Back to Journals » International Journal of Nanomedicine » Volume 9 » Issue 1
Development and characterization of magnetic iron oxide nanoparticles with a cisplatin-bearing polymer coating for targeted drug delivery
Authors Unterweger H ![]() , Tietze R, Janko C
, Tietze R, Janko C ![]() , Zaloga J, Lyer S
, Zaloga J, Lyer S ![]() , Dürr S, Taccardi ON, Goudouri M, Hoppe A, Eberbeck D, Schubert D, Boccaccini A, Alexiou C
, Dürr S, Taccardi ON, Goudouri M, Hoppe A, Eberbeck D, Schubert D, Boccaccini A, Alexiou C ![]()
Received 4 March 2014
Accepted for publication 9 April 2014
Published 5 August 2014 Volume 2014:9(1) Pages 3659—3676
DOI https://doi.org/10.2147/IJN.S63433
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Video abstract presented by Unterweger H.
Views: 4872
Harald Unterweger,1 Rainer Tietze,1 Christina Janko,1 Jan Zaloga,1 Stefan Lyer,1 Stephan Dürr,1 Nicola Taccardi,2 Ourania-Menti Goudouri,3 Alexander Hoppe,3 Dietmar Eberbeck,4 Dirk W Schubert,5 Aldo R Boccaccini,3 Christoph Alexiou1
1ENT Department, Section of Experimental Oncology and Nanomedicine (SEON), Else Kroener-Fresenius-Stiftung-Professorship, University Hospital Erlangen, 2Chair of Chemical Engineering I (Reaction Engineering), 3Institute of Biomaterials, Department of Materials Science and Engineering, University Erlangen-Nuremberg, Erlangen, 4Physikalisch-Technische Bundesanstalt, Berlin, 5Institute of Polymer Materials, Department of Materials Science and Engineering, University Erlangen-Nuremberg, Erlangen, Germany
Abstract: A highly selective and efficient cancer therapy can be achieved using magnetically directed superparamagnetic iron oxide nanoparticles (SPIONs) bearing a sufficient amount of the therapeutic agent. In this project, SPIONs with a dextran and cisplatin-bearing hyaluronic acid coating were successfully synthesized as a novel cisplatin drug delivery system. Transmission electron microscopy images as well as X-ray diffraction analysis showed that the individual magnetite particles were around 4.5 nm in size and monocrystalline. The small crystallite sizes led to the superparamagnetic behavior of the particles, which was exemplified in their magnetization curves, acquired using superconducting quantum interference device measurements. Hyaluronic acid was bound to the initially dextran-coated SPIONs by esterification. The resulting amide bond linkage was verified using Fourier transform infrared spectroscopy. The additional polymer layer increased the vehicle size from 22 nm to 56 nm, with a hyaluronic acid to dextran to magnetite weight ratio of 51:29:20. A maximum payload of 330 µg cisplatin/mL nanoparticle suspension was achieved, thus the particle size was further increased to around 77 nm with a zeta potential of –45 mV. No signs of particle precipitation were observed over a period of at least 8 weeks. Analysis of drug-release kinetics using the dialysis tube method revealed that these were driven by inverse ligand substitution and diffusion through the polymer shell as well as enzymatic degradation of hyaluronic acid. The biological activity of the particles was investigated in a nonadherent Jurkat cell line using flow cytometry. Further, cell viability and proliferation was examined in an adherent PC-3 cell line using xCELLigence analysis. Both tests demonstrated that particles without cisplatin were biocompatible with these cells, whereas particles with the drug induced apoptosis in a dose-dependent manner, with secondary necrosis after prolonged incubation. In conclusion, combination of dextran-coated SPIONs with hyaluronic acid and cisplatin represents a promising approach for magnetic drug targeting in the treatment of cancer.
Keywords: magnetic drug targeting, superparamagnetic iron oxide nanoparticles, dextran, hyaluronic acid
Introduction
Magnetic drug targeting is one of the most promising approaches for the treatment of cancer.1–3 Its basic principle is to physically direct a loaded magnetic drug carrier system to a specific organ or tissue with an externally applied magnetic field for drug accumulation.4,5 Compared with conventional drug administration, this strategy leads, on the one hand, to a reduced drug concentration in the body compartments overall, thus decreasing systemic side effects, including nausea, hair loss, and neurotoxicity;6–8 on the other hand, the concentration in the affected tissue is effectively increased, resulting in an enhanced therapeutic effect.9,10
Magnetic particles commonly used for magnetic drug targeting include superparamagnetic iron oxide nanoparticles (SPIONs), eg, magnetite (Fe3O4), maghemite (γ-Fe2O3), and other ferrites.11–13 These particles are biocompatible and have superparamagnetic characteristics depending on their size, which has led to their widespread use in bioseparation, hyperthermia, magnetic resonance imaging, and drug targeting.9,14–19 Particularly in terms of lymph node detection, SPIONs when used as contrast agents in magnetic resonance imaging offer a considerable improvement over other imaging techniques.20,21 SPIONs have a large surface area to volume ratio, so tend to agglomerate in order to reduce their surface energy.22 To prevent this behavior, stabilization agents consisting of single molecules or polymers have to be used. Both have in common the ability to induce repulsion and counteract the magnetic attraction between particles by steric and/or electrostatic means. However, the stabilization approach with polymers is not liable to ligand exchange, and therefore provides a more stable shell when compared with single molecules. This can be achieved in situ by using the coprecipitation technique in the presence of polymer surface-complexing agents.23–25
In this project, dextran was deployed to stabilize fabricated SPIONs. Dextran is a biocompatible, biodegradable, and water-dispersible branched polysaccharide.23,26 The hydroxyl groups of this polymer can be used to add various functionalities, eg, interlinking with hyaluronic acid (HA). HA is a naturally occurring linear polysaccharide composed of β-1,4-D-glucuronic acid-β-1,3-D-N-acetylglucosamine, a repeating disaccharide. HA is biodegradable, immunoneutral, and has excellent biocompatibility.27 In the human body, it can be found as one of the glycosaminoglycan components of the extracellular matrix, in the synovial fluid of joints, and in the vitreous body.28,29 Further, it has been reported that interactions between HA and proteins play a critical role in cell-cell and cell-matrix adhesion, cell growth, and cell migration, as well as in wound healing.25,30 Because of the interaction between HA and the CD44 receptor, which is expressed in the high affinity state by many tumor-derived cells, active targeting strategies can be achieved. A major advantage of HA is that it can form polymer-metal complexes than can incorporate drugs, eg, cis-diamminedichloroplatinum(II), also known as cisplatin.
Cisplatin is an anticancer agent that is used extensively for various types of tumors, including head and neck, testicular, ovarian, bladder, and lung cancers.6,31,32 Once inside the cytoplasm, cisplatin is hydrolyzed to its cationic monoaqua form, which is able to react with negatively charged DNA and other intracellular components.33 Significant bending and distortion of the helical DNA structure then occurs, resulting in an apoptotic cell response.34 Nevertheless, because of its lack of selectivity, cisplatin can have severe side effects, such as those mentioned above.35 Therefore, it is highly desirable to develop a drug delivery system that can transport this drug to the desired organ or tissue, in order to reduce its side effects and increase its local efficacy. In this paper, we describe a novel approach, where such a drug vehicle was designed based on a combination of SPIONs for magnetic drug targeting and HA for active targeting.
Materials and methods
Materials
Chemicals for synthesis of the iron oxide nanoparticles, and 4-(2-hydroxyethyl)piperazine-1-ethanesulfonic acid (HEPES), and sterile and unsterile Rotilabo® syringe filters with a cellulose mixed ester membrane were provided by Carl Roth (Karlsruhe, Germany) and Merck (Darmstadt, Germany), respectively. HA sodium salt from Streptococcus equi (molar mass of HA ≈1.5 mDa), hyaluronidase from bovine testes type I-S (HAse, 400–1,000 units/mg), dextran from Leuconostoc spp. (molar mass 6 kDa), epichlorohydrin, N-methyl-morpholine, 2-chloro-4,6-dimethoxy-1,3,5-triazine, amberlite resin IR120 (hydrogen form), cisplatin powder, propidium iodide, Triton X-100®, trisodium citrate dihydrate, trypan blue, and fluorescein isothiocyanate (FITC) were purchased from Sigma-Aldrich (Hamburg, Germany). Sulfosalicylic acid 20% solution was obtained from AppliChem (Darmstadt, Germany). Roswell Park Memorial Institute 1640 medium, F-12K medium, fetal calf serum, L-glutamine, penicillin-streptomycin, Hoechst 33342, and hexamethylindodicarbocyanine iodide dye (DiI) were purchased from Life Technologies (Carlsbad, CA, USA). Ringer’s solution containing 0.9% NaCl (w/v) was supplied by DeltaSelect (Rimbach, Germany). Annexin V was supplied by Responsif (Erlangen, Germany) and labeled with FITC according to the manufacturer’s instructions. Spectra/Por® 6 dialysis tubing with a molecular weight cut-off of 10 kDa and a diameter of 29 mm was purchased from Serva Electrophoresis (Heidelberg, Germany). Vivaspin 20 ultrafiltration units with a 100 kDa molecular weight cut-off were supplied by Sartorius Stedim Biotech (Goettingen, Germany). Water used in all experiments was of double-distilled quality.
Preparation of dextran-coated SPIONs
The preparation of dextran-coated SPIONs (SEONDEX) was performed according to the protocol described by Kamat et al.25 In brief, FeCl3·6H2O, FeCl2·4H2O in molar ratios (Fe3+/Fe2+ =2) as well as specific amounts of dextran (1.0–4.5 g, in increments of 0.5 g) were dissolved in 10 mL of water. This solution was filtered through a 0.22 μm cellulose mixed ester membrane, before its volume was increased to 15 mL with water; next it was cooled to 0°C–4°C, while being continuously stirred under an argon atmosphere. Subsequently, 5 mL of ice-cold 25% NH3 was added dropwise to the solution, leading to a greenish suspension of SPIONs. After 5 minutes, the reaction mixture was heated and kept at 75°C for a further 40 minutes, which changed the color of the suspension from greenish to brownish. Upon completion, the reaction mixture was cooled to room temperature and dialyzed (molecular weight cut-off 10 kDa) against water for 24 hours, with five water exchanges to remove excess NH4OH. Using ultrafiltration (molecular weight cut-off 100 kDa) in a 5430R Eppendorf centrifuge (Hamburg, Germany) at 6,500 rpm for 20 minutes (five repetitions), the mixture was then cleared of excess dextran and concentrated to a total volume of 20 mL. To stabilize the coating on the SPIONs, crosslinking of dextran was established by adding 10 mL of 5 M NaOH, followed by dropwise addition of 4 mL of epichlorohydrin to the nanoparticle suspension, which was then vigorously stirred for 24 hours. After this step, the solution was dialyzed against water, with five exchanges over 24 hours, as well as concentrated by ultrafiltration to a final volume of 30 mL, leading to colloidally stable SEONDEX.
Degradation of HA
HA with a high molar mass (molecular weight 1.5 mDa) was degraded to low molar mass HA using the procedure described by Jeong et al.36 Two grams of HA were dissolved in 500 mL of phosphate-buffered saline (with 137 mM NaCl, 2.7 mM KCl, 10 mM Na2HPO4, and 2 mM KH2PO4 adjusted to pH 6.5 with 1 N HCl) and then 28.6 mg HAse was added. This mixture was immediately stirred for 5 minutes at room temperature to carry out the degradation, followed by vigorous stirring at 95°C for 15 minutes in an oil bath. After this step, the solution was transferred into dialysis tubes (molecular weight cut-off 10 kDa) and dialyzed against water for 72 hours with 20 water exchanges. The purified solution was filtered through a 0.22 μm cellulose mixed ester membrane, frozen, and then lyophilized using a Christ Alpha 1–2 LDplus apparatus (Osterode am Harz, Germany).
The success of degradation was examined quantitatively by measuring the intrinsic viscosity [η] using an Ubbelohde viscometer (ViscoSystem® AVS 470; Schott Instruments, Mainz, Germany).37,38 For this approach, HA was dissolved in 200 mM NaCl solution and diluted to concentrations of 1, 0.75, 0.5, 0.25, and 0.125 mg/mL. In the first step, the Ubbelohde viscometer equipped with a CC-130A Visco 3 water bath (Huber SE, Berching, Germany) at 25°C was used to measure the run time (tc) of the samples through the capillary (diameter 0.63 mm). From these values, [η] was determined as:
|
|
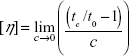
where t0 is the run time of the solvent, c is the HA concentration of the samples, and 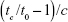 is referred to as reduced viscosity. The molar mass Mw (viscosity average) of the degraded HA was then calculated according to the Mark–Houwink relationship:
is referred to as reduced viscosity. The molar mass Mw (viscosity average) of the degraded HA was then calculated according to the Mark–Houwink relationship:
|
|
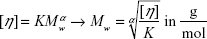
with α =0.716 and K =5.075·10−2 mL/g being the Mark–Houwink coefficients for HA dissolved in 200 mM NaCl.37 The experiment was repeated three times, and the results were averaged.
Formation of HA coating and drug loading
Before the HA coating could be applied, the SEONDEX had to be aminated by 4 mL of NH3 at 37°C for 36 hours. After functionalization, the mixture was dialyzed against water for 2 days, with ten water exchanges, to obtain SEONDEX-NH2. The HA was prepared by dissolving 100 mg in water (10 mL) and mixing the solution with amberlite resin until pH 3 was reached. The solution was stirred for 4 hours to convert HA into its carboxylic acid form. Next, the resin was filtered off and the water was removed by lyophilization. Under an argon atmosphere, the solid was then redissolved in 6.5 mL of 0.2 M NaCl solution. Next, 50 μL of N-methyl-morpholine and 30 mg of 2-chloro-4,6-dimethoxy-1,3,5-triazine were mixed with the polymer solution. After stirring for 1 hour, 10 mL of SEONDEX-NH2 were added, followed by further stirring for 3 hours at room temperature. Upon completion, the mixture was dialyzed against water, with ten water exchanges over 24 hours. In the following text, the particles without drug are referred to as SEONDEX-HA. To incorporate the drug into the HA coating of the particles, 10 mg of cisplatin were added and the mixture was stirred slowly for 3 days in the dark. Finally, the resulting colloid was dialyzed against water for 6 hours, with three water exchanges. In the following text, the particles with drug are referred to as SEONDEX-HA*CPt. The drug content and loading efficiency were calculated as follows:
|
|
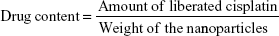
|
|
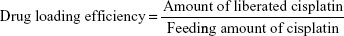
The experiment was performed in triplicate, and the results were averaged.
Particle size and zeta potential measurement
The hydrodynamic size (Z-average size) of the nanoparticles was acquired by dynamic laser scattering with a Zetasizer Nano ZS (Malvern Instruments, Herrenberg, Germany) operated in backscattering mode at 173° with a He-Ne laser beam (λ =632.8 nm). For the zeta potential measurements, which were performed at 25°C with a scattering angle of 90°, the particles were dispersed in aqueous solution. The experiment was repeated three times, and the results were averaged.
Transmission electron microscopy
Transmission electron microscopy (TEM) images were taken using a CM300 UltraTWIN instrument (Philips, Eindhoven, The Netherlands) operating at 300 kV in imaging mode, which results in a wavelength of 1.97 pm. Specimens were prepared by drop casting of a 10 μL aliquot of a dilute NP solution on an Athene® grid (Plano GmbH, Wetzlar, Germany). The size distribution was determined by examination of an overview TEM image using ImageJ software (version 1.48; National Institutes of Health, Bethesda, MD, USA).
X-ray diffraction
In this study, a D8 Advance (Bruker, Billerica, MA, USA) X-ray diffraction device was used to perform a θ/2θ measurement in order to determine the interplanar distances (dhkl) and therefore the crystalline phases present in the specimens. The samples were prepared by dispersing lyophilized particles in ethanol and drop casting of this dispersion onto a sample holder. The Cu Kα1 beam with a wavelength λ =0.154056 nm was used as the X-ray source, and the angular range was 25°–70° with a step size of 0.014°/sec. Afterwards, the interplanar spacing was determined by Bragg’s law (5) and the lattice parameter (a) by equation 6:39
|
|
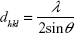
|
|
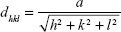
with θ as the half diffraction angle and h, k, and l as the Miller indices of the diffraction plane.
According to the Debye–Scherrer formula, the crystallite size dXRD for the sample is given by
|
|
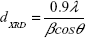
where β is the full width at half maximum value of the X-ray diffraction lines.39
Determination of iron content
The iron content of the particles was determined by photometric assay.40 Firstly, an iron stock solution was prepared by dissolving FeCl2 in diluted HCl, resulting in an iron concentration of 0.28 mg/mL. Next, 100 μL of the particle sample with 0, 10, 20, 40, 60, and 80 μL of the stock solution were transferred into 2 mL tubes. Then, 50 μL of HCl were added to each tube and the volume was increased to 970 μL with water. The solutions were heated for 10 minutes at 90°C. After cooling to room temperature, 200 μL of sulfosalicylic acid, 80 μL of 10% hydroxylammonium chloride, and 500 μL of NH3 were added, resulting in a yellow color. The specimens were pipetted into a 96-well plate (TPP, Sigma-Aldrich), and light absorption at 405 nm was determined using a multimode microplate reader (Filtermax F5, Molecular Devices, Sunnyvale, CA, USA). The achieved absorbance was used to calculate the iron content of the particle samples. Afterwards, the iron content can be used to determine the magnetite content by taking into account the different molar masses. The experiment was performed in triplicate, and the results were averaged.
Magnetization measurements
Magnetization was measured using a superconducting quantum interference device (SQUID)-based susceptometer (QD-MPMS-XL-5, Quantum Design Inc, San Diego, CA, USA). The field was increased from 0 to 4,000 kA/m and afterwards decreased to −50 kA/m. For further details, see Eberbeck et al.41
Fourier transform infrared spectroscopy
The chemical composition of the lyophilized nanoparticle specimens was determined using a Nicolet Impact 420 Fourier transform infrared spectroscopy device (Thermo Fisher Scientific, Waltham, MA, USA). The spectra were collected in ATR-D mode in the mid-infrared region from 4,000 cm−1 to 530 cm−1, with a step size of 0.5 cm−1.
Determination of cisplatin content
Inductively coupled plasma atomic emission spectroscopy was used to determine the cisplatin content. A Spectro Ciros-CCD device (Spectro Analytical Instruments GmbH, Kleve, Germany) was used to determine the emission line of platinum at 214.423 nm. The final cisplatin concentration was then calculated by taking into account the dilution factor of the samples and the different molar masses of platinum and cisplatin.
Drug release study
The release of cisplatin from SEONDEX-HA*CPt in physiological saline at 37°C was evaluated by the dialysis method.42 First, 5 mL of the particles were transferred into a dialysis bag, which was placed into a flask and filled with 95 mL of phosphate-buffered saline. The flask was put into a rotary incubator shaker at 250 rpm and 37°C. After 0.5, 2, 4, 8, 24, and 48 hours, 2 mL of the phosphate-buffered saline was removed and replaced by fresh phosphate-buffered saline. The platinum content in the removed aliquot of phosphate-buffered saline was determined by inductively coupled plasma atomic emission spectroscopy. The amount of cisplatin removed in one step was taken into account in the following steps. In order to see the effect of HAse on the release kinetics, this study was repeated with addition of 1 mg of HAse into the dialysis bag. Five milliliters of pure cisplatin dissolved in phosphate-buffered saline with the same drug concentration as SEONDEX-HA*CPt were used as a control.
In vitro toxicity
Cell lines and culture conditions
Two different cell lines were used to investigate the in vitro toxicity of SEONDEX-HA and SEONDEX-HA*CPt. The first was a Jurkat cell line (ACC 282, DSMZ, Braunschweig, Germany) of nonadherent human T-cell leukemia cells, cultured in Roswell Park Memorial Institute 1640 medium supplemented with 10% fetal calf serum, 1% glutamine, 1% penicillin-streptomycin, and 1% HEPES (10 mM, pH 7.2). The second was a human prostate cancer cell line, PC-3 (CRL-1435, American Type Culture Collection, Wesel, Germany), cultured in F-12K medium (supplemented with 10% fetal calf serum and 1% glutamine) that was used as the adherent cell line. Both cell lines were cultured in a CO2 incubator (INCOmed, Schwabach, Memmert, Germany) at 37°C in 95% humidified air and 5% CO2.
Analysis of Jurkat cells using flow cytometry
The Jurkat cells were counted and adjusted to a cell density of 250,000 cells per mL in cell culture medium. First, 1 mL of cell suspension was seeded into each well of a 48-well plate (TPP, Sigma-Aldrich). Next, 30.4 μL of different dilutions of sterile-filtered SEONDEX-HA*CPt in water as well as of cisplatin in phosphate-buffered saline were pipetted to the cells, to match the overall cisplatin concentration as shown in Table 1. In order to examine the toxicity of the nanoparticles without the drug, sterile filtered SEONDEX-HA dilutions with the same iron concentrations as the SEONDEX-HA*CPt dilutions were added to the cells. The treated cells were then incubated at 37°C in 95% humidified air and 5% CO2. After 0, 24, 36, and 48 hours, 75 μL aliquots were taken from the cell suspension and incubated with 300 μL of freshly prepared staining solution containing 1 μg/mL Annexin V-FITC, 20 μg/mL propidium iodide, 1 μg/mL Hoechst 33342, and 5.1 μg/mL DiI in Ringer’s solution for 20 minutes at 4°C. A further 75 μL of the cell suspension were incubated with 450 μL of PIT solution (1 mg/mL sodium citrate, 0.1% Triton X-100, and 50 μg/mL propidium iodide in water) for at least 12 hours at 4°C, to observe the cell cycle and DNA degradation during apoptosis.
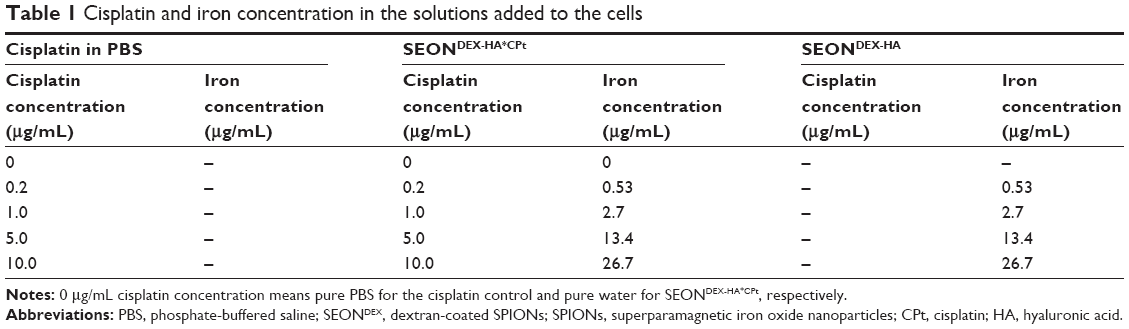 | Table 1 Cisplatin and iron concentration in the solutions added to the cells |
Flow cytometry was performed employing a Gallios cytofluorometer (Beckman Coulter, Pasadena, CA, USA). The excitation and emission wavelengths of the utilized fluorescence dyes are summarized in Table 2. Electronic compensation was used to eliminate bleed through fluorescence. The data analysis was performed with Kaluza software version 2.0 (Beckman Coulter). All flow cytometry experiments were conducted in triplicate, and the results were averaged. Tests for statistical significance was carried out using the Student’s t-test in Excel (Microsoft Corporation, Redmond, WA, USA) (*P<0.05, **P<0.005, and ***P<0.0005).
 | Table 2 Excitation and emission wavelengths of fluorescence dyes used |
Analysis of PC-3 cells using xCELLigence
The in vitro toxicity of adherent PC-3 cells was performed using an xCELLigence RTDP system (ACEA Biosciences, San Diego, CA, USA).43 This device is a label-free, real time cell analysis system that measures the impedance change of an electrode when cells are adhering, spreading, or dying on it. First, 500, 1,000, 1,500, 2,000, 2,500, and 3,000 cells/well were seeded into E-plates and incubated at 37°C in 95% humidified air and 5% CO2. After 24 hours, the medium was removed and replaced with 90 μL of fresh medium, followed by addition of 10 μL of SEONDEX-HA and SEONDEX-HA*CPt to achieve iron concentrations of 10, 30, and 50 μg/mL (corresponding to cisplatin concentrations of 3.7, 11.2, and 18.7 μg/mL) in the wells. As a control, 10 μL of water was added to the cell suspensions. Subsequently, the E-plates were incubated for a further 7 days, during which the cell index was measured. The cell index is a dimensionless parameter and derived from the relative change in the measured electrical impedance to represent cell status. All experiments were performed in triplicate, and the results were averaged.
Results and discussion
Structural characterization of SEONDEX
The first intermediate was dextran-coated SPIONs (SEONDEX), which was the prerequisite for formation of SEONDEX-HA. The dextran content during coprecipitation had an influence on the Z-average, which was acquired with dynamic light scattering and is shown in Figure 1. It is noteworthy that a dextran content of less than 2.0 g (in a total volume of 20 mL) was not sufficient to prevent formation of unstable agglomerates, at least with the given concentrations and precipitation parameters, such as stirring velocity, temperature, and input speed of the base. Therefore, formation of a stable colloid was first achieved with 2.0 g of dextran and had a Z-average of 37.5±0.6 nm. Basically, a further increase in dextran content led to a decrease in the hydrodynamic size until a saturation value of about 21.1±0.9 nm was reached for 4.5 g dextran. This observation makes sense because dextran adsorbs on the particle surface and confines the space for magnetite particle growth as well as agglomeration. The more polymer present in the solution, the more the confinement, until no agglomeration of magnetite particles can take place and only individual particles are dispersed in the dextran matrix. This hypothesis is strengthened with the assortment of TEM images shown in Figure 2. The overview image for SEONDEX 2.0 (number after SEONDEX applies to the dextran content used during coprecipitation; in this case, 2.0 means 2.0 g in a total volume of 20 mL) shows an agglomeration of roundish magnetite particles embedded in a polymer matrix. Figure 3 shows magnetite particle size distributions for SEONDEX 2.0 (Figure 3A) and 4.5 (Figure 3B) derived from measuring 250 particles in the TEM figures (data not shown) with ImageJ software. For both samples, the sizes ranged from 3.0 nm to 6.6 nm and had a mean value of 4.6±0.8 nm for SEONDEX 2.0 and 4.3±0.9 nm for SEONDEX 4.5, respectively. Therefore, it can be concluded that the core size distributions for all SEONDEX samples were quite narrow and in the same order of magnitude, independent of the dextran content. The narrow distribution can be explained by the fact that during coprecipitation the iron salts as well as the added ammonia were quickly and homogenously distributed within the dextran matrix due to the high rotary speed of the stirrer. In the TEM images for SEONDEX 2.0 with a higher magnification, it is possible to see the lattice planes of individual particles, which indicate that the individual magnetite particles were monocrystalline. In order to further examine the crystalline phases of SEONDEX, an X-ray diffraction pattern was recorded for SEONDEX 2.0 and SEONDEX 4.5 (Figure 4). The SEONDEX 2.0 sample shows typical peaks for the face-centered cubic spinel structure of magnetite, with the major peak at 35.3° corresponding to the (311) plane.44 Because of the small crystallites, other peaks for magnetite at 30.3° (220), 43.3° (400), 57.2° (511), and 62.5° (440) are less distinctive.45 In the case of the sample with the highest dextran content during coprecipitation, SEONDEX 4.5, the (311) peak almost vanishes, indicating a lower crystallinity.
 | Figure 1 Z-average of the particles decreased with increasing dextran content during coprecipitation. |
 | Figure 2 Overview TEM image of SEONDEX 2.0 particles shows agglomerates of SPIONs in a polymer matrix (A). Overview TEM image of SEONDEX 4.5 particles shows individual SPIONs embedded in a polymer matrix (B). Higher magnification of SEONDEX 2.0 particles, with visible lattice planes (C). |
 | Figure 3 Magnetite particle distributions derived from measuring 50 particles of the TEM images with ImageJ software. |
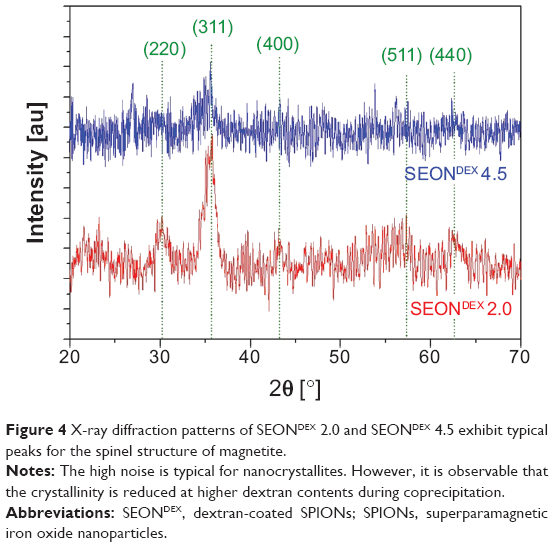 | Figure 4 X-ray diffraction patterns of SEONDEX 2.0 and SEONDEX 4.5 exhibit typical peaks for the spinel structure of magnetite. |
Additionally, magnetite as well as maghemite crystallizes in a cubic spinel structure, with the difference that maghemite possesses 21/3 vacancies confined to octahedral interstitial sites.46 As a consequence, the peak positions only differ slightly from each other, which means that it is not possible to decide from the given DEXPION 2.0 X-ray diffraction pattern, whether maghemite, magnetite, or a mixture of both, was created. Nevertheless, the use of Bragg’s law (5) with the peak position for the (311) plane resulted in dhkl =0.2540 nm, ie, the interplanar spacing between those planes. This value applied in (6) leads to the lattice parameter a =0.8425 nm. It is very close to that of magnetite (amagnetite =0.8396), whereas it is farther away from maghemite (amaghemite =0.8347), indicating that the coprecipitation under inert gas favored formation of magnetite.46 The shift between the theoretical and the measured value could be due to the presence of the polymer as well as the small crystallites and the associated broadening of the peaks. These were also the reasons why the presence of maghemite cannot be fully excluded. Additionally, the full width at half maximum of the (311) peak can be used to calculate the crystallite size according to the Debye–Scherrer formula (7). The obtained value of dXRD =4.3 nm for SEONDEX 2.0 coincides with the mean core particle size of dTEM =4.6±0.8 nm. This proves the aforementioned assumption that the individual magnetite particles were monocrystalline.
Comparisons with the literature revealed that the values for magnetite size derived from coprecipitation in the presence of dextran were in the same range and also narrowly distributed. Pardoe et al for instance, reported a magnetite particle size of 4.11±0.9 nm.24 Magnetite particles by Bogdanov et al had a diameter of 4.6±1.2 nm and an agglomerate size of 20±4 nm.47 In contrast with these values, bare SPIONs precipitated without the presence of a polymer exhibit a higher mean particle size and a broader size distribution in the range of 8±2 nm.48 This difference can be easily explained because, if no polymer is present, Ostwald ripening will be more distinct during coprecipitation, resulting in a larger diameter.
The magnetic properties of SEONDEX are also very important because after administration of the drug delivery vehicles into the body, they are positioned in the tumor tissue with the aid of an external magnetic field. The magnetization curves for SEONDEX were recorded by means of SQUID measurements, which are depicted in Figure 5. All samples show superparamagnetic behavior, with a saturation magnetization dependent on the dextran concentration during precipitation (Table 3). The sample with the amount of lowest dextran showed a saturation magnetization of 454±23 kA/m, whereas the sample with the highest content showed a value of 186±10 kA/m. The value for SEONDEX 2.0 is comparable with that of bulk magnetite, the saturation magnetization of which is 480 kA/m at room temperature.46 In general, the decrease can be explained with the results outlined above. The X-ray diffraction patterns show lesser crystallinity with increasing dextran content, which is why the saturation magnetization is reduced. The curves also showed no hysteresis or residual magnetization. This is very important because once the applied field is removed from the particles, the lack of residual magnetization reduces the probability of agglomeration, thereby avoiding uptake by phagocytes as well as the danger of thrombosis.12
 | Figure 5 Magnetization curves for different SEONDEX after normalization to the volume fraction of the magnetic phase. |
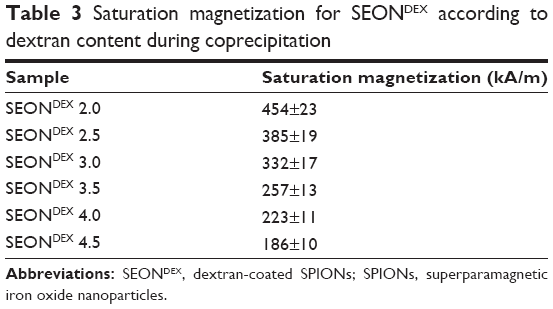 | Table 3 Saturation magnetization for SEONDEX according to dextran content during coprecipitation |
Structural characterization of SEONDEX-HA
The SEONDEX 4.5 sample was chosen for further experiments involving HA and drug loading because their agglomerates were the smallest ones and simultaneously had sufficient magnetic properties. Particle size matters, since only sterile filtration remains as a readily accessible option, which is why the hydrodynamic size of the entire system has to be smaller than 200 nm.
Before deploying HA as a surfactant for SEONDEX, it has to be degraded enzymatically to overcome its poor water solubility. The success of degradation was evaluated by measuring the intrinsic viscosity with an Ubbelohde viscometer. Figure 6 shows the reduced viscosity as a function of HA concentration in 200 mM NaCl. The data points have a linear behavior in the chosen concentration window. On these grounds, [η] can be obtained from the y-intercept of a linear fit. This leads to an [η] of 147.48 mL/g and in combination with (2) to a molar mass of 69±1 kDa, which is in good agreement with the values reported in the literature.36,38
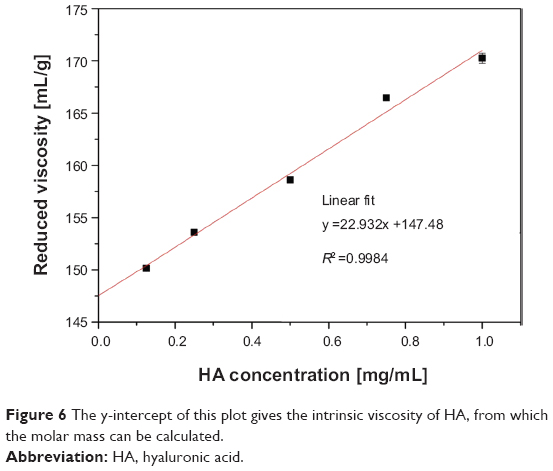 | Figure 6 The y-intercept of this plot gives the intrinsic viscosity of HA, from which the molar mass can be calculated. |
Prior to esterification, the amination of SEONDEX led to SEONDEX-NH2 with a size of 22±0.2 nm. The functionalization changed the zeta potential from −2.5±0.8 mV to 25.8±0.8 mV at pH 7.5 in an aqueous solution. Esterification with pure HA resulted in SEONDEX-HA with a size of 56.0±4.6 nm, as depicted in Figure 7A. Taking the iron concentration and the change in solid content into account, the particles consisted of 51% HA, 29% dextran, and 20% magnetite. Due to the anionic character of HA, the zeta potential was reversed to −33.9±0.7 mV in water at pH 7.5 (Figure 7B), which enabled electrostatic stabilization. SEONDEX-HA did not precipitate after weeks of storage. After incorporation of the drug, the particle size increased further to 76.7±5.6 nm and the zeta potential changed to −45.0±0.3 mV (in water at pH 7.5). The reason behind this increase was the formation of a polymer-metal complex between the carboxylic acid of HA and the platinum atom of cisplatin, which changed the folding and conformation of the polymer. The SEONDEX-HA*CPt were also stable in water. This approach was efficient in terms of drug encapsulation efficacy (43.2%±3%) and in terms of the total amount of incorporated drug (330±10 μg/mL).
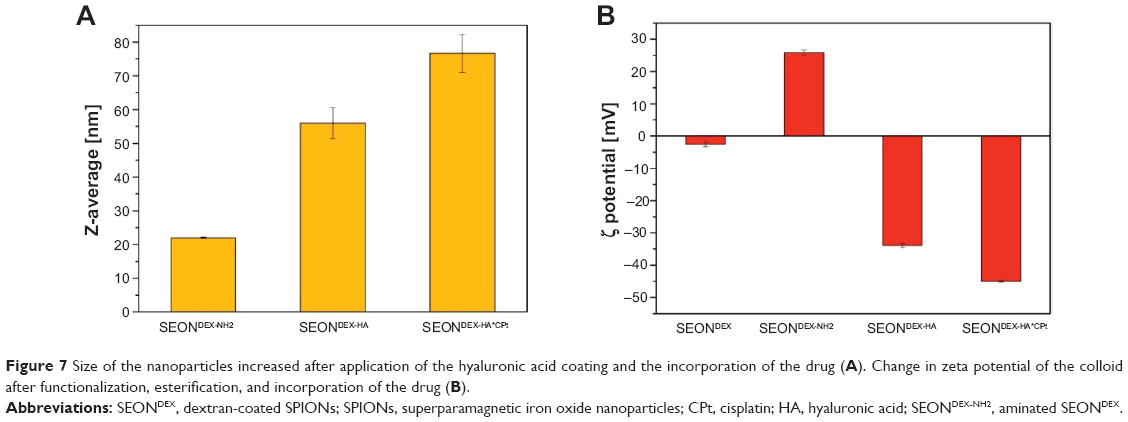 | Figure 7 Size of the nanoparticles increased after application of the hyaluronic acid coating and the incorporation of the drug (A). Change in zeta potential of the colloid after functionalization, esterification, and incorporation of the drug (B). |
In order to analyze the chemical structure of the different particle surfaces, FTIR spectra were obtained and are presented in Figure 8. After amination, the SEONDEX-NH2 should have characteristic bands of N-H deformation. Unfortunately, those bands are usually in the region of 3,200 cm−1 and were in this case overlapped by the broad O-H peak of physically adsorbed water and residual hydroxyl groups in the dextran chain.23 However, the success of the functionalization can be observed in the relative intensity change of the α-glucopyranose ring deformation modes. The nitrogen atom has a lower electronegativity and therefore alters the electron density distribution in the α-glucopyranose ring. This alteration led to a slightly different deformation and vibration behavior of the ring, which caused the change of FTIR spectrum in this region. Since HA has an amide bond in its structure, it is not easy to distinguish between this structural amide and the amide that caused the linkage between SEONDEX-NH2 and dextran. Nevertheless, indications for the existence of this linkage amide were given on closer examination of the wave number region between 1,250 cm−1 and 1,750 cm−1 for SEONDEX-HA and pure HA. The relative intensities between those peaks as well as the peak positions slightly differed from each other. This alteration might be caused by the different frameworks of the amide bond, as the linkage between dextran and HA the amide bond is rather fixed, while in pure HA it is located at the second carbon atom of the N-acetylglucosamine and is therefore more flexible. As a result, the FTIR measurements are in agreement with the changes in size and zeta potential of the particles and because of that are further proof for the modification of the particle surface.
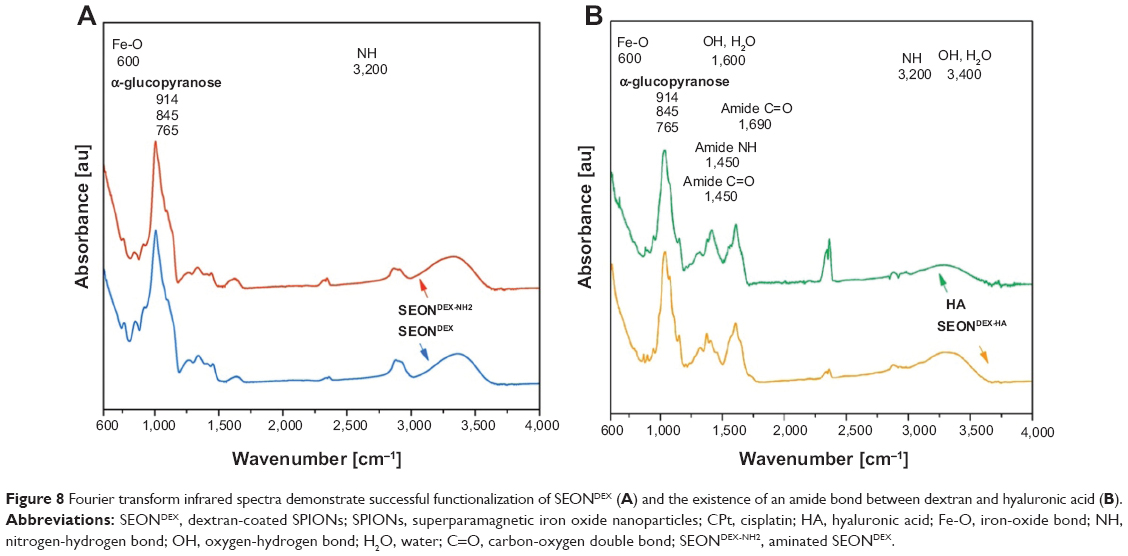 | Figure 8 Fourier transform infrared spectra demonstrate successful functionalization of SEONDEX (A) and the existence of an amide bond between dextran and hyaluronic acid (B). |
After the structural and chemical analysis, it can be concluded that the fabrication of superparamagnetic particles with a cisplatin-bearing HA coating, which can be controlled using magnetic forces (see Figure 9), was successful.
 | Figure 9 Fabricated SEONDEX-HA*CPt can be controlled with a neodymium magnet. |
Biological characterization of SEONDEX-HA
Before the biological impact on cells was evaluated, the drug release kinetics of SEONDEX-HA*CPt were investigated in physiological saline at 37°C. Figure 10 depicts the time-dependent cumulative release of cisplatin, which shows two stages. Firstly, an initial burst release (26.2%) occurred within 30 minutes, which is typical for a surface-bound drug. Afterwards, the kinetics were dominated by a continuous release of cisplatin until 48 hours due to inverse ligand substitution of chloride atoms in phosphate-buffered saline. Pure cisplatin dissolved in phosphate-buffered saline was used as a control in order to investigate how long it took the drug to escape the dialysis tube after its liberation. The initial release of the control was 60% and after 1 hour around had 90% diffused out of the tube. This comparison illustrates that SEONDEX-HA*CPt successfully retarded release of the drug. This is an important feature because liberation of cisplatin should be minimal during transport of the drug vehicle to the target site. High levels of HAse activity have been found in various tumors.49,50 Its presence in dialysis tubes increased the cisplatin release rate, due to enzymatic degradation of HA. Consistently, the initial release within the first 30 minutes increased from 26.2% to 42.4% and the whole drug was released rapidly within 24 hours. Since SEONDEX-HA*CPt dispersed in an aqueous solution did not release a measurable amount of cisplatin within 48 hours, it can be concluded that the release mechanism in vivo and in vitro is determined by a combination of HA degradation and inverse ligand substitution and diffusion through the polymer shell.
 | Figure 10 Release kinetics of cisplatin from SEONDEX-HA*CPt in PBS at 37°C in comparison with pure cisplatin dissolved in PBS. |
The influence of various SEONDEX-HA*CPt, SEONDEX-HA, and pure cisplatin in phosphate-buffered saline (referred to as “cisplatin fluid” in the following text) concentrations on Jurkat cells was determined using Annexin V/propidium iodide as a marker for apoptosis (Annexin V positive, propidium iodide negative) and necrosis (Annexin V positive, propidium iodide positive) in flow cytometer analysis. The basic principle for this measurement is the interaction of Annexin V-FITC with phosphatidylserine, which is one of the components of cell membranes.51 In viable cells, phosphatidylserine is located in the inner leaflet of the plasma membrane, and during apoptosis it is exposed on the outer leaflet of the still intact plasma membrane. In this apoptotic state Annexin V-FITC binds to phosphatidylserine and can then be recognized in the flow cytometer. In contrast to apoptotic cells, necrotic cells are characterized by a ruptured plasma membrane. Propidium iodide, as a normally membrane-impermeable dye, can in the case of such membrane morphology penetrate into the cells and intercalate into the DNA, giving rise to a fluorescence signal. The results for these Annexin V/propidium iodide measurements are presented in Figure 11. Treatment of Jurkat cells with SEONDEX-HA (without drug) for 48 hours demonstrated that no cell death was induced in the given iron concentration range and the cells had a viability of around 90%, comparable with that of the untreated controls. Cisplatin concentrations of 0.2 μg/mL for cisplatin fluid and SEONDEX-HA*CPt did not cause a significant increase of cell death. However, cisplatin concentrations from 1 μg/mL onwards induced apoptosis in a dose-dependent manner, with secondary necrosis after prolonged incubation for 48 hours. The reason for this behavior is the formation of interstrand and intrastrand crosslinks caused by cisplatin inside the DNA.52,53 Thus, the cells treated with SEONDEX-HA*CPt showed a slower increase of apoptotic/necrotic cells in comparison with cisplatin fluid. This observation makes sense, since the cisplatin had to be released from its carrier in the first place before it could react with the DNA in the cell.
 | Figure 11 Death of Jurkat cells determined by Annexin V/propidium iodide staining after treatment with cisplatin fluid, SEONDEX-HA*CPt, and SEONDEX-HA. SEONDEX-HA without the drug did not induce cell death, whereas cisplatin fluid and SEONDEX-HA*CPt induced apoptosis in a dose-dependent manner, with secondary necrosis after prolonged incubation. |
The impact of the particles and fluid cisplatin is also shown in the light microscope images recorded after 30 hours of treatment (Figure 12). The controls and the particles without drug displayed Jurkat cells with viable morphology. In comparison, the cisplatin fluid and SEONDEX-HA*CPt samples exhibited blebbing and shredding of the plasma membrane, starting from 1 μg/mL cisplatin and increasing in severity with increasing concentration. In addition to Annexin V/propidium iodide measurement, the mitochondrial membrane potential of the cells was analyzed by Hoechst 33342/DiI staining. The principle for this method is that healthy cells have a proton gradient in the inner mitochondrial plasma membrane, generating the conversion from adenosine diphosphate to adenosine triphosphate. A decrease in this mitochondrial potential is an indicator of apoptosis and necrosis and can be detected with DiI, a compound that penetrates the cytosol of cells and accumulates in active mitochondria.51 After treatment of the cells with SEONDEX-HA for 48 hours, the mitochondrial membrane potential was not significantly affected (Figure S1). The results for fluid cisplatin and SEONDEX-HA*CPt are in good agreement with the Annexin V/propidium iodide measurements, showing a dose-dependent increase in membrane potential reduction and also a delayed effect of SEONDEX-HA*CPt compared with fluid cisplatin.
 | Figure 12 Light microscope images of Jurkat cells after 30 hours of treatment with cisplatin fluid. |
Lastly, PIT measurements were performed to observe the cell cycle and DNA degradation during apoptosis. The cell cycle of proliferating cells can be categorized into the following phases:54 G1-phase (postmitotic, production of organelles), S-phase (DNA synthesis), G2-phase (premitotic, preparation for cell division), and M-phase (mitosis, cell division). During apoptosis, the DNA is degraded by DNases into smaller fragments, producing the subG1-phase. Consistent with the aforementioned results, the SEONDEX-HA did not influence the cell cycle or DNA degradation when compared with the controls, even after 48 hours of incubation (Figure S2). In contrast with this, the cells treated with cisplatin fluid and SEONDEX-HA*CPt produced a high amount of subG1-phase, reflecting DNA degradation due to induction of cell death, as expected from the previous results. Taking all the flow cytometry results into account, it can be concluded that the cytotoxic impact of SEONDEX-HA*CPt on nonadherent Jurkat cells resulted solely from the released cisplatin and not from other chemical components or characteristics of the particles.
The second method by which the in vitro toxicity of the fabricated particles was investigated involved use of the xCELLigence analysis system. This device was used to measure the impedance change of an electrode, on which different concentrations of PC-3 cells were incubated for 7 days. Proliferation of cells results in increased impedance, whereas cell death is characterized by reduced impedance, reflecting dissolution of the adherent cells from the electrode. The impedance changes for different particle treatments are shown in Figure 13. Cells treated with SEONDEX-HA did not show a decrease compared with the controls, implying normal proliferation of PC-3 cells with no influence from the particles. With an increase in cell concentration per well, the mean value of the impedance change increased as expected. In contrast with this, the impedance of SEONDEX-HA*CPt-treated cells was reduced, which is an indicator of cell death. This analysis showed the same trend as the flow cytometry analysis, with the result that SEONDEX-HA were not harmful to cells, at least in the two cell lines tested. These findings also confirmed that the cytotoxic effect of SEONDEX-HA*CPt derived solely from the release of cisplatin.
 | Figure 13 With an xCELLigence system, the impedance change was measured for 7 days during incubation of PC-3 cells, while they were treated with SEONDEX-HA and SEONDEX-HA*CPt. |
Conclusion
In this project, SPIONs with a dextran and a cisplatin-bearing HA coating were successfully synthesized as a novel cisplatin drug delivery system. This is the first time to the authors’ knowledge that SPIONs have been concurrently combined with HA and cisplatin. The particles showed superparamagnetic behavior with a saturation magnetization dependent on the dextran content during coprecipitation. In addition, they showed no signs of agglomeration in water or in phosphate-buffered saline over several weeks. Their biological activity was investigated using nonadherent Jurkat cells in flow cytometry experiments and for adherent PC-3 cells in xCELLigence experiments. On the one hand, both tests demonstrated that particles without drug were biocompatible with these cells and showed no cytotoxic effects. On the other hand, particles with incorporated cisplatin induced apoptosis in a dose-dependent manner, with secondary necrosis after prolonged incubation. An interesting question for future analyses is whether cells expressing CD44 receptors have a higher particle uptake due to interactions with HA when compared with cells that do not express them. In conclusion, the combination of dextran-coated SPIONs with HA and cisplatin represents a promising approach for use in magnetic drug targeting for cancer therapy.
Acknowledgments
The authors are grateful to the Deutsche Forschungsgmeinschaft (DFG) (AL 552/5-1 and TR 408/4-1), to the Bavarian State Ministry of the Environment and Consumer Protection (74-U8793-2012/7-35), to the Emerging Fields Initiative of the University of Erlangen-Nuremberg (EFI), to the DFG Cluster of Excellence – Engineering of Advanced Materials (EAM), and to the Else Kröner-Fresenius-Stiftung, Bad Homburg vor der Höhe, Germany, for their support. We further acknowledge support by Deutsche Forschungsgemeinschaft and Friedrich-Alexander-Universität Erlangen-Nürnberg within the Open Access Publishing funding program. We also would like to thank Professor Hermann (Department for Internal Medicine 3, University Hospital Erlangen, Germany) for providing Annexin V-FITC.
Disclosure
The authors report no conflicts of interest in this work.
References
Tietze R, Lyer S, Durr S, Alexiou C. Nanoparticles for cancer therapy using magnetic forces. Nanomedicine (Lond). 2012;7(3):447–457. | ||
Alexiou C, Tietze R, Schreiber E, et al. Cancer therapy with drug loaded magnetic nanoparticles – magnetic drug targeting. J Magn Magn Mater. 2011;323(10):1404–1407. | ||
Tietze R, Jurgons R, Lyer S, et al. Quantification of drug-loaded magnetic nanoparticles in rabbit liver and tumor after in vivo administration. J Magn Magn Mater. 2009;321(10):1465–1468. | ||
Alexiou C, Diehl D, Henninger P, et al. A high field gradient magnet for magnetic drug targeting. IEEE Appl Supercond. 2006;16(2): 1527–1530. | ||
Seliger C, Jurgons R, Wiekhorst F, et al. In vitro investigation of the behaviour of magnetic particles by a circulating artery model. J Magn Magn Mater. 2007;311(1):358–362. | ||
Cheng L, Jin C, Lv W, Ding Q, Han X. Developing a highly stable PLGA-mPEG nanoparticle loaded with cisplatin for chemotherapy of ovarian cancer. PLoS One. 2011;6(9):e25433–e25433. | ||
Tietze R, Lyer S, Durr S, et al. Efficient drug-delivery using magnetic nanoparticles-biodistribution and therapeutic effects in tumour bearing rabbits. Nanomedicine. 2013;9(7):961–971. | ||
Janko C, Dürr S, Munoz L, et al. Magnetic drug targeting reduces the chemotherapeutic burden on circulating leukocytes. Int J Mol Sci. 2013; 14(4):7341–7355. | ||
Bealle G, Di CR, Kolosnjaj-Tabi J, et al. Ultra magnetic liposomes for mr imaging, targeting, and hyperthermia. Langmuir. 2012;28(32): 11834–11842. | ||
Alexiou C, Tietze R, Schreiber E, Lyer S. [Pharmakotherapie mittels Nanomedizin]. Der Onkologe. 2011;17(5):405–412. German. | ||
Dorniani D, Hussein MZ, Kura AU, Fakurazi S, Shaari AH, Ahmad Z. Preparation of Fe3O4 magnetic nanoparticles coated with gallic acid for drug delivery. Int J Nanomedicine. 2012;7:5745–5756. | ||
Wahajuddin, Arora S. Superparamagnetic iron oxide nanoparticles: magnetic nanoplatforms as drug carriers. Int J Nanomedicine. 2012;7: 3445–3471. | ||
Zhao Y, Dunnill CW, Zhu Y, et al. Low-temperature magnetic properties of hematite nanorods. Chem Mater. 2007;19(4):916–921. | ||
Shi S, Jia JF, Guo XK, et al. Toxicity of iron oxide nanoparticles against osteoblasts. J Nanopart Res. 2012;14(9):1091. | ||
Lyer S, Tietze R, Jurgons R, et al. Visualisation of tumour regression after local chemotherapy with magnetic nanoparticles – a pilot study. Anticancer Res. 2010;30(5):1553–1557. | ||
Alexiou C, editor. Nanomedicine: Basic and Clinical Applications in Diagnostics and Therapy. Volume 2. Basel, Switzerland: Karger; 2011. | ||
Lyer S, Tietze R, Dürr S, et al. Diagnostic imaging in cancer therapy with magnetic nanoparticles. In: Buzug TM, Borgert J, editors. Magnetic Particle Imaging. Volume 140. Heidelberg, Germany: Springer Verlag; 2012. | ||
Brunke O, Odenbach S, Jurgons R, Alexiou C, Hilger I, Beckmann F. Determination of the magnetic particle distribution in tumour tissue by means of x-ray tomography. J Phys Condens Matter. 2006; 18(38):S2903. | ||
Tietze R, Rahn H, Lyer S, et al. Visualization of superparamagnetic nanoparticles in vascular tissue using XmuCT and histology. Histochem Cell Biol. 2011;135(2):153–158. | ||
Harisinghani MG, Barentsz J, Hahn PF, et al. Noninvasive detection of clinically occult lymph-node metastases in prostate cancer. N Engl J Med. 2003;348(25):2491–2499. | ||
Kau RJ, Alexiou C, Stimmer H, Arnold W. Diagnostic procedures for detection of lymph node metastases in cancer of the larynx. ORL J Otorhinolaryngol Relat Spec. 2000;62(4):199–203. | ||
Kim DK, Zhang Y, Voit W, Rao KV, Muhammed M. Synthesis and characterization of surfactant-coated superparamagnetic monodispersed iron oxide nanoparticles. J Magn Magn Mater. 2001;225(1–2):30–36. | ||
Carmen Bautista M, Bomati-Miguel O, del Puerto Morales M, Serna CJ, Veintemillas-Verdaguer S. Surface characterisation of dextran-coated iron oxide nanoparticles prepared by laser pyrolysis and coprecipitation. J Magn Magn Mater. 2005;293(1):20–27. | ||
Pardoe H, Chua-anusorn W, St Pierre TG, Dobson J. Structural and magnetic properties of nanoscale iron oxide particles synthesized in the presence of dextran or polyvinyl alcohol. J Magn Magn Mater. 2001;225(1–2):41–46. | ||
Kamat M, El-Boubbou K, Zhu DC, et al. Hyaluronic acid immobilized magnetic nanoparticles for active targeting and imaging of macrophages. Bioconjug Chem. 2010;21(11):2128–2135. | ||
Lee YT, Woo K, Choi KS. Preparation of water-dispersible and biocompatible iron oxide nanoparticles for MRI agent. IEEE Trans Nanotechnol. 2008;7(2):111–114. | ||
Lapcik L Jr, Lapcik L, De Smedt S, Demeester J, Chabrecek P. Hyaluronan: preparation, structure, properties, and applications. Chem Rev. 1998;98(8):2663–2684. | ||
Laurent TC, Fraser JR. Hyaluronan. FASEB J. 1992;6(7):2397–2404. | ||
Laurent TC, Laurent UB, Fraser JR. Functions of hyaluronan. Ann Rheum Dis. 1995;54(5):429–432. | ||
Raman R, Sasisekharan V, Sasisekharan R. Structural insights into biological roles of protein-glycosaminoglycan interactions. Chem Biol. 2005;12(3):267–277. | ||
Noro R, Yoshimura A, Yamamoto K, et al. Alternating chemotherapy with amrubicin plus cisplatin and weekly administration of irinotecan plus cisplatin for extensive-stage small cell lung cancer. Anticancer Res. 2013;33(3):1117–1123. | ||
Sunwoo JB. A cisplatin-resistant subpopulation of mesenchymal-like cells in head and neck squamous cell carcinoma. Cell Cycle. 2011; 10(17):2834–2835. | ||
Ratanapharn A. New insights into cellular responses to cisplatin. In: Kojima T, Morita Y, editors. Cisplatin: Pharmacology, Clinical Uses and Adverse Effects. Hauppauge, NY, USA: Nova Science Publishers Inc.; 2012. | ||
Wang G, Reed E, Li QQ. Molecular basis of cellular response to cisplatin chemotherapy in non-small cell lung cancer. Oncol Rep. 2004;12(5): 955–965. | ||
Carrick S, Ghersi D, Wilcken N, Simes J. Platinum containing regimens for metastatic breast cancer. Cochrane Database Syst Rev. 2004;2:CD003374. | ||
Jeong YI, Kim ST, Jin SG, et al. Cisplatin-incorporated hyaluronic acid nanoparticles based on ion-complex formation. J Pharm Sci. 2008;97(3):1268–1276. | ||
Gura E, Hückel M, Müller PJ. Specific degradation of hyaluronic acid and its rheological properties. Polym Degrad Stabil. 1998;59(1–3): 297–302. | ||
Lee HG, Cowman MK. An agarose gel electrophoretic method for analysis of hyaluronan molecular weight distribution. Anal Biochem. 1994;219(2):278–287. | ||
Cullity BD, Stock SR. Elements of X-Ray Diffraction. Upper Saddle River, NJ, USA: Prentice Hall; 2001. | ||
Dokuzovic L. Kolorimetrie-Photometrie. Available from: http://www.lickl.net/doku/photo.pdf. Accessed June 8, 2014. | ||
Eberbeck D, Wiekhorst F, Wagner S, Trahms L. How the size distribution of magnetic nanoparticles determines their magnetic particle imaging performance. Appl Phys Lett. 2011;98:182502.1–182502.3. | ||
Nishiyama N, Okazaki S, Cabral H, et al. Novel cisplatin-incorporated polymeric micelles can eradicate solid tumors in mice. Cancer Res. 2003;63(24):8977–8983. | ||
Dürr S, Lyer S, Mann J, et al. Real-time cell analysis (RTCA) of human cancer cell lines after chemotherapy with functionalized magnetic nanoparticles. Anticancer Res. 2011;31(5):1966–1967. | ||
Salazar-Alvarez G, Muhammed M, Zagorodni AA. Novel flow injection synthesis of iron oxide nanoparticles with narrow size distribution. Chem Eng Sci. 2006;61(14):4625–4633. | ||
Mohapatra S, Pramanik N, Ghosh SK, Pramanik P. Synthesis and characterization of ultrafine poly(vinylalcohol phosphate) coated magnetite nanoparticles. J Nanosci Nanotechnol. 2006;6(3):823–829. | ||
Cornell RM, Schwertmann U. Crystal structure. In: The Iron Oxides: Structure, Properties, Reactions, Occurences and Uses. John Wiley & Sons: New York, NY, USA; 2004. | ||
Bogdanov AA Jr, Martin C, Weissleder R, Brady TJ. Trapping of dextran-coated colloids in liposomes by transient binding to aminophospholipid: preparation of ferrosomes. Biochim Biophys Acta. 1994; 1193(1):212–218. | ||
Morales MP, Veintemillas-Verdaguer S, Montero MI, et al. Surface and internal spin canting in γ-Fe2O3 nanoparticles. Chem Mater. 1999;11(11):3058–3064. | ||
Murai T, Kawashima H. A simple assay for hyaluronidase activity using fluorescence polarization. Biochem Biophys Res Commun. 2008;376(3): 620–624. | ||
Sugahara KN, Hirata T, Hayasaka H, Stern R, Murai T, Miyasaka M. Tumor cells enhance their own CD44 cleavage and motility by generating hyaluronan fragments. J Biol Chem. 2006;281(9):5861–5868. | ||
Munoz LE, Maueroder C, Chaurio R, Berens C, Herrmann M, Janko C. Colourful death: six-parameter classification of cell death by flow cytometry – dead cells tell tales. Autoimmunity. 2013;46(5): 336–341. | ||
Raber J, Zhu C, Eriksson LA. Theoretical study of cisplatin binding to DNA:the importance of initial complex stabilization. J Phys Chem B. 2005;109(21):11006–11015. | ||
Zwelling LA, Filipski J, Kohn KW. Effect of thiourea on survival and DNA cross-link formation in cells treated with platinum(II) complexes, L-phenylalanine mustard, and bis(2-chloroethyl)methylamine. Cancer Res. 1979;39(12):4989–4995. | ||
Riccardi C, Nicoletti I. Analysis of apoptosis by propidium iodide staining and flow cytometry. Nat Protoc. 2006;1(3):1458–1461. |
Supplementary materials
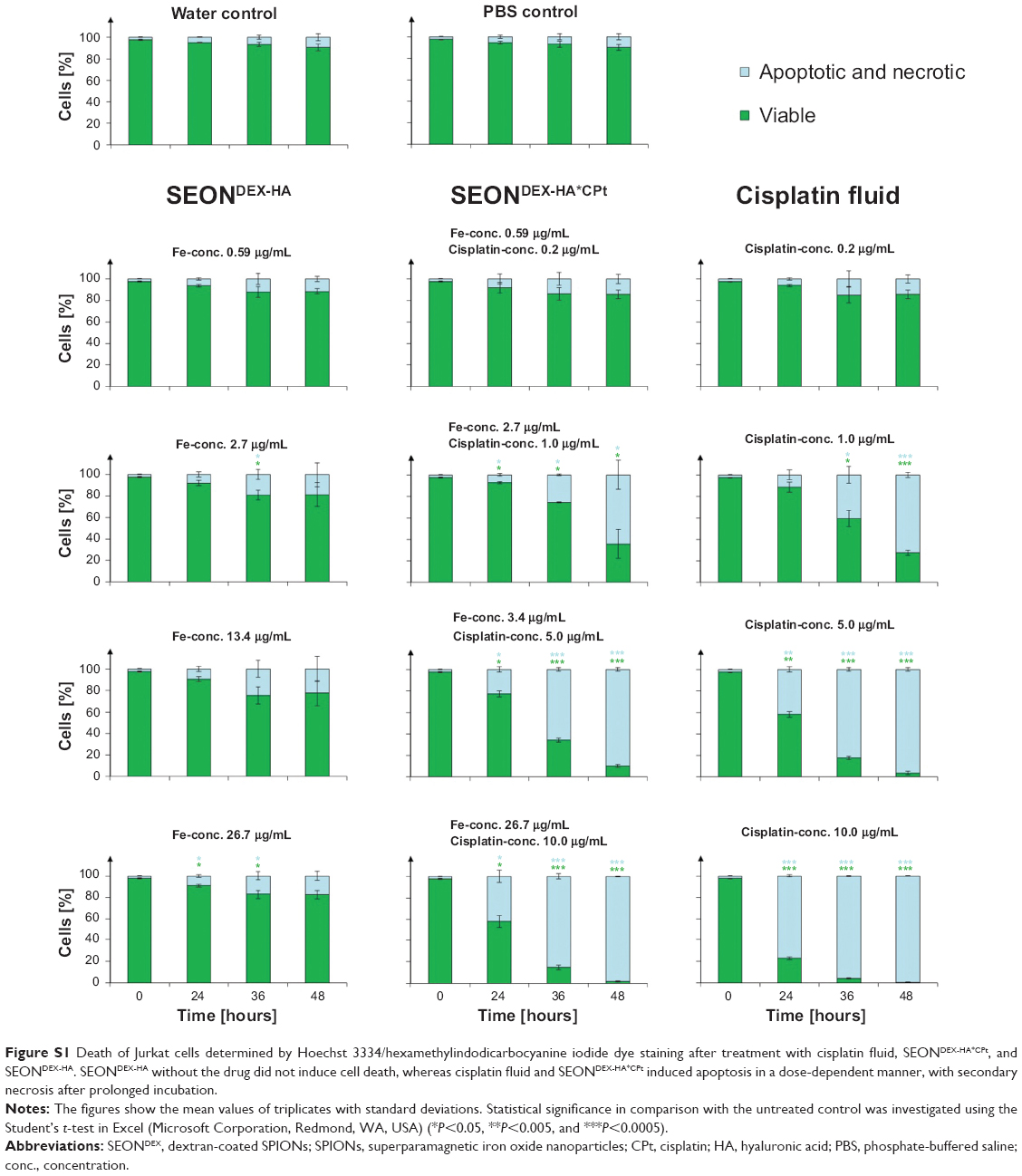 | Figure S1 Death of Jurkat cells determined by Hoechst 3334/hexamethylindodicarbocyanine iodide dye staining after treatment with cisplatin fluid, SEONDEX-HA*CPt, and SEONDEX-HA. SEONDEX-HA without the drug did not induce cell death, whereas cisplatin fluid and SEONDEX-HA*CPt induced apoptosis in a dose-dependent manner, with secondary necrosis after prolonged incubation. |
 | Figure S2 Cell cycle and DNA degradation of Jurkat cells determined by PIT staining after treatment with cisplatin fluid, SEONDEX-HA*CPt, and SEONDEX-HA. SEONDEX-HA without drug did not induce DNA degradation (increase in subG1-phase), whereas cisplatin fluid and SEONDEX-HA*CPt induced DNA degradation in a dose and time dependent manner. |
 © 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.