polysaccharide complex extracted from porcine intestinal mucosa, is a blend of two glycosaminoglycan (GAG) entities, namely a fast-moving heparin (HP) fraction and a dermatan sulfate (DS; 20%) component. The compound is unique among HP-like substances in that it is biologically active by both the parenteral and oral routes. A main feature of the agent is to undergo extensive absorption by the vascular endothelium. For this reason, in preclinical studies, SDX administered parenterally displays an antithrombotic action similar to that of HPs but associated with fewer alterations of the blood clotting mechanisms and tests, thus being much less conducive to bleeding risk than HPs. When given orally, SDX is associated with minimal changes in classic coagulation tests, but maintains a number of important effects on the structure and function of endothelial cells (EC), and the intercellular matrix. These activities include prevention or restoration of the integrity and permeability of EC, counteraction versus chemical, toxic or metabolic EC injury, regulation of EC–blood cell interactions, inhibition of microvascular inflammatory and proliferative changes, and other similar effects, thus allowing oral SDX to be considered as an endothelial-protecting agent. The best available clinical evidence of the efficacy of SDX administered orally with or without an initial parenteral phase is the following: alleviation of symptoms in chronic venous disease and especially acceleration of healing of venous leg ulcers; prevention of cardiovascular events in survivors after acute myocardial infarction; marked improvement of intermittent claudication in patients with peripheral occlusive arterial disease; and abatement of proteinuria in patients with diabetic nephropathy that may contribute to the amelioration or stabilization of kidney function. Although further clinical trials are warranted, SDX is presently widely accepted in many countries as an effective and safe long-term, endothelial-protecting drug.
Keywords: sulodexide, glycosaminoglycans, chronic venous disease, cardiovascular disease, diabetic nephropathy
Glycosaminoglycans: biochemical structure and functions
Generalities
Sulodexide (SDX) belongs to a class of substances known as glycosaminoglycans (GAGs). Also known as mucopolysaccharides because of their viscous properties, GAGs are a family of natural anionic polycarbohydrates found in mammalian tissues, where they play a crucial role in different biological processes. In fact, they regulate the activity of a wide range of proteins (including chemokines, cytokines, growth factors, enzymes, morphogens, and adhesion molecules), involved both in physiological and in pathological situations.1 In this regard, GAGs, by mediating cell-to-cell and cell-to-matrix interactions, are essential in the development and correct functioning of multicellular organisms.2 GAGs are large, linear, frequently sulfated, negatively charged polysaccharides with a molecular weight ranging 1–2,000 kDa. They are organized as repetitions of disaccharide units of an uronic acid (D-glucuronic acid [GlcA] or L-iduronic acid [IdoA]) and an acetylated amino sugar (N-acetylgalactosamine or N-acetylglucosamine) either sulfated or non-sulfated. The position of sulfation on the sugar backbone of GAGs is widely variable depending on their cell and tissue localization; moreover they exist both as part of proteoglycans and as free chains (Figure 1).1,3 Because of their variability in the composition of the disaccharide sequences, sulfation, and geometry of the glycosidic linkage, GAGs can be divided into non-sulfated GAGs (including hyaluronic acid [HA]) and sulfated GAGs (including chondroitin sulfate [CS], dermatan sulfate [DS], keratan sulfate [KS], heparin [HP], and heparan sulfate [HS]; Figure 1).
 | Figure 1 Disaccharide units and carbohydrate sequences of the six main types of GAGs. The characterizing uronic acid and hexosamine are specified. Positions of sulfate groups are identified, showing that sulfate groups are more numerous in HP, followed by HS, CS, DS, and KS, and are absent in HA. The latter does not attach to proteins to form proteoglycans. |
Although in human tissues all the abovementioned types of GAGs exist, in this review we will focus on the two GAG compounds mainly represented in the complex natural drug SDX, which only includes DS and HP.
Dermatan sulfate
Among sulfated GAGs, a crucial role is played by the CS family, that includes CSs (CS–A and CS–C) and DS (also known as chondroitin sulfate B, CS–B), possessing the same underlying structure of repeating disaccharide units.
All CSs (CS–A, –B, and –C) are anionic linear polymeric carbohydrates consisting of alternating disaccharide units of GlcA and D-N-acetylgalactosamine, and can be variously modified by sulfate groups replacing one, or more, of the OH groups at C–4 (to form CS–A) and/or C–6 (to form CS–C) of N-acetylgalactosamine and/or C–2 of glucuronic acid, thus making these compounds highly anionic. These post-translational modifications can generate 16 isomers of the repeating disaccharides.4 All three CS compounds are linked to serine (Ser) residues of core proteins through a tetrasaccharidic complex consisting of xylose, two galactose molecules, and glucuronic acid (Figure 1). All chondroitin chains vary in size up to 100 or more disaccharide repeating units. The biosynthesis of CS chains on core proteins may result in the formation of peculiar CS proteoglycans, such as aggrecan, versican, decorin, and biglycan, which play crucial roles in cell biology, signal transduction, embryogenesis, stem cell regulation, and differentiation, cancer proliferation, etc.5
DS (or CS–B) derives from the action of 5-epimerase on the CS chain, resulting in an epimerization of the glucuronic acid into iduronic acid. DS (CS-B) can also be 2-O-sulfated, thus differing from both CS–A and CS–C. The substantial difference between CS and DS lies in the relationship between the two uronic acids (iduronic and glucuronic acid) and in the degree of sulfation of the chain.
For what concerns tissue localization and biological functions, CS–A isoforms are found in significant amounts in cartilage, bone, and corneas, whereas CS–C is expressed in cartilage, tendons, heart valves, and the nucleus pulposus of the intervertebral discs. Besides its structural role in osteoarticular and connective tissues, CS is also involved in the development of the central nervous system, in wound healing, infections, signaling pathways, in cell division, and morphogenesis. DS, on the other hand, is mainly found in the skin, dermis, heart valves, and blood vessel walls. Thanks to its ability to bind the tropocollagen fibrils, DS may play an important role in directing and ordering the components of fibrillar collagen, especially at the level of the dermis where it is present in high amounts. All these functions seem to be due to specific interactions of saccharide domains with a wide variety of molecules (such as growth factors, cytokines, chemokines, adhesion molecules, and lipoproteins).4,5 Moreover, DS exerts an anticoagulant activity by inhibiting Factor X and thrombin activation through the natural inhibitor HP cofactor II (HC II). This pathway works as an accessory anticoagulant system towards the main physiological system based on antithrombin, and its activation by DS may be of clinical value in certain conditions.6
Heparan sulfate and Heparins
The HSs, a family of GAGs including HP, are polydisperse polymers7 made up by repeating disaccharide units consisting of α-D-glucosamine coupled with uronic acids: 90% GlcA and 10% IdoA. Heparan sulfate is characterized by an average of less than one sulfate per disaccharide; and is predominantly composed of GlcA linked to glucosamine (Figure 1). The sulfated domains of HS closely resemble those of HP and share its binding properties to proteins. HS chains are generally longer than those of HP with an average molecular weight of 29 kDa, ranging 5–50 kDa.
The length of the HP chain can also vary, with an average molecular weight of 13 kDa, ranging 3–30 kDa. The combination of HP polydispersity and microheterogeneity makes this compound structurally very complex.8 HP shows the maximum degree and heterogeneity of sulfation both at uronic acid and at glucosamine, and is therefore the substance with the greatest number of negative charges known so far. Although its most common structure is the tri-sulfated disaccharide, consisting of iduronic acid with one sulfate group at C–2 and glucosamine with two sulfate groups, about 12 structural variants of the sulfated disaccharide unit exist, making HP a highly heterogeneous substance.
HP is physiologically synthesized by mast cells in the connective tissue of several mammalian species, where it has a crucial anticoagulant/antithrombotic effect due to its ability to enhance the activity of the natural occurring serine-protease inhibitor, antithrombin (AT). HP thereby catalyzes the inhibition of all serine-proteases of the intrinsic coagulation pathway such as Factors IXa, XIa, XIIa, and also of those of the common pathway, such as thrombin and Factor Xa. Besides their known activities in coagulation, HP and HS bind growth factors, cytokines, and morphogens, as well as other enzymes and molecules involved in cell signaling and growth, cell adhesion, migration, differentiation, and angiogenesis.9 HP, as commonly used in therapy, is presently extracted from porcine intestinal mucosa and shows similarity to the human endogenous HP.
Efforts to develop HP congeners endowed with optimal antithrombotic effects coupled with lesser anticoagulant and pro-hemorrhagic activity resulted in the preparation of low molecular weight HPs (LMWHs), obtained by chemical or enzymatic depolymerization or fractionation of HP, thus providing compounds with shorter polysaccharide chains and lower molecular weight (3,000–6,500 Da) and degree of sulfation.10 LMWHs are characterized, versus (vs) unfractionated heparin (UFH), by a more predictable dose-response, prevalent effect on Factor X over Factor II activation, and a reduced pro-hemorrhagic to antithrombotic ratio. The first generation LMWHs contained 25%–50% of fragments with 18 or more disaccharides (molecular weight ≥6,000 D), while newer agents (second generation or ultra-LMWHs) contain a much higher percentage of short chains (molecular weight <3,000 D) with a high number of chains containing a specific pentasaccharide with high affinity and exclusive activity on Factor X. Fondaparinux is a synthetic compound mimicking this specific ‘core’ pentasaccharide of HPs.10
SDX and its glycosaminoglycan composition
A highly purified GAG, SDX is extracted from the porcine intestinal mucosa by a patented process,11 and differs from HPs as being composed of two distinct fractions. This natural mixture contains 80% iduronylglycosaminoglycan sulfate (IGS, best known as fast-moving HP [FMH] because of its electrophoretic mobility in the barium propanediamine system), and 20% DS (or CS–B). One of the main characteristics of SDX is absorption through the intestinal mucosa for both the entire compound and for its fractions.12 Due to the presence of two different GAG fractions, SDX simultaneously potentiates the anti-protease activities of both AT and HP cofactor II (HC II).6 Thrombin inhibition induced by SDX is therefore the result of an additive or possibly synergistic effect of both components.13
The fast-moving Heparin fraction in SDX
The IGS or FMH fraction of SDX has a mean molecular weight of about 7 kDa. This fraction of SDX has properties somewhat closer to those of LMWHs when compared to UFH, and has less effect on global coagulation tests and a lower degree of pro-hemorrhagic effects. The FMH fraction confers to SDX other major differences from UFH, including longer half-life and especially oral bioavailability.12
The Dermatan sulfate fraction in SDX
DS, made up of many various disaccharide units with a mean molecular mass of 25 kDa, has been demonstrated to inhibit thrombus formation and growth in different experimental models of venous thrombosis. At variance with HPs, there is a paucity of experimental data available on the effect of DS on arterial thrombus formation.14 However, in a model of arterial thrombosis in rats, the effects of a low and high molecular weight DS, of SDX, and of low and high molecular weight HP, were compared: results suggested that DSs also are effective inhibitors of arterial thrombosis in rats, without inducing bleeding complications.14 DS was also clinically active in the prevention of postoperative DVT in humans.15 Moreover, recent evidence highlighted a novel biological action of DS, namely inhibition of matrix metalloproteinases (MMP), which play a key role in extracellular matrix (ECM) remodeling, thus also conferring protective effects to SDX against vascular wall damage and inflammation in chronic venous diseases (CVD).16,17 More recently, it has also been found that DS may act as an adjuvant factor for initiating and accelerating wound healing.18
Thus, in SDX the two coexistent GAGs may act in an additional, possibly synergistic fashion, in regard to both the antithrombotic, and the endothelial-protecting and wound-healing properties.
Pharmacology of SDX
Pharmacokinetics
SDX (VesselTM, Alfa Wasserman, Bologna, Italy) is prepared in the pharmaceutical forms of intravenous (iv) or intramuscular (im) ampoules of 600 lipasemic units (LSU) equivalent to 60 mg, or oral capsules of 250 LSU (25 mg). It is presently marketed as a drug in Italy, Spain, Eastern Europe, and in the countries of South America and Asia.
After iv administration of 50–100 mg of SDX, the plasma peak concentrations amount to 8–20 mg/L shortly after injection. These levels sharply decline within 1–4 hours to 0.5–1 mg/L and gradually approach the null concentration after 18 hours.19 By the oral route, SDX is absorbed within 1–2 hours and behaves essentially as a monocompound despite its dual nature. The comparative bioavailability vs the iv. OK route is between 20% and 60%, with a median value of 40%.12,19 It is noteworthy that with the same doses as above (50–100 mg), orally induced concentrations range 0.7–1.5 mg/L and the time to peak is about 4 hours. Thereafter, similar concentrations are maintained at least up to 18 hours and remain noninferior to those seen with iv administration 4 hours after the peak. Practically, in a steady state condition, and regardless of the administration route, 50 mg of SDX (2 pills or about 1 ampoule) will yield a plasma concentration of 1 mg/L, while with 100 mg, the plasma level rises to 1.5 mg/L.
The distribution volume of SDX is very large, due to a higher affinity of the substance for the extensive surface area of the endothelium rather than for plasma proteins.20 Because of this property, effects on the classic global coagulation tests are seen only with iv SDX, during and shortly after the initial peak; on the contrary, after oral administration, an anticoagulant effect with the current global tests is never apparent.20 Catabolism of SDX is based on N-desulfation. Metabolism is liver dependent and excretion is mostly kidney dependent.20
Pharmacodynamics
In vitro studies
SDX has peculiar anticoagulant activity. In vitro and ex vivo, the anticoagulant potency of the compound expressed as concentration capable of doubling the clotting time is maximal (0.4 μg/mL) in the Heptest (a test suitable for HP-like agents) and the thrombin clotting time (TCT, 0.5 μg/mL). These potencies appear about six times lower in the setting of activated partial thromboplastin time (APTT) and 50 times lower in that of prothrombin time (PT).
SDX inhibits Factor Xa and, more potently, Factor II. In fact, the IC50 is reported to be 0.20 μg/mL for Factor Xa inhibition and 0.10 μg/mL for anti-factor IIa.21 Consequently, SDX delays the onset of prothrombin activation to thrombin, although it is somewhat weaker in this respect than UFH and LMWH. In fact, though immediately less potent than HP, SDX can over time effectively inhibit thrombin-mediated amplification of blood coagulation.21 Moreover, in a recent ex vivo study,22 direct supplementation of human whole blood or plasma with equal gravimetric amounts of SDX or enoxaparin (enoxa), resulted in a stronger anticoagulant effect of SDX in the settings of PT, APTT, TT, and Heptest assays. In the APTT, the stronger effect of SDX was especially significant at the concentrations of 6.25 μg/mL and 12.5 μg/mL. These figures, similar to those observed at peak after iv or oral administration, are approximately compatible with therapeutic concentrations. SDX was also superior to enoxa in the inhibition of other phenomena induced by tissue factor (TF) as microparticle formation, P-selectin expression, and TF-induced platelet aggregation. SDX was therefore shown to be a strong and balanced inhibitor of the intrinsic, extrinsic, and common pathways of the clotting mechanism and related platelet involvement.22
These in vitro results have been repeatedly confirmed in in vivo studies measuring thrombin formation and other coagulation parameters. However, when SDX was administered in vivo by the parenteral route, the global plasma anticoagulant activity was consistently lower than that measured after HPs (UFH and LMWH) given at equipotent in vitro doses.21,23 This phenomenon is attributed to the quoted higher affinity of SDX for the extensive vascular and microvascular endothelium rather than for plasma proteins, and is in line with the concept, which is well accepted for low-dose UFH and LMWHs, that HP-like compounds can be effective at cellular levels even in the absence of overt anticoagulant activity in plasma.
Human preclinical pharmacodynamics of the oral preparation
The pharmacodynamic activities of oral SDX can be summarized as follows: coagulation is not affected in terms of global tests, but more subtle markers such as the prothrombin activation fragment (F1+2) and thrombin–antithrombin (TAT) complex show a low-grade persistent inhibition of prothrombin to thrombin activation and hence of blood coagulation.24 Release of TFPI has also been demonstrated with the oral route,25 and fibrinogen has been found to be decreased in several clinical studies.26 Fibrinolytic activators are increased (tissue plasminogen activator [TPA], and urokinase plasminogen activator [UPA]) whereas the main inhibitor (plasminogen activator inhibitor [PAI] 1) decreases.27 Thus, oral SDX induces a mild but sustained shifting of the coagulolytic balance toward anticoagulation and fibrinolysis.28
A number of other activities have also been demonstrated with oral SDX such as the anti-inflammatory, anti-proliferative, and endothelial protective properties, which further extend the properties of the compound and will be discussed later.
SDX in venous thrombogenesis
SDX is an effective antithrombotic agent in several models of venous thrombosis. The active doses of SDX for venous thrombogenesis in rabbits are 20 mg/kg iv for the effective dose (ED)50, and 125 mg/kg for the ED100. Within these doses, only the Heptest was prolonged but APTT and TT were unchanged.21,23
It is also noteworthy that the antithrombotic effect obtained with iv SDX compared well with that of a dose of HP of double potency in terms of plasma AT units titrated in vitro.29 Explanations forwarded for this apparent paradox are: the presence in SDX of the DS component providing concomitant activation of the HC II pathway; preferential localization of SDX activity at the thrombus surface; higher release by SDX from the vascular wall of TF pathway inhibitor (TFPI), which is the major inhibitor of the extrinsic pathways capable of inactivating complexes of TF with Factor VIIa or Xa at cell surfaces.21,24
SDX in arterial thrombogenesis and atherogenesis
SDX was studied in an acute model of carotid artery thrombosis induced by electrical stimulation, and also in chronic animal models.30,31 In both conditions, SDX compared well with HP and with ASA when appropriate. Equivalence in antithrombotic activity in the chronic model was seen at 8 mg/kg for HP and 10 mg/kg for SDX. At equally active antithrombotic doses of the two agents, bleeding time was prolonged by 100% with HP and by only 25% with SDX.31 The presence of arterial antithrombotic activity is even more important by considering that SDX is also able to effectively release lipoprotein lipase activity by any administration route. In fact, SDX reduces cholesterol blood levels and aortic accumulation in cholesterol-fed rabbits32 and increases lipoprotein catabolism23 in the liver both in normal and hypertriglyceridemic animals. In this way, SDX displays a complex anti-atherosclerotic and antithrombotic activity that can be considered an interesting property of this compound.
SDX, fibrinolysis, and thrombolysis
SDX intravenously administered in rats induces acceleration of spontaneous fibrinolysis-thrombolysis of preformed thrombi.33 This property is in accord with an increase in plasminogen activators – TPA and UPA in primates and other animals. These results were also confirmed in humans by an increase in TPA and decrease in plasminogen activator inhibitor (PAI),34–36 especially in patients with diabetes.28
Effects on blood cells, inflammation, and proliferation
SDX inhibits leukocyte activation and their adhesion to endothelial cells (EC). Release of P-selectin, cathepsin G, cytokines, tumor necrosis factor (TNF) and platelet aggregation factor from polymorphonuclear leukocytes is also diminished by SDX. Platelet and platelet–leukocyte aggregation induced by proteases, such as cathepsin G and thrombin is similarly reduced.37 More recently, a potent inhibitory effect on MMPs, a family of proteolytic enzymes responsible for the degradation of the ECM, was shown by one of us.16,17,38 In a recent ex vivo study in human blood and in a leukocyte cell line, the effect of SDX in reducing excretion and plasma levels of several forms of MMPs (eg. MMP9) was observed, thus suggesting a therapeutic role for SDX in chronic inflammatory vascular disease with destruction of the ECM, as for instance chronic venous insufficiency (CVI).38 Other agents of the inflammatory process are also inhibited, such as interleukin 6 and other cytokines. The complex action of SDX on inflammation also includes the inhibition of transforming growth factor ² 1 (TGF-²1), as well as of the intercellular adhesion molecule 1 (ICAM-1).39 The anti-inflammatory effect of SDX is associated with an antiproliferative action, through regulation of several growth factors.40 Release of the hepatocyte growth factor (HGF) after injection following daily oral administration in humans has also been shown.41
Endothelial protection effects
The endothelial cellular lining is composed of cells endowed with multiple functions influencing mechanisms such as: blood coagulation (protein C, protein S, thrombomodulin system, HP cofactor II, and the TFPI system); fibrinolysis (activators and inhibitors such as TPA and PAIs); platelet adhesion and aggregation (activators such as thromboxane A2 and von Willebrand factor, and inhibitors such as prostacyclin); interactions with platelets and other blood cells (by expression of selectin, other integrins, and cytokines); and finally regulation of vascular tone especially in the microcirculation, by release of several vasoactive substances. Thus, the endothelium is a signaling center interconnecting circulating blood cells with structures of the vascular wall such as the subendothelial matrix and the subintimal and medial smooth muscle cells.
Protection of these complex endothelial structures is essential in order to maintain blood fluidity by allowing homeostasis; to prevent thrombosis in the macro- and micro-circulation through fast removal of excess fibrin; and to modulate vascular tone by maintaining both vasoconstricting and vasodilating abilities.
Functional efficiency of the endothelium and resistance to endothelial damage is secured by the morphofunctional integrity of the fundamental substance or ‘matrix’ of the connective tissue, mainly composed of GAGs. The matrix also coats the endothelium with fine intercellular surface material, the so called ‘glycocalix’, a filament-like GAG lining that regulates permeability and selectivity of the EC. In several experimental conditions, SDX has been shown to be able to maintain or restore the integrity of the endothelial glycocalix-protecting structures that are mainly composed of GAGs.42 At this regard, in streptozotocin-induced diabetes in rats, SDX was able to protect the vascular endothelium by improving its multiple functions43 and restoring the related intimal morphologic alterations.44
Other activities of SDX
A lipoprotein lipase stimulating activity was among the first effects observed with the substance: in fact, the agent, besides its gravimetric expression in milligrams, can also be titrated as lipoprotein lipase releasing units (LRU) or LSU. In patients with peripheral arterial disease (PAD) and hyperdyslipidemia submitted to im and oral cycles of SDX in a double-blind, crossover design,45 a significant decrease in total and VLDL-triglycerides and in VLDL-cholesterol but not in total or LDL-cholesterol was observed, with a concomitant rise in antiatherogenic fractions such as HDL-cholesterol and apolipoprotein A1. These changes are relevant especially in relation to the peculiar dyslipidemia of diabetes and the metabolic syndrome.
An effect of SDX on blood viscosity has been described,46 mainly mediated by the lowering effect of the agent on fibrinogen and hence plasma viscosity, with some contribution by lowered chylomicrons and VLDL. The hemorheologic effect is particularly important in protecting the endothelial integrity in the microcirculation.47
Therefore, besides its recognized antithrombotic effect, SDX has antiatherosclerotic potential, as also demonstrated in earlier studies32 and further validated by more recent experimental results showing inhibition of smooth muscle cells and neointimal proliferation in rat carotid arteries.48
Clinical applications of SDX
Chronic venous disease
Pathophysiology
The definition ‘chronic venous disease’ (CVD) encompasses a syndrome consisting of multiple morphological and/or functional changes in the venous system, particularly of the lower limbs. This condition is characterized by varicosities of different degrees and caliber, ranging from telangiectasias and reticular veins to multinodular and troncular varices of different sizes. Varicosities may be associated with edema and skin changes, such as pigmentation, eczema, and lipodermatosclerosis (white atrophy), possibly leading to skin ulcerations. CVD usually presents with symptoms such as itching and heaviness and moderate-to-severe pain in the standing position, which is alleviated during walking.
There exist several forms of CVD with venous insufficiency,49 namely the primary varicose syndromes (PVS), post-thrombotic syndrome (PTS), and, to a minor degree, some congenital venous disorders. Regarding the two main forms, PVS covers two-thirds and PTS one-third of all cases of CVD.50
Epidemiological data are very variable according to the criteria adopted. In the recent Edinburgh and Bonn epidemiological studies51,52 based on the CEAP (clinical, etiological, anatomical, pathophysiological) classification,53 the mildest form, isolated telangiectasia, is reported to affect 59% of the population; venous edema amounts to 14.8%; and venous ulceration to 0.7%. In general, epidemiological studies show a higher prevalence of any degree of venous disease in subjects over 60 and in the female sex.
The etiology of CVD recognizes a number of factors such as familiarity, lifestyle, occupations involving prolonged standing position, obesity, multiple pregnancies, and a family history of varices, or deep, or superficial vein thrombosis (DVT, SVT), especially if recurrent.
The main mechanism responsible for chronic venous insufficiency and its progression is venous hypertension,54 resulting from blood stagnation and reflux into the superficial venous system in the PVS, and from thrombotic residues, valvular damage, and consequent reflux into both the deep and the superficial venous systems in the PTS. Compensatory mechanisms by the recruitment of collateral veins and increased lymphatic drainage may fail over time. The consequences of chronic venous hypertension are microcirculatory congestion, capillary failure, endothelial damage, and biochemical changes affecting permeability, leading to chronic skin changes, edema, and in severe cases, cutaneous ulcerations. Leukocyte activation with expression of proinflammatory cytokines, leukocyte adhesion and trapping in microvessels, and possibly deposition of intra- and peri-capillary fibrin have been described as important pathogenetic steps.54
Treatment of CVI and rationale for SDX
Patients with chronic venous conditions, although obtaining significant relief from vascular surgery when appropriate, need long-term medical therapy during the lifelong course of their disease. Compression therapy is the foundation of treatment; its effectiveness is widely known in the treatment of moderate-to-severe CVI even with edema and ulcerations.55 Pharmacological therapy has an adjuvant role on top of compression and is especially useful in relieving edema and pain.56 Evidence of benefit has been obtained with the micronized, purified flavonoid fraction containing diosmin and hesperidin (MPF), oxerutin and analogs, and some extractive GAGs, principally SDX.
Focusing on the mechanism of action of SDX in venous insufficiency, some properties of the agent discussed above seem especially important, such as the protective effect on EC, restoration of intimal anionic charges, the improvement of altered capillary permeability, and profibrinolytic, hemorheologic, and antiproliferative effects, as well as the inhibition of blood cell adhesion to endothelium.57 Recently, a counteracting effect of SDX on venous neointimal hyperplasia induced in rats by arterialization of the femoral vein has been observed, and attributed to an inhibitory activity of the agent on the angiopoietin-2 system. This observation could be important in the prevention of venous disorders as well as of restenosis of venous grafts.58
In an early, randomized double-blind, placebo-controlled study59 on patients with CVI due to PVS or PTS, SDX was shown to significantly alleviate a number of ailments and symptoms such as edema, pain, paresthesia, and cramps. A parallel reduction in the limb venous pressure was also documented in these patients. Quite recently, the collaborative clinical practice guidelines of the American Society for Vascular Surgery and the American Venous Forum,60 included SDX in a selected group of venoactive drugs to be used in addition to compression in patients with varicose veins and related conditions, especially if suffering from edema and pain.
SDX in the post-thrombotic syndrome
Little is known about the influence of venoactive drugs, and also of SDX, on the natural history of CVI. There are few follow-up studies in primary CVI, while there has been some approach of this kind in post-DVT patients; more data are expected from an ongoing trial. Although PTS may occur even after a single DVT episode, DVT recurrences are a major risk factor for this condition.61 Prevention of DVT in surgical and medical risk circumstances is an important measure, but would not cover the idiopathic forms of DVT that carry a much higher risk of recurrences and are prone to produce more cases of PTS and related CVI.62
Early diagnosis and correct medical therapy of acute DVT is undoubtedly important. However, after termination of oral anticoagulant treatment generally lasting 6–12 months, a residual risk of DVT recurrences as high as 8% per year persists, especially after idiopathic DVTs, a risk that slowly declines after the first 2 years.62 Traditional anticoagulation either at adjusted or at fixed low doses, if continued beyond the predefined termination time, would expose the patient to a risk of major bleeding that can be evaluated to approximately 2%–3% per year.63 Moreover, a residual thrombotic risk would still reappear after extended anticoagulation. The problem has been successfully approached with the new oral anticoagulants (dabigatran, rivaroxaban, apixaban) in the so-called DVT extended studies.64 However, neither the traditional nor the new oral anticoagulants seem fit for indefinite duration of treatment, as they maintain a bleeding risk, which is bound to increase with age. In this indication, low-dose aspirin has often been prescribed by practitioners, but only recently was found to be effective on DVT recurrences65 or on total vascular events including DVT.66 However, it is known that severe bleeding complications can occur with aspirin, although in rare cases (1 in 1,000 patients/year).67 Moreover, especially in Mediterranean countries, many patients have poor gastric tolerance to low-dose aspirin.
From this evidence, it seems appropriate to evaluate a drug like SDX, which displays by the oral route a low but sustained antithrombotic activity with negligible hemorrhagic risk, together with profibrinolytic and endothelial-protecting effects potentially useful in this clinical setting.
The safety of SDX in the treatment of DVT was suggested in a Spanish open controlled prospective study68 in which the agent was compared with adjusted dose acenocoumarol in the post-acute phase of DVT, just after HP treatment. After a standard period of 3 months, no difference in DVT recurrences with less hemorrhagic complications was observed with SDX.
A systematic experience with SDX in the extended treatment of DVT [post-vitamin K antagonists (VKA) phase] was collected in an Italian registry (the SanVal Registry)69 of about 400 patients with documented DVT who, after completing their predefined course of oral anticoagulant therapy were all treated with adequate elastic compression and randomized to receive or not receive oral SDX. After 6, 12, and particularly 24 months of SDX treatment, a significant difference in the incidence of DVT recurrences was recorded in SDX vs control patients (at 24 months 7.4% in SDX vs 17.9% in the control group). The efficacy of SDX in this instance deserves however, to be confirmed in a large multicenter randomized placebo-controlled trial.
In fact, the SURVET study started in 2010 and presently under way70 will probably answer the question of whether SDX can be a reasonable option for extended prevention of DVT recurrences after a regular course of oral anticoagulants. Indeed, the excellent safety and tolerability profile of SDX may supply the agent with a competitive advantage over other options. It should finally be mentioned that oral SDX might safely be associated with aspirin, as shown in our study of intermittent claudication in which about 70% of the SDX-treated patients were currently on antiplatelet (mainly aspirin) treatment.26
Treatment of venous ulcers
Pathophysiology
The main cause of venous skin ulcerations is decompensated CVI. Increased filtration of fibrinogen and proteins and recruitment of inflammatory cells in the lower districts of the limb, associated with tissue infiltration by blood products such as hemosiderin, various proteins, and cytokines, are the main factors liable to induce inflammatory changes in the skin and subcutaneous layers. The consequences of this situation are the formation of pericapillary fibrin cuffs, and/or leukocyte accumulation and trapping in the microvascular bed, both leading to capillary microthrombi formation.71 In certain sites characterized by maximal venular stasis and hypertension, as in skin areas drained by an incompetent collateral vein or affected by high gravitational or external pressure, microvascular hypoxia may induce skin destruction and necrosis, resulting in an open wound, namely the venous ulcer. The occurring inflammatory process involves the activation and release of cytokines, TNF, and various proteolytic enzymes, especially the MMPs, which are increased in the wound exudate and in the back-flowing blood. This inflammatory and proteolytic pattern is indeed a necessary step for the repair process, but in the case of excessive and persistent proteolytic activation or superimposed infection, it may severely hamper the healing of venous leg ulcers (VLU).17
The basis of VLU treatment, especially of the ‘difficult’ ones with scarce propensity to heal, is a correct wound bed preparation (WBP), including the maintenance of adequate moisture, antisepsis, and compression bandaging.72 A detailed description of these measures is beyond the scope of this review. It must also be made clear that VLU systemic pharmacologic treatment, although important, has mainly an adjuvant role. However, considering that despite optimal WBP, local care and adequate compression, only about 30% of VLU will be healed after 1 year from diagnosis,73 even a moderate advantage offered by systemic adjuvant pharmacologic agents would be highly beneficial.
Pharmacologic treatment of venous ulcers and SDX
Among agents tested for systemic treatment in patients with VLU, we quote aspirin, pentoxifylline, stanozolol, purified micronized flavonoids (MPF), and rutosides.56 The use of sulfated GAGs such as SDX seems highly appropriate given the endothelial protective, anti-inflammatory, and reparative properties of the agent. Thus, after a pilot study74 with promising results, in its venous arm, the SUAVIS (SDX arterial venous Italian study)75 showed that SDX administered in addition to standardized local and compression treatment was associated with more frequent and faster healing of VLU (Figure 2). In this trial, a group of 230 patients were randomized to receive in double-blind conditions either placebo or SDX administered orally after a short iv phase. Complete ulcer repair was reached in 35% of patients at 2 months and in 52% at 3 months, vs 21% and 33% with placebo, respectively, with both differences being highly significant. With SDX, the rate of decrease over time of ulcer surface areas and the interaction of drug treatment and compression were both significant. A significant decrease in fibrinogen levels was also seen.75
 | Figure 2 Percentage of patients healed from chronic venous ulcers during 3-month treatment with SDX (120 patients) or placebo (110 patients) on top of standardized local and compression therapy. A double-blind trial, intention-to-treat evaluation. |
The results of the SUAVIS study were soon confirmed by a study of a Polish group76 focused on large venous ulcers.
That GAG compounds can be beneficial in the healing process of VLU was also shown in a study77with a similar, although not identical extractive sulfated GAG, mesoglycan. The total rate of healing was 97% for treated vs 82% for non-treated patients (P = 0.05).
The group responsible for clinical evidence in the area of VLU is periodically reviewing various drugs likely to be active in accelerating VLU healing in the presence of compression therapy. In the last available analysis,78 a high level of evidence was attributed only to SDX and to pentoxifylline, over micronized flavonoids, mesoglycan, and others (Table 1). Another guideline79 judged the evidence for the first three drugs at grade 2B and recommended further trials.
 | Table 1 Adjuvant effect of systemic drugs on healing rates of venous ulcers on top of adequate compression and topical therapy |
Thus, in patients with VLU adequately treated with correct WBP and compression, SDX showed beneficial results by accelerating the rate of wound healing and increasing the proportion of ulcers healed at a predefined time. SDX therefore stands as a valuable adjuvant agent in the treatment of ‘difficult’ venous ulcers.
SDX in arterial and cardiovascular disease
Pathophysiology and rationale
The above described properties of SDX prompted clinical research with this agent in arterial disorders. In fact, many of the effects described here, such as the anticoagulant, antithrombotic, and profibrinolytic effects; its ability of inducing release of the TFPI; the fibrinogen and viscosity-lowering actions; and the lipoprotein-lipase–stimulating activity, warranted specific trials in arterial vascular disease. Moreover, in recent times, the effects of SDX on the structure and function of the endothelium; its inhibiting effect on endothelial blood cell interactions, including leukocytes and platelets; and the ‘glycocalix’ theory42 fostered further studies on the compound in the atherothrombotic process, especially based on the role of endothelial injury as a triggering factor of the atherothrombotic plaque and its evolution.
Clinical trials
Post-myocardial infarction long-term treatment
Many early and small studies with SDX on atherosclerotic conditions and complications can be found in the literature over 1980–2000.21,23,57
In patients surviving an acute myocardial infarction (AMI) there is a high risk of AMI recurrence and cardiovascular death, especially during the first year after the event. A great number of studies and meta-analyses have established the efficacy of antiplatelet drugs, such as low-dose aspirin and the thienopyridines (ticlopidine, clopidogrel) in lowering this risk, more significantly for nonfatal than fatal events.80 However, up to the mid-90s, it was still acceptable to test SDX in comparison with a standard therapy not including aspirin or other antithrombotic drugs.
The IPO-V2, a landmark trial for SDX in the cardiovascular area, was a multicenter, randomized study aimed at investigating the efficacy of SDX in preventing cardiovascular and thrombotic events or death by any cause during the first year after an AMI.81 Almost 4,000 patients entered the study and were randomized within 7–10 days from the acute episode to receive or not receive SDX, given first parenterally for 1 month and thereafter orally, for a total of at least 1 year of treatment and follow-up. All patients underwent standard pharmacological treatment with the exclusion of antiplatelet and anticoagulant drugs. At the study’s end, significant reductions of deaths (32%), re-infarctions (28%), and occurrence of left ventricular thrombi (53%) were recorded in the SDX group. Long-term SDX treatment started early after AMI was therefore effective in preventing atherothrombotic recurrent events, with an order of efficacy similar to that of classic anticoagulant or antiplatelet agents.
Peripheral arterial disease
Another important field of application was identified in PAD. The main symptom of PAD is intermittent claudication, but it is common knowledge that many cases of PAD are asymptomatic despite the presence of overt atherothrombotic plaques and stenoses. Early diagnosis and treatment of patients with PAD are necessary not only for preventing severe stages of limb ischemia (critical leg ischemia), but especially for limiting diffusion of the atherothrombotic process to vital arterial areas.82 In fact, PAD patients are at high risk of AMI, stroke, and cardiovascular mortality, and consequently control of disease progression and diffusion is essential.83
A first pathophysiological approach to SDX in PAD showed beneficial effects of the agent on blood rheology and fibrinogen.84 Further clinical studies suggesting a benefit of SDX on intermittent claudication were evaluated in a meta-analysis by Gaddi et al.85 In this systematic review, 18 studies of SDX in PAD, with a total of 1,159 patients, qualified for inclusion. SDX treatment was associated with an increase of the pain-free walking distance by 36%. This effect was accompanied by a lowering of fibrinogen, plasma viscosity, and triglycerides, with an increase of HDL-cholesterol. Thus, at that time, there was already a solid suggestion of the efficacy of SDX in modifying the symptoms and risk factors of PAD. However, a randomized controlled study of adequate numerosity was necessary to confirm the value of SDX in this condition.
In the SUAVIS – arterial arm study,26 a randomized double-blind, placebo-controlled trial on 286 patients with intermittent claudication adequately instructed for progressive walking exercise, SDX was administered first parenterally for 20 days, and then orally for 6 months. Doubling of the pain-free walking distance at end-treatment was achieved by 23.8% of patients in the SDX group vs 9.2% in the placebo group. The average pain-free walking distance increased from baseline by 65% with SDX vs 28% with placebo and the increase in maximum walking distance was even greater (Figure 3). Results were equivalent for patients with or without diabetes and a concurrent decrease in fibrinogen levels was observed with SDX. All quoted differences were significant. Although the study was not designed to disclose effects on cardiovascular events, it is interesting to note that only four patients under SDX experienced major vascular events vs eleven with placebo. Thus, a further trial of SDX focused on evolution and the outcomes of PAD-related major cardiovascular events now seems warranted. A further comparative study on intermittent claudication also suggested a superiority of SDX over pentoxifylline (PTX).86
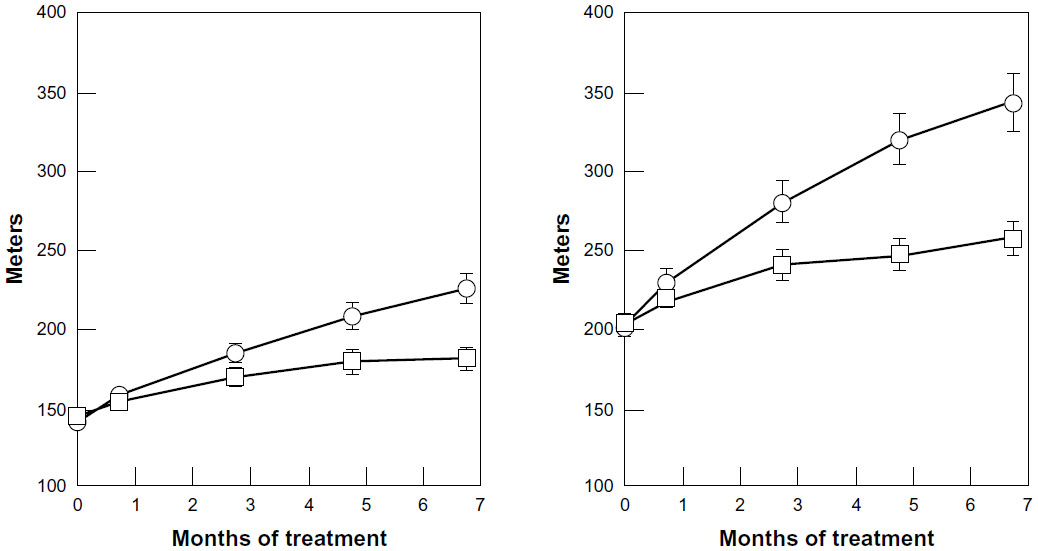 | Figure 3 Mean (± SEM) pain-free (left panel) and maximum walking distance (right panel) during treatment with sulodexide (n = 141) or placebo (n = 143) in patients with peripheral arterial obstructive disease (P = 0.001 in both comparisons). Also, considering the total increase (in meters), a statistically significant difference was reached for both pain-free walking distance and maximum walking distance (P = 0.001 for both comparisons). |
 = sulodexide;
= sulodexide;  = placebo. Copyright © 2002. Reproduced with permission from Oxford University Press. Coccheri S, Scondotto G, Agnelli G, Palazzini E, Zamboni V. Sulodexide in the treatment of intermittent claudication. Results of a randomized, double blind, multicentre, placebo-controlled study. Eur Heart J. 2002;23(13):1057–1065.
= placebo. Copyright © 2002. Reproduced with permission from Oxford University Press. Coccheri S, Scondotto G, Agnelli G, Palazzini E, Zamboni V. Sulodexide in the treatment of intermittent claudication. Results of a randomized, double blind, multicentre, placebo-controlled study. Eur Heart J. 2002;23(13):1057–1065.The efficacy of SDX in PAD was recently revisited by Lasierra-Cirujeda et al,87 who commented on the beneficial effects of SDX on fibrinogen, lipids, hemorheology, and fibrinolysis, stressing also the absence of interaction between SDX and most other drugs used in the long-term treatment of PAD, including aspirin.
Cerebrovascular disorders
Regarding cerebrovascular diseases, a number of early studies will not be quoted here because of small size or weak design. The efficacy of treatment with oral SDX for 6 months was compared with that of ticlopidine after carotid endarterectomy.88 Differences in the outcome of intimal thickening (initial restenosis) were not significant, but SDX compared well with ticlopidine.
It is also important here to mention vascular dementia (VaD), a condition of cognitive impairment in which macro- and especially microvascular alterations in the brain may play a crucial role. In the VADISS (VaD Italian SDX study), a multicenter, double-blind trial, 86 patients with ascertained VaD were randomly treated with oral SDX or PTX for 6 months.89 Clinical efficacy assessed by means of a validated rating scale for dementia (GBS) was in favor of SDX vs PTX in terms of the amelioration of motor, cognitive, and emotional functions. This study represents an unusual application of SDX that may deserve further extension.
Diabetic nephropathy and other diabetic complications
DN and proteinuria
Diabetic nephropathy (DN) is one of the main causes of morbidity and mortality in patients with diabetes. DN is conducive to end-stage renal disease in about 10% of cases of type 2 diabetes, and also increases the risk of fatal and nonfatal cardiovascular events, which are particularly common in diabetes.90
The onset of DN is often anticipated by, and thereafter associated with proteinuria, indicated as micro-albuminuria for 20–200 mg/dL and as macro-albuminuria when exceeding 200 mg/dL. The initial pathologic substrate of DN consists of thickening and proliferation of the mesangial matrix. The structural alteration of basement membranes consists in decreased synthesis, depolymerization, and depletion of the main anionic membrane GAG, HS.91 The process is associated with the induction and activation of heparanase-1 (HPR1), a specific enzyme degrading HP-like substances.92 Mesangial expansion also occurs, likely due to increased levels of TGF-²1.39 The consequent changes in the glomerular membrane electrostatic charge allow leaking of albumin into the glomerular filtrate. The high protein concentration in the filtrate damages tubular cells and stimulate interstitial fibrosis, thus leading to chronic renal damage and consequently, renal insufficiency.91
Biological background for SDX in diabetic nephropathy
Among its endothelial-protecting properties, in preclinical investigations, SDX showed a specific protection effect on endothelial alterations, including those linked to diabetes. A number of toxic and pro-inflammatory effects occurring in cultured EC after glucose supplementation, such as increased free radicals, monocyte chemotactic protein and interleukin 6 production, were effectively inhibited by exposure to SDX in the culture medium.93 SDX administered in vivo in diabetic rats inhibited hyperoxidative processes in the kidney tissue, as shown by enhancement of superoxide dismutase and catalase activities and the reduction of malondialdehyde production.94 Correspondingly, glomerular basement membrane thickening was prevented and albumin excretion reduced in comparison with untreated diabetic rats. In streptozotocin-induced DN in mice, SDX treatment reduced proteinuria and improved renal function, although it had multiple and differential effects on several signaling pathways that are yet to be understood.95 It was also recently demonstrated that long-term treatment with oral SDX in diabetic rats, besides reducing albuminuria, suppresses synthesis of vascular endothelial growth factor, and the renal expression of profibrotic molecules.96 A renoprotective effect was also seen with SDX in rats with doxorubicin-induced nephropathy: the related podocyte alterations as well as proteinuria were reduced and the increased expression of heparanase, usually occurring in this toxic nephropathy, was also inhibited.97
The inhibitory effect of SDX on HPR1, the heparan sulfate cleaving enzyme, was in fact confirmed in a recent in vitro study on proximal tubular cell lines.92 This effect of SDX is a specific property of the FMH component of SDX. Likewise, SDX counteracts mechanisms of tubular fibrosis by inhibiting overexpression of mesenchymal markers and hence the mesenchymal transition of epithelial tubular cells, thus likely opposing the progression to tubular fibrosis and end-stage renal failure.92
Certain limitations of the protective effects of SDX were observed by Rossini et al,98 in their experimental work in rats that were genetically diabetic or with radiation-induced nephropathy. In both conditions, SDX administration was associated with a reduction in early proteinuria, but no effect was recorded on late proteinuria, and mesangial matrix expansion was not prevented.
The above mentioned results confirm the nephroprotective effect of SDX, also considering that this agent, by preventing HS degradation in the microvascular wall, favors restoration of the ‘permselectivity’ of basement membranes.99 However, the concomitant effects of GAGs and angiotensin II on HS degradation and resynthesis could possibly dull the antiproteinuric effect of SDX. In fact, an experimental study on subtotally nephrectomized rats comparing the effects of SDX, irbesartan (IRB), and their association, showed that increased serum creatinine was reduced by SDX in early phases, and only later by IRB. SDX or IRB appeared equivalent in reversing the reduced expression of endothelial nitric oxide synthase. Overall, SDX had similar effects to IRB but no further advantage could be gained by their association.100
These concepts may help the clinician in the interpretation of some controversial results obtained with SDX in diabetic nephropathy.
Clinical pharmacology and trials
In a small human pharmacology study,42 the effect of oral SDX on the endothelial glycocalyx was studied in patients with type 2 diabetes. The baseline glycocalyx dimension in the retinal and sublingual microvasculature was decreased in diabetics and restored by SDX. The increased transcapillary albumin excretion rate (AER) usually found in diabetic subjects also showed an evident trend to reduction. Similarly, the elevated plasma hyaluronidase, a marker of accelerated HA metabolism during diabetes, was also restored to lower values.
After a number of small studies, a multicenter randomized trial, the DiNAS (DN albuminuria SDX), published in 2002,101 showed that oral SDX given for 4 months at the doses of 50, 100, or 200 mg vs placebo to patients with type 2 diabetes, significantly decreased their AER. The effect was dose-dependent and appeared maximal with the 200 mg dose, was maintained for at least 4 months, and was not different according to the presence of macro- or micro-albuminuria, type 1 or type 2 diabetes, and concomitant treatment or no treatment with angiotensin converting enzyme inhibitors (ACEIs). The authors concluded that, SDX was effective and safe in the treatment of diabetic proteinuria. Similar results were obtained of a study102 of a cohort of 237 diabetic patients (90% type 2) treated for 6 months with a low dose of oral SDX. By analogy, in a common renal condition also characterized by glomerular microvascular damage, the immunoglobulin-A nephropathy, a double-blind randomized study103 showed that oral SDX achieved a 50% reduction of urinary protein excretion in a number of patients (21.4% vs 12.5% in the controls) and significantly reduced their proteinuria/creatinine ratio.
After the DiNAS study, SDX gained the interest of many nephrologic groups as a possible supportive agent in patients with incomplete response or persisting albuminuria despite treatment with ACEIs or angiotensin receptor blocker (ARBs).
A pilot study104 indicated that oral SDX at 200 mg daily was more often successful, although not significantly, than placebo in inducing at least 50% reduction of proteinuria in patients with type 2 diabetes treated with maximal allowed doses of ACE inhibitors or ARBs. The subsequent large, multicenter, double-blind trial of SDX in 1,056 type 2 diabetics with microalbuminuria failed however, to demonstrate an advantage of SDX added on top of maximal ACEI/ARB treatment.105 A parallel study in type 2 diabetes patients with more severe renal impairment and macro-albuminuria, also treated with the maximal allowed doses of ACEI/ARB agents, was prematurely stopped and could not demonstrate any effect of additional SDX on counteracting the marked increase in serum creatinine and occurrence of end-stage renal failure.106
After the publication of these studies, and especially after the publication of an editorial107 that sounded a capital sentence for SDX in this indication, we forwarded several objections to some aspects of the two trials.108 In fact, patients enrolled in the study by Lewis et al 105 were obviously more severe (mostly chronic kidney disease, CKD, stage >3) than the majority of those in the DiNAS. Even more heavily affected were those included in the parallel evolution study.106 A further concern was that the requirement of a maximal and protracted treatment with ACE or ARB inhibitors may have resulted in under-evaluation of the real levels of proteinuria, and may have left little or no allowance for a superimposed effect of SDX. As previously reported, there is also some experimental indication that maximal anti-angiotensin treatment could dull the antiproteinuric effect of SDX.100 Thus, studies with a specific design may be necessary in order to clarify whether the two treatments (ie, angiotensin inhibition and SDX) can be advantageously associated.
In our opinion, the results of the two above trials cannot deny the antiproteinuric efficacy of SDX, and dismissing SDX from this indication is unjustified, as was also pointed out in other commentaries.109,110 The agent is likely more effective in patients at the initial phases of type 2 diabetes or diabetic nephropathy, not necessarily treated with maximal doses of ACEIs or ARBs, and deserves further evaluation in more appropriate conditions.
Diabetic retinopathy and macular edema
Beneficial results have been reported with SDX in early diabetic non-proliferative retinopathy in studies111–113 in which oral treatment with SDX was associated with a reduction in retinal microvascular abnormalities, hard exudates, and hemorrhages; regression of associated macular edema was also observed.
Diabetic neuropathy and diabetic foot
SDX has recently been shown to counteract peripheral nerve damage caused by microvascular dysfunction in streptozotocin-induced diabetes in rats,114 but only preliminary clinical data are available on diabetic foot.115
Other clinical perspectives
Preliminary results have also been obtained in retinal vein occlusion. In a controlled study,116 significantly greater visual and fluoroangiographic recovery were shown following a therapeutic protocol that included SDX. Also, in cases of senile macular degeneration, slowing of the evolution or slight improvement of visual acuity were reported.117
In the VascVert study on vascular vertigo, oral SDX appeared more effective than anti-aggregating agents (aspirin or ticlopidine).118 Similarly, in a controlled study of patients with tinnitus, all treated with melatonin to restore the sleep–wake equilibrium, supplementation with oral SDX was associated with improvements of acufenometry and specific quality of life.119
Conclusion
SDX, for a long time considered a ‘minor’ antithrombotic agent similar to other heparin-like substances, has gained renewed interest especially during the last decade. In fact, after oral administration, SDX displays new biological properties, such as the ability to regulate endothelium blood cell interactions; to counteract vascular inflammatory and proliferative changes; and to protect and restore structures and functions of the injured endothelium, all this while maintaining to a mild degree those inherent antithrombotic and profibrinolytic activities that prevail after parenteral administration.
Clinical evidence of the efficacy of SDX by the oral route have been obtained in chronic venous disease; adjuvant treatment of venous ulcers; prevention of cardiovascular events after myocardial infarction; relief of intermittent claudication in patients with peripheral arterial disease; and reduction of proteinuria in diabetic nephropathy. A collaborative trial is presently under way on the prevention of thromboembolic recurrences during extended treatment after deep vein thrombosis. Concerning diabetic nephropathy, new trials specifically designed for the prevention of renal damage in recent onset diabetic subjects rather than for treatment of overt or severe diabetic nephropathy are highly desirable.
Acknowledgments
The authors wish to acknowledge the significant contributions of: Dr Donatella Orlando (Bologna) for her invaluable collaboration in the organization and preparation of the manuscript; Dr Daniela Ligi (Urbino) who contributed to the biological chapters and figure composition; and Dr Giuseppe D’Ambrosio (Milan) who kindly supplied internal material from Alfa-Wassermann.
Disclosure
The authors report no conflicts of interest in this work.
References
Afratis N, Gialeli C, Nikitovic D, et al. Glycosaminoglycans: key players in cancer cell biology and treatment. FEBS J. 2012;279:1177–1197. | |
Gandhi NS, Mancera RL. The structure of glycosaminoglycans and their interactions with proteins. Chem Biol Drug Des. 2008;72(6):455–482. | |
Rudd TR, Skidmore MA, Guimond SE, et al. Glycosamino-glycan origin and structure revealed by multivariate analysis of NMR and CD spectra. Glycobiology. 2009;19(1):52–67. | |
Asimakopoulou AP, Theocharis AD, Tzanakakis GN, Karamanos NK. The biological role of chondroitin sulfate in cancer and chondroitin-based anticancer agents. In Vivo. 2008;22(3):385–389. | |
Lauder RM. Chondroitin sulphate: a complex molecule with potential impacts on a wide range of biological systems. Complement Ther Med. 2009;17:56–62. | |
Tollefsen DM. Vascular dermatan sulphate and heparin cofactor II. Prog Mol Biol Transl Sci. 2010;93:351–372. | |
Malavaki CJ, Theocharis AD, Lamari FN, et al. Heparan sulfate: biological significance, tools for biochemical analysis and structural characterization. Biomed Chromatogr. 2011;25(1–2):11–20. | |
Mulloy B. Structure and physicochemical characterisation of heparin. Handb Exp Pharmacol. 2012;(207):77–98. | |
Dreyfuss JL, Regatieri CV, Jarrouge TR, Cavalheiro RP, Sampaio LO, Nader HB. Heparan sulfate proteoglycans: structure, protein interactions and cell signaling. An Acad Bras Cienc. 2009;81(3):409–429. | |
Bick RL, Frenkel EP, Walenga J, Fareed J, Hoppensteadt DA. Unfractionated heparin, low molecular weight heparins, and pentasaccharide: basic mechanism of actions, pharmacology and clinical use. Hematol Oncol Clin North Am. 2005;19(1):1–51. | |
Bianchini P, inventor; Opocrin SRL, assignee. Method for preparing glucoronyl-glucosamino-glycan sulphates exhibiting antilipasaemic activity. United States patent US 3936351. February 3, 1976. | |
Silvestro L, Lanzarotti E, Marchi E, et al. Human pharmacokinetics of glycosaminoglycans using deuterium-labeled and unlabeled substances: evidence for oral absorption. Semin Thromb Hemost. 1994;20: 281–292. | |
Cosmi B, Cini M, Legnani C, Pancani C, Calanni F, Coccheri S. Additive thrombin inhibition by fast moving heparin and dermatan sulfate explains the anticoagulant effect of sulodexide, a natural mixture of glycosaminoglycans. Thromb Res. 2003;109(5–6):333–339. | |
Iacoviello L, D’Adamo MC, Pawlak K, et al. Antithrombotic activity of dermatan sulphates, heparins and their combination in an animal model of arterial thrombosis. Thromb Haemost. 1996;76(6):1102–1107. | |
Di Carlo V, Agnelli G, Prandoni P, et al. Dermatan sulphate for the prevention of postoperative venous thromboembolism in patients with cancer. DOS (Dermatan sulphate Oncolologic Surgery) Study Group. Thromb Haemost. 1999;82(1):30–34. | |
Mannello F, Raffetto JD. Matrix metalloproteinase activity and glycosaminoglycans in chronic venous disease: the linkage among cell biology, pathology and translational research. Am J Transl Res. 2011;3(2):149–158. | |
Mattana P, Mannello F, Ferrari P, Agus GB. Vascular pathologies and inflammation: the antinflammatory properties of sulodexide. J Vasc Endovasc Surg. 2012;19:1–7. | |
Plichta JK, Radek KA. Sugar coating wound repair: a review of FGF10 and DS in wound healing and their potential application in burn wounds. J Burn Care Res. 2012;33:299–310. | |
Milani MR, Busutti L, Breccia A, et al. Pharmacokinetics of sulodexide evaluation from 131-labelled fast-moving heparin after single intravenous and oral administration on man at different doses. Brit J Clin Res. 1992;3:161–178. | |
Marchi E, Barbanti M, Milani R, et al. Organ glycosaminoglycan distribution after intravenous and oral administration in rats. Semin Thromb Hemost. 1994;20:297–300. | |
Ofosu FA. Pharmacological actions of sulodexide. Semin Thromb Hemost. 1998;24:127–137. | |
Adiguzel C, Iqbal O, Hoppensteadt D, et al. Comparative anticoagulant and platelet modulatory effects of enoxaparin and Sulodexide. Clin Appl Thromb Hemost. 2009;15(5):501–511. | |
Harenberg J. Review of pharmacodynamics, pharmacokinetics, and therapeutic properties of sulodexide. Med Res Rev. 1998;18:1–20. | |
Borawski J, Dubowski M, Rydzewska-Rosolowska A, Mysliwiec M. Intravenous and oral Sulodexide versus coagulation activation markers in humans. Clin Appl Thromb Hemost. 2009;15(5):596–598. | |
Borawski J, Naumnik B, Dubowski M, Mysliwiec M. Full-length TFPI release by heparinoid Sulodexide. Clin Appl Thromb Hemost. 2010;16(4):485–487. | |
Coccheri S, Scondotto G, Agnelli G, Palazzini E, Zamboni V. Sulodexide in the treatment of intermittent claudication. Results of a randomized, double blind, multicentre, placebo-controlled study. Eur Heart J. 2002;23(13):1057–1065. | |
Callas DD, Hoppensteadt DA, Jeske W, et al. Comparative pharmacologic profile of a glycosaminoglycan mixture, Sulodexide, and a chemically modified heparin derivative, Suleparoid. Semin Thromb Hemost. 1993;19 Suppl 1:49–57. | |
Ceriello A, Quatrano A, Marchi E, Barbanti M, Giugliano D. Impaired fibrinolytic response to increased thrombin activation in type 1 diabetes mellitus: effects of the glycosaminoglycan sulodexide. Diabetes Metab. 1993;19(2):225–229. | |
Buchanan MR, Liao P, Smith LJ, et al. Prevention of thrombus formation and growth by antithrombin III and heparin cofactor II-dependent thrombin inhibitors:: importance of heparin cofactor II. Thromb Res. 1994;74:463–475. | |
Hornstra G, Vendelmans-Starrenburg A. Induction of experimental arterial occlusive thrombi in rats. Atherosclerosis. 1973;17(3):369–382. | |
Andriuoli G, Mastacchi R, Barbanti M. Antithrombotic activity of a glycosaminoglycan (sulodexide) in rats. Thromb Res. 1984;34(1):81–86. | |
Tarugi P, Tiozzo-Costa, Barbanti M, et al. Effect of sulodexide, a heparin-like compound, on experimental atherosclerosis in the rabbit. Med Sci Res. 1987;15:1071–1072. | |
Barbanti M, Guizzardi S, Calanni F, Marchi E, Babbini M. Antithrombotic and thrombolytic activity of Sulodexide in rats. Int J Clin Lab Res. 1992;22(3):179–184. | |
Agrati AM, Mauro M, Savasta C, et al. A double-blind, cross-over, placebo-controlled study of the profibrinolytic and antithrombotic effects of oral sulodexide. Adv Ther. 1992;9:147–155. | |
Mauro M, Ferraro G, Palmieri GC. Profibrinolytic and antithrombotic effects of sulodexide oral administration: a double-blind, cross-over, placebo-controlled study. Curr Ther Res. 1992;51:342–350. | |
Mannarino E, Pasqualini L, Ciuffetti G, et al. Effect of oral administration of sulodexide on fibrinolysis and plasma viscosity: a pilot study. Drug Invest. 1992;4:346–350. | |
Cerletti C, Rajtar G, Marchi E, et al. Interaction between glycosaminoglycans, platelets and leukocytes. Semin Thromb Hemost. 1994;20: 245–253. | |
Mannello F, Medda V, Ligi D, Raffetto JD. Glycosaminoglycan Sulodexide inhibition of MMP-9 gelatinase secretion and activity: possible pharmacological role against collagen degradation in vascular chronic diseases. Curr Vasc Pharmacol. 2013;11(3):354–365. | |
Borawski J, Dubowski M, Pawlak K, et al. Effect of Sulodexide on plasma transforming growth factor ß1 in healthy volunteers. Clin Appl Thromb Hemost. 2010;16:60–65. | |
Tiozzo R, Cingi MR, Pietrangelo A, et al. Effect of heparin-like compounds on the in vitro proliferation and protein synthesis of various cell types. Arzneimittelforschung. 1989;39(1):15–20. | |
Borawski J, Dubowski M, Pawlak K, Mysliwiec M. Sulodexide induces hepatocyte growth factor release in humans. Eur J Pharmacol. 2007;558(1–3):167–171. | |
Broekhuizen LN, Lemkes BA, Mooij HL, et al. Effect of sulodexide on endothelial glycocalyx and vascular permeability in patients with type 2 diabetes mellitus. Diabetologia. 2010;53:2646–2655. | |
Kristova V, Liskova S, Sotnikova R, et al. Sulodexide improves endothelial dysfunction in streptozotocin-induced diabetes in rats. Physiol Res. 2008;57:491–494. | |
Vasquez J, Mathison Y, Romero-Vecchione E, et al. Effect of sulodexide on aortic vasodilation capacity and associated morphological changes in rats with streptozotocin-induced diabetes. Invest Clin. 2010;51(4):467–477. Spanish. | |
Crepaldi G, Fellin R, Calabrò A, et al. Double-blind multicenter trial on a new medium molecular weight glycosaminoglycan. Current therapeutic effects and perspectives for clinical use. Atherosclerosis. 1990;8(13):233–243. | |
Lunetta M, Salanitri T. Lowering of plasma viscosity by the oral administration of the glycosaminoglycan sulodexide in patients with peripheral vascular disease. J Int Med Res. 1992;20(1):45–53. | |
Cospite M, Ferrara F, Cospite V, et al. Sulodexide and the microcirculatory component in microphlebopathies. Curr Med Res. 1992;13(1):56–60. | |
Park HY, Kang S, Kim GY, et al. Inhibition of neointimal proliferation of rat carotid artery by sulodexide. J Korean Med Sci. 1997;12(3):210–214. | |
Bergan JJ, Schmid-Schonbein GW, Smith PD, et al. Chronic venous disease. N Engl J Med. 2006;355(5):488–498. | |
Andreozzi GM. Sulodexide in the treatment of chronic venous disease. Am J Cardiovasc Drugs. 2012;12(2):73–81. | |
Evans CJ, Fowkes FG, Ruckley CV, et al. Prevalence of varicose veins and chronic venous insufficiency in men and women in the general population: Edinburgh Vein Study. J Epidemiol Community Health. 1999;53(3):149–153. | |
Maurins U, Hoffman BH, Losch C, et al. Distribution and prevalence of reflux in the superficial and deep venous system in the general population: results from the Bonn Vein Study, Germany. J Vasc Surg. 2008;48(3):680–687. | |
Eklof B, Rutherford RB, Bergan JJ, et al. Revision of the CEAP classification for chronic venous disorders: consensus statement. J Vasc Surg. 2004;40(6):1248–1252. | |
Bergan JJ, Pascarella L, Schmid-Schoenbein GW. Pathogenesis of primary chronic venous disease: Insights from animal models of venous hypertension. J Vasc Surg. 2008;47(1):183–192. | |
Cohen JM, Akl EA, Khan SR. Pharmacologic and compression therapies for post thrombotic syndrome. Chest. 2012;141(2):308–320. | |
Marinovic´ KuliŠic´ S, Lupi D. Pharmacological treatment in patients with chronic venous disease. Acta Dermatovenerol Croat. 2012;20(3):197–200. | |
Lauver AD, Lucchesi BR. Sulodexide: a renewed interest in this glycosaminoglycan. Cardiovasc Drug Rev. 2006;24(3–4):214–226. | |
Lei Y, Zheng Z, Wang Y, et al. Sulodexide may alleviate neointimal hyperplasia by inhibiting angiopoietin-2 in an arteriovenous fistula model. Mol Med Rep. 2013;7(3):831–835. | |
Saviano M, Maleti O, Liguori L. Double-blind, double-dummy, randomized, multicentre clinical assessment of the efficacy, tolerability and dose-effect relationship of Sulodexide in chronic venous insufficiency. Curr Med Res Opin. 1993;13(2):76–108. | |
Gloviczki P, Camerota AJ, Dalsing MC, et al. The care of patients with varicose veins and associated chronic venous diseases: clinical practice guidelines of the American Society for Vascular Surgery and the American Venous Forum. J Vasc Surg. 2011;53(5S):2S–28S. | |
Kahn SR. The post-thrombotic syndrome: prognosis and pitfalls. Br J Haematol. 2006;134(4):357–365. | |
Palareti G, Legnani C, Cosmi B, et al. Predictive value of D-dimer test for recurrent venous thromboembolism after anticoagulation withdrawal in subjects with a previous idiopathic event and carriers of congenital thrombophilia. Circulation. 2003;108(39):313–318. | |
Palareti G, Leali N, Coccheri S, et al. Bleeding complications of oral anticoagulant treatment: an inception-cohort, prospective collaborative study (ISCOAT). Italian Study on Complications of Oral Anticoagulant Therapy. Lancet. 1996;348:423–428. | |
Connors JM. Extended treatment of venous thromboembolism. N Engl J Med. 2013;368(8):767–769. | |
Becattini C, Agnelli G, Schenone A, et al. WARFASA Investigators. Aspirin for preventing the recurrence of venous thromboembolism. N Engl J Med. 2012;366(1):1959–1967. | |
Brighton TA, Eikelboom JW, Mann K, et al. ASPIRE Investigators. Low-dose aspirin for preventing recurrent venous thromboembolism. N Engl J Med. 2012;367(21):1979–1987. | |
Garcia Rodriguez LA, Lin KJ, Hernandez-Diaz S, Johansson S. Risk of upper gastrointestinal bleeding with low-dose acetylsalicylic acid alone and in combination with clopidogrel and other medications. Circulation. 2011;123:1108–1115. | |
Cirujeda JL, Granado PC. A study on the safety, efficacy, and efficiency of sulodexide compared with acenocoumarol in secondary prophylaxis in patients with deep vein thrombosis. Angiology. 2010;57(1):53–64. | |
Errichi BM, Cesarone MR, Belcaro G, et al. Prevention of recurrent deep venous thrombosis with sulodexide: the SanVal registry. Angiology. 2004;55(3):243–249. | |
Andreozzi GM, Palareti G. Rationale and study design of the SURVET trial (Sulodexide in Secondary Prevention of Recurrent Deep Vein Thrombosis). Proceedings of the 21st International Congress on Thrombosis; July 6–9, 2010; Milan, Italy. Abstract; p302. | |
Nicolaides AN. Chronic venous disease and the leukocyte-endothelium interaction: from symptoms to ulceration. Angiology. 2005;56: S11–9. | |
Falanga V, Saap LJ, Ozonoff A. Wound bed score and its correlation with healing of chronic wounds. Dermatol Ther. 2006;19(6):383–390. | |
Kurz X, Kahn SR, Abenhaim L, et al. Chronic venous disorders of the leg: epidemiology, outcomes, diagnosis and management. Summary of an evidence-based report of the VEINES task force. Venous Insufficiency Epidemiologic and Economic Studies. Int Angiol. 1999;18(2):83–102. | |
Scondotto G, Aloisi D, Ferrari P, et al. Treatment of venous leg ulcers with Sulodexide. Angiology. 1999;50(1):883–889. | |
Coccheri S, Scondotto G, Agnelli G, Aloisi D, Palazzini E, Zamboni V. (SUAVIS group). Randomised, double blind, multicentre, placebo controlled study of sulodexide in the treatment of venous leg ulcers. Thromb Haemost. 2002;87(6):947–952. | |
Kucharzewski M, Franek A, Koziolek H. Treatment of venous leg ulcers with Sulodexide. Phlebologie. 2003;32:115–120. | |
Arosio E, Ferrari G, Santoro L, Gianese F, Coccheri S; Mesoglycan Venous Insufficiency Group. A placebo-controlled, double blind study of mesoglycan in the treatment of chronic venous ulcers. Eur J Vasc Endovasc Surg. 2001;22(4):365–372. | |
Nelson EA. Venous leg ulcers. Clin Evid. 2011;12:1902. | |
Kearon C, Kahan SR, Agnelli G, et al. Antithrombotic therapy for venous thromboembolic disease: American College of Chest Physicians Evidence-Based Clinical Practice Guidelines (8th ed). Chest. 2008;133: 454S–545S. | |
Baigent C, Blackwell L, Collins R, et al. Aspirin in the primary and secondary prevention of vascular disease: collaborative meta-analysis of individual participant data from randomised trials. Lancet. 2009;373: 1849–1860. | |
Condorelli M, Chiariello M, Dagianti A, et al. IPO-V2: a prospective, multicenter, randomized, comparative clinical investigation of the effects of sulodexide in preventing cardiovascular accidents in the first year after acute myocardial infarction. J Am Coll Cardiol. 1994;23(1):27–34. | |
Coccheri S, Palareti G. The cardiovascular risk burden of intermittent claudication. Eur Heart J. 2002;4(Suppl B):B46–B49. | |
Criqui MH, Denenberg JO, Langer RD, Fronek A. The epidemiology of peripheral arterial disease: importance of identifying the population at risk. Vasc Med. 1997;2:221–226. | |
Cicco G, Stingi GD, Vicenti P, Tarallo MS, Pirrelli A. Hemorheology and tissue oxygenation in hypertensives with lipoidoproteinosis and peripheral occlusive arterial disease (POAD) treated with sulodexide and pravastatine and evaluated with laser assisted optical rotational red cell analyzer (LORCA) and transcutaneous oxymetry. Min Cardioangiol. 1999;47:351–359. | |
Gaddi A, Galletti C, Illuminati B, et al. Meta-analysis of some results of clinical trials on sulodexide therapy in peripheral occlusive arterial disease. J Int Med Res. 1996;24:389–406. | |
Shustov SB. Controlled clinical trial on the efficacy and safety of oral sulodexide in patients with peripheral occlusive arterial disease. Curr Med Res Opin. 1997;13(19):573–582. | |
Lasierra-Cirujeda J, Coronel P, Aza MJ, Gimeno M. Use of sulodexide in patients with peripheral vascular disease. J Blood Med. 2010;1: 105–115. | |
Stella A, Tarantini G, Palareti G, et al. Pharmacological therapy after carotid endoarterectomy: heparin, sulodexide, ticlopidine. G Ital Chir Vasc. 1999;6:161–179. | |
Parnetti L, Mari D, Abate G, et al. Vascular dementia Italian sulodexide study (VA.D.I.S.S.). Clinical and biological results. Thromb Res. 1997;87(2):225–233. | |
Coccheri S. Approaches to prevention of cardiovascular complications and events in diabetes mellitus. Drugs. 2007;67(7):997–1026. | |
Weiss R, Niecestro R, Raz I. The role of Sulodexide in the treatment of diabetic nephropathy. Drugs. 2007;67(18):2681–2696. | |
Masola V, Onisto M, Zaza G, Lupo A, Gambaro G. A new mechanism of action of Sulodexide in diabetic nephropathy: inhibits heparanase-1 and prevents FGF-2 induced renal epithelial mesenchymal transition. J Transl Med. 2012;10:213. | |
Ciszewicz M, Polubinska A, Antoniewicz A, Suminska-Janinska K, Breborowicz A. Sulodexide suppresses inflammation in human endothelial cells and prevents glucose cytoxicity. Transl Res. 2009;153(3):118–123. | |
Shu J, Zeng LY, Lin KY, et al. Renal protective effects of Sulodexide in diabetic rats and its antioxidative mechanism. Nan Fang Yi Ke Da Xue Xue Bao. 2009;29(4):778–780. Chinese. | |
Yung S, Chau MK, Zhang CZ, Chan TM. Sulodexide decreases albuminuria and regulates matrix protein accumulation in C57BL/6 mice with streptozotocin-induced type I diabetic nephropathy. PLoS One. 2013;8(1):e54501. | |
Cha JJ, Kang YS, Hyun YY, et al. Sulodexide improves renal function through reduction of vascular endothelial growth factor in type 2 diabetic rats. Life Sci. 2013;92(23):118–124. | |
Chen S, Fang Z, Zhu Z, Deng A, Liu J, Zhang C. Protective effect of sulodexide on podocyte injury in adriamycin nephropaty rats. J Huazhong Univ Sci Technolog Med Sci. 2009;29(6):715–719. | |
Rossini M, Naito T, Yang H, et al. Sulodexide ameliorates early but not late kidney disease in models of radiation nephropaty and diabetic nephropaty. Nephrol Dial Transplant. 2010;25(6):1803–1810. | |
Gaddi AV, Cicero AF, Gambaro G. Nephroprotective action of glycosaminoglycans: why the pharmacological properties of sulodexide might be reconsidered. Int J Nephrol Renovasc Dis. 2010;3: 99–105. | |
Li P, Ma LL, Xie RJ, et al. Treatment of 5/6 nephrectomy rats with sulodexide: a novel therapy for chronic renal failure. Acta Pharmacol Sin. 2012;33(5):644–651. | |
Gambaro G, Kinalska I, Oksa A, et al. Oral sulodexide reduces albuminuria in microalbuminuric and macroalbuminuric type 1 and type 2 diabetic patients: the Di.N.A.s: randomized trial. J Am Soc Nephrol. 2002;13(6):1615–1625. | |
Blouza S, Dakhii S, Abid H, Aissaoui M, et al; DAVET (Diabetic Albuminuria Vessel Tunisia) Study Investigators. Efficacy of low-dose oral Sulodexide in the management of diabetic nephropathy. J Nephrol. 2010;23(4):415–424. | |
Bang K, Chin HJ, Chae DW, et al. Anti-proteinuric effect of sulodexide in immunoglobulin a nephropathy. Yonsei Med J. 2011;52(4):588–594. | |
Heersprink HL, Greene T, Lewis JB, et al. Collaborative Study Group. Effect of Sulodexide in patients with type 2 diabetes and persistent albuminuria. Nephrol Dial Transplant. 2008;23:1946–1954. | |
Lewis EJ, Lewis JB, Greene T, et al; Collaborative Study Group. Sulodexide for kidney protection in type 2 diabetes patients with macroalbuminuria: a randomized controlled trial. Am J Kidney Dis. 2011;58(5):729–736. | |
Packham DK, Wolfe R, Reutens AT, et al; Collaborative Study Group. Sulodexide fails to demonstrate renoprotection in overt type 2 diabetic nephropathy. J Am Soc Nephrol. 2012;23:123–130. | |
House AA, Weir MA. Sulodexide for diabetic nephropathy: another one bites the dust. Am J Kidney Dis. 2011;58(5):692–694. | |
Coccheri S. Game not over for sulodexide. Am J Kidney Dis. 2012; 59(3):467. | |
Gaddi AV, Benedetto D. Surrogate versus clinical end points and venture capital companies: the doubts of a clinician. Am J Kidney Dis. 2012;60(1):168–171. | |
Gambaro G. Discounting the efficacy of sulodexide in diabetic nephropathy is premature. Am J Kidney Dis. 2012;69(1):169–170. | |
Rubbi F, Caramazza R, Boccia S, et al. The effects of sulodexide on diabetic retinopathy. Minerva Cardioangiol. 2000;48(Suppl 1):81–82. | |
Karimov KT, Shakhmaliyeva AM. Effect of sulodexide on non-proliferative diabetic retinopathy. Azerbaijan Med J. 2002;1:72–76. | |
Chin HS, Kang SW, Kawak HW, et al. Effect of sulodexide in diabetic patients with mild to moderate nonproliferative retinopathy. AAO 2011 – Annual Meeting of the American Academy of Ophtalmology. Abstract 498; p221. | |
Jin HY, Lee KE, Song SK, et al. Sulodexide prevents peripheral nerve damage in streptozotocin induced diabetic rats. Eur J Pharmacol. 2012;674(2–3):217–226. | |
Koblik T, Sieradzki J, Sendur R, et al. The effect of insulin and sulodexide (Vessel Due F) on diabetic foot syndrome. Pilot study in elderly patients. J Diabetes Complications. 2001;15(2):69–74. | |
Anfossi DG, Bella GM, Chiriotti S. Comparative study of two different protocols in the treatment of retinal vein occlusion. Minerva Opthalmol. 1992;34:29–36. | |
Szafilk J, Kaminska A. Usefulness of Vessel Due F (sulodexide) in treatment of patients with diabetic retinopathy, senile macular degeneration and retinal vein occlusion. Okulistyka. 2000;3:1–4. | |
Guidetti G. Treatment of vascular vertigo in outpatients: multicentre experience (VascVert study). Otorinolaringol. 2005;55:237–246. | |
Neri G, Baffa C, De Stefano A, et al. Management of tinnitus: oral treatment with melatonin and sulodexide. J Biol Regul Homeost Agents. 2009;23(2):103–110. |
 © 2013 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2013 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.