Back to Journals » International Journal of Nanomedicine » Volume 11
Development and characterization of self-assembling lecithin-based mixed polymeric micelles containing quercetin in cancer treatment and an in vivo pharmacokinetic study
Authors Chen L, Chen Y, Su C, Hong C, Ho H ![]() , Sheu M
, Sheu M ![]()
Received 5 January 2016
Accepted for publication 7 March 2016
Published 15 April 2016 Volume 2016:11 Pages 1557—1566
DOI https://doi.org/10.2147/IJN.S103681
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Professor Lei Yang
Ling-Chun Chen,1,* Ying-Chen Chen,1,* Chia-Yu Su,1 Chung-Shu Hong,1 Hsiu-O Ho,1 Ming-Thau Sheu1,2
1School of Pharmacy, College of Pharmacy, 2Clinical Research Center and Traditional Herbal Medicine Research Center, Taipei Medical University Hospital, Taipei Medical University, Taipei, Taiwan
*These authors contributed equally to this work
Abstract: Quercetin (Que) is known to have biological benefits including an anticancer effect, but low water solubility limits its clinical application. The aim of this study was to develop a lecithin-based mixed polymeric micelle (LMPM) delivery system to improve the solubility and bioavailability of Que. The optimal Que-LMPM, composed of Que, lecithin, Pluronic® P123, and 1,2-distearoyl-sn-glycero-3-phosphoethanolamine-N-methoxy[poly(ethylene glycol)-2000] in a proportion of 3:1:17.5:2.5 (w/w), was prepared by a thin-film method. The average size, polydispersion index, encapsulating efficiency, and drug loading of Que-LMPM were 61.60±5.02 nm, 0.589±0.198, 96.87%±9.04%, and 12.18%±1.11%, respectively. The solubility of Que in the Que-LMPM system increased to 5.81 mg/mL, compared to that of free Que in water of 0.17–7.7 µg/mL. The Que-LMPM system presented a sustained-release property in vitro. The in vitro cytotoxicity assay showed that the 50% inhibitory concentration values toward MCF-7 breast cancer cells for free Que, blank LMPMs, and Que-LMPMs were >200, >200, and 110 µM, respectively, indicating the nontoxicity of the LMPM carrier, but the LMPM formulation enhanced the cytotoxicity of Que against MCF-7 cells. A cellular uptake assay also confirmed the intake of Que-LMPM by MCF-7 cells. An in vivo pharmacokinetic study demonstrated that Que-LMPMs had higher area under the concentration–time curve and a longer half-life, leading to better bioavailability compared to a free Que injection. Due to their nanosize, core–shell structure, and solubilization potential, LMPMs were successfully developed as a drug delivery system for Que to improve its solubility and bioavailability.
Keywords: quercetin, micelle, lecithin, Pluronic®, DSPE-PEG, mixed micelles, bioavailability
Introduction
Breast cancer is a leading cause of cancer deaths in women in not only Taiwan but also the whole world.1 Large-scale epidemiological cohort studies reported that breast cancer is associated with cigarettes, and one study focused on nicotine (Nic) because >80% of Nic is absorbed after smoking.2,3 Another study mentioned the role of the nicotinic acetylcholine receptor (nAChR) in carcinogenesis, and Nic is known to be a high-affinity nAChR agonist.4 Therefore, inhibition of nAChR-mediated signals represents a potential strategy for breast cancer treatment. Quercetin (Que), an nAChR antagonist, was found to inhibit the proliferation of human breast cancer cells through blockage of Nic receptors and nAChR subunit expression.5 Que is a component of most edible fruits and vegetables, with the highest concentrations found in onions, apples, and red wine.6,7 Although the anticancer mechanisms are not yet fully understood, current evidence demonstrates that Que is beneficial for improving breast cancer chemotherapy and is a potential chemopreventive agent.5 However, the poor water solubility of Que leads to its minimal absorption in the gastrointestinal tract, and its oral bioavailability (BA) is <17% in rats and only 1% in men.8,9 Que was previously applied in early-stage clinical trials as an anticancer agent; however, it required the use of solvents such as dimethyl sulfoxide (DMSO) or ethanol.10 In addition, chemical modifications were attempted to improve Que’s solubility, but resulted in a loss of drug efficacy.11 The low hydrophilicity of Que thus affects its biological activity; as a result, clinical applications of Que are greatly restricted. However, two recent studies demonstrated that the antioxidant activity and antiproliferative effects of Que could be enhanced by encapsulating in PLGA nanoparticles and absorption on nanodiamond, respectively.12,13 This encouraged to utilize alternative strategies based on pharmaceutical technologies to improve the water solubility and BA of Que.
Polymeric micelle-based drug delivery systems made from amphiphilic polymers, which self-assemble into structures of hydrophobic cores and hydrophilic shells, have been widely applied for delivering poorly soluble drugs.14,15 The hydrophobic core of micelles can serve as a cargo space for encapsulating various poorly soluble therapeutic agents and is responsible for the drug stability and release patterns, while the outer hydrophilic shell protects against attack from the reticuloendothelial system (RES) and beneficially modifies the pharmacokinetics and biodistribution behavior, overall resulting in an increase in BA. The small size of micelles can achieve a favorable biodistribution; moreover, the nanosize of micelles permits their extravasation and accumulation in tumor sites, which is passive targeting by the enhanced permeability and retention effect.16–21 An additional advantage of micelles from a practical point of view is that they can simply be reproduced and are easy to prepare on a large scale.22
One limitation with traditional micelles made with an amphiphilic polymer is that the solubilization is determined by the total number of micelles in the system. To improve the solubilization ability, additional hydrophobic materials are added to form a novel delivery system, that is, mixed polymeric micelles (MPMs). The rationale of MPMs is to increase the volume of the hydrophobic core of each micelle by incorporating hydrophobic materials, and hence, MPMs provide more space in which a hydrophobic drug can be solubilized. MPMs retain all the advantages of traditional micelles described earlier, while further increasing the solubilization capacity of poorly soluble drugs. Krishnadas et al23 prepared a paclitaxel micelle system using 1,2-distearoyl-sn-glycero-3-phosphoethanolamine-N-methoxy[poly(ethylene glycol)-2000] (DSPE-PEG2K) only or a mixture of DSPE-PEG2K and egg-phosphatidylcholine (ePC), and they concluded that the latter (the mixture of DSPE-PEG2K and ePC) showed increased solubilization compared to the former one (DSPE-PEG2K). Similarly, Wei et al22 developed MPMs for paclitaxel using Pluronic® P123 and F127 (BASF, Hanover, Germany), and the 50% inhibitory concentration (IC50) of micelles against human lung adenocarcinoma A-549 cells was lower than that of a Taxol® injection (Bristol-Myers Squibb Company, Princeton, NJ, USA) (the commercial product of paclitaxel) and free paclitaxel. Notably, doxorubicin-loaded MPMs composed of Pluronic L61 and F127 were successfully evaluated in a Phase II study in patients with advanced esophageal carcinoma.24 Zhao et al25 prepared MPMs using Pluronic P123 and d-alpha tocopheryl polyethylene glycol succinate (TPGS), and the solubility of Que dramatically increased to 5.56 mg/mL (a 2,738-fold increase compared to free Que). Therefore, MPMs offer synergistic properties such as increased drug stability, loading efficiencies, and efficacy that are superior to those of traditional micelles using individual components.26
Lecithin is a hydrophobic mixture of naturally occurring phospholipids, including phosphatidylcholine, phosphatidylethanolamine, and phosphatidylinositol. It is widely utilized in food and pharmaceutical applications and is considered a safe and biocompatible excipient. Phospholipids, as important components of cell membranes, can maintain membrane fluidity and help in easy absorption of drugs, so lecithin-based formulations are a method to increase BA.26 Lecithin-based nanoparticles of docetaxel were reported to enhance the antitumor effects and be biocompatible after an intravenous injection.27 Hu et al28 also showed that lecithin-based nanoparticles displayed a sustained-release profile, with ~80% of docetaxel released within 72 hours, and there was a higher oral BA compared to that of a docetaxel solution (8.75% vs 2.40%). Li et al29 prepared lecithin-based nanoparticles for Que, and in vivo pharmacokinetic (PK) studies showed it had a higher BA (more than fivefold) compared to a Que suspension, and the time of maximum concentration observed (Tmax) and the mean residence time (MRT) were both delayed. Therefore, lecithin-based formulations were able to improve the therapeutic efficacy of drugs with poor oral absorption.30
The aim of this study was to develop novel lecithin-based MPMs (LMPMs) for Que to increase its solubility and BA. Que-LMPMs were prepared with lecithin and amphiphilic polymers (TPGS, DSPE-PEG, Pluronic, and Cremophor [BASF, Hanover, Germany]®) by a thin-film method. We attempted to create micelles with a particle size of <100 nm, an encapsulation efficiency (EE) of >90%, and drug loading (DL) of >10%. The optimal formulation was further characterized in terms of its physicochemical properties (morphological observations and in vitro drug release), in vitro cytotoxicity and uptake, and in vivo pharmacokinetics.
Materials and methods
Chemicals and reagents
Granular l-α-lecithin was supplied by Acros (Thermo Fisher Scientific, Waltham, MA, USA). Pluronic series F87, F127, and F68, TPGS, and Cremophor (ELP and RH40) were purchased from BASF (Hanover, Germany). Que and Pluronic series L121, F108, and P123 were purchased from Sigma-Aldrich Co. (St Louis, MO, USA). DSPE-PEG2K was from NOF (Tokyo, Japan). Heparin 5,000 IU/mL was provided by China Chemical & Pharmaceutical (Hsinchu, Taiwan). Dulbecco’s Modified Eagle’s Medium, fetal bovine serum, and glutamine were purchased from Biowest (Nuaillé, France). All reagents for the high-performance liquid chromatographic (HPLC) analysis were of HPLC grade, and other reagents were of analytical grade.
Preparation of Que-LMPMs
Que-LMPMs were prepared by a thin-film method as described earlier.31 Briefly, Que was dissolved in acetone and lecithin was dissolved in dichloromethane (CH2Cl2), and then mixed at a predetermined ratio with a fixed amount of Que at 6 mg. Next, different ratios of various polymers (Pluronic series F87, F127, F68, L121, F108, and P123, TPGS, Cremophor RH40 and ELP, and DSPE-PEG2K) were added and mixed in a round bottom flask. The mixture was subsequently evaporated by rotary evaporation (rotavapor R124; Buchi, Flawil, Switzerland) at 40°C±1°C under reduced pressure to obtain a thin film, and then 1 mL of deionized water was added, followed by gentle shaking until the thin film was dissolved. Unincorporated Que aggregates were removed by passing through a 0.22 μm filter (EMD Millipore, Billerica, MA, USA), and Que-LMPMs were obtained and further evaluated.
Characterization of Que-LMPMs
The average particle size and size distribution of Que-LMPMs were measured with an N5 submicron particle size analyzer (Beckman Coulter, Brea, CA, USA) at room temperature. The surface morphology was observed with transmission electron microscopy (TEM; Hitachi H-600, Hitachi Ltd., Tokyo, Japan).
Quantification of Que
Que was analyzed by HPLC (Pump PU-980; Jasco, Tokyo, Japan) by a method from Hertog et al32 with little modification. The Que concentration was determined by a Vercopak (GL Sciences, Tokyo, Japan) Inertsil 6 ODS-3 column (6 μm, 150×4.6 mm; Tokyo, Japan). The mobile phase was a mixture of methanol/0.025 M phosphoric acid (6/4, v/v) at a flow rate of 1.0 mL/min at 30°C. The column effluent was monitored by an ultraviolet detector (UV-975, Jasco) at a wavelength of 375 nm. The HPLC method was validated to have an acceptable coefficient of variation for accuracy and precision. Once the concentration of Que was known from the validated calibration curve, the EE and DL were calculated according to Equations 1 and 2, respectively:
|
|
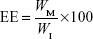
and
|
|
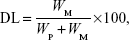
where WM is the weight of drug in the micelles, WI is the weight of initial feeding drug, and WP is the weight of initial feeding polymers.
In vitro release studies
The drug released from the Que-LMPMs was captured in the medium of a 0.5% Tween 80 aqueous solution using the dialysis bag method.25 The Que-LMPM solution containing 2 mg Que or 2 mL free Que solution (1 mg/mL in propylene glycol) was put into a separate dialysis bag (MWCO 3,500, Cellu-Sep® T1 [Orange Scientific, Seguin, TX, USA]; Seguin, TX, USA). The bag was placed in a conical flask immersed in 100 mL of dissolution medium at 37°C with 100 rpm shaking. At each time point, the dialysis bag was relocated in a new dissolution medium to maintain a sink condition. The concentration of Que released from the dialysis bag was analyzed by the HPLC method as described in the “Quantification of Que” section. All measurements were carried out in triplicate. For comparison, the release of free Que from the propylene glycol solution was conducted under the same conditions.
Cellular uptake study
MCF-7 breast cancer cells were seeded on 24-well plates at a density of 3×104 cells/0.5 mL/well. Twenty-four hours later, coumarin 6 (3-(2′-benzothiazolyl)-7-diethylaminocoumarin)-loaded Que-LMPMs were added to the wells and incubated for 48 hours. After removing the coumarin 6-loaded Que-LMPMs and washing the wells with phosphate-buffered saline, cell nuclei were stained with Hoechst 33342. Cells were then observed with an inverted fluorescence microscope linked with a confocal imaging system (Leica TCS SP5; Leica Microsystems, Wetzlar, Germany) using an fluorescein isothiocyanate filter. The presence of coumarin 6 was detected by excitation at 450 nm and emission at 510 nm. Coumarin 6 is hydrophobic, so it could be encapsulated with Que in the inner hydrophobic area of micelles when preparing Que-LMPMs in the same way, and thus, it could demonstrate the fate of Que-LMPMs. The Laboratory Animal Center of Taipei Medical University (Taipei, Taiwan, ROC) approved the use of human cell lines in cell uptake study and animal studies (approval number LAC-103-0126).
Cell viability assays
The cytotoxicity of the optimal Que-LMPMs, blank LMPMs (without Que), and a free Que solution against MCF-7 breast cancer cells was evaluated by a tetrazolium salt MTT assay. MCF-7 cells were expanded in Dulbecco’s Modified Eagle’s Medium with 10% fetal bovine serum, 10% l-glutamine, and 10% penicillin–streptomycin. Cells were seeded at a density of 3×104 cells per well in 24-well plates. After 48 hours of treatment at 37°C with 5% CO2, 300 μL of MTT (3 mg/mL) was added to each well for 3 hours. Then, the medium was removed, and 200 μL DMSO was added to dissolve the formed purple formazan crystals. Cell viability was measured with a spectrophotometer at an absorbance of 570 nm (Bio-Tek, Winooski, VT, USA). Cells with no treatment were used as a control group. The IC50 was defined as the concentration of treatment that inhibited cell growth by 50%.
In vivo PK studies of intravenous administration
Male Sprague Dawley rats, 8–10 weeks old, were used to investigate the PK profile of the optimal Que-LMPMs and free Que solution (Que was dissolved in a solvent of DMSO/PEG400=3/7). Rats were given a single intravenous administration of 10 mg/kg of Que-LMPMs or the free Que solution (three rats per group). Blood samples were collected into heparinized tubes from the jugular vein at 5, 10, 15, 30, 60, 90, 120, 240, 360, 480, and 720 minutes after administration. All blood samples were immediately centrifuged at 3,000 rpm for 15 minutes to obtain plasma. The plasma was stored at −30°C before the HPLC analysis. PK parameters were represented as the value of mean and standard deviation from individual rats in each group and estimated through a noncompartmental analysis. The terminal elimination rate constant (Ke) was estimated from the slope of the log-linear phase of the declining plasma concentration of Que versus time graph. The half-life (T1/2) was calculated using the following equation: T1/2 = ln 2/Ke. The area under the concentration time curve from the beginning to the last time point (AUC0→last) was calculated using the trapezoidal method. Summation of AUC0→last and the concentration at the last measured point divided by Ke yielded AUC0→∞. Clearance (CL) was calculated by dividing the dose by AUC0→∞ and the volume of distribution (V) by dividing CL by Ke. The animal study was approved by the Laboratory Animal Center of Taipei Medical University (Taipei, Taiwan, ROC) (approval number LAC-101-0277).
Statistical analysis
Statistical analysis of all results was performed by Student’s t-test assuming unequal variance. Two-tailed P-values of <0.05 were regarded as statistically significant differences. Tabulated data are presented as mean ± standard deviation.
Results
Preparation and characterization of Que-LMPMs
Lecithin is soluble in CH2Cl2 and chloroform (CHCl3), while Que is not soluble in these two solvents, but is only very slightly soluble in methanol, ethanol, and acetone. Preliminarily, Que was dissolved in either methanol or ethanol or acetone; the solvent for lecithin was CH2Cl2 or CHCl3, and then the solutions of Que and lecithin were mixed at various ratios to determine the optimal solvent mixture capable of dissolving both Que and lecithin at reasonable quantities. Results demonstrated that >10 mg/mL of lecithin was able to dissolve when the ratios of CH2Cl2/acetone, CH2Cl2/methanol, and CH2Cl2/ethanol of 2/8, 6/4, and 8/2, respectively, were exceeded and when the ratios of CHCl3/acetone, CHCl3/methanol, and CHCl3/ethanol were 8/2, 5/5, and 5/5, respectively. A higher ratio of CH2Cl2 or CHCl3 increased the solubility of lecithin; however, it simultaneously reduced the solubility of Que. By considering safety and the optimization of solubility, we chose a ratio of acetone/CH2Cl2 of 7/3 to mix Que and lecithin for the following experiments.
We attempted to produce micelles with a particle size of <100 nm, an EE of >90%, and a DL of >10%. The major impact factors for LMPM fabrication are the type of polymers and the proportions of drug and polymers. So, various amphiphilic polymers (TPGS, DSPE-PEG, Pluronic series, and Cremophor) and different ratios of Que, lecithin, and amphiphilic polymers were studied.33 Table 1 lists the particle size, DL, and EE of optimal formulations for each polymer. With the use of Cremophor, TPGS, DSPE-PEG, Pluronic F68, F108, F87, F127, or L121, none of these formulations met the earlier criteria for particle size, DL, and EE, regardless of the ratio used. When Que:lecithin:PP123 was in a weight ratio of 3:1:20, the particle size, EE, and DL were 78.7 nm, 88.07%, and 11.01%, respectively. Furthermore, when we added DSPE-PEG2K and adjusted the ratio of Que:lecithin:PP123:DSPE-PEG2K to 3:1:17.5:2.5 (w/w), the particle size, EE, and DL were optimized to 61.60±5.02 nm, 96.87%±9.04%, and 12.18%±1.11%, respectively, and it was selected as the optimal Que-LMPM for the following evaluations.
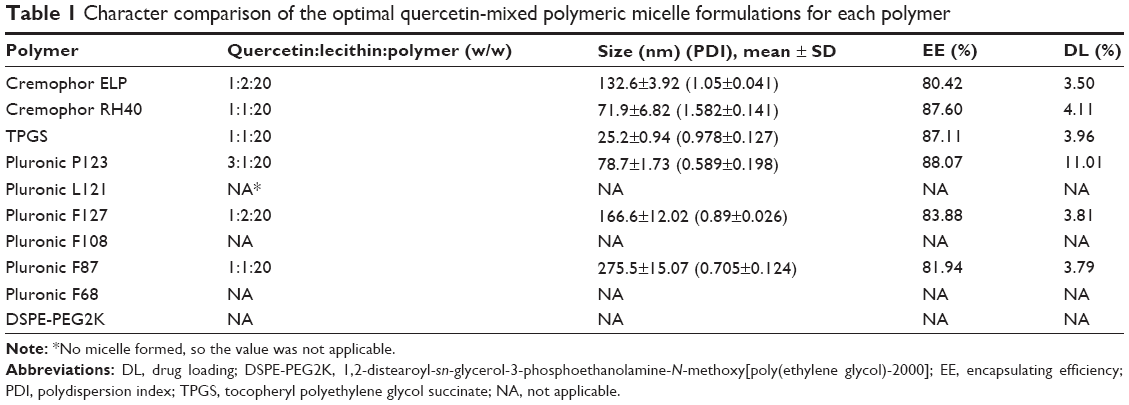 | Table 1 Character comparison of the optimal quercetin-mixed polymeric micelle formulations for each polymer |
Transmission electron micrographs
The morphology of optimum Que-LMPMs was observed on TEM images. Figure 1 reveals that micelles displayed a spherical shape with uniform particles; particle sizes measured from TEM images were ~60–70 nm and were in good agreement with those measured with the N5 submicron particle size analyzer.
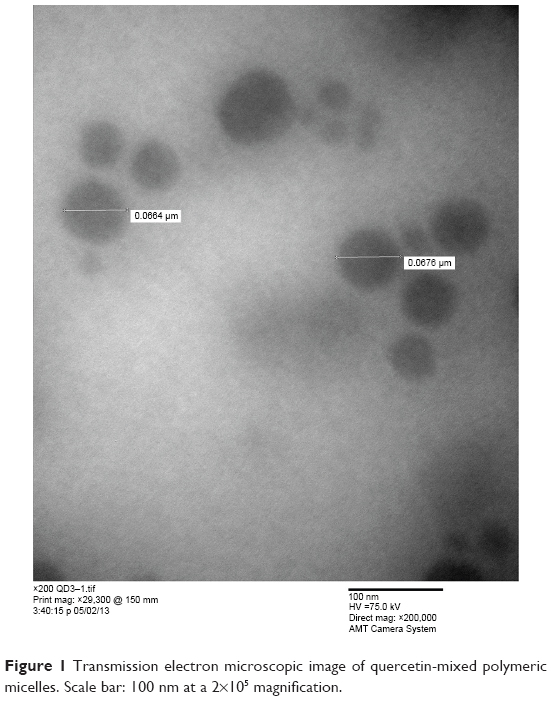 | Figure 1 Transmission electron microscopic image of quercetin-mixed polymeric micelles. Scale bar: 100 nm at a 2×105 magnification. |
In vitro release studies
The in vitro release of Que from micellar formulations under a sink condition was investigated by a dialysis method. As shown in Figure 2, only 8% of Que was released from optimal Que-LMPMs within the first 12 hours, while 60% of Que was released from the free Que solution. At the end of the study, ~30% of Que initially incorporated in the micelles had been released. Even at 60 hours, the release of free Que could not reach 100%, because Que was able to dissolve in propylene glycol; however, after 10 hours of dialysis, the propylene glycol had been dialyzed out, which caused the precipitation of Que in the dialysis bag.
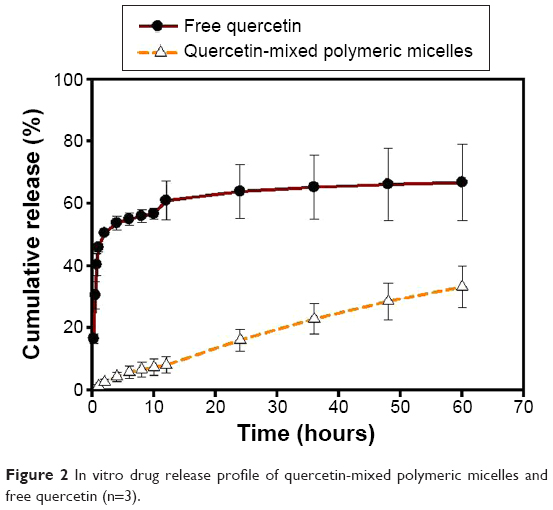 | Figure 2 In vitro drug release profile of quercetin-mixed polymeric micelles and free quercetin (n=3). |
Cellular uptake study and in vitro cytotoxicity
Figure 3A shows the bright field of MCF-7 cancer cells. Figure 3B and C shows that the nuclei of MCF-7 cancer cells were stained with Hoechst 33342 as blue, while Que-LMPMs with coumarin 6 stained green. This demonstrates that Que-LMPMs were taken up by cells as shown in Figure 3D and confirms its cytotoxic effect.
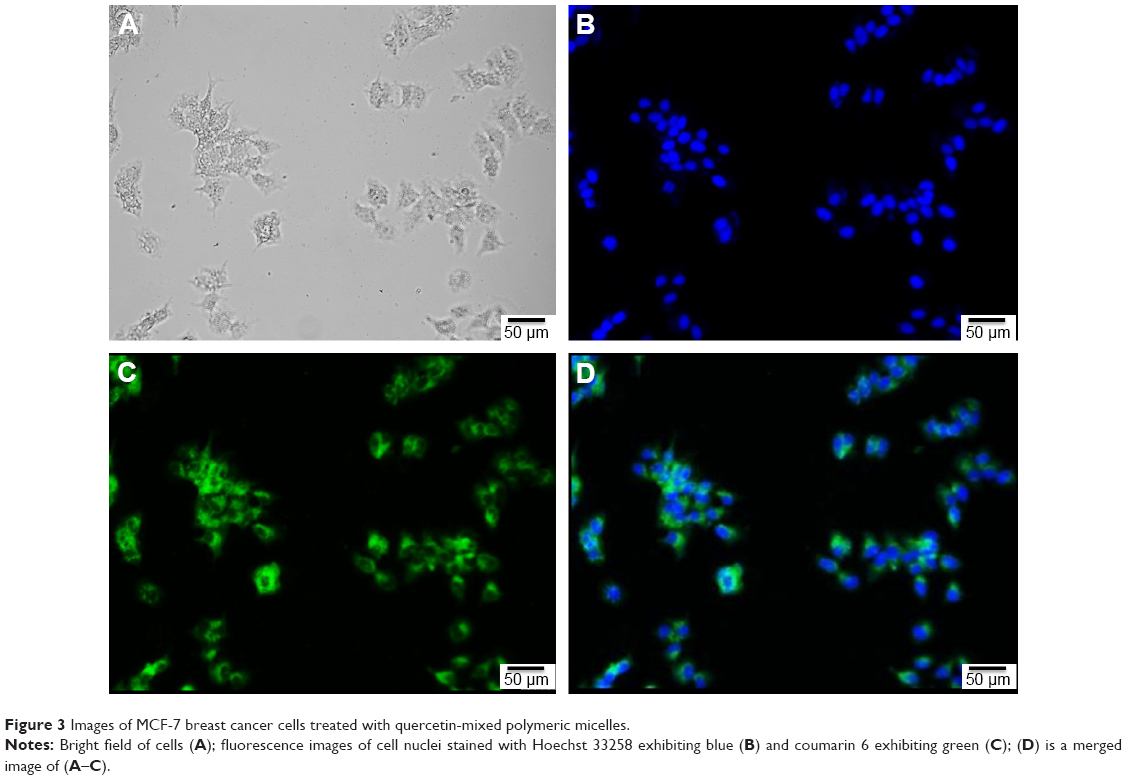 | Figure 3 Images of MCF-7 breast cancer cells treated with quercetin-mixed polymeric micelles. |
Figure 4 demonstrates the cell viability after treatment of Que, Que-MPMs, and blank MPMs. The IC50 value of blank LMPM (without Que) developed in this study was >200 μM, while that of optimal Que-LMPM was 110 μM.
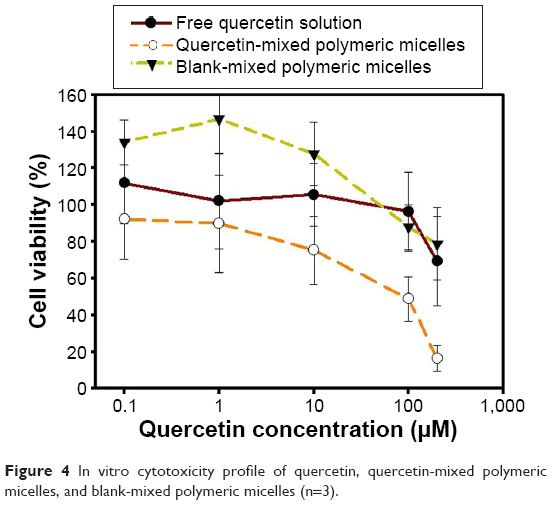 | Figure 4 In vitro cytotoxicity profile of quercetin, quercetin-mixed polymeric micelles, and blank-mixed polymeric micelles (n=3). |
In vivo PK study of Que-LMPMs
Figure 5 shows the plasma concentration of Que versus time curve in Sprague Dawley rats after intravenous administration of a free Que solution or Que-LMPMs at a dose of 10 mg/kg. Both Que formulations were well tolerated by the animals. The PK parameters are shown in Table 2. The value of AUC0→∞ of Que-LMPMs and the half-life were higher than those of free Que, while CL of Que-LMPMs was slower. Compared to the free Que solution, the AUC0→∞ increased 2.4-fold.
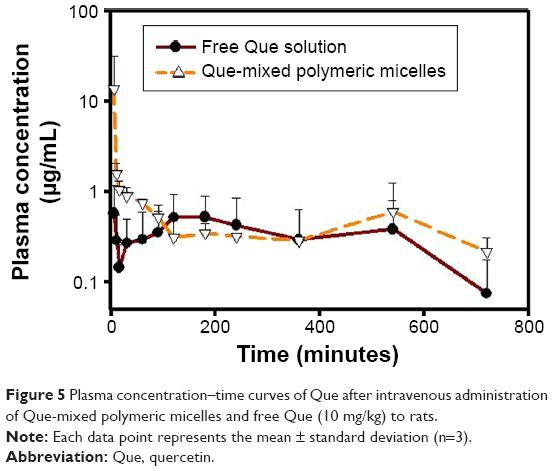 | Figure 5 Plasma concentration–time curves of Que after intravenous administration of Que-mixed polymeric micelles and free Que (10 mg/kg) to rats. |
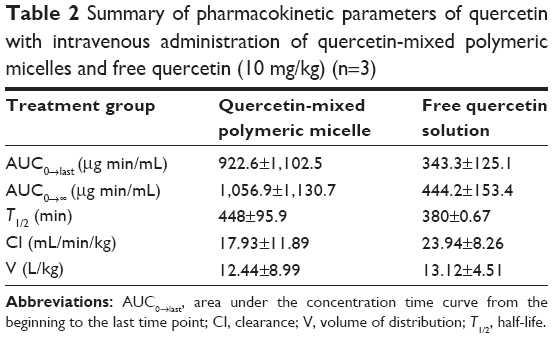 | Table 2 Summary of pharmacokinetic parameters of quercetin with intravenous administration of quercetin-mixed polymeric micelles and free quercetin (10 mg/kg) (n=3) |
Discussion
Que is known to have biological benefits; but in clinical settings, it is mostly hampered by its low water solubility and poor absorption in the body.34 Thus, it is imperative to develop alternative delivery systems for Que, and it was the objective of the present study to develop a lecithin-based MPM system that could increase the solubility and BA of Que, and potentially improve its anticancer activity.35
The major factors affecting the particle size, DL, and EE of LMPMs are the nature and amount of the polymers. When using DSPE-PEG2K as an amphiphilic polymer, the particle sizes of all formulations were >2,000 nm, regardless of the ratios of Que, lecithin, and DSPE-PEG2K. The value of the hydrophilic–lipophilic balance for DSPE-PEG2K is 14 and the hydrophilicity is relatively high; therefore, it might have a lower ability to incorporate a hydrophobic drug and form micelles. This reason could also explain the results of the formulations with Pluronic F68, F108, F87, and F127.
With a ratio of Que:lecithin:PF87 of 1:0:20, the particle size was >5,000 nm. After the addition of lecithin, the particle size decreased to 237.90 nm at the ratio of 1:1:20. The insertion of lecithin into the micelles formed LMPMs, with a possible transition of polyoxyethylene–polyoxypropylene chains from a brush to a mushroom conformation as suggested previously, thus decreasing the particle size.36 Lecithin is a mixture of phospholipids and is regarded as a well-tolerated and nontoxic compound, making it suitable for long-term use in high amounts. It is totally biodegradable and is an integral part of biological membranes. In this study, lecithin was thus used as a hydrophobic material for the micelles. In PL121 formulations, EE increased with an increasing amount of lecithin because the addition of lecithin into the micellar system increased the volume of the hydrophobic region of the micelle and, hence, provided more space for the hydrophobic drug to be solubilized.37 Similarly, Gao et al38 prepared polyethylene glycol-phosphatidylethanolamine micelles, and ~15 mg paclitaxel was encapsulated. Furthermore, the addition of ePC increased the paclitaxel load to 33 mg because ePC created particles with higher hydrophobic contents.37 However, the high hydrophilic–lipophilic balance value and the short hydrophilic chain of PL121 allowed the micelles to easily aggregate.
Each of these polymers has unique advantages for drug delivery. So, an appropriate polymer should be chosen to achieve specific goals. Formulations of PP123 met the criteria for particle sizes. PP123 is PEO20–PPO68–PEO20, where PEO is poly(ethylene oxide) and PPO is poly(propylene oxide).39 A unique feature of PP123 is that it can self-assemble into spherical micelle structures constructed by PEO as a hydrophilic outer shell to maintain the dispersion stability and PPO as a hydrophobic inner core to serve as a “pool” to incorporate the hydrophobic agents.40 We speculated that the triblock structure of PP123 inserted well into the hydrophobic core, which provided more space to incorporate as much Que as possible and offered steric hindrance to prevent the micelles from adhering to each other. Also, using PP123 could serve as an inert carrier and also modify the biological response.41 PP123 was reported to inhibit the efflux actions of P-glycoproteins. The overexpression of P-glycoproteins in cancer tissues often reduces the accumulation of therapeutic agents at tumor sites by pumping them out, thereby reducing the efficacy and, hence, causing multidrug resistance to a variety of chemotherapies.42–44 Thus, PP123 micelles have garnered much attention as they can increase the solubility, improve the circulation time, and suppress multidrug resistance.
Furthermore, the addition of DSPE-PEG2K optimized the particle size, EE, and DL. Drug release is affected by the particle size; smaller particles usually have a larger surface area and faster drug release, while encapsulated drugs slowly diffuse out of larger particles. On the contrary, smaller particles have a greater risk of aggregation; so, it is always a challenge to formulate micelles of a small size while maintaining maximum stability. Because DSPE-PEG2K is amphiphilic, it accumulates at the surface of these hydrophobic Que particles and reduces the surface tension between the drug and buffer interface. The insertion of DSPE-PEG2K prevents particle–particle interactions and further growth of micelles by providing a steric barrier. In addition, the PEG chain on the surface of the micelles renders them sterically stabilized, avoiding opsonization and RES uptake.
Zhao et al25 used TPGS and PP123 to prepare Que micelles, and the particle size was 18.43 nm, EE was 88.94%±3.71%, and DL was 10.59%±0.38%. Compared to the results of Zhao et al, our larger particle size may have been due to the addition of lecithin, which formed a larger hydrophobic core. But with it, a greater amount of Que was entrapped, resulting in higher EE and DL values. The solubility of Que is 0.17–7.7 μg/mL, but through the solubilization of Que-LMPMs, it increased to 5.81 mg/mL (a 754.8–34,189.4-fold increase).45
For successful drug delivery, it is important that drug-loaded micelles retain the drug during a certain time period after administration. Que is brilliant yellow and cannot be dissolved in pure water, as was also confirmed by previous observations of a turbid yellow slurry. So, Que was dissolved in a propylene glycol solution as a free Que solution in the drug release study.25 Most Que in the propylene glycol solution was released within 12 hours, and this suggests that Que could freely diffuse through the dialysis membrane. During the same time period, 8% of Que was released from Que-LMPMs, which was much slower than the free Que solution. This result shows that the micelle carrier not only solubilized the poorly soluble drug but also sustained Que release. These results were similar to those of many studies reporting that drug-loaded LMPMs provide a sustained-release pattern.
The anticancer activity of Que was significantly improved over the free drug through the vehicle of LMPMs, as determined by the in vitro cell viability assay. Nevertheless, nondrug-loaded LMPMs did not possess obvious toxicity to MCF-7 breast cancer cells, indicating that the micellar system was safe, biocompatible, and nontoxic. The self-assembling LMPMs are capable of solubilizing a wide range of poorly water-soluble molecules, and they act as a safe carrier to deliver a drug without the use of potentially harmful surfactants and excipients. On the other hand, the IC50 of the optimum Que-LMPM was higher than that fabricated by Zhao et al25 with TPGS and PP123 (110 vs 23.6 μM). The higher cytotoxicity of the Que-LMPMs of Zhao et al may have been due to the carrier system itself because the IC50 of the blank LMPM was already 100 μM. Zhao et al25 mentioned that the cytotoxicity of TPGS/PP123 micelles was higher than that of PP123 empty micelles due to the use of TPGS. However, in this study, the LMPMs prepared with PP123 and lecithin were less cytotoxic.
The in vivo PK study demonstrated that Que-LMPMs slowly released Que, resulting in a higher AUC, a longer half-life, and lower CL. The increase in the AUC with Que-LMPMs usually indicates a higher BA, which might be attributed to the following reasons: 1) lecithin, a mixture of phospholipids, is an important component of cell membranes and allowed Que to be easily absorbed; 2) the sustained-release property of the micelles increased the circulation time and exposure of Que in the systematic circulation; 3) the encapsulation of Que into LMPMs might have protected Que from degradation, and together with the effect of DSPE-PEG2K, protected Que from being recognized by the RES; and 4) the small particle size of the micelles reduced the uptake by the RES. Moreover, it also allowed the extravasation of the carriers, minimized the risks of embolism in capillaries, and permitted sterilization of the product to be easily carried out by filtration, which is beneficial for practical large-scale manufacture, once clinically available.46 In addition, the small size characteristic provides efficient passive tumor-targeting ability by an enhanced permeability and retention effect, leading to passive tumor-targeting ability for future anticancer studies.47–49
Conclusion
In this study, self-assembling LMPMs containing Que were developed by a thin-film method. Due to their nanosize, core–shell structure, and solubilization potential, the LMPMs proved to be a successful drug delivery system for Que in cancer treatment. They could be a promising nanomedicine technology platform for potential applications to other poorly soluble anticancer drugs.
Acknowledgment
Sincere thanks are given for the financial support of the Ministry of Science and Technology of ROC (NSC102-2320-B-038-022-MY3 and NSC100-2623-B-038-001-MY3).
Disclosure
The authors report no conflicts of interest in this work.
References
Torre LA, Bray F, Siegel RL, Ferlay J, Lortet-Tieulent J, Jemal A. Global cancer statistics, 2012. CA Cancer J Clin. 2015;65(2):87–108. | ||
Lin Y, Kikuchi S, Tamakoshi K, et al; Japan Collaborative Cohort Study Group for Evaluation of Cancer Risk. Active smoking, passive smoking, and breast cancer risk: findings from the Japan Collaborative Cohort Study for Evaluation of Cancer Risk. J Epidemiol. 2008;18(2):77–83. | ||
Armitage AK, Dollery CT, George CF, Houseman TH, Lewis PJ, Turner DM. Absorption and metabolism of nicotine from cigarettes. Br Med J. 1975;4(5992):313–316. | ||
Schuller HM. Nitrosamines as nicotinic receptor ligands. Life Sci. 2007;80(24–25):2274–2280. | ||
Shih YL, Liu HC, Chen CS, et al. Combination treatment with luteolin and quercetin enhances antiproliferative effects in nicotine-treated MDA-MB-231 cells by down-regulating nicotinic acetylcholine receptors. J Agric Food Chem. 2010;58(1):235–241. | ||
Guo W, Kong E, Meydani M. Dietary polyphenols, inflammation, and cancer. Nutr Cancer. 2009;61(6):807–810. | ||
Huang WY, Cai YZ, Zhang Y. Natural phenolic compounds from medicinal herbs and dietary plants: potential use for cancer prevention. Nutr Cancer. 2010;62(1):1–20. | ||
Fearn RA, Hirst BH. Predicting oral drug absorption and hepatobiliary clearance: Human intestinal and hepatic in vitro cell models. Environ Toxicol Pharmacol. 2006;21(2):168–178. | ||
Gugler R, Leschik M, Dengler HJ. Disposition of quercetin in man after single oral and intravenous doses. Eur J Clin Pharmacol. 1975;9(2–3):229–234. | ||
Ferry DR, Smith A, Malkhandi J, et al. Phase I clinical trial of the flavonoid quercetin: pharmacokinetics and evidence for in vivo tyrosine kinase inhibition. Clin Cancer Res. 1996;2(4):659–668. | ||
Yuan ZP, Chen LJ, Fan LY, et al. Liposomal quercetin efficiently suppresses growth of solid tumors in murine models. Clin Cancer Res. 2006;12(10):3193–3199. | ||
Pool H, Quintanar D, Figueroa JdD, et al. Antioxidant effects of quercetin and catechin encapsulated into PLGA nanoparticles. J Nanomater. 2012;2012:86–86. | ||
Gismondi A, Reina G, Orlanducci S, et al. Nanodiamonds coupled with plant bioactive metabolites: a nanotech approach for cancer therapy. Biomaterials. 2015;38:22–35. | ||
Gaucher G, Dufresne M-H, Sant VP, Kang N, Maysinger D, Leroux J-C. Block copolymer micelles: preparation, characterization and application in drug delivery. J Control Release. 2005;109(1–3):169–188. | ||
Torchilin VP. Structure and design of polymeric surfactant-based drug delivery systems. J Control Release. 2001;73(2–3):137–172. | ||
Gong J, Chen M, Zheng Y, Wang S, Wang Y. Polymeric micelles drug delivery system in oncology. J Control Release. 2012;159(3):312–323. | ||
Lu Y, Park K. Polymeric micelles and alternative nanonized delivery vehicles for poorly soluble drugs. Int J Pharm. 2013;453(1):198–214. | ||
Jones M, Leroux J. Polymeric micelles – a new generation of colloidal drug carriers. Eur J Pharm Biopharma. 1999;48(2):101–111. | ||
Yokoyama M. Polymeric micelles as a new drug carrier system and their required considerations for clinical trials. Expert Opin Drug Deliv. 2010;7(2):145–158. | ||
Maeda H, Wu J, Sawa T, Matsumura Y, Hori K. Tumor vascular permeability and the EPR effect in macromolecular therapeutics: a review. J Control Release. 2000;65(1–2):271–284. | ||
Maeda H, Sawa T, Konno T. Mechanism of tumor-targeted delivery of macromolecular drugs, including the EPR effect in solid tumor and clinical overview of the prototype polymeric drug SMANCS. J Control Release. 2001;74(1–3):47–61. | ||
Wei Z, Hao J, Yuan S, et al. Paclitaxel-loaded Pluronic P123/F127 mixed polymeric micelles: formulation, optimization and in vitro characterization. Int J Pharm. 2009;376(1–2):176–185. | ||
Krishnadas A, Rubinstein I, Onyüksel H. Sterically stabilized phospholipid mixed micelles: in vitro evaluation as a novel carrier for water-insoluble drugs. Pharm Res. 2003;20(2):297–302. | ||
Kabanov AV, Batrakova EV, Alakhov VY. Pluronic block copolymers as novel polymer therapeutics for drug and gene delivery. J Control Release. 2002;82(2–3):189–212. | ||
Zhao L, Shi Y, Zou S, Sun M, Lil L, Zhail G. Formulation and in vitro evaluation of quercetin loaded polymeric micelles composed of pluronic P123 and D-a-tocopheryl polyethylene glycol succinate. J Biomed Nanotechnol. 2011;7(3):358–365. | ||
Jin X, Zhang ZH, Sun E, Tan XB, Zhu FX, Jia XB. A novel drug-phospholipid complex loaded micelle for baohuoside I enhanced oral absorption: in vivo and in vivo evaluations. Drug Dev Ind Pharm. 2013;39(9):1421–1430. | ||
Yanasarn N, Sloat BR, Cui Z. Nanoparticles engineered from lecithin-in-water emulsions as a potential delivery system for docetaxel. Int J Pharm. 2009;379(1):174–180. | ||
Hu K, Cao S, Hu F, Feng J. Enhanced oral bioavailability of docetaxel by lecithin nanoparticles: preparation, in vitro, and in vivo evaluation. Int J Nanomedicine. 2012;7:3537–3545. | ||
Li H, Zhao X, Ma Y, Zhai G, Li L, Lou H. Enhancement of gastrointestinal absorption of quercetin by solid lipid nanoparticles. J Control Release. 2009;133(3):238–244. | ||
Yanyu X, Yunmei S, Zhipeng C, Qineng P. The preparation of silybin-phospholipid complex and the study on its pharmacokinetics in rats. Int J Pharm. 2006;307(1):77–82. | ||
Gao Y, Li LB, Zhai G. Preparation and characterization of Pluronic/TPGS mixed micelles for solubilization of camptothecin. Colloids Surf B Biointerfaces. 2008;64(2):194–199. | ||
Hertog MGL, Hollman PCH, Venema DP. Optimization of a quantitative HPLC determination of potentially anticarcinogenic flavonoids in vegetables and fruits. J Agric Food Chem. 1992;40(9):1591–1598. | ||
Sezgin Z, Yuksel N, Baykara T. Preparation and characterization of polymeric micelles for solubilization of poorly soluble anticancer drugs. Eur J Pharm Biopharm. 2006;64(3):261–268. | ||
Tan BJ, Liu Y, Chang KL, Lim BK, Chiu GN. Perorally active nanomicellar formulation of quercetin in the treatment of lung cancer. Int J Nanomedicine. 2012;7:651–661. | ||
Chiappetta DA, Sosnik A. Poly(ethylene oxide)-poly(propylene oxide) block copolymer micelles as drug delivery agents: improved hydrosolubility, stability and bioavailability of drugs. Eur J Pharm Biopharm. 2007;66(3):303–317. | ||
Rex S, Zuckermann MJ, Lafleur M, Silvius JR. Experimental and Monte Carlo simulation studies of the thermodynamics of polyethyleneglycol chains grafted to lipid bilayers. Biophys J. 1998;75(6):2900–2914. | ||
Alkan-Onyuksel H, Ramakrishnan S, Chai HB, Pezzuto JM. A mixed micellar formulation suitable for the parenteral administration of taxol. Pharm Res. 1994;11(2):206–212 | ||
Gao Z, Lukyanov AN, Singhal A, Torchilin VP. Diacyllipid-polymer micelles as nanocarriers for poorly soluble anticancer drugs. Nano Lett. 2002;2(9):979–982. | ||
Jansson J, Schillén K, Olofsson G, Cardoso da Silva R, Loh W. The interaction between PEO-PPO-PEO triblock copolymers and ionic surfactants in aqueous solution studied using light scattering and calorimetry. J Phys Chem B. 2004;108(1):82–92. | ||
Nakanishi T, Fukushima S, Okamoto K, et al. Development of the polymer micelle carrier system for doxorubicin. J Control Release. 2001;74(1–3):295–302. | ||
Kabanov AV, Batrakova EV, Miller DW. Pluronic block copolymers as modulators of drug efflux transporter activity in the blood-brain barrier. Adv Drug Deliv Rev. 2003;55(1):151–164. | ||
Batrakova EV, Li S, Elmquist WF, Miller DW, Alakhov VY, Kabanov AV. Mechanism of sensitization of MDR cancer cells by pluronic block copolymers: selective energy depletion. Br J Cancer. 2001;85(12):1987–1997. | ||
Alakhov V, Moskaleva E, Batrakova EV, Kabanov AV. Hypersensitization of multidrug resistant human ovarian carcinoma cells by pluronic P85 block copolymer. Bioconjug Chem. 1996;7(2):209–216. | ||
Venne A, Li S, Mandeville R, Kabanov A, Alakhov V. Hypersensitizing effect of pluronic L61 on cytotoxic activity, transport, and subcellular distribution of doxorubicin in multiple drug-resistant cells. Cancer Res. 1996;56(16):3626–3629. | ||
Gao Y, Wang Y, Ma Y, et al. Formulation optimization and in situ absorption in rat intestinal tract of quercetin-loaded microemulsion. Colloids Surf B Biointerfaces. 2009;71(2):306–314. | ||
des Rieux A, Fievez V, Garinot M, Schneider YJ, Preat V. Nanoparticles as potential oral delivery systems of proteins and vaccines: a mechanistic approach. J Control Release. 2006;116(1):1–27. | ||
Allemann E, Gurny R, Doelker E. Drug-loaded nanoparticles – Preparation methods and drug targeting issues. Eur J Pharm Biopharm. 1993;39(5):173–191. | ||
van Vlerken LE, Amiji MM. Multi-functional polymeric nanoparticles for tumour-targeted drug delivery. Expert Opin Drug Deliv. 2006;3(2):205–216. | ||
Nasongkla N, Bey E, Ren J, et al. Multifunctional polymeric micelles as cancer-targeted, MRI-ultrasensitive drug delivery systems. Nano Lett. 2006;6(11):2427–2430. |
 © 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.