Back to Journals » International Journal of Nanomedicine » Volume 9 » Issue 1
Detection of PLGA-based nanoparticles at a single-cell level by synchrotron radiation FTIR spectromicroscopy and correlation with X-ray fluorescence microscopy
Authors Pascolo L, Bortot B, Benseny-Cases N, Gianoncelli A, Tosi G, Ruozi B, Rizzardi C ![]() , De Martino E, Vandelli MA, Severini GM
, De Martino E, Vandelli MA, Severini GM
Received 4 December 2013
Accepted for publication 17 January 2014
Published 7 June 2014 Volume 2014:9(1) Pages 2791—2801
DOI https://doi.org/10.2147/IJN.S58685
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 4
Lorella Pascolo,1 Barbara Bortot,1 Nuria Benseny-Cases,2 Alessandra Gianoncelli,3 Giovanni Tosi,4 Barbara Ruozi,4 Clara Rizzardi,5 Eleonora De Martino,1 Maria Angela Vandelli,4 Giovanni Maria Severini1
1Institute for Maternal and Child Health, Istituto di Ricovero e Cura a Carattere Scientifico Burlo Garofolo, Trieste, Italy; 2European Synchrotron Radiation Facility, Polygone Scientifique Louis Néel, Grenoble, France; 3Elettra-Sincrotrone Trieste, Area Science Park, Basovizza, Trieste, Italy; 4Department of Life Sciences, University of Modena and Reggio Emilia, Modena, Italy; 5Department of Anatomical Pathology, Department of Pathology and Forensic Medicine, University of Trieste, Trieste, Italy
Abstract: Poly-lactide-co-glycolide (PLGA) is one of the few polymers approved by the US Food and Drug Administration as a carrier for drug administration in humans; therefore, it is one of the most used materials in the formulation of polymeric nanoparticles (NPs) for therapeutic purposes. Because the cellular uptake of polymeric NPs is a hot topic in the nanomedicine field, the development of techniques able to ensure incontrovertible evidence of the presence of NPs in the cells plays a key role in gaining understanding of their therapeutic potential. On the strength of this premise, this article aims to evaluate the application of synchrotron radiation-based Fourier transform infrared spectroscopy (SR-FTIR) spectromicroscopy and SR X-ray fluorescence (SR-XRF) microscopy in the study of the in vitro interaction of PLGA NPs with cells. To reach this goal, we used PLGA NPs, sized around 200 nm and loaded with superparamagnetic iron oxide NPs (PLGA-IO-NPs; Fe3O4; size, 10–15 nm). After exposing human mesothelial (MeT5A) cells to PLGA-IO-NPs (0.1 mg/mL), the cells were analyzed after fixation both by SR-FTIR spectromicroscopy and SR-XRF microscopy setups. SR-FTIR-SM enabled the detection of PLGA NPs at single-cell level, allowing polymer detection inside the biological matrix by the characteristic band in the 1,700–2,000 cm-1 region. The precise PLGA IR-signature (1,750 cm-1 centered pick) also was clearly evident within an area of high amide density. SR-XRF microscopy performed on the same cells investigated under SR-FTIR microscopy allowed us to put in evidence the Fe presence in the cells and to emphasize the intracellular localization of the PLGA-IO-NPs. These findings suggest that SR-FTIR and SR-XRF techniques could be two valuable tools to follow the PLGA NPs’ fate in in vitro studies on cell cultures.
Keywords: PLGA-NPs, cell targeting, SR-FTIR, SR-XRF, imaging
Introduction
Polymeric biodegradable nanoparticles (NPs) are of great interest in the field of nanomedicine because of their capability to encapsulate, protect, control, and target the active molecules toward a specific site (organ, tissue, cells).1 These properties allowed the polymeric NPs of highly biocompatible materials to become a versatile platform for delivering and targeting a large variety of compounds, ranging from small molecules to larger macromolecules, such as proteins and oligonucleotides.2–5
Among the available polymeric materials, the most widely used are the aliphatic polyesters, specifically poly(lactic acid) and poly(glycolic acid) and their copolymer [poly(lactide-co-glycolide), or PLGA]. PLGA is a material widely used in drug delivery systems because of its biocompatibility and biodegradability into glycolic acid and lactic acid. In general, the preparation methods of PLGA NPs are affected by several factors, such as the chemicophysical characteristics of the drug to be loaded, the possibility of using organic solvents, or the choice of obtaining matrix or reservoir systems. The most applied technologies are the single-emulsion,6–8 the double-emulsion,9–13 and the nanoprecipitation procedures.14–18 Recent advances in the design of colloidal drug carriers included the modifications of the NPs’ surface capable of improving the stability, prolonging the circulation in the bloodstream, and targeting the active molecules toward specific cell populations or extracellular matrix components.1,7
As in vitro studies are needed for toxicological and preclinical studies, an important research field is identifying suitable techniques capable of allowing the investigation of in vitro interaction of NPs with the cells. In fact, conventional techniques usually display a limited applicability with nanomaterials, whereas high-performing microscopy approaches request complicated sample preparation procedures. Among the different emerging imaging and microscopy techniques, synchrotron radiation X-ray fluorescence (SR-XRF) spectromicroscopy represents a very attractive approach because of its capability to map the NPs’ distribution in the cells and to determine and quantify their chemical composition with submicron lateral resolution.19 Because of the opportunity to apply the SR-XRF technique on simply fixed cells, elemental mapping allows us to photograph the sample without introducing artifacts. This method has already been successfully used at the single-cell level to reveal the presence and effects of nanomaterials and NPs.20,21
Synchrotron-based Fourier transform infrared (SR-FTIR) microscopy is another advanced technique that is emerging as a useful tool for label-free investigation of biological samples, although the intrinsic limited spatial resolution of IR spectroscopy could, in principle, reduce its applicability to follow NPs at single-cell level. IR spectroscopy is based on the absorption of IR beams as a result of resonance with vibrational motions of functional molecular groups. The major components of biological tissues are proteins, nucleic acids, carbohydrates, and lipids, all of which have specific absorption bands in the IR frequency domain.22 FTIR microspectroscopy combines IR spectroscopy and microscopy to determine chemical composition in a small sample area. The application of SR as a high-brightness source of IR photons has enabled the technique to achieve analysis at the diffraction limit (typically, half the wavelength of the vibrational frequency)23 while preserving a high spectral quality. Recent reports support the applicability of FTIR not only in clinical diagnosis, by detecting disease-related alterations of the mayor components in tissues,22 but also in in vitro cell uptake studies of drugs.24
As reported in a large number of articles, in biomedical fields, IO NPs are used in both diagnostics; for example, as a contrast agent medium in radiological imaging techniques25 and in magnetic resonance imaging as contrast agents,26 especially for liver and spleen. However, the relatively high toxicity of magnetic NPs restricts the use of these materials to human beings.27,28
Taking into account this evidence and considering that the detection of PLGA NPs at a cellular level is quite difficult, we therefore tried to “protect” IO NPs inside a polymeric shield (ie, PLGA NPs) and to achieve a combination of the advantages of IO NPs, in terms of high performance in imaging techniques,29 and those of PLGA nano-encapsulation, in terms of drug delivery and targeting features.30–32
In this article, superparamagnetic IO (Fe3O4; size, 10–15 nm) was therefore loaded into PLGA NPs (size, 200 nm) by a simple emulsion preparation process, and we exploited the ability of the application of the SR-FTIR microscopy to follow the fate of IO-loaded NPs in vitro at the single-cell level. By using PLGA NPs loaded with IO NPs, we report on a correlative microscopic approach by combining SR-FTIR and SR-XRF spectromicroscopy on the same cells.
Materials and methods
PLGA (50:50 lactide glycolide molar ratio) RG 503H (inherent viscosity, 0.38 dL/g) was used as received from the manufacturer (Boehringer-Ingelheim, Ingelheim am Rhein, Germany). According to the experimental titration results of the carboxylic end of the polymers (4.94 mg KOH/g polymer), the molecular weight of RG 503H was calculated to be 11,000 Da. Superparamagnetic IO NPs (IO size, 10–15 nm; saturation magnetization, 43.8 emu/g) were purchased from SkySpring Nanomaterials (Houston, TX, USA). A Milli-Q water system (Millipore, Bedford, MA, USA), supplied with distilled water, provided high-purity water (18 Ω) for the NPs’ preparation. All the solvents were of analytical grade, and all other chemicals were obtained commercially and used without further purification.
NP preparation
NPs were prepared by simple emulsion method8–10 with some modifications in the preparation procedures. Thus, briefly, to obtain PLGA NPs loaded with superparamagnetic Fe3O4 (PLGA-IO-NPs), PLGA 503H (100 mg) was dissolved in CH2Cl2 (3 mL). Separately, an exact weighed amount of IO (10 mg) was suspended in CH2Cl2 (1 mL), using a sonication bath (2–3 minutes; Branson Ultrasonics Co., Danbury, CT, USA). The PLGA solution and IO suspension were mixed under sonication. Then, a polyvinyl alcohol (PVA, molecular weight, 15,000 Da; Sigma Aldrich, Milan, Italy) water solution (3% weight/volume [w/v], 8 mL) was added under sonication at 55 W for 60 seconds. The obtained emulsion was then poured into 25 mL of a PVA water solution (0.3% w/v) and maintained under stirring with a magnetic stirrer (1,000 rpm) for 2–3 hours until the organic solvent was completely evaporated. The NPs were purified by five centrifugations (15,000 rpm for 10 minutes at 4°C; J21; Beckman, Palo Alto, CA, USA) to remove the PVA residuals. The purified suspension was freeze-dried (Lyovac GT-2; Leybold-Heraus, Hanau, Germany) at −50°C for 48 hours and 5×10−3 Torr as pressure, using trehalose (1:1 weight/weight [w/w], Sigma Aldrich) as cryoprotectant.
NP characterization
Both the size and surface charge of the PLGA-IO-NPs were assessed by photon correlation spectroscopy and laser Doppler anemometry, using a Zetasizer Nano ZS (Malvern Instruments, Malvern, United Kingdom; laser 4 mW He–Ne, 633 nm, automatic lLaser attenuator, transmission from 100% to 0.0003%, Avalanche photodiode detector, quantum efficiency >50% at 633 nm; t=25°C). The results were normalized with respect to a polystyrene standard solution.
The morphological observations of the PLGA-IO-NPs were performed with a scanning electron microscope (SEM; XL-40; Philips, Eindhoven, The Netherlands) operating at 8 kV. The PLGA-IO-NPs were washed several times with Milli-Q water, and then a drop of the final water suspension was placed onto a SEM sample holder and dried under vacuum (10−2 mmHg). The dried samples were coated under argon atmosphere with a 10-nm gold palladium thickness (Emitech K550 Super Coated; Emitech Ltd, Ashford, United Kingdom) to increase their electrical conductivity. The PLGA-IO-NPs were then processed for the evaluation of morphology and shape by analyzing images at different magnifications (from 13,000× to 16,000×).
Moreover, negative staining transmission electron microscopy (TEM) was used for the evaluation of the inner morphology of all batches of NPs. In particular, a drop of a water-diluted suspension of the purified NPs (about 0.05 mg/mL) was placed on a 200-mesh formvar copper grid (TABB Laboratories Equipment, Berks, United Kingdom), which allowed to it adsorb, and the surplus was removed by filter paper. A drop of 2% (w/v) aqueous solution of uranyl acetate was added and left in contact with the sample for 5 minutes (initially, we tested different concentrations of uranyl acetate solution; the condition applied in this study was the most suitable for maintaining the integrity of the sample during the preparation). The surplus water was removed, and the sample was dried at room conditions before NPs were imaged with a TEM operating at an acceleration voltage of 200 KV (model JEM 2010; JEOL, Peabody, MA, USA).
X-ray energy-dispersive spectroscopy (EDS) was used for elemental composition analysis, and samples were prepared on a carbon-coated copper grid (Sample Grid CarbonOnly®, 200 mesh [Ted Pella, Inc., Redding, CA, USA]) and kept under vacuum desiccation for 6 hours before loading them onto a specimen holder. To determine the elemental composition and purity of the sample by atom percentage of metal, elemental analysis on single particles was carried out using EDS attachment equipped with TEM.
Moreover, we have measured direct current magnetization as a function of an applied magnetic field of both PLGA-IO-NPs and IO. Measurements were performed at room temperature by means of a Physical Property Measurement System AC/DC susceptometer (Quantum Design, San Diego, CA, USA).
Evaluation of the IO content in the NPs
To determine the amount of IO loaded into PLGA-IO-NPs, a weighed amount of PLGA-IO-NPs was treated with 5 mL dimethyl sulfoxide for 30 minutes.33 Then 10 mL HCl 6N was added, maintaining the reaction up to 2 hours. The resultant solution was diluted with an aqueous solution of HCl (1% w/w). The iron concentration in the solution was determined by atomic absorption spectrophotometry measurements at 248 nm (SpectrAA-10 plus; Varian, Palo Alto, CA, USA). A calibration curve was obtained by treating an acidic solution of FeCl3 (1 g/L) in the same conditions.
IO loading efficiency (%) and IO content (% w/w) were calculated by Equations 1 and 2, respectively:IO loading
IO loading efficiency (%) = (mass of IO in PLGA-IO-NPs/mass of IO used in formulation) × 100; | [1] |
IO content(% w/w) = (mass of IO in PLGA-IO-NPs/massof PLGA-IO-NPs) × 100; | [2] |
Cell culture and sample preparation
MeT5A, nonmalignant transformed human pleural mesothelial cells, were cultured in Roswell Park Memorial Institute 1640 medium with 10% (volume/volume [v/v]) heat-inactivated fetal calf serum, 2 mM glutamine, 100 U/mL penicillin, and 100 μg/mL streptomycin (all from Invitrogen, Milan, Italy) in a 5% CO2 incubator at 37°C.
To perform in vitro treatment experiments with PLGA-IO-NPs, MeT5A cells were seeded at a concentration of 8.7×104 cell/mL onto silicon nitrite (Si3N4) 100 nm thick windows (Silson Ltd., Northampton, United Kingdom) contained in 24 multiwell plates. The day after seeding, culture medium was replaced with fresh medium containing PLGA-IO-NPs at a concentration of 0.1 mg/mL, and MeT5A cells were incubated at 37°C for 16 hours. MeT5A cells similarly grown onto silicon nitrite windows, but not exposed to PLGA-IO-NPs (untreated cells), were used as control.
After incubation, samples were fixed at room temperature with 4% paraformaldehyde aqueous solution (Sigma Aldrich) for 20 minutes. Then, samples were washed with Milli-Q water before the analysis.
SR-based FTIR spectromicroscopy
Spectra of single cells were recorded at the ID21 beamline synchrotron (European Synchrotron Radiation Facility),34 using the bending and edge radiations from a bending magnet. Spectra were recorded on a Nicolet Continuum XL microscope (Thermo Fisher Scientific, Waltham, MA, USA) equipped with a 50 mm liquid nitrogen-cooled mercury cadmium telluride “A” detector, a 32X/NA0.65 Schwarzschild objective, a motorized knife-edge aperture, and an XYZ motorized stage and coupled with a Nicolet 5700 spectrometer (Thermo Fisher). Spectra of a minimum of about five individual cells were recorded in transmission between 800 and 4,000 cm−1, with 128 scans at 4 cm−1 resolution, 15×15 μm aperture dimension. At least three cells for each condition were mapped with an 8×8 μm aperture dimension and 6×6 μm step size.
Chemical maps were created and analyzed with the Omnic software (Thermo Fisher) by measuring and plotting the intensity of the specific bands of the second derivative of the spectra: PLGA, 1,770–1,740 cm−1;35 amide I, 1,670–1,625 cm−1; lipids, 2,970–2,950 cm−1. PyMCA (European Synchrotron Radiation Facility) and Origin 8.0 (OriginLab, Northampton, MA, USA) were used for data analysis.
SR-XRF microscopy
The absorption and phase contrast images, together with the low-energy XRF elemental maps, were collected at the TwinMic beamline (Elettra Sincrotrone, Trieste, Italy),36 where the incoming monochromatic beam is focused on the specimen by means of zone plate diffractive optics. The sample is raster-scanned across the X-ray probe, and the transmitted X-rays are collected through a fast-readout charge-coupled device camera (Andor Technology, Belfast, Northern Republic of Ireland), providing absorption and phase contrast images.37,38 Simultaneously, the X-ray fluorescence photons emitted by the specimen were acquired by 8 silicon drift detectors located in front of the specimen in an annular geometry.39,40 The incident energy was chosen as 1.1 keV to provide optimal detection of Fe, Na, O, and C, whereas the spot size was 600 nm in diameter, which was good enough to detect PLGA-IO-NP clusters and provide information at a subcellular level. The collected XRF spectra were fitted with the PyMCA software.41 Both cells, exposed and not exposed to PLGA-IO-NPs, were analyzed by low-energy XRF microscopy.
Results and discussion
NP characterization and IO content
PLGA-IO-NPs were characterized in their physicochemical properties in terms of size (211±10 nm) and surface charge (−11±2 mV). PLGA-IO-NPs showed a good homogeneity, as confirmed by the low value of the polydispersity index (0.11±0.01). The SEM photomicrographs of the PLGA-IO-NPs showed well-structured and well-formed particles with a smooth surface without strong evidence of formation of aggregates (Figure 1A).
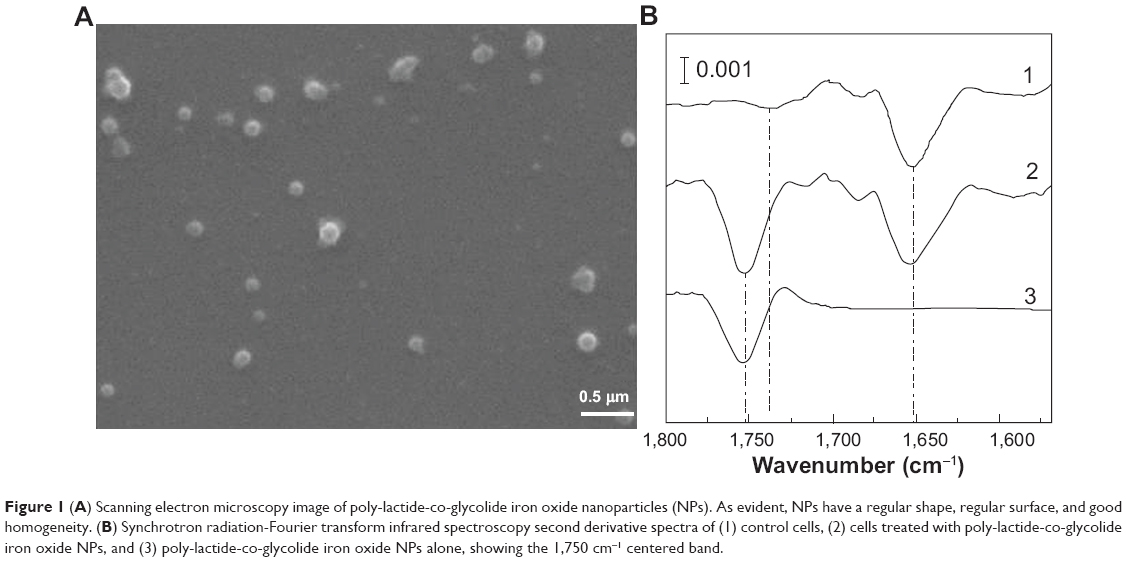 | Figure 1 (A) Scanning electron microscopy image of poly-lactide-co-glycolide iron oxide nanoparticles (NPs). As evident, NPs have a regular shape, regular surface, and good homogeneity. (B) Synchrotron radiation-Fourier transform infrared spectroscopy second derivative spectra of (1) control cells, (2) cells treated with poly-lactide-co-glycolide iron oxide NPs, and (3) poly-lactide-co-glycolide iron oxide NPs alone, showing the 1,750 cm−1 centered band. |
The results obtained from the study on the encapsulation efficiency with the atomic absorption spectrophotometry procedure described earlier showed that the loading efficiency was close to 14% (±2%), which corresponded to about 1.5 (±0.8) mg IO per 100 mg PLGA-IO-NPs. This result is in line with other previous reports42 demonstrating the ability of PLGA NPs to encapsulate and stabilize the IO suspension.
Moreover, TEM measurements were used to study the morphological confirmation of iron NPs. A TEM micrograph (Figure 2) showed homogeneous distribution of nearly spherical NPs with diameters ranging between 8–12 nm (Figure 2, left panel). The composition of the Fe3O4 (IO) was confirmed by X-ray EDS microanalysis of the analyzed point (10 nm of beam; Figure 2, right panel). The presence of Fe and O can be observed, with iron abundance higher than oxygen signals. Moreover, together with Fe and O ones, the copper signal is present because of the grids used for sample preparation, in which traces of Cu are present, as reported by the manufacturer.
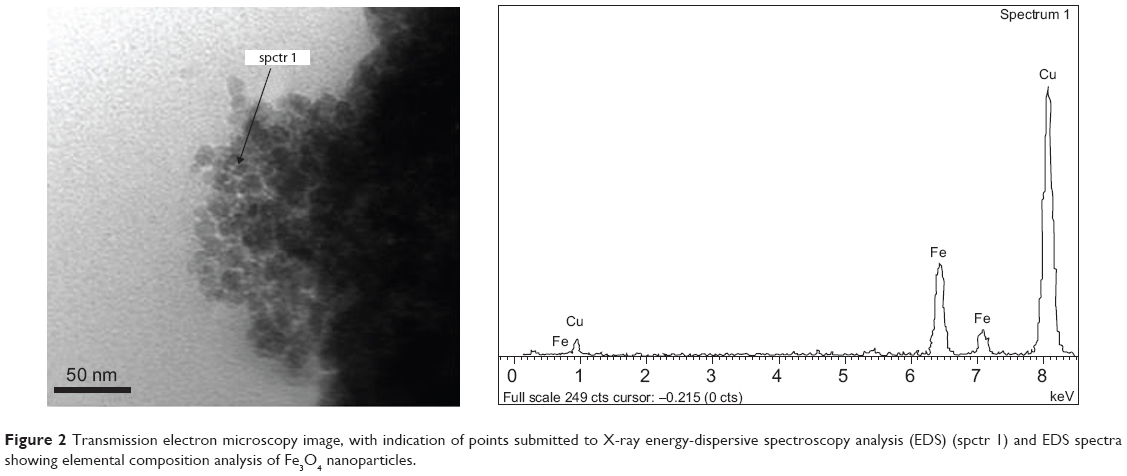 | Figure 2 Transmission electron microscopy image, with indication of points submitted to X-ray energy-dispersive spectroscopy analysis (EDS) (spctr 1) and EDS spectra showing elemental composition analysis of Fe3O4 nanoparticles. |
In addition, PLGA-IO-NP morphology was evaluated by TEM imaging (Figure 3). As evident, these polymeric NPs were spherical in shape, with delimited contours, but were partially superimposed and difficult to analyze. The average diameter of PLGA-IO-NPs obtained analyzing TEM images was about 250 nm, similar to the result from photon correlation spectroscopy measurement shown before. Notably, the influence of surface charge, slightly negative, led to possible aggregation of NPs, as seen in TEM images. This is a result of the almost neutral surface charge of NPs and the lack of added surfactant (ie, PVA). Thus, it could be hypothesized that small aggregates of NPs can interact with the cells. In this view, the uptake mechanisms by cells could be different with respect to those of single, nonaggregated NPs.
 | Figure 3 Transmission electron microscopy images of poly-lactide-co-glycolide iron oxide nanoparticles. In particular, two different exemplificative transmission electron microscopy images are representative of the whole sample in (A, B). Panel C is referring to enlargement of area in the red rectangle in panel B, with higher magnification. The points submitted to EDS analysis are indicated with spctr 1, spctr 2 and spctr 3. |
Considering the microscopical technique adopted, the metallic iron and the polymer can be distinguished from the corresponding color contrast in TEM images. In fact, TEM instruments are able to distinguish the elements with higher atomic numbers as signals darker than those with lower atomic numbers.43 In particular, in our images (Figure 4), NPs appeared to be formed by lighter and darker regions, randomly concentrated. In some PLGA-IO-NPs, we observed regions with a much higher dark contrast, suggesting an important concentration of the metal. Then, IO NPs seemed stably distributed into PLGA NPs, but they seemed to display a tendency to form clusters into the polymeric matrix with a consequent heterogeneous distribution. Punctual EDS analysis confirmed this architecture with IO NPs loaded into the polymeric matricial structure. In fact, close to the signal of carbon, forming the frame of polymer constituting the nanocarriers (together with the oxygen and hydrogen), EDS spectra indicated the presence of Fe and O. Also in this case, the copper peak was detected as an interference signal.
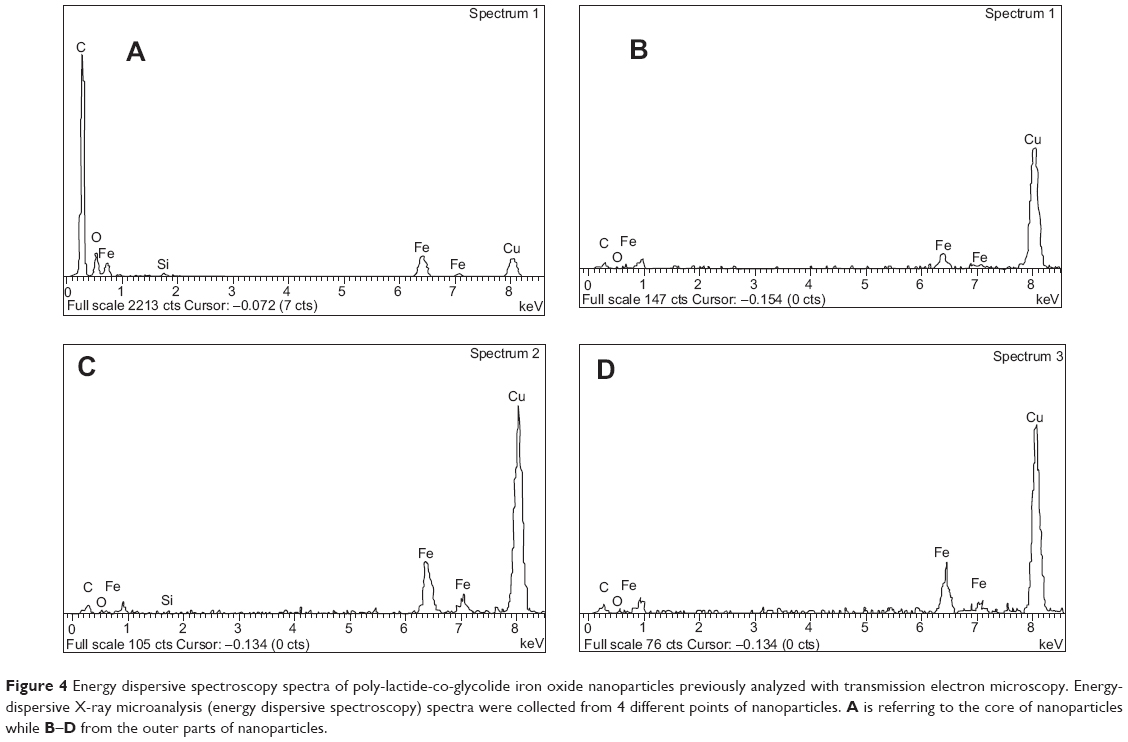 | Figure 4 Energy dispersive spectroscopy spectra of poly-lactide-co-glycolide iron oxide nanoparticles previously analyzed with transmission electron microscopy. Energy-dispersive X-ray microanalysis (energy dispersive spectroscopy) spectra were collected from 4 different points of nanoparticles. A is referring to the core of nanoparticles while B–D from the outer parts of nanoparticles. |
The measurements aiming to assess direct current magnetization as a function of an applied magnetic field of the IO encapsulated into PLGA-IO-NPs were compared with an equivalent sample of bare IO. The results are reported in Figure 5, showing that magnetic behavior of PLGA-IO-NPs is not substantially affected by encapsulation into PLGA matrix and is not different from the behavior of the bare IO NPs.
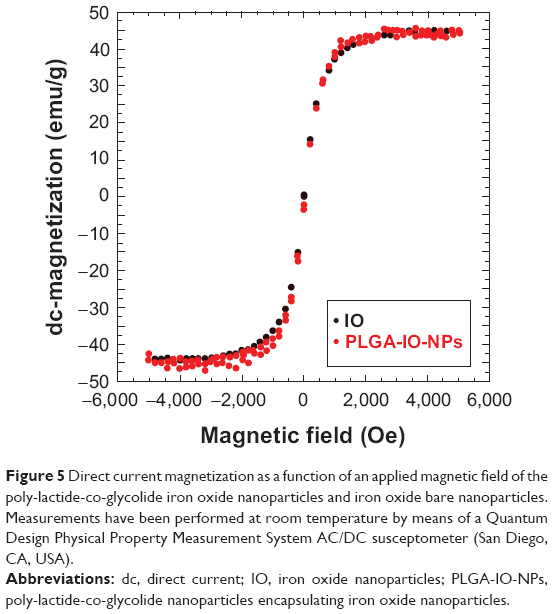 | Figure 5 Direct current magnetization as a function of an applied magnetic field of the poly-lactide-co-glycolide iron oxide nanoparticles and iron oxide bare nanoparticles. Measurements have been performed at room temperature by means of a Quantum Design Physical Property Measurement System AC/DC susceptometer (San Diego, CA, USA). |
These results strongly agree with previous reports. In particular, as recently discovered,44 the encapsulation processes did not significantly affect the chemicophysical properties of both carriers and IO NPs. Experiments on magnetic relaxation, magnetic hysteresis loop measurements, and viscosity properties of IO-loaded PLGA NPs45 confirmed that the encapsulation in PLGA NPs did not negatively influence the magnetic features of IO NPs.
SR-FTIR spectromicroscopy
MeT5a cells grown onto silicon nitride windows and incubated with PLGA-IO-NPs (0.1 mg/mL) were analyzed under the SR-FTIR set-up by collecting single-point spectra for cells (aperture dimension of 15×15 μm). Figure 1B shows the second derivative analysis of a control cell (spectrum 1), of a cell treated with PLGA-IO-NPs (spectrum 2), and of a PLGA-IO-NP solution alone (spectrum 3) placed onto a transparent silicon nitride window. PLGA-IO-NPs clearly showed a specific band at 1,750 cm−1 corresponding to the carbonyl ester bond of the PLGA already reported in the literature.33 In the controls, a band centered at 1,740 cm−1, because of the absorption of the carbonyl of the phospholipid, is present. This band clearly differs from the PLGA-IO-NPs’ carboxyl band, which is absorbing at higher wave numbers with a much higher intensity.
MeT5A cells, incubated with PLGA-IO-NPs at a concentration of 0.1 mg/mL, were mapped using SR-FTIR (Figure 6), and local SR-FTIR spectra were recorded at the subcellular level (Figure 6B–D, F–H). Chemical maps at the single-cell level were generated by measuring and plotting the intensity of a specific band for protein (amide I), lipids, and PLGA-IO-NPs. Maps were plotted using the second derivative of the spectra (maximum absorption is represented in blue, and minimum absorption in red). The maximal intensities of amide I and lipid plot allowed us to recognize the bulge corresponding to the nucleus, both in the treated and untreated cells. The characteristic signature of the PLGA band (Figure 1B, 3) was found only in the cells treated with PLGA-IO-NPs, suggesting a plasma membrane and/or cytoplasmic localization. These signals were not observed in untreated cells (Figure 6D).
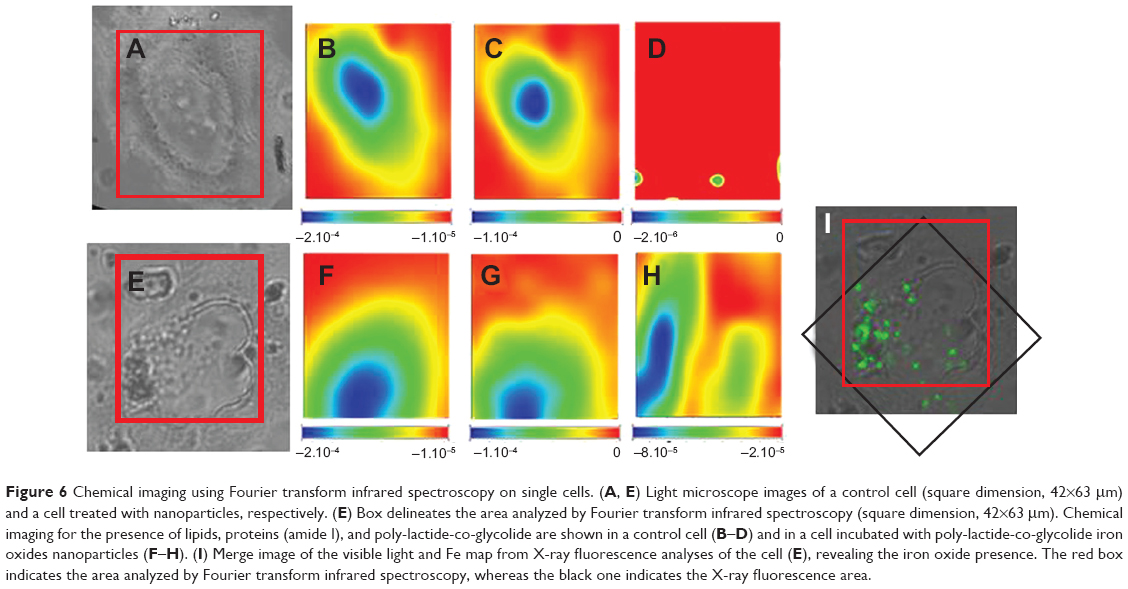 | Figure 6 Chemical imaging using Fourier transform infrared spectroscopy on single cells. (A, E) Light microscope images of a control cell (square dimension, 42×63 μm) and a cell treated with nanoparticles, respectively. (E) Box delineates the area analyzed by Fourier transform infrared spectroscopy (square dimension, 42×63 μm). Chemical imaging for the presence of lipids, proteins (amide I), and poly-lactide-co-glycolide are shown in a control cell (B–D) and in a cell incubated with poly-lactide-co-glycolide iron oxides nanoparticles (F–H). (I) Merge image of the visible light and Fe map from X-ray fluorescence analyses of the cell (E), revealing the iron oxide presence. The red box indicates the area analyzed by Fourier transform infrared spectroscopy, whereas the black one indicates the X-ray fluorescence area. |
SR-XRF spectromicroscopy
Figure 7 shows the results of SR-XRF microscopy analyses of two representative cells: a control cell (Figure 7A) and a cell exposed to PLGA-IO-NPs (Figure 7B). The selected energy allowed us to map light elements, including characteristic constituents of the cell material, such as C, O, and Na atoms, and to compare their distribution with that of Fe atoms, loaded into the PLGA-IO-NPs. Indeed, the elemental map of Fe of the control cells does not show a significant signal and distribution, suggesting that Fe concentration in this sample is below the detection limits of the TwinMic LE-XRF system. In contrast, the Fe signal is clearly visible in the cells exposed to PLGA-IO-NPs, and its distribution nicely correlates with some dark clusters appearing in the corresponding absorption image. O and Na signals have a quite similar distribution, whereas the C signal seems to have a slight increase in some high-absorbing regions of the nucleus. In the cells exposed to PLGA-IO-NPs, some high spots in C and O atoms could correspond to high amounts of PLGA-IO-NPs, although they cannot be used to precisely localize the PLGA-IO-NPs. The distribution and intensity of Na do not seem to be affected by the presence of the PLGA-IO-NPs. Figure 6K reports the Fe map of the cell in Figure 6E. The XRF analyses have been performed after the FTIR experiments. The iron signal is clearly compatible with the PLGA map and supports the presence of the NPs on the cell membrane and possibly intracellularly.
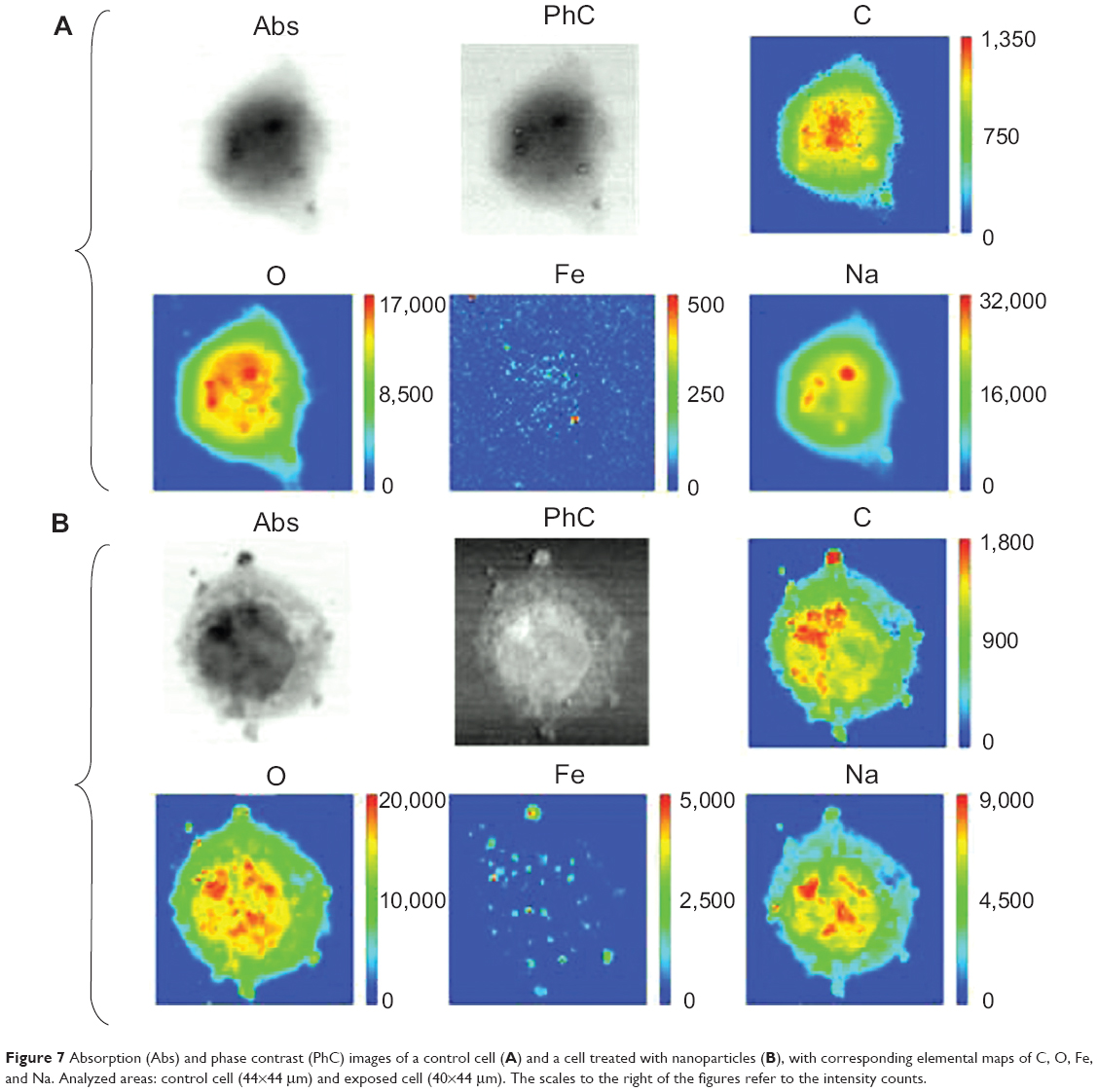 | Figure 7 Absorption (Abs) and phase contrast (PhC) images of a control cell (A) and a cell treated with nanoparticles (B), with corresponding elemental maps of C, O, Fe, and Na. Analyzed areas: control cell (44×44 μm) and exposed cell (40×44 μm). The scales to the right of the figures refer to the intensity counts. |
Conclusion
In vitro studies are fundamental to demonstrating the cell-specificity of engineered PLGA-NPs. However, conventional techniques usually display a limited applicability, requiring specific procedures for the sample preparation, which could lead to artifacts. As SR-based FTIR spectromicroscopy could be considered an attractive technique for bioimaging at a cellular level, in this article, we combined IR spectromicroscopy with XRF microscopy to determine the chemical composition in a small sample area, thus allowing the intracellular identification of nanocarriers. As shown, we loaded PLGA NPs with IO NPs (10–15 nm), obtaining PLGA-IO-NPs, which allowed us to take advantage of both SR-FTIR spectromicroscopy and SR-XRF microscopy.
As recently reported,46 PLGA NPs encapsulating magnetic IO represent a significant advancement in the aim of translating magnetic resonance imaging-based cell tracking to the clinic. Several benefits of this strategy could be described as the large amounts of IO that NPs encapsulate and the advantage of having a US Food and Drug Administration-approved polymer matrix. In this view, the idea of combining the best aspects of magnetic NPs with biodegradable, biocompatible, and nanosized PLGA NPs would represent an innovative strategy for coupling both magnetic and drug delivery aims. Remarkably, the possible double use of PLGA NPs encapsulating IO NPs plus a pharmacologically active drug (ie, albumin or anticancer agents)42,47,48 could open the way to new insight and therapeutic/imaging approaches with good translatability.
The results from SR-FTIR and SR-XRF analyses obtained after exposure of MeT5A to PLGA-IO-NPs allowed us to retrieve useful correlative outputs on NPs’ intracellular fate on the same single cell. The two synchrotron methods are attractive for biological studies. They are both label-free techniques requiring only minimal treatment of the sample during its preparation. SR-FTIR imaging is completely nondestructive but is limited to micrometric resolution, as the smallest synchrotron beam size is in the range of 3–5 mm. For higher-resolution requirements, SR-XRF can provide the elemental distribution at submicron spatial resolutions, but because it is based on illuminating the sample with X-ray photons, it can induce radiation damage. This damage mainly appears on biological samples as mass loss and shrinkage, especially when exposed to high radiation doses. More important, the main drawback of the imaging synchrotron techniques is the limited access to the synchroton facilities due to overbooking applications, which does not allow large series of samples to be analyzed. However, the combined approach we present serves to provide information that could be the base for more-conventional analyses.
Even if pharmaceutical nanotechnology (and its application to medicine) is aiming both to individuate novel therapeutic approaches for difficult-to-treat pathologies, such as central nervous system disorders,49,50 and to implement the possibility of diagnosis or detection,51–53 an important role has to be dedicated to the intracellular dynamics and the intracellular trafficking of nanocarriers, as it is a hot topic in the nanomedicine field. Aiming to highlight the intracellular fate of nanocarriers requires novel and specific technologies for assessing nanoscopic grades of analysis and detection; therefore the findings of this research underlined the usefulness and potentiality of the application of these advanced spectroscopic and imaging techniques (particularly the nondestructive FTIR technique) to study NP localization in vitro.
Acknowledgment
The authors gratefully thank Professor Marco Affronte, University of Modena and Reggio Emilia, for supporting direct current magnetization analysis.
Disclosure
N B-C had a postdoctoral fellowship funded by Fundacion Alfonso Martin Escudero. The authors report no other conflicts of interest in this work.
References
Nicolas J, Mura S, Brambilla D, Mackiewicz N, Couvreur P. Design, functionalization strategies and biomedical applications of targeted biodegradable/biocompatible polymer-based nanocarriers for drug delivery. Chem Soc Rev. 2013;42(3):1147–1235. | ||
Bala I, Hariharan S, Kumar MN. PLGA nanoparticles in drug delivery: the state of the art. Crit Rev Ther Drug Carrier Syst. 2002;1(5):387–422. | ||
Danhier F, Ansorena E, Silva JM, Coco R, Le Breton A, Préat V. PLGA-based nanoparticles: an overview of biomedical applications. J Control Release. 2012;161(2):505–522. | ||
Moghimi SM, Hunter AC, Murray JC. Nanomedicine: current status and future prospects. FASEB J. 2005;19(3):311–330. | ||
Debbage P. Targeted drugs and nanomedicine: present and future. Curr Pharm Des. 2009;15(2):153–172. | ||
Jaiswal J, Gupta SK, Kreuter J. Preparation of biodegradable cyclosporine nanoparticles by high-pressure emulsification-solvent evaporation process. J Control Release. 2004;96(1):169–178. | ||
Galindo-Rodriguez S, Allémann E, Fessi H, Doelker E. Physicochemical parameters associated with nanoparticle formation in the salting-out, emulsification-diffusion, and nanoprecipitation methods. Pharm Res. 2004;21(8):1428–1439. | ||
Alonso MJ, Gaspar MM, Blanco MD, et al. Development of poly(lactic/glycolic)acid nanoparticles containing a high loading of l-asparaginase. Proceedings of the 23rd International Symposium Controlled Release Bioactive Materials. 1996. | ||
Zambaux MF, Bonneaux F, Gref R, et al. Influence of experimental parameters on the characteristics of poly(lactic acid) nanoparticles prepared by a double emulsion method. J Control Release. 1998;50(1–3):31–40. | ||
Vrignaud S, Benoit JP, Saulnier P. Strategies for the nanoencapsulation of hydrophilic molecules in polymer-based nanoparticles. Biomaterials. 2011;32(33):8593–8604. | ||
Mundargi RC, Babu VR, Rangaswamy V, Patel P, Aminabhavi TM. Nano/micro technologies for delivering macromolecular therapeutics using poly(D,L-lactide-co-glycolide) and its derivatives. J Control Release. 2008;125(3):193–209. | ||
Fessi H, Devissaguet JP, Puisieux F, et al. Process for the preparation of dispersible colloidal systems of a substance in the form of nanoparticles. United States patent US 5118528 A. 1992 Jun 2. | ||
Kumari A, Yadav SK, Yadav SC. Biodegradable polymeric nanoparticles based drug delivery systems. Colloids Surf B Biointerfaces. 2010; 75(1):1–18. | ||
Barichello JM, Morishita M, Takayama K, Nagai T. Encapsulation of hydrophilic and lipophilic drugs in PLGA nanoparticles by the nanoprecipitation method. Drug Dev Ind Pharm. 1999;25(4):471–476. | ||
Ueda M, Iwara A, Kreuter J. Influence of the preparation methods on the drug release behaviour of loperamide-loaded nanoparticles. J Microencapsul. 1998;15(3):361–372. | ||
Quintanar-Guerrero D, Allémann E, Fessi H, Doelker E. Preparation techniques and mechanisms of formation of biodegradable nanoparticles from preformed polymers. Drug Dev Ind Pharm. 1998; 24(12):1113–1128. | ||
Lü JM, Wang X, Marin-Muller C, et al. Current advances in research and clinical applications of PLGA-based nanotechnology. Expert Rev Mol Diagn. 2009;9(4):325–341. | ||
Grabrucker AM, Garner CC, Boeckers TM, et al. Development of novel Zn2+ loaded nanoparticles designed for cell-type targeted drug release in CNS neurons: in vitro evidences. PLoS One. 2011;6(3):e17851. | ||
Bohic S, Cotte M, Salomé M, et al. Biomedical applications of the ESRF synchrotron-based microspectroscopy platform. J Struct Biol. 2012;177(2):248–258. | ||
Marmorato P, Ceccone G, Gianoncelli A, et al. Cellular distribution and degradation of cobalt ferrite nanoparticles in Balb/3T3 mouse fibroblasts. Toxicol Lett. 2011;207(2):128–136. | ||
Lewis DJ, Bruce C, Bohic S, et al. Intracellular synchrotron nanoimaging and DNA damage/genotoxicity screening of novel lanthanide-coated nanovectors. Nanomedicine (Lond). 2010;5(10):1547–1557. | ||
Miller LM, Dumas P. From structure to cellular mechanism with infrared microspectroscopy. Curr Opin Struct Biol. 2010;20(5):649–656. | ||
Carr GL. Resolution limits for infrared microspectroscopy explored with synchrotron radiation. Rev Sci Instrum. 2001;7:1613. | ||
Clède S, Lambert F, Sandt C, et al. Synchrotron radiation FTIR detection of a metal-carbonyl tamoxifen analog. Correlation with luminescence microscopy to study its subcellular distribution. Biotechnol Adv. 2013;31(3):393–395. | ||
Weissleder R, Hahn PF, Stark DD, et al. MR imaging of splenic metastases: ferrite-enhanced detection in rats. AJR Am J Roentgenol. 1987;149(4):723–726. | ||
Tiefenauer LX, Tschirky A, Kühne G, Andres RY. In vivo evaluation of magnetite nanoparticles for use as a tumor contrast agent in MRI. Magn Reson Imaging. 1996;14(4):391–402. | ||
Quintanar-Guerrero D, Allémann E, Doelker E, Fessi H. Preparation and characterization of nanocapsules from preformed polymers by a new process based on emulsification-diffusion technique. Pharm Res. 1998;15(7):1056–1062. | ||
Magdolenova Z, Drlickova M, Henjum K, et al. Coating-dependent induction of cytotoxicity and genotoxicity of iron oxide nanoparticles. Nanotoxicology. Epub 2013 Nov 14. | ||
Li L, Jiang W, Luo K, et al. Superparamagnetic iron oxide nanoparticles as MRI contrast agents for non-invasive stem cell labeling and tracking. Theranostics. 2013;3(8):595–615. | ||
Chan M, Schopf E, Sankaranarayanan J, Almutairi A. Iron oxide nanoparticle-based magnetic resonance method to monitor release kinetics from polymeric particles with high resolution. Anal Chem. 2012;84(18):7779–7784. | ||
Hwang JH, Noh YW, Choi JH, et al. In vivo imaging of islet transplantation using PLGA nanoparticles containing iron oxide and indocyanine green. Magn Reson Med. Epub 2013 May 2. | ||
Douziech-Eyrolles L, Marchais H, Hervé K, et al. Nanovectors for anticancer agents based on superparamagnetic iron oxide nanoparticles. Int J Nanomedicine. 2002;(4):541–550. | ||
Okassa LN, Marchais H, Douziech-Eyrolles L, et al. Optimization of iron oxide nanoparticles encapsulation within poly(D,L-lactide-co-glycolide) sub-micron particles. Eur J Pharm Biopharm. 2007;67(1):31–38. | ||
Susini J, Cotte M, Scheidt K, Chubar O, Polack F, Dumas F. Technical Report: The FTIR Spectro-Microscopy End-Station at the ESRF-ID21 Beamline. Synchrotron Radiat News. 2007;20:13–16. | ||
Källrot M, Edlund U, Albertsson AC. Surface functionalization of degradable polymers by covalent grafting. Biomaterials. 2006; 27(9):1788–1796. | ||
Kaulich B, Bacescu D, Susini J, et al. TwinMic – A European Twin X-ray Microscopy Station Commissioned at ELETTRA. In: Aoki S, Kagoshima Y, Suzuki Y, editors. Proceedings of the 8th International Conference on X-ray Microscopy. Vol. 7. Tokyo: IPAP; 2006: 22–25. | ||
Morrison GR, Gianoncelli A, Kaulich B, Bacescu D, Kovac J. Fast-readout CCD System for Configured-Detector Imaging in STXM. In: Aoki S, Kagoshima Y, Suzuki Y, editors. Proceedings of the 8th International Conference on X-ray Microscopy. Vol. 7. Tokyo: IPAP; 2006:377–379. | ||
Gianoncelli A, Morrison GR, Kaulich B, Bacescu D, Kovac J. Scanning transmission x-ray microscopy with a configurable detector. Appl Phys Lett. 2006;89(25):251117–251117–3. | ||
Gianoncelli A, Kaulich B, Alberti R, et al. Simultaneous soft X-ray transmission and emission microscopy. Nuclear Instruments Methods Physics Res Sect A. 2009;608(1):195–198. | ||
Gianoncelli A, Kourousias G, Stolfa A, Kaulich B. Recent developments at the TwinMic beamline at ELETTRA: an 8 SDD detector setup for low energy X-ray fluorescence. J Physics Conf Ser. 2013;425:182001. | ||
Sole A, Papillon E, Cotte M, Walterb PH, Susinia J. A multiplatform code for the analysis of energy-dispersive X-ray fluorescence spectra. Spectrochim Acta Part B: Atomic Spectroscopy. 2007;62(1):63–68. | ||
Schleich N, Sibret P, Danhier P, et al. Dual anticancer drug/superparamagnetic iron oxide-loaded PLGA-based nanoparticles for cancer therapy and magnetic resonance imaging. Int J Pharm. 2013; 447(1–2):94–101. | ||
Yuvakkumar R, Elango V, Rajendran V, Kannan N. Preparation and characterization of zero valent iron nanoparticles. Digest J Nanomater Biostructures. 2011;6(4):1771–1776. | ||
Gun S, Edirisinghe M, Stride E. Encapsulation of superparamagnetic iron oxide nanoparticles in poly-(lactide-co-glycolic acid) microspheres for biomedical applications. Mater Sci Eng C Mater Biol Appl. 2013;33(6):3129–3137. | ||
Lee SJ, Jeong JR, Shin SC, et al. Magnetic enhancement of iron oxide nanoparticles encapsulated with poly(D,L-latide-co-glycolide). Colloids Surf A: Physiochem Eng Aspects. 2005;255(1–3):19–25. | ||
Tang KS, Hashmi SM, Shapiro EM. The effect of cryoprotection on the use of PLGA encapsulated iron oxide nanoparticles for magnetic cell labeling. Nanotechnology. 2013;24(12):125101. | ||
Shubhra QT, Feczkó T, Kardos AF, et al. Co-encapsulation of human serum albumin and superparamagnetic iron oxide in PLGA nanoparticles: part II. Effect of process variables on protein model drug encapsulation efficiency. J Microencapsul. 2014;31(2):156–165. | ||
Ling Y, Wei K, Zou F, Zhong S. Temozolomide loaded PLGA-based superparamagnetic nanoparticles for magnetic resonance imaging and treatment of malignant glioma. Int J Pharm. 2012;430(1–2):266–275. | ||
Tosi G, Fano RA, Bondioli L, et al. Investigation on mechanisms of glycopeptide nanoparticles for drug delivery across the blood–brain barrier. Nanomedicine (Lond). 2011;6(3):423–436. | ||
Tosi G, Ruozi B, Belletti D. Nanomedicine: the future for advancing medicine and neuroscience. Nanomedicine (Lond). 2012;7(8):1113–1116. | ||
Tosi G, Rivasi F, Gandolfi F, Costantino L, Vandelli MA, Forni F. Conjugated poly(D,L-lactide-co-glycolide) for the preparation of in vivo detectable nanoparticles. Biomaterials. 2005;26(19):4189–4195. | ||
Tosi G, Bondioli L, Ruozi B, et al. NIR-labeled nanoparticles engineered for brain targeting: in vivo optical imaging application and fluorescent microscopy evidences. J Neural Transm. 2011;118(1):145–153. | ||
Ruozi B, Belletti D, Tombesi A, et al. AFM, ESEM, TEM, and CLSM in liposomal characterization: a comparative study. Int J Nanomedicine. 2011;6:557–563. |
 © 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.