Back to Archived Journals » Oncolytic Virotherapy » Volume 7
Development and applications of oncolytic Maraba virus vaccines
Authors Pol JG ![]() , Atherton MJ, Bridle BW
, Atherton MJ, Bridle BW ![]() , Stephenson KB, Le Boeuf F, Hummel JL
, Stephenson KB, Le Boeuf F, Hummel JL ![]() , Martin CG, Pomoransky J, Breitbach CJ, Diallo JS
, Martin CG, Pomoransky J, Breitbach CJ, Diallo JS ![]() , Stojdl DF, Bell JC, Wan Y, Lichty BD
, Stojdl DF, Bell JC, Wan Y, Lichty BD
Received 30 July 2018
Accepted for publication 18 October 2018
Published 26 November 2018 Volume 2018:7 Pages 117—128
DOI https://doi.org/10.2147/OV.S154494
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Chae-Ok Yun
Jonathan G Pol,1–5 Matthew J Atherton,6 Byram W Bridle,7 Kyle B Stephenson,8 Fabrice Le Boeuf,9,10 Jeff L Hummel,6,11 Chantal G Martin,8 Julia Pomoransky,8 Caroline J Breitbach,8 Jean-Simon Diallo,9,10 David F Stojdl,8,12 John C Bell,8–10 Yonghong Wan,6 Brian D Lichty6,8
1Gustave Roussy Comprehensive Cancer Institute, Villejuif, France; 2Institut National de la Santé Et de la Recherche Médicale (INSERM), U1138, Paris, France; 3Team 11 labelled Ligue Nationale contre le Cancer, Cordeliers Research Center, Paris, France; 4Université Paris Descartes/Paris V, Sorbonne Paris Cité, Paris, France; 5Sorbonne Universités/Université Pierre et Marie Curie/Paris VI, Paris, France; 6Department of Pathology and Molecular Medicine, McMaster Immunology Research Centre, McMaster University, Hamilton, ON, Canada; 7Department of Pathobiology, Ontario Veterinary College, University of Guelph, Guelph, ON, Canada; 8Turnstone Biologics, Ottawa, ON, Canada; 9Centre for Innovative Cancer Research, Ottawa Hospital Research Institute, Ottawa, ON, Canada; 10Department of Biochemistry, Microbiology and Immunology, University of Ottawa, Ottawa, ON, Canada; 11Clinical Trial Division, CANSWERS, Georgetown, ON, Canada; 12Children’s Hospital of Eastern Ontario Research Institute, Ottawa, ON, Canada
Abstract: Oncolytic activity of the MG1 strain of the Maraba vesiculovirus has proven efficacy in numerous preclinical cancer models, and relied not only on a direct cytotoxicity but also on the induction of both innate and adaptive antitumor immunity. To further expand tumor-specific T-cell effector and long-lasting memory compartments, we introduced the MG1 virus in a prime-boost cancer vaccine strategy. To this aim, a replication-incompetent adenoviral [Ad] vector together with the oncolytic MG1 have each been armed with a transgene expressing a same tumor antigen. Immune priming with the Ad vaccine subsequently boosted with the MG1 vaccine mounted tumor-specific responses of remarkable magnitude, which significantly prolonged survival in various murine cancer models. Based on these promising results, we validated the safety profile of the Ad:MG1 oncolytic vaccination strategy in nonhuman primates and initiated clinical investigations in cancer patients. Two clinical trials are currently under way (NCT02285816; NCT02879760). The present review will recapitulate the discoveries that led to the development of MG1 oncolytic vaccines from bench to bedside.
Keywords: Maraba MG1, oncolytic virus, tumor antigen, cancer vaccine, MAGE-A3
Background
In 2015, oncolytic virotherapy officially joined the armamentarium of cancer immunotherapies in Western countries following the clinical approval of the herpes simplex virus (HSV) T-VEC against melanoma by the US Food and Drug Administration (FDA) and European Medicines Agency (EMA).1 Since, more than 60 Phase I/II trials evaluating oncolytic viruses (OVs) have been initiated (source: https://www.clinicaltrials.gov/). Among them, we have initiated two first-in-man studies to evaluate the safety and efficacy of an oncolytic vaccine based on the Maraba virus in patients with advanced solid tumors (clinicaltrials.gov identifiers: NCT02285816, NCT02879760).
Maraba virus was first isolated from Amazonian phlebotomine sand flies in Brazil and has not been detected outside South America to date.2 Phylogenetically, Maraba virus belongs to the vesiculovirus genus of the Rhabdoviridae family and is genetically distinct from but shares some homology with the prototypical vesicular stomatitis virus (VSV).3 The virion consists of a bullet-shaped enveloped particle (Ø: 70 nm × L: 170 nm) that harbors an 11-kb single-stranded negative-sense RNA genome (NCBI reference: NC_025255). Its genome contains a 3′ leader sequence and a 5′ trailer sequence separated by five open reading frames, each encoding one viral protein: nucleocapsid (N), phosphoprotein (P), matrix (M), glycoprotein (G), and polymerase (L). Vesiculoviruses infect a wide range of mammalian hosts (eg, horses, cattle, pigs, mules, rodents) where they are responsible for a benign acute disease that mostly manifests with oral vesicles or ulcers. Reported cases of VSV infection in humans mainly concerned laboratory workers, farmers and veterinarians, and remained largely asymptomatic. Rare clinical manifestations included fever and subsequent flu-like symptoms.4 Regarding Maraba virus, no virus-related pathogenicity has been reported in humans and only one case of seroconversion against viral antigens has been documented in the literature.2 For these reasons, the nonpathogenic Maraba arbovirus constitutes an agent of interest for therapeutic purpose.3
The present review will recapitulate the preclinical studies that have demonstrated Maraba virus’ superior propensity to induce cancer cell death, to promote and adjuvantize antitumor immunity, to boost the latter when exploited as a cancer vaccine vector, and to instate immune memory that protects from disease recurrence.
Direct oncolysis of Maraba virus-infected malignant entities
Among the numerous viral strains demonstrating oncolytic activity,5–9 our group and others characterized the ability of vesiculoviruses to preferentially infect, replicate in, and kill malignant cells. VSV paved the way in the early 2000s.10–14 More recently, 20 strains of rhabdoviruses have been screened for oncolytic property, among which Maraba virus showed the broadest oncotropism. The virus was the only candidate to complete a lytic cycle in all human and murine cell lines tested and derived from a variety of cancer types (ie, breast, brain, colon, skin, lung, ovarian, mammary, prostate, and renal cancers).3 Of note, Maraba virus (like VSV) exploits, yet not exclusively, the ubiquitous low-density lipoprotein receptor (LDLR) for its entry in the target cells providing one explanation for the wide range of malignant cell hosts infected.15,16 Consistent with this, reduced expression of LDLR was associated with a decreased susceptibility to Maraba virus entry and killing in some cell lines derived from ovarian cancer patient ascites.16
In order to enhance and focus Maraba virus replication in malignant cells, its genome has been genetically engineered. Two single mutations have been introduced which translated into the L123W and Q242R substitutions in the sequence of the M and G proteins, respectively. In vitro, the resulting strain, named MG1, demonstrated a faster replication, a larger burst size, and an increased killing potency in tumor cells, in comparison to the wild-type (wt) and to other mutant strains of Maraba virus. Inversely, MG1 was strongly attenuated in normal primary cells validating its oncoselectivity.3 Abortive replication in normal cells was mostly due to the inability of MG1 to block type I interferon [IFN]-mediated antiviral immunity, thus restraining its productive cycle to cells deficient/defective in the IFN signaling pathway; an abnormality frequently acquired during oncogenesis.3
In vitro, MG1 has now validated an oncolytic activity against multiple adherent cancer cell lines of human, canine, and murine origins (Table 1).3,15–20 Additionally, Tong et al also evaluated the killing activity of MG1 against a four-dimensional culture model that mimics the successive stages leading to peritoneal carcinomatosis in ovarian cancer, namely 1) monolayer of adherent tumor cells, 2) detached cells in suspension, 3) suspended three-dimensional spheroids, and 4) reattached spheroids.15 Interestingly, MG1 was able to infect, replicate, and induce cell death in ovarian cancer cells, regardless of the stage, be either adherent or suspended, individualized or organized in a spheroid structure. In comparison, two oncolytic poxviruses, namely myxomavirus (MYXV) and double-deleted vaccinia virus (vvDD), were also able to eliminate adherent ovarian tumor cells but to a lesser extent than MG1. Although MYXV and vvDD were also capable of infecting and spreading into suspended spheroids, they lost their cytolytic activity in the final stages of the lytic cycle unless the spheroids readhered. Altogether, this study highlighted the rather unique oncolytic activity of MG1 against spheroids, a structure that can be detected in the peritoneal cavity of ovarian cancer patients and that precedes metastatic implantation in the peritoneal wall.15 Comparative evaluation has also been performed against sarcoma cell lines where MG1 also outcompeted referral OV strains embodied by VSVΔM51, HSV-1 N212, vvDD, and type 3 Dearing reovirus.17
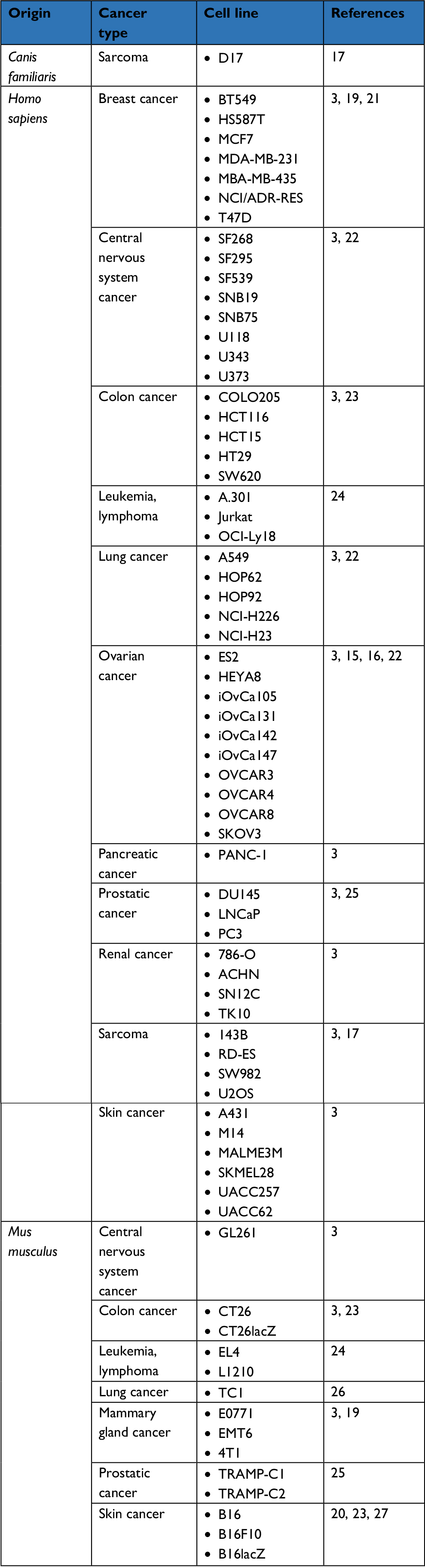 | Table 1 List of cancer cell lines susceptible to Maraba MG1 oncolysis in vitro |
Ex vivo, the MG1 strain of the Maraba virus has been evaluated against various primary human cancer biopsies, where it also displayed productive infection and significant cytopathic effect. Resected tissues were originating from prostate cancer, head and neck squamous cell carcinoma, or sarcomas.17,18,25 These latter are of poor prognosis and patients affected with such bone and soft tissue malignancies remain in an urgent need for alternative treatments. In line with this, MG1 successfully infected and replicated in no less than six subtypes of sarcoma collected from 21 distinct patients and including osteosarcoma, undifferentiated pleomorphic sarcoma, chondrosarcoma, leiomyosarcoma, and rhabdomyosarcoma. Thus, OV therapy could represent a promising approach for the care of these highly morbid diseases.17
In vivo, MG1 can be safely delivered systemically, opening a window of opportunity for the treatment of not only localized but also disseminated cancerous lesions. In immunocompetent mice, the maximum tolerated dose of intravenous MG1 reaches a high dose of 109 plaque-forming units (pfu); 2 logs higher than the parental Maraba virus.3 When repeatedly administered to rodents bearing syngeneic subcutaneous CT26 colorectal carcinoma, MG1 successfully infected the tumor bed and replicated in situ, leading to complete tumor regression and durable cures. Similarly, MG1 was as impressively efficient at clearing CT26 lung metastases. In comparison to its prototypical cousin VSV, MG1 showed superior therapeutic efficacy.3 Ultimately, the oncolytic activity of MG1 was confirmed in multiple additional syngeneic murine tumor models (Table 2).17 Potent oncolytic activity was also demonstrated in xenograft models using human cancer cell lines or patient-derived tumors implanted in immunodeficient mice (Table 2).3,17,19,21,23 To unlock the access to enhanced oncotoxicity, multimodal combination regimens have been developed.19,22 For instance, co-treatment of mice bearing syngeneic subcutaneous breast tumors with MG1 (intratumoral) together with the microtubule stabilizer paclitaxel (intraperitoneal) demonstrated synergistic benefit.19 While each agent employed on its own barely slowed tumor growth, their combination translated into a significant extension of survival. Mechanistically, paclitaxel impaired IFN-β production, thus increasing MG1 replication and oncolysis in syngeneic breast tumors as well as patient-derived triple-negative breast cancer xenografts in mice.19
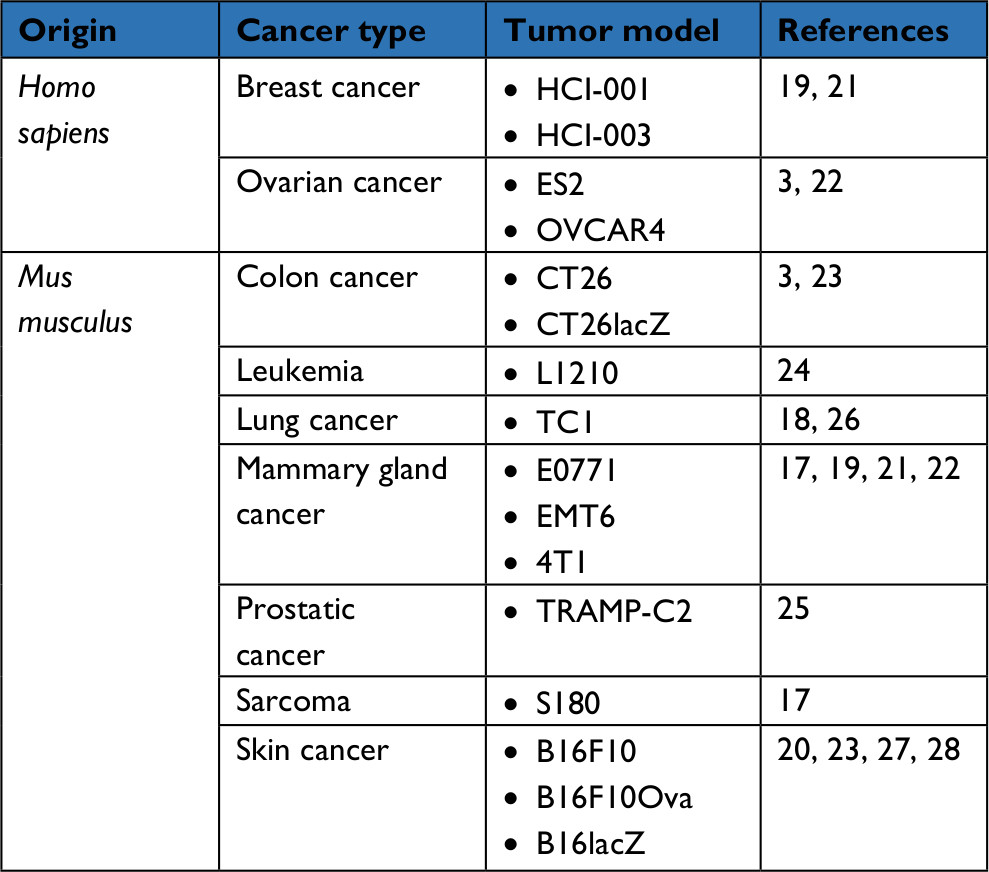 | Table 2 List of tumor xenografts or syngeneic transplants in which MG1-enrolling treatments demonstrated efficacy in mice |
Immunotherapeutic activity of Maraba virus
On top of its potent oncolytic activity, MG1 therapeutic efficacy also relies on its intrinsic ability to induce antitumor immunity. For instance, murine B16 melanoma responded to MG1 independently of its replication cycle.27 Indeed, both parental and replication-incompetent minimally UV-inactivated MG1 performed well in clearing lung metastases. Of note, this held true only at high doses as the efficacy of nonreplicating MG1 gradually dropped with doses lower than 108 pfu, unlike its replicative counterpart that preserves its full potency. Within 24 hours following systemic delivery of MG1, mice showed a splenomegaly, which resulted from a local increase of dendritic cells (DCs) and natural killer lymphocytes (NKs). Acute NK cell activation lasted up to 5 days post-MG1 administration and was accompanied by a remarkable enhancement of effector NKs secreting IFN-γ or granzyme B. MG1-mediated NK cell activation required an interaction between the virus and conventional DCs, and appeared critical for the therapeutic efficacy, whereas selective depletion of NKs abolished tumor growth control.
Such propensity to stimulate an antitumor immunity appeared of particular interest in a neoadjuvant setting.21,27 Indeed, stress associated with surgery is profoundly immunosuppressive and promotes cancer progression.29 As an illustration, untreated tumor-bearing mice undergoing surgery displayed shorter survival than unoperated controls, with the number of malignant lesions flaring postoperation. However, presurgery administration of MG1, be it replication-competent or attenuated, prevented a negative outcome by maintaining the anti-neoplastic immune pressure.27 Neoadjuvant MG1 also demonstrated benefit against murine triple-negative breast tumor models.21 Intratumoral administration of MG1 prior to resection was indeed instating a systemic adaptive antitumor immunity that protected 20% of the animals against distant postoperative tumor rechallenge. This propensity was dependent upon viral replication as UV-inactivated MG1 did not provide protection against tumor recurrence. Moreover, intravenous delivery of MG1 appeared more efficient than intralesional injection leading to 40% disease-free animals following cancer rechallenge. The T-cell compartment was involved in the antineoplastic activity and engaged into memory as 90% of the mice cured from the postoperative rechallenge completely rejected a second one later on.21 A transcriptomic analysis revealed that pathways linked to immune responses were enriched during MG1 infection. Increased expression of chemokines (eg, chemokine C-C motif ligand 5 [CCL5], C-X-C motif chemokine ligand 11 [CXCL11]) was mirrored at the protein level, as well as activation of signal transducer and activator of transcription 1 (STAT1), of the nuclear factor κB (NFκB) subunit p65 and of interferon regulatory factor 3 (IRF3). Induction of these proinflammatory factors was requiring the presence of pattern recognition receptors (PRRs) involved in rhabdovirus intracellular sensing. For instance, the stimulation of Ccl5, Cxcl10, and Il6 was depending on retinoic acid-inducible gene I (RIG-I) as well as on myeloid differentiation primary response 88 (MYD88) in some cell lines.21 In vivo, MG1-infected tumors showed greater infiltration by T-cells at the time of surgery, a migration that was depending on CXCR3, the target receptor of the Th1/Tc1-associated chemokines CXCL9, 10, and 11. Ex vivo, restimulation of splenocytes confirmed the genesis of tumor-specific reactivity in MG1-treated mice and revealed that production of IFN-γ upon reactivation was reliant on the receptor IFN-αR1. Last but not least, Maraba MG1 treatment happened to sensitize 4T1 breast tumors to checkpoint blockade immunotherapy.21 Indeed, MG1 delivery upregulated programmed death ligand 1 (PD-L1) protein level in three breast tumor models evaluated. Such observation suggests that cancer-specific T-cells may ultimately show mitigated cytotoxic activity against tumor cells. In this environment, adjunction of the immune checkpoint inhibitors anti-cytotoxic T lymphocyte-associated protein 4 (CTLA4) and anti-programmed cell death protein 1 (PD-1) to neoadjuvant MG1 significantly extended survival of breast tumor-bearing animals, eradicating the disease in 60%–90% of the animals depending on the model.21 Altogether, these preclinical findings based on MG1 demonstrate a potential benefit for neoadjuvant virotherapy.30,31
Furthermore, the immunogenic property of MG1 has been exploited to improve the efficacy of cancer cell vaccines against leukemia24 and metastatic solid cancers23 in preclinical murine models. While MG1 could infect several leukemia and lymphoma cell lines in vitro (Table 1), its therapeutic imprint was null in mice harboring L1210 leukemia.24 However, infusion of MG1-infected γ-irradiated leukemia cells (iLOV) to leukemic mice led to complete responses in 60% of the animals. In a prophylactic setting, iLOV infusion conferred complete protection against leukemia challenge. Thus, MG1 infection converted leukemia cells into a potent cancer vaccine. As an aside, this property was shared with its rhabdoviral cousin VSVΔM51. Increased immunogenicity of infected malignant cells was not solely the result of activation of antiviral defense pathways. Ex vivo, MG1 infection also induced the overexpression of some immunostimulatory molecules (ie, CD40 and OX40L) at levels at least comparable to those reached after incubation with the toll-like receptor (TLR) agonists lipopolysaccharide and poly I:C. However, administration of irradiated leukemia cells together with the TLR agonists could not recapitulate the protective efficacy of iLOV. Similarly, injecting uninfected irradiated viable, apoptotic or necrotic leukemia cells, with or without concurrent rhabdovirus administration was ineffective. Thus, the molecular features of rhabdovirus-induced cancer immunogenic cell death (ICD) (ie, tumor antigen presentation and spread, surface exposure or release of pathogen- and damage-associated molecular patterns [PAMPs and DAMPs]) appeared essential for effective iLOV but best achieved in ex vivo confined conditions. Further clinical trials will test autologous iLOV-based therapeutic vaccines in patients with acute lymphoblastic leukemia. A similar approach has been evaluated against metastatic solid tumor models.23 In this instance, a recombinant MG1 expressing interleukin (IL)-12 has been utilized. The cytokine IL-12 stimulates proliferation, IFN-γ production, as well as cytotoxicity of both NK and T-lymphocytes.32 Secretion of large amounts of IL-12 was validated following infection of B16 melanoma and CT26 colon carcinoma cell lines with MG1-IL12. Intravenous delivery of an MG1-IL12-infected cell vaccine (MG1-IL12-ICV) in mice resulted in a considerable increase of activated/cytotoxic NK cells in the lungs. Treatment of mice bearing melanoma lung metastases with MG1-IL12-ICV impressively controlled the expansion of pulmonary lesions.23 In comparison, control MG1-ICV enhanced the proportion of effective NKs in the lungs to a much lesser extent than its IL12-secreting counterpart and with a reduced ability to eradicate lung metastases. MG1-IL12-ICV was then tested against two peritoneal carcinomatosis models in which B16 and CT26 tumor cells were seeded intraperitoneally in immunocompetent mice. On the one hand, MG1-IL12-ICV significantly extended long-term survival of the animals harboring B16 peritoneal metastases. Efficacy of the MG1-IL12-ICV was dependent on both NK cells and CD8+ T lymphocytes as their selective depletion abolished antitumor activity. Ex vivo assays highlighted a major role of DC-secreted CXCL10 in enhancing NK-cell migratory behavior and activation upon coculture with B16 melanoma cells infected with MG1-IL12. Importantly, these observations were validated with the SW620 human colon carcinoma cell line infected with MG1-IL12 and cocultured with PBMCs of cancer patients. On the other hand, MG1-IL12-ICV successfully eradicated peritoneal metastases of CT26 colon carcinoma, curing 100% animals when repeatedly administered intraperitoneally.23 In comparison, cure rate of the unarmed MG1-ICV did not exceed 40% of the mice. Interestingly, the co-injection of purified IL-12 together with MG1-ICV offered no benefit. Knowing that systemic administration of cytokines is often accompanied with severe adverse events, it appeared that locally elevated levels of IL-12 in the tumor microenvironment were not only safe but also critical to support effective antitumor immunity. In conclusion, MG1-IL12-ICV offers great promise for the treatment of patients diagnosed with peritoneal carcinomatosis, the most problematic site of metastasis for abdominal malignancies such as gastrointestinal and ovarian cancers.
Maraba virus as a vector for oncolytic vaccination
To further consolidate the immunotherapeutic activity of oncolytic vesiculoviruses, we and others have equipped their genome with transgenes overexpressing tumor antigens.18,20,25,26,33–42 The resulting so-called “oncolytic vaccine” aims at potentiating the adaptive arm of antitumor immunity.
Proof of concept for oncolytic MG1 vaccination was demonstrated against syngeneic murine melanoma.20 Dopachrome tautomerase (DCT), an enzyme involved in melanogenesis, was chosen as the tumor-associated antigen for its well-characterized immunogenicity. The DCT transgene was inserted between the G and L genes of the rhabdoviral genome, a strategic locus for preserving the relative expression levels of the different viral factors while allowing a reasonable translation of the tumor antigen. When administered as a sole agent, MG1-DCT did not prime detectable adaptive T-cell responses against the melanoma antigen.20 As previously revealed, this lack of immune priming efficiency by vesiculovirus-based vaccines coincided with a sustained T-cell reactivity against foreign viral antigens (particularly the N protein) which skewed immune effector specificity away from the autologous tumor antigen.35,39 To counterbalance the reactivity between virus and cancer antigens, we opted for a prime-boost strategy that introduces a serotype five adenoviral (Ad) vaccine as an immune primer. The replication-defective (E1/E3 deleted) Ad vector expressing DCT was delivered intramuscularly, both for safety and efficient immunization purposes. At a dose of 2 × 108 pfu, Ad-DCT induced a moderate DCT-specific adaptive response with 5% of spleen and blood CD8+ T cells reacting against the immunodominant epitope of the melanoma antigen. Subsequent administration of 2 × 109 pfu of a vesiculovirus-based vaccine expressing the same antigen impressively boosted the DCT response.20,39 Out of total circulating CD8+ T-lymphocytes, 30% turned to react against the DCT epitope following MG1-DCT infusion, a boost significantly stronger than with VSV-DCT which peaked at 20%.20 Antiviral CD8+ T-cell response underwent a concurrent threefold decrease relative to animals that did not receive Ad-DCT prime prior to vesiculovirus vaccine boost: 15% reactivity against the N factor when the oncolytic vesiculovirus was given as a priming vector vs 5% when employed as a booster.39 Importantly, the prime-boost immunization also stimulated reactivity against tumor antigens not encoded by the vaccines (ie, the melanoma-associated antigen gp100), thus supporting antigen spreading and cross-presentation upon treatment-induced oncotoxicity.39 Studies on the Ad-DCT prime:VSV-DCT boost also revealed that this vaccination strategy not only enhanced the quantity of CD8+ T cells reacting to tumor antigens but also improved their quality. Precisely, boosting with an oncolytic vesiculovirus vaccine increased 1) the proportion of polyfunctional cancer-specific T lymphocytes (double positivity for IFN-γ and the tumor necrosis factor [TNF]-α) and 2) of cytotoxic effectors (production of granzyme B), 3) their avidity to tumor epitopes, as well as 4) their functional memory.37 The intravenous route for delivering MG1-DCT vaccine booster was preferred for its unique ability to induce both CD8+ and CD4+ T-cell responses, robust enough to be readily observable in the blood, as well as to facilitate the access of the oncolytic agent to tumor site(s). Regarding the interval between vaccine prime and boost, a well-established dogma states that the longer the better for prophylactic vaccination protocols, with immunizations commonly spread several weeks or months apart.43 Specifically, the immune boost aims at mobilizing highly proliferative central memory T cells (TCM) after primed effector cells (TEFF) have declined. With that said, delaying vaccine recall would also prevent patrolling primed TEFF to recognize as a target to neutralize the cells that “catch” the booster vaccine and present de novo the target antigen, an undesired event that would limit cross-presentation of the antigen by DCs to the memory compartments. To our surprise, MG1 vaccine potently boosted DCT responses as early as 9 days post-Ad-DCT prime.20 This beneficial property of oncolytic vesiculovirus vaccines was dependent upon the infection of follicular B cells in the spleen following systemic delivery.38 As TEFF cannot traffic through follicular areas, these infected B cells serve as a secured source of tumor antigens for neighboring DCs within the follicle. The latter can then engage TCM even during the peak of the TEFF response.38,44 By authorizing short intervals between prime and boost, MG1-based cancer vaccination favors a rapid and effective immunotherapy of the disease, a critical advantage to prevent disease progression in advanced stages. When evaluated against melanoma metastatic models, the Ad-DCT:MG1-DCT prime-boost not only extended median survivals in comparison to Ad-DCT-treated animals but also cured 20% and 30% of the animals bearing brain and lung metastases, respectively. In the lung metastatic model, Ad:MG1 prime-boost was compared with Ad:VSV and demonstrated superior efficacy with median survivals reaching 75 vs 55 days, respectively. Despite stimulating both tumor-specific CD8+ and CD4+ T-cell responses, therapeutic activity was largely mediated by the CD8+ T effectors. Indeed, selective depletion of the latter completely abolished the antitumor activity, whereas an equivalent intervention on the CD4+ T-cell compartment did not affect outcome.20 Finally, the Ad:MG1 prime-boost vaccination induced long-term antitumor memory that protected cured animals from tumor rechallenge.20
The prime-boost vaccination strategy involving an oncolytic MG1 vaccine has since been translated to the treatment of human papillomavirus (HPV)-positive tumors and prostate cancer.18,25,26 To this purpose, the replication-defective Ad and the oncolytic MG1 virus were armed either with a tetravalent transgene expressing the E6 and E7 antigens of the HPV serotypes 16 and 18,18 or with a transgene encoding the human six-transmembrane antigen of the prostate (hSTEAP).25 Preclinical evaluations were performed against murine syngeneic models of TC1 lung carcinoma, an HPV16-E6/E7+ cell line, or TRAMP-C2 prostate cancer. Prime-boost oncolytic vaccination with Ad-E6/E7:MG1-E6/E7 mounted a weak response against HPV-E6 but a robust one against HPV-E7 with 1% and 60% of circulating CD8+ T-cells reacting against the antigen, respectively.18 Importantly, a substantial number of these reactive lymphocytes demonstrated a polyfunctional activity in the blood and spleen, with coproduction of the proinflammatory cytokines IFN-γ, TNF-α, and IL-2. Meanwhile, MG1-E6/E7 reached the tumor and exerted direct oncolytic activity. Twenty-four hours after intravenous delivery, MG1-E6/E7 infection was associated with a local increase in the expression of genes involved in antigen presentation (eg, B2m, H2-K1, Tap1, Tapbp), antiviral innate immunity (eg, Ddx28, Pml, Irf7, Stat1, and IFN-inducible factors such as Irgm, Ifi35, and Ifi204) or T-cell activation (eg, Cd28, Il2ra), in comparison to untreated tumors. Overall, treatment demonstrated a remarkable efficacy with complete tumor regression documented in 75% of the hosts and led to the establishment of long-term immune memory.18 Regarding oncolytic vaccination with Ad-hSTEAP:MG1-hSTEAP, strong CD8+ T cell responses were detected in blood samples against the human STEAP, expressed from the vaccines, as well as against the endogenous murine STEAP (40% vs 10% reactivity among circulating CD8+ T cells, respectively), thus suggesting that immunization with the human xenoantigen was breaching mouse peripheral immune tolerance. Consistent with this, Ad:MG1 vaccination against the prostate antigen was as efficient in females as in males. When applied to the treatment of subcutaneous TRAMP-C2 syngeneic prostate tumors, Ad-hSTEAP:MG1-hSTEAP significantly slowed tumor growth and extended median survival. Transcriptional profiling and immunohistochemical analysis of immune-related markers were performed on tumor samples collected at the peak of the boost response (ie, 6 days post-MG1) and compared with untreated tumors.25 Altogether, T-cell infiltration and activation was illustrated in treated tumors by an upregulation of 1) T-cell surface markers (eg, CD2, CD3, CD5, CD7, CD8), 2) receptors/ligands involved in T-cell migration (eg, CXCR3, CXCL9), and 3) surface (eg, CD27 and the immune checkpoints ICOS, TIGIT, PD-1), cytoplasmic (eg, NFAT, ZAP70, LCK, EOMES), or secreted (eg, Granzyme B) factors related to T-cell activation and effector function. Genes involved in antigen processing and presentation were also enriched following oncolytic vaccination (eg, Ciita, H2-K1, H2-DMa, Tap1).25
Overall, these preclinical data highlighted the ability of the Ad:MG1 oncolytic vaccination to generate massive tumor-specific effector CD8+ T-cell populations with remarkable therapeutic efficacy as well as immune memory protecting from cancer recurrence.
Transitioning the Ad:MG1 oncolytic vaccination from mice to the clinic
The aforementioned preclinical observations encouraged the evaluation of the Ad:MG1 oncolytic vaccination approach in the clinic. Prior to initiation of investigations in humans, a toxicology study of the MG1 oncolytic vaccine to enter first-in-man trial was executed in healthy tumor-free outbred nonhuman primates.45 Additionally, with the aim of extending the Ad:MG1 vaccination to the treatment of veterinary cancer patients, we characterized its safety and tolerability profiles in healthy tumor-free outbred cats.33
Five cats were enrolled to evaluate the safety of the Ad prime:MG1 boost strategy.33 Animals received an intramuscular injection of 1 × 1010 pfu of replication-defective E1/E3-deleted Ad5 encoding the human placenta-specific 1 (hPLAC1), a cancer/testis antigen reexpressed in multiple malignancies such as breast/mammary tumors.46 Twenty-one days post-Ad-hPLAC1 prime, the felines were boosted intravenously with one high dose of 2.5 × 1011 pfu of MG1-hPLAC1. Vaccination with MG1 caused salivation during infusion, likely attributable to nausea, followed by acute mild pyrexia that vanished within 24 hours. All cats showed transient inappetence after MG1-hPLAC1 injection leading to an average weight loss of 370 g at 24 hours. Body weight returned to normal within 8 days. Complete blood count was monitored prior and after vaccine prime and boost. Intramuscular Ad5 had no impact while systemic delivery of MG1 caused transient leukopenia (n=2/5), lymphopenia (n=4/5), thrombocytopenia (n=2/5), and neutrophilia (n=1/5), which all resumed within a week. Cats boosted with MG1 displayed neither viremia nor shedding of the virus. Indeed, MG1 genomes could be detected in plasma (n=3/5), urine (n=2/5), or fecal (n=1/5) samples within the week that followed infusion, but no replicative viral particles were recovered. At the end of the study (18 days post-MG1 treatment), the five cats did not demonstrate relevant clinical signs or abnormal behavior. Postmortem histopathological evaluations revealed spleen (n=5/5) and lymph node (n=3/5) hyperplasia. Other findings were made in three cats and included mild myocardial and hepatic congestion in one cat, coronary arteropathy in a second one, and focal myocardial fibrosis as well as mild cholangitis and pyelitis in the third cat. MG1 genomes were detected in the spleen, heart, and lungs, in the absence of replicative viral particles. Altogether, these preclinical data validated the tolerability and nonpathogenicity of the Ad:MG1 vaccination, thus supporting their use to treat feline patients.33
Twenty cynomolgus macaques (Macaca fascicularis) were immunized with the Ad and MG1 vaccines expressing the human melanoma-associated antigen-A3 (MAGE-A3), a cancer/testis antigen aberrantly expressed in melanoma, lung carcinoma, and other malignancies.47,48 Priming was performed via intramuscular delivery of 1 × 1010 pfu of an Ad vaccine expressing MAGE-A3 (Ad-MAGEA3). Boost was mediated by two infusions, three days apart, of MG1-MAGEA3. Two doses of the MG1 vaccine were evaluated: 1 × 1010 (n=8/20) or 1 × 1011 (n=12/20) pfu. Moreover, three intervals between Ad prime and MG1 boost were tested: 2 weeks (n=8/20), 4 weeks (n=4/20), and 6 weeks (n=8/20). Some primates experienced anorexia (50%), constipation (25%), or emesis (17%) that resolved within 72 hours. Grade 1/2 weight loss and transient mild pyrexia were noted in 25% and 35% of the macaques, respectively. The count of circulating neutrophils transiently dropped in all subjects, with neutropenia observed in six macaques, following the first infusion of MG1-MAGEA3. Decline in the count of blood lymphocytes was frequent but less pronounced. Maraba MG1 genomes were detected in blood samples of all macaques and in the majority of secondary lymphoid tissues (spleen and lymph nodes). Viral genome was absent in feces samples but detected in 15% and 50% of urine and saliva samples, respectively. However, no replicative MG1 was detected in any of the samples analyzed and no lesions of pathologic significance were revealed by histological analyses at necropsy. In addition to determining the innocuity of the prime-boost strategy, the study was also an opportunity to evaluate the efficiency of the Ad:MG1 vaccination in primates.45 Ad-MAGEA3:MG1-MAGEA3 mounted detectable humoral and cellular immunities against epitopes of the human MAGE-A3. The magnitude of CD4+ and CD8+ T-cell responses were remarkable enough to be quantified in the blood, regardless of the MG1 dose or prime-boost interval. Five percent of total circulating CD8+ T lymphocytes were reactive against human MAGE-A3 on average, approaching an impressive 15% in the best responder. The quality of the reactive CD8+ T cell population was remarkable with 60% dual production of the IFN-γ and TNF-α cytokines. Interestingly, immunization with the xenoantigen human MAGE-A3 raised responses against epitopes of the cynomolgus homolog, validating the ability of the strategy to overcome host peripheral tolerance. Overall, Ad-hMAGEA3:MG1-hMAGEA3 vaccination was safe, well tolerated, and highly immunogenic in naive outbred tumor-free nonhuman primates.45
Following on the study in nonhuman primates, two clinical trials were initiated evaluating Ad:MG1-MAGEA3 in human patients with MAGE-A3-positive solid tumors. The first Phase I/II trial is recruiting participants with incurable MAGE-A3-expressing solid tumors (clinicaltrials.gov reference: NCT02285816/CCTG IND.I214).49 A first-dose MTD of 1 × 1011 pfu MG1-MAGEA3 was defined. Patients received two infusions (three days apart) of MG1-MAGEA3 alone (Arm A), or were administered with intramuscular 1 × 1010 pfu of Ad-MAGEA3 alone (Arm B), or were treated with Ad-MAGEA3 followed two weeks later with systemic MG1-MAGEA3 (Arm C). Dose-limiting toxicities (hypoxia/dyspnea, vomiting, headache) occurred in four subjects, two in each Arm A and C. Treatment-related toxicities in Arm C occurred a few hours to days after MG1-MAGEA3 and mostly constituted of diarrhea, nausea, vomiting, anorexia, chills, fatigue, fever, flu-like symptoms, hypophosphatemia, headache, and hypotension. Transcriptomic analyses of tumor biopsies revealed a modulation of numerous pro-inflammatory genes. Markers of NK lymphocytes and activated antigen-presenting cells were detected (ie, CD56, CD68, CD80, HLA-A, HLA-B, TLR3). Several chemokines and cytokines supporting inflammation were induced, such as CCL2, CCL5, CX3CL1, CXCL10, IL-6, and TNF while the immunosuppressive TGF-β appeared downregulated. Detection of MG1-MAGEA3 genomes in blood samples of patients was witnessed two weeks post-input dose clearance, thus confirming the ability of the virus to replicate in humans. Importantly, antitumor immunity was evidenced in three out of the six patients evaluated, with over 1% of total circulating CD8+ T cells reacting against MAGE-A3 in one participant.49 A second Phase I/II clinical trial will evaluate the safety and efficacy of the Ad-MAGEA3:MG1-MAGEA3 treatment, combined with the PD-1 immune checkpoint inhibitor pembrolizumab, in patients with previously treated metastatic non-small-cell lung cancer (NCT02879760). In conclusion, available clinical data validate the feasibility of the Ad:MG1 oncolytic vaccination and its ability to stimulate adaptive antitumor cell response in cancer patients. Furthermore, a trial evaluating the Ad:MG1 approach against HPV-positive malignancies has just been open in the US and Canada.
Concluding remarks
Maraba virus appears to be a powerful multifunctional tool in the fight against cancer. First, many tumors are susceptible to MG1-mediated oncolysis. Furthermore, MG1-induced cell death is highly immunogenic, likely ascribed to tumor and viral antigen spreading together with the local release of DAMPs that act as adjuvants, among which features the type I IFN response with a demonstrated critical role. Consequently, MG1 oncotoxicity stimulates both innate and adaptive arms of antitumor immunity. Such immunotherapeutic activity demonstrated benefit in association with surgery, chemotherapy, and other immunotherapies, including immune checkpoint inhibitors and cancer vaccines. Cancer vaccines incarnate a promising approach for cancer treatment as they instate 1) polyfunctional and polyantigenic immune effector responses that can lyze malignant cells spared by direct oncolysis and 2) an immune memory that will prevent cancer recurrence. Two kinds of cancer vaccinations have proven successful in preclinical studies: MG1-infected cancer cell vaccines and MG1-based prime-boost vaccination. Reinforcing adaptive antitumor responses using the Ad prime-MG1 boost vaccination resulted in a remarkable extension of the survival in several murine models. As an aside, recent studies highlighted the somehow universal boosting efficacy of MG1 vaccine.26,28 Indeed, alternative priming agents, substituting the Ad vaccine and embodied by purified tumor-associated peptides or Listeria monocytogenes armed with the same cancer antigen, also raised substantial antitumor immune activity.26,28 The Ad:MG1-MAGEA3 prime-boost approach is currently being evaluated in cancer patients affected with advanced MAGE-A3+ cancers such as melanoma. Disclosed results validated a modulation of the tumor immune microenvironment and the generation of an antitumor immunity in early-phase clinical testing.
In addition to Maraba MG1, other OVs are undergoing Phase I/II clinical trials against advanced melanoma. They include the two herpesviruses T-VEC and HF10, the reovirus type 3 Reolysin, the coxsackievirus type A21 Cavatak, and the Ad5 OBP-301.6 Current and future investigations will determine their relative safety and efficacy. More generally, considering the plethora of strains available, a therapeutic benefit of OVs (as a monotherapy or in combination regimens) is anticipated against a wide range of malignant histotypes. To achieve this goal, pros and cons of their respective biology will have to be carefully considered, including their tropism and immunogenicity, among other criteria that will be briefly discussed here. For instance, Maraba MG1 penetrates into cells through LDLR, which is expressed by most nucleated cells. In contrast, some OVs exploit non-ubiquitous cell entry receptors. As an illustration, Cavatak binds to the intercellular adhesion molecule 1 (ICAM-1) whose expression is limited to normal endothelial and immune cells and to several tumors (eg, melanoma, colorectal, breast, and lung cancers).50 Consequently, MG1’s broad tropism is an undeniable asset to treat virtually any malignancy. However, it comes along with infection of normal tissues that may raise safety concerns. To prevent off-target infection, OV cell tropism can be narrowed, for instance, through pseudotyping of viral surface proteins. Alternatively, off-target replication can be abrogated via mutation of virulence factors and/or insertion of regulatory sequences in the OV genome.6 The latter genetic manipulations have been applied to Maraba MG1-MAGEA3 (mutation of the M protein normally responsible for inhibiting the antiviral response),3 as well as to T-VEC (deletion of the virulence factors ICP34.5 and ICP47)1 and OBP-301 (preferential expression of the E1 virulence factors in malignant cells through promoter engineering).51 In contrast, HF10, Reolysin, and Cavatak consist of naturally occurring strains. Consequently, MG1 off-target infection has minimal adverse consequences because of its exquisite sensitivity to the IFN response (abortive viral life cycle in healthy cells). A risk of potential reversion of the MG1 mutations in patients cannot be ignored. If unlikely (considering the rapid clearance of the virus by the host), the reverted wild-type strain would remain susceptible to innate cell immunity and could, if needed, be shut down pharmacologically through administration of purified type I IFNs. Neutralization of the virion by the immune system represents an additional obstacle for the delivery and spread of viruses whose seroprevalence in patients is elevated, such as HSV-1 (eg, T-VEC, HF10) or Ad5 (eg, OPB-301). For this reason, these highly immunogenic viruses are essentially administered intratumorally rather than systemically. However, in an absence of virus-induced abscopal effect, local delivery would result in no efficacy against metastatic lesions. Encouragingly, some OVs like MG1, Reolysin, or Cavatak demonstrated an ability to circulate in the blood flow, then reach tumor sites and/or mediate diffuse antitumor activity.6 Interestingly, for viruses like MG1 or Reolysin, the systemic route also allows an increased interaction with immune cells/lymphoid organs that supports their immunotherapeutic activity.20,38,52 To improve their efficacy, OVs can be armed with transgenes. In this perspective, the cloning capacity of the viral genome dictates the number and size of the inserts. For instance, the cloning capacity of the MG1 genome is on the order of 5–6 kb, allowing for the addition of multiple transgenes or fusion proteins. In contrast, large viruses like the recombinant HSV-1 T-VEC harbor numerous nonessential open-reading frames that could be substituted with transgenes. Genetic inserts have demonstrated significant value, particularly in enhancing the immunotherapeutic activity of oncolytic agents. As an illustration, expression of the chemokine granulocyte-macrophage colony-stimulating factor (GM-CSF) from T-VEC and of tumor antigens from MG1-based vaccines promotes the recruitment of antigen-presenting cells, antigen cross-presentation and, ultimately, T-cell activation. With strategies like the Ad:MG1 prime-boost cancer vaccination, the ensuing T-cell antitumor response can reach a strong magnitude. As a downside, intensifying immune reactivity against non-mutated antigens can overcome peripheral self-tolerance and potentially induce autoimmunity. This consequence has been observed preclinically in immunocompetent mice bearing melanoma which developed vitiligo following Ad:MG1 vaccination targeting the melanocyte antigen DCT.20,36,39 Similarly, immunotherapy with autologous TCR-engineered T-cells targeting one defined MAGE-A3 epitope demonstrated neurotoxicity or cardiotoxicity in melanoma and myeloma patients due to supposed cross-reactivity against the brain isoform MAGE-A12 or the muscle protein Titin.53–55 Such a dramatic consequence has not been witnessed in the 20 tumor-free macaques immunized with the Ad:MG1 MAGE-A3 vaccine but is being closely monitored in cancer patients. Current Phase II investigations will determine if the contained expansion of a broad spectrum of MAGE-A3-specific T-cell clones by Ad and MG1 vaccines (which express the full-length antigen) is safe and tolerated. Finally, only a few strategies are currently aimed at stimulating the early phases of oncolytic viro-immunotherapy, namely the induction of 1) innate cell immunity and 2) cancer ICD. On the one hand, innate sensing of viral antigens and PAMPs has long been considered as a hindrance for efficient OV therapy. Although the contribution of viral molecules to the overall immunogenicity of infected tumors remains incompletely understood, evidence is accumulating in favor of a critical role in OV-mediated immunotherapeutic activity.30 This holds true for MG1 whose detection by PRRs (particularly RIG-I and the adapter component MyD88) contributes to its adjuvant property.21 In line with this, overexpression of PRR-interacting factors from oncolytic vaccinia viruses (ie, TIR-domain-containing adapter-inducing interferon-β [TRIF] or DNA-dependent activator of IRFs [DAI]) successfully enhanced antitumor activity.56,57 On the other hand, cancer ICD has been mainly studied utilizing chemotherapy. Together with tumor antigen spreading, well-established hallmarks of ICD include the surface exposure of calreticulin following the induction of an ER stress and the release/secretion of high-mobility group box 1 protein (HMGB1), ATP, annexin A1, and type I IFNs. These DAMPs act as adjuvants by favoring DC recruitment, homing, antigen cross-presentation via phagocytosis, and sustained T-cell activation.58 However, the profile of DAMPs produced upon infection by a given OV has been scarcely investigated.59 Ecto-calreticulin, extracellular ATP, and HMGB1 release have been described following Ad and coxsackieviruses-mediated oncolysis.60,61 In contrast, ICD markers ascribed to treatment with oncolytic rhabdoviruses like MG1 have not been reported. Ultimately, the characterization and comparison of the immunogenic signature of the oncolytic viral candidates, from innate sensing and ICD to the activation and exhaustion of cytotoxic lymphoid cells, should drive the development of improved treatment strategies. Follow-up of long-term adverse events, such as autoimmunity, and their management will remain critical, particularly in association with immune checkpoint inhibitors.
In conclusion, MG1-Maraba virus is a promising oncolytic vaccine already shown to be safe and immunogenic in early Phase I studies. Results of these studies as well as imminent studies targeting other antigens in various malignancies will become available over the next several years.
Disclosure
JGP was supported by the Seerave Foundation. BDL was supported by the Terry Fox Foundation, the Ontario Institute for Cancer Research, BioCanRx, and Turnstone Biologics. JGP, MJA, BWB, DFS, JCB, and BDL share ownership of patents for cancer vaccination involving oncolytic Maraba MG1. BDL, JCB, and DFS are cofounders, equity holders, and on the Board of Directors of Turnstone Biologics. KBS, CGM, JP, and CJB are employees of Turnstone Biologics. JLH is the founder and clinical science director of CANSWERS. The authors report no other conflicts of interest in this work.
References
Pol J, Kroemer G, Galluzzi L. First oncolytic virus approved for melanoma immunotherapy. Oncoimmunology. 2016;5(1):e1115641. | ||
Travassos da Rosa AP, Tesh RB, Travassos da Rosa JF, Herve JP, Main AJ Jr. Carajas and Maraba viruses, two new vesiculoviruses isolated from phlebotomine sand flies in Brazil. Am J Trop Med Hyg. 1984;33(5):999–1006. | ||
Brun J, McManus D, Lefebvre C, et al. Identification of genetically modified Maraba virus as an oncolytic rhabdovirus. Mol Ther. 2010;18(8):1440–1449. | ||
Johnson JE, Nasar F, Coleman JW, et al. Neurovirulence properties of recombinant vesicular stomatitis virus vectors in non-human primates. Virology. 2007;360(1):36–49. | ||
Pol JG, Rességuier J, Lichty BD. Oncolytic viruses: a step into cancer immunotherapy. Virus Adapt Treat. 2011;4(1):1–21. | ||
Pol JG, Lévesque S, Workenhe ST. Trial Watch: oncolytic viro-immunotherapy of hematologic and solid tumors. OncoImmunology. 2018:1–32. | ||
Pol J, Buqué A, Aranda F, et al. Trial Watch-Oncolytic viruses and cancer therapy. Oncoimmunology. 2016;5(2):e1117740. | ||
Pol J, Bloy N, Obrist F, et al. Trial Watch:: Oncolytic viruses for cancer therapy. Oncoimmunology. 2014;3:e28694. | ||
Breitbach CJ, Lichty BD, Bell JC. Oncolytic Viruses: Therapeutics With an Identity Crisis. EBioMedicine. 2016;9:31–36. | ||
Stojdl DF, Abraham N, Knowles S, et al. The murine double-stranded RNA-dependent protein kinase PKR is required for resistance to vesicular stomatitis virus. J Virol. 2000;74(20):9580–9585. | ||
Stojdl DF, Lichty B, Knowles S, et al. Exploiting tumor-specific defects in the interferon pathway with a previously unknown oncolytic virus. Nat Med. 2000;6(7):821–825. | ||
Stojdl DF, Lichty BD, Tenoever BR, et al. VSV strains with defects in their ability to shutdown innate immunity are potent systemic anti-cancer agents. Cancer Cell. 2003;4(4):263–275. | ||
Lichty BD, Power AT, Stojdl DF, Bell JC. Vesicular stomatitis virus: re-inventing the bullet. Trends Mol Med. 2004;10(5):210–216. | ||
Lichty BD, Stojdl DF, Taylor RA, et al. Vesicular stomatitis virus: a potential therapeutic virus for the treatment of hematologic malignancy. Hum Gene Ther. 2004;15(9):821–831. | ||
Tong JG, Valdes YR, Barrett JW, et al. Evidence for differential viral oncolytic efficacy in an in vitro model of epithelial ovarian cancer metastasis. Mol Ther Oncolytics. 2015;2:15013. | ||
Tong JG, Valdes YR, Sivapragasam M, et al. Spatial and temporal epithelial ovarian cancer cell heterogeneity impacts Maraba virus oncolytic potential. BMC Cancer. 2017;17(1):594. | ||
Le Boeuf F, Selman M, Son HH, et al. Oncolytic Maraba Virus MG1 as a Treatment for Sarcoma. Int J Cancer. 2017;141(6):1257–1264. | ||
Atherton MJ, Stephenson KB, Pol J, et al. Customized Viral Immunotherapy for HPV-Associated Cancer. Cancer Immunol Res. 2017;5(10):847–859. | ||
Bourgeois-Daigneault MC, St-Germain LE, Roy DG, et al. Combination of Paclitaxel and MG1 oncolytic virus as a successful strategy for breast cancer treatment. Breast Cancer Res. 2016;18(1):83. | ||
Pol JG, Zhang L, Bridle BW, et al. Maraba virus as a potent oncolytic vaccine vector. Mol Ther. 2014;22(2):420–429. | ||
Bourgeois-Daigneault MC, Roy DG, Aitken AS, et al. Neoadjuvant oncolytic virotherapy before surgery sensitizes triple-negative breast cancer to immune checkpoint therapy. Sci Transl Med. 2018;10(422): eaao1641. | ||
Mahoney DJ, Lefebvre C, Allan K, et al. Virus-tumor interactome screen reveals ER stress response can reprogram resistant cancers for oncolytic virus-triggered caspase-2 cell death. Cancer Cell. 2011;20(4):443–456. | ||
Alkayyal AA, Tai LH, Kennedy MA, et al. NK-Cell Recruitment Is Necessary for Eradication of Peritoneal Carcinomatosis with an IL12-Expressing Maraba Virus Cellular Vaccine. Cancer Immunol Res. 2017;5(3):211–221. | ||
Conrad DP, Tsang J, Maclean M, et al. Leukemia cell-rhabdovirus vaccine: personalized immunotherapy for acute lymphoblastic leukemia. Clin Cancer Res. 2013;19(14):3832–3843. | ||
Atherton MJ, Stephenson KB, Tzelepis F, et al. Transforming the prostatic tumor microenvironment with oncolytic virotherapy. Oncoimmunology. 2018;7(7):e1445459. | ||
Atherton MJ, Stephenson KB, Nikota JK, et al. Preclinical development of peptide vaccination combined with oncolytic MG1-E6E7 for HPV-associated cancer. Vaccine. 2018;36(16):2181–2192. | ||
Zhang J, Tai LH, Ilkow CS, et al. Maraba MG1 virus enhances natural killer cell function via conventional dendritic cells to reduce postoperative metastatic disease. Mol Ther. 2014;22(7):1320–1332. | ||
Aitken AS, Roy DG, Martin NT, Sad S, Bell JC, Bourgeois-Daigneault MC. Brief Communication; A Heterologous Oncolytic Bacteria-Virus Prime-Boost Approach for Anticancer Vaccination in Mice. J Immunother. 2018;41(3):125–129. | ||
Kim R. Effects of surgery and anesthetic choice on immunosuppression and cancer recurrence. J Transl Med. 2018;16(1):8. | ||
Gujar S, Pol JG, Kim Y, Lee PW, Kroemer G. Antitumor Benefits of Antiviral Immunity: An Underappreciated Aspect of Oncolytic Virotherapies. Trends Immunol. 2018;39(3):209–221. | ||
Gujar S, Pol JG, Kroemer G. Heating it up: Oncolytic viruses make tumors ‘hot’ and suitable for checkpoint blockade immunotherapies. Oncoimmunology. 2018;7(8):e1442169. | ||
Croxford AL, Kulig P, Becher B. IL-12-and IL-23 in health and disease. Cytokine Growth Factor Rev. 2014;25(4):415–421. | ||
Hummel J, Bienzle D, Morrison A, et al. Maraba virus-vectored cancer vaccines represent a safe and novel therapeutic option for cats. Sci Rep. 2017;7:15738. | ||
Boisgerault N, Kottke T, Pulido J, et al. Functional cloning of recurrence-specific antigens identifies molecular targets to treat tumor relapse. Mol Ther. 2013;21(8):1507–1516. | ||
Bridle BW, Boudreau JE, Lichty BD, et al. Vesicular stomatitis virus as a novel cancer vaccine vector to prime antitumor immunity amenable to rapid boosting with adenovirus. Mol Ther. 2009;17(10):1814–1821. | ||
Bridle BW, Chen L, Lemay CG, et al. HDAC inhibition suppresses primary immune responses, enhances secondary immune responses, and abrogates autoimmunity during tumor immunotherapy. Mol Ther. 2013;21(4):887–894. | ||
Bridle BW, Clouthier D, Zhang L, et al. Oncolytic vesicular stomatitis virus quantitatively and qualitatively improves primary CD8+ T-cell responses to anticancer vaccines. Oncoimmunology. 2013;2(8):e26013. | ||
Bridle BW, Nguyen A, Salem O, et al. Privileged Antigen Presentation in Splenic B Cell Follicles Maximizes T Cell Responses in Prime-Boost Vaccination. J Immunol. 2016;196(11):4587–4595. | ||
Bridle BW, Stephenson KB, Boudreau JE, et al. Potentiating cancer immunotherapy using an oncolytic virus. Mol Ther. 2010;18(8):1430–1439. | ||
Kottke T, Errington F, Pulido J, et al. Broad antigenic coverage induced by vaccination with virus-based cDNA libraries cures established tumors. Nat Med. 2011;17(7):854–859. | ||
Pulido J, Kottke T, Thompson J, et al. Using virally expressed melanoma cDNA libraries to identify tumor-associated antigens that cure melanoma. Nat Biotechnol. 2012;30(4):337–343. | ||
Rommelfanger DM, Wongthida P, Diaz RM, et al. Systemic combination virotherapy for melanoma with tumor antigen-expressing vesicular stomatitis virus and adoptive T-cell transfer. Cancer Res. 2012;72(18):4753–4764. | ||
Ledgerwood JE, Zephir K, Hu Z, et al; VRC 310 Study Team. Prime-boost interval matters: a randomized phase 1 study to identify the minimum interval necessary to observe the H5 DNA influenza vaccine priming effect. J Infect Dis. 2013;208(3):418–422. | ||
Zhang L, Bridle BW, Chen L, et al. Delivery of viral-vectored vaccines by B cells represents a novel strategy to accelerate CD8(+) T-cell recall responses. Blood. 2013;121(13):2432–2439. | ||
Pol JG, Acuna SA, Yadollahi B, et al. Preclinical evaluation of a MAGE-A3 vaccination utilizing the oncolytic Maraba virus currently in first-in-human trials. Oncoimmunology. 2018;7(7):1–15. | ||
Yuan H, Chen V, Boisvert M, Isaacs C, Glazer RI. PLAC1 as a serum biomarker for breast cancer. PLoS One. 2018;13(2):e0192106. | ||
Drake CG, Lipson EJ, Brahmer JR. Breathing new life into immunotherapy: review of melanoma, lung and kidney cancer. Nat Rev Clin Oncol. 2014;11(1):24–37. | ||
Gjerstorff MF, Andersen MH, Ditzel HJ. Oncogenic cancer/testis antigens: prime candidates for immunotherapy. Oncotarget. 2015;6(18):15772–15787. | ||
Jonker DJ, Hotte SJ, Abdul Razak AR, et al. Phase I study of oncolytic virus (OV) MG1 maraba/MAGE-A3 (MG1MA3), with and without transgenic MAGE-A3 adenovirus vaccine (AdMA3) in incurable advanced/metastatic MAGE-A3-expressing solid tumours: CCTG IND.214. J Clin Oncol. 2017;35(15 Suppl):e14637. | ||
Reina M, Espel E. Role of LFA-1 and ICAM-1 in Cancer. Cancers (Basel). 2017;9(11):E153. | ||
Fujiwara T, Shirakawa Y, Kagawa S. Telomerase-specific oncolytic virotherapy for human gastrointestinal cancer. Expert Rev Anticancer Ther. 2011;11(4):525–532. | ||
Prestwich RJ, Ilett EJ, Errington F, et al. Immune-mediated antitumor activity of reovirus is required for therapy and is independent of direct viral oncolysis and replication. Clin Cancer Res. 2009;15(13):4374–4381. | ||
Morgan RA, Chinnasamy N, Abate-Daga D, et al. Cancer regression and neurological toxicity following anti-MAGE-A3 TCR gene therapy. J Immunother. 2013;36(2):133–151. | ||
Cameron BJ, Gerry AB, Dukes J, et al. Identification of a Titin-derived HLA-A1-presented peptide as a cross-reactive target for engineered MAGE A3-directed T cells. Sci Transl Med. 2013;5(197):ra03. | ||
Linette GP, Stadtmauer EA, Maus MV, et al. Cardiovascular toxicity and titin cross-reactivity of affinity-enhanced T cells in myeloma and melanoma. Blood. 2013;122(6):863–871. | ||
de Graaf JF, de Vor L, Fouchier RAM, van den Hoogen BG. Armed oncolytic viruses: A kick-start for anti-tumor immunity. Cytokine Growth Factor Rev. 2018;41:28–39. | ||
Hirvinen M, Capasso C, Guse K, et al. Expression of DAI by an oncolytic vaccinia virus boosts the immunogenicity of the virus and enhances antitumor immunity. Mol Ther Oncolytics. 2016;3:16002. | ||
Galluzzi L, Buqué A, Kepp O, Zitvogel L, Kroemer G. Immunogenic cell death in cancer and infectious disease. Nat Rev Immunol. 2017;17(2):97–111. | ||
van Vloten JP, Workenhe ST, Wootton SK, Mossman KL, Bridle BW. Critical Interactions between Immunogenic Cancer Cell Death, Oncolytic Viruses, and the Immune System Define the Rational Design of Combination Immunotherapies. J Immunol. 2018;200(2):450–458. | ||
Miyamoto S, Inoue H, Nakamura T, et al. Coxsackievirus B3 is an oncolytic virus with immunostimulatory properties that is active against lung adenocarcinoma. Cancer Res. 2012;72(10):2609–2621. | ||
Diaconu I, Cerullo V, Hirvinen ML, et al. Immune response is an important aspect of the antitumor effect produced by a CD40L-encoding oncolytic adenovirus. Cancer Res. 2012;72(9):2327–2338. |
 © 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.