")
Back to Journals » International Journal of Nanomedicine » Volume 13
Detection of Mycobacterium tuberculosis purified ESAT-6 (Rv3875) by magnetic bead-coupled gold nanoparticle-based immuno-PCR assay
Authors Singh N, Dahiya B, Radhakrishnan VS, Prasad T, Mehta PK
Received 21 July 2018
Accepted for publication 26 September 2018
Published 12 December 2018 Volume 2018:13 Pages 8523—8535
DOI https://doi.org/10.2147/IJN.S181052
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Thomas Webster
Netrapal Singh,1,2 Bhawna Dahiya,1 Venkatraman Srinivasan Radhakrishnan,3 Tulika Prasad,3 Promod K Mehta1,4
1Centre for Biotechnology, Maharshi Dayanand University, Rohtak, Haryana, India; 2Institute of Synthetic Biology (iSynBio), Shenzhen Institutes of Advanced Technology, Chinese Academy of Sciences, Shenzhen, China; 3Advanced Instrumentation Research & Facility (AIRF) and Special Centre for Nanoscience (SCNS), Jawaharlal Nehru University (JNU), New Delhi, Delhi, India; 4Microbiology Department, Central University of Haryana, Mahendergarh, India
Purpose: Immuno-PCR (I-PCR), an ultrasensitive method, combines the versatility of ELISA with the exponential amplification capacity of PCR. Coupling of detection antibodies with the reporter DNA is a critical step of I-PCR. Gold nanoparticles (GNPs) and magnetic beads (MBs) are relatively easy to attach with the antibodies and DNA. Therefore, we designed MB-coupled GNP-based I-PCR (MB-GNP-I-PCR) assay for the detection of Mycobacterium tuberculosis antigen.
Methods: GNPs were synthesized by chemical reduction and seed-mediated synthesis. Functionalized GNPs were prepared by coupling GNPs with the detection antibodies and reporter DNA and were characterized. Detection limit of M. tuberculosis-specific purified early secreted antigenic target-6 (ESAT-6) (Rv3875) was determined by MB-GNP-I-PCR.
Results: Transmission electron microscopy revealed spherical and slightly polydispersed GNPs of ~20 and ~60 nm size. Coupling of antibodies to GNPs was indicated by a shift in absorption maxima from 524 to 534 nm, which was confirmed by transmission electron microscopy. A color reaction with ELISA and the presence of 76 bp product by PCR further validated the coupling of detection antibodies and signal DNA to the functionalized GNPs. Also, attachment of capture antibodies with MBs was confirmed by magneto-ELISA. Detection limit of purified ESAT-6 by MB-GNP-I-PCR was determined to be 10 fg/mL, 105-fold lower than analogous ELISA. Notably, no sample matrix effect was observed in the saliva samples of healthy individuals spiked with the purified ESAT-6.
Conclusion: Unlike conventional I-PCR (solid format), MB-GNP-I-PCR (liquid format) is relatively simple with the reduced background signals, which can be further exploited for the clinical diagnosis of tuberculosis.
Keywords: MB-GNP-I-PCR, functionalized GNPs, ELISA, LOD
Introduction
Tuberculosis (TB) is the leading cause of death worldwide from a single infectious agent, which ranks above HIV infection. In 2016, there were an estimated 1.3 million TB deaths among HIV-negative people and an additional 0.37 million deaths among HIV-positive individuals.1 Rapid and accurate diagnosis of TB is crucial to initiate an early anti-tubercular therapy so as to reduce the risk of disease transmission and avoid unnecessary morbidity and mortality. During the last two decades, an unprecedented interest has been emerged for designing new TB diagnostic tools but still a reliable and accurate test for rapid diagnosis for all forms of TB, ie, pulmonary, extrapulmonary, TB-HIV coinfection and latent TB is lacking. We developed immuno-PCR (I-PCR) assay based on the detection of array of Mycobacterium tuberculosis antigens, ie, Ag85B (Antigen 85B, Rv1886c), ESAT-6 (early secreted antigenic target-6), cord factor (trehalose 6,6′-dimycolate) and PstS1 (phosphate-specific transporter lipoprotein, Rv0934),2–4 which certainly revealed better sensitivity than ELISA. ESAT-6 encoded by region of difference (RD1) is M. tuberculosis complex specific and is considered as a potential biomarker for the diagnosis of TB by ELISA and I-PCR, either alone or in combination with other RD1 and RD2 antigens such as CFP-10 (culture filtrate protein-10, Rv3874) and MPT64 (mycobacterial protein from species tuberculosis-64, Rv1980c).5,6 In fact, ESAT-6 is an immunodominant T-cell stimulatory antigen recognized by specific interferon-γ secreting T cells, which are present in higher numbers in TB patients with active infection than in uninfected individuals.7 Strikingly, I-PCR could detect a significantly higher number of samples from smear-negative pulmonary and paucibacillary extrapulmonary TB patients3,8 and thus showed superiority in sensitivity over the routine diagnostic methods, ie, smear, histopathology and nucleic acid amplification tests such as PCR. Coupling of detection antibodies with the reporter DNA is a crucial step of I-PCR, and various strategies are employed such as streptavidin-protein A, streptavidin-biotin setup, succinimidyl-4-(N-maleimidomethyl)cyclohexane-1-carboxylate (SMCC) system and recombinant phage particle where surface displayed single-chain variable fragments and phage DNAs themselves act as detection antibodies and DNA tag, respectively, in phage display-mediated I-PCR.8–10
Over the last two decades, nanobiotechnology has emerged as the most promising tool for clinical diagnostics.11 In particular, due to small size (1–100 nm), corresponding high surface-to-volume ratio and unusual target binding properties, nanoparticles (NPs) have been used as signal amplification tools.12,13 One such example is the NP-based bio-barcode amplification assay (BCA), which was initially introduced by Nam et al14 for protein and nucleic acid detection with ultrahigh sensitivity using multiple types of NPs, such as gold nanoparticles (GNPs), silica, iron oxide and magnetic particles (MPs). Hundreds of barcode DNAs released from each GNP could elicit 2–3 log amplification in detection sensitivity for each captured targets.15–17 Each barcode DNA could be interpreted by nucleic acid detection methods such as light scattering measurement, NP probe technology based on hybridization to microarray, colorimetric method or fluorescent approach for recognizing the targets.18–20 NP-based I-PCR assays have been developed for the rapid and efficient diagnosis of various viral and parasitic infections, including foot and mouth disease viruses, Hantaan virus nucleocapsid protein, respiratory syncytial virus and Giardia lamblia cysts, with improved detection limit of biomarkers;12,21–23 however, no report is yet available to detect mycobacterial antigens using NP-based I-PCR. GNPs have the ease of modification to provide various chemistries for attachment with the antibodies and DNA and can overcome the complicated preparation of antibody-DNA conjugates.12,24 The large ratio of DNA to antibody (usually 100–300:1) on the functionalized GNPs can further improve the sensitivity of I-PCR. Therefore, we aimed to design NP- based sensitive I-PCR comprising magnetic bead (MB)-coupled GNP-I-PCR assay (MB-GNP-I-PCR) for detection of purified M. tuberculosis ESAT-6.
Materials and methods
Materials
Purified recombinant ESAT-6 (NR-14868), rabbit anti-ESAT-6 polyclonal antibody (pAb, NR-13803) and guinea pig anti-M. tuberculosis CDC1551 pAb (NR-13818) were received as generous gifts from BEI Resources, ATCC (Manassas, VA, USA). Goat anti-rabbit IgG and goat anti-rabbit IgG-alkaline phosphatase were purchased from Merck (Bengaluru, India). Bovine serum albumin (BSA), sulfo-SMCC and 1,4-dithiothreitol (DTT) were procured from Merck KGaA (Darmstadt, Germany). Trisodium citrate dihydrate and gold chloride (chloroauric acid) were obtained from Finar Pvt Ltd (Ahmedabad, India). Sodium chloride, Tween-20 and hydroquinone were purchased from Hi-Media Laboratories (Mumbai, India). MagnaBind Amine Derivated Beads were procured from Thermo Fisher Scientific (Waltham, MA, USA). NAP-5 columns were purchased from GE Healthcare UK Ltd (Little Chalfont, UK).
Oligonucleotides and primer sequences
Oligonucleotides and primer sequences used in this work are shown in Table 1.
 | Table 1 Oligonucleotides and primer sequences for MB-GNP-I-PCR assay |
Preparation of GNPs
GNPs (~20 nm size) were prepared as described previously25,26 with slight modifications. Briefly, 0.5 mL of 200 mM gold chloride solution was added to a conical flask (250 mL) containing 99.5 mL of MilliQ water, which was heated with stirring. When gold chloride solution started boiling, 10 mL of 38.8 mM trisodium citrate dihydrate was quickly added followed by continuous vigorous stirring. The change in color from pale yellow to deep red wine in a minute indicated the formation of colloidal GNPs. The heating was turned off but the stirring continued till the solution reached room temperature (RT). GNPs were collected in a clean glass container and stored at RT.
GNPs of ~60 nm size were formed by seed-mediated synthesis as detailed by Kumar et al.27 Briefly, 2 mL of ~20 nm GNPs (prepared by the method of Turkevich et al25) was mixed with 1 mL of 34 μM trisodium citrate and 1 mL of 25.4 μM gold chloride solution at RT (~18°C). To this mixture, 0.5 mL of 22.7 μM hydroquinone was added and the final volume was made to 100 mL using distilled water and by continuously stirring at RT for 1 hour.
Characterization of GNPs and MBs
To verify the preparation of GNPs by chemical reduction process, the colloidal solution was scanned between 400 and 700 nm in an ultraviolet-visible (UV-vis) absorption spectrophotometer (UV-1800; Shimadzu Corporation, Kyoto, Japan) and the optical properties were recorded as detailed by Potůčková et al.28
The size and shape of the GNPs and MBs were analyzed using 200 kV transmission electron microscope (TEM; JEOL make TEM JEM-2100F, Tokyo, Japan) and the mean particle size was determined using Image Tool Software (Multipoint image database software for grain and particle analysis; Dietermann & Heuser Solution GmbH, Greifenstein, Germany). The surface morphologies of GNPs and MBs were analyzed under high vacuum using 20 kV scanning electron microscope (SEM; Carl Zeiss E®AG-EVO®40 SEM; Oberkochen, Germany) and the elemental composition was determined by energy-dispersive X-ray spectroscopy (EDS; E-sprit 1.8 X-ray microanalysis, Quantax 200, Bruker Nano GmbH, Berlin, Germany). Samples were prepared for electron microscopy by placing few drops of GNPs/MBs on a copper grid and subsequently dried before imaging.
Functionalization of GNPs with antibody and DNA
About 1 nM solution of thiolated DNA sequence was used as capture DNA for hybridization with signal DNA. The disulfide bond in thiolated DNA was cleaved by 100 mM DTT and 0.1 M phosphate buffer (pH 8.3) by incubating at RT for 30 minutes. The activated thiolated DNA sequence was desalted and purified using centrifugal filters.13 The purified capture DNA was stored at −20°C until further use.
Functionalization of GNPs (~20 nm) with antibodies and DNA was performed according to previously described method by Perez et al12,13 with little modifications. GNPs were used at a concentration of ~1×1011 particles/mL in 0.1 mM phosphate buffer and pH was adjusted to 9.3 with 1 M NaOH. To it, 0.5 mL of rabbit anti-ESAT-6 pAb (1:500) was added and incubated at RT for 45 minutes with gentle shaking. Then, 100 μL of activated capture DNA was added and incubated at RT for 30 minutes again with gentle shaking. Salt-aging procedure was then followed by adding 10 μL of 10% Tween-20 and 40 μL of 2 M NaCl in 10 mM PBS. Subsequently, 50 μL of 10% BSA solution was added and incubated at RT for 30 minutes to stabilize the particles followed by cold centrifugation at 12,000 rpm for 15 minutes. Supernatant was removed and 0.5 mL of PBS was added to resuspend the particles. This was followed by the addition of 100 μL of 5 pM signal DNA at 37°C for 1 hour to allow hybridization with the capture DNA. The unbound signal DNA was removed by centrifugation at 12,000 rpm for 15 minutes. After thorough washing, functionalized GNPs were resuspended in PBS and stored at 4°C until further use.
Coupling of capture antibody to MBs
Sulfo-SMCC was used to activate the MBs.12 Briefly, 300 μL of coupling buffer was added to 200 μL of MB solution (~50 mg/mL) in a microcentrifuge tube. External magnetic field was applied to pull MBs to the side of a microcentrifuge tube. The supernatant was removed and MBs were resuspended in coupling buffer. After washing with PBST, 50 μL of 1 mM sulfo-SMCC was added to the MBs. The solution was mixed and incubated at RT for 1 hour. MBs were washed to remove excess sulfo-SMCC. The activation of MBs and antibody reduction were performed simultaneously at the same time. For antibody reduction, 500 μL of guinea pig anti-M. tuberculosis pAb (1:500) was mixed with 500 μL of 40 mM DTT and incubated at RT for 1 hour. The reduced antibody was purified from DTT using a NAP-5 column. Equal parts of purified reduced antibody and the activated MBs were mixed in a microcentrifuge tube and incubated at RT for 1 hour for coupling reaction. The reaction was stopped by adding 10 μL of 100 mM β-mercaptoethanol followed by incubation at RT for 1 hour. After washing, MBs were resuspended in 500 μL PBS and stored at 4°C until further use.
Characterization of functionalized GNPs and MBs coupled with antibodies
UV-vis absorption spectroscopy
The optical properties of the GNPs and functionalized GNPs were monitored using UV-vis absorption spectroscopy. The absorbance maxima of GNPs and functionalized GNPs were recorded after scanning between 400 and 700 nm.28
Transmission electron microscope
Functionalized GNPs and MBs coupled with antibodies were negatively stained with uranyl acetate and subsequently dried on a copper grid for assessing their size. TEM analysis of functionalized GNPs was performed at a magnification of 200 KX, whereas analysis of MBs coupled with antibodies was carried out at 3 KX magnification. To further validate the attachment of antibodies with GNPs, 20 nm GNPs were conjugated with rabbit anti-ESAT-6 pAb (1:500) followed by incubation with secondary goat anti-rabbit IgG (1:500) conjugated with 60 nm GNPs. After washing, GNPs were analyzed by TEM. Controls included 20 nm GNPs conjugated with 1% BSA followed by incubation at RT for 1 hour with 60 nm GNPs coupled with secondary goat anti-rabbit IgG as well as 20 nm GNPs (with no pAb) and 60 nm GNPs (with no secondary goat anti-rabbit IgG). In addition, another control was run with 20 nm GNPs (with rabbit anti-ESAT-6 pAb) incubated with 60 nm GNPs (with no secondary goat anti-rabbit IgG).
ELISA
Indirect ELISA was performed as described by Singh et al29 with little modifications to validate the conjugation of GNPs with rabbit anti-ESAT-6 pAb in functionalized GNPs. One hundred microliters of purified ESAT-6 (1 μg/mL in coating buffer) was coated in a microtitre plate by incubating overnight at 4°C followed by washing with PBS containing 0.05% Tween-20 (PBST) and the addition of 200 μL of blocking buffer (5% BSA in PBS) at RT for 2 hours. After washing, 100 μL of functionalized GNPs (1:50) coupled with rabbit anti-ESAT-6 pAb was added and incubated at RT for 1 hour. After thorough washing with PBST, 100 μL of goat anti-rabbit IgG alkaline phosphatase (1:1,000) was added and the plate was incubated at RT for 1 hour, which was followed by the addition of 100 μL of p-nitrophenyl phosphate substrate solution. The plate was incubated in the dark at RT for 20 minutes. The appearance of color was monitored by taking optical density (OD) at 405 nm in an ELISA reader. Controls included for the experiment were positive control (GNPs coupled with rabbit anti-ESAT-6 pAb) and negative controls, ie, 1) ESAT-6 coated + unbound GNPs (without anti-ESAT-6 pAb); 2) no ESAT-6 coated + functionalized GNPs; 3) no ESAT-6 coated + GNPs (with anti-ESAT-6 pAb); and 4) no ESAT-6 coated + GNPs (alone) were run to ensure the validation of functionalized GNPs. In addition, ELISA-positive control (no GNPs, ESAT-6-coated, rabbit anti-ESAT-6 pAb) and ELISA-negative control (no GNPs, ESAT-6 coated, no rabbit anti-ESAT-6 pAb) were included.
PCR
PCR was performed to validate the conjugation of capture DNA (partially complementary with the signal DNA) with the functionalized GNPs. Robostrip wells were coated with 100 μL of purified ESAT-6 (1 μg/mL). After washing, 100 μL of functionalized GNPs (1:50) was added and incubated at RT for 1 hour. Again after washing, the unbound DNA (capture/signal) was removed by the addition of 100 μL of MilliQ water followed by heating at 95°C for 15 minutes to release the signal DNA, which was amplified by PCR using primers for signal DNA. The presence of a 76 bp product in 3.5% agarose gel electrophoresis revealed the attachment of signal DNA on functionalized GNPs. PCR-positive control (signal DNA), PCR-negative controls (no template DNA; PCR grade water only) and I-PCR-negative controls (ESAT-6 + GNPs coupled with rabbit anti-ESAT-6 pAb; ESAT-6 + signal DNA; no ESAT-6 + functionalized GNPs; no ESAT-6 + GNPs coupled with anti-ESAT-6 pAb; no ESAT-6 + signal DNA) were also included to ensure the validation of coupling of signal DNA in functionalized GNPs.
Magneto-ELISA
To validate the coupling of capture antibodies with MBs, magneto-ELISA was performed in a microcentrifuge tube as detailed by Wacker et al30 with some modifications. MBs were coupled with 100 μL of guinea pig anti-M. tuberculosis pAb (1:1,000), which were washed with PBST using an external magnetic field followed by the subsequent addition of 200 μL of 5% BSA solution, 100 μL each of purified ESAT-6 (100 ng/mL), rabbit anti-ESAT-6 pAb (1:1,000) and goat anti-rabbit IgG alkaline phosphatase (1:1,000) after thorough washing at each step using an external magnetic field. Presence of color was observed after the addition of p-nitrophenyl phosphate substrate solution and the OD was taken at 405 nm in ELISA reader. Controls included 1) no capture antibody, 2) no ESAT-6 coated and 3) no ESAT-6, no capture antibody.
MB-GNP-I-PCR for purified ESAT-6 detection
The method of Perez et al12 was followed with little modifications. Fifty microliters of MBs coupled with guinea pig anti-M. tuberculosis pAb (1:1,000, capture antibody) was taken in a microcentrifuge tube. To it, 50 μL of ten-fold serial dilution of ESAT-6 ranging from 10 mg/mL to 1 attogram (ag)/mL in PBS were added followed by washing and the addition of 200 μL of 5% BSA solution. The reaction mixtures were incubated at RT for 1 hour with constant shaking to avoid the settling of the MBs. After incubation at RT for 1 hour, MBs were washed with PBST by applying external magnetic field and the unbound antigen was removed. Then, 50 μL of functionalized GNPs (of ~20 nm, 1×1011 particles/mL) was added and incubated at RT for 1 hour with constant shaking. MBs were washed, resuspended in 100 μL of MilliQ water and heated at 95°C for 10 minutes. External magnetic field was applied and 10 μL of supernatant was taken in an Eppendorf tube for PCR analysis. Both PCR-positive control (signal DNA at 1 ng/mL) and PCR-negative controls (no template DNA, only PCR grade water) were included. To demonstrate the specificity of functionalized GNPs coupled with anti-ESAT-6 pAb, different RD antigens such as CFP-10, ESAT-6, CFP-21 and MPT64 as well as Ag85B (100 ng/mL) were added separately (in triplicates) in microcentrifuge tubes and were subjected to MB-GNP-I-PCR assay.
This study involved human subjects; therefore, prior approval was obtained from the Institute Human Ethics Committee of MD University, Rohtak (IHEC/2017/108). Saliva samples (n=6) were collected from healthy individuals after taking their prior written informed consent and were pretested at various dilutions for the absence of ESAT-6. Samples were spiked with the purified recombinant ESAT-6 (at a concentration of 1 μg/mL) and the detection limit was determined by I-PCR and ELISA to study the sample matrix effect. MB-GNP-I-PCR assay has been schematically represented in Figure 1.
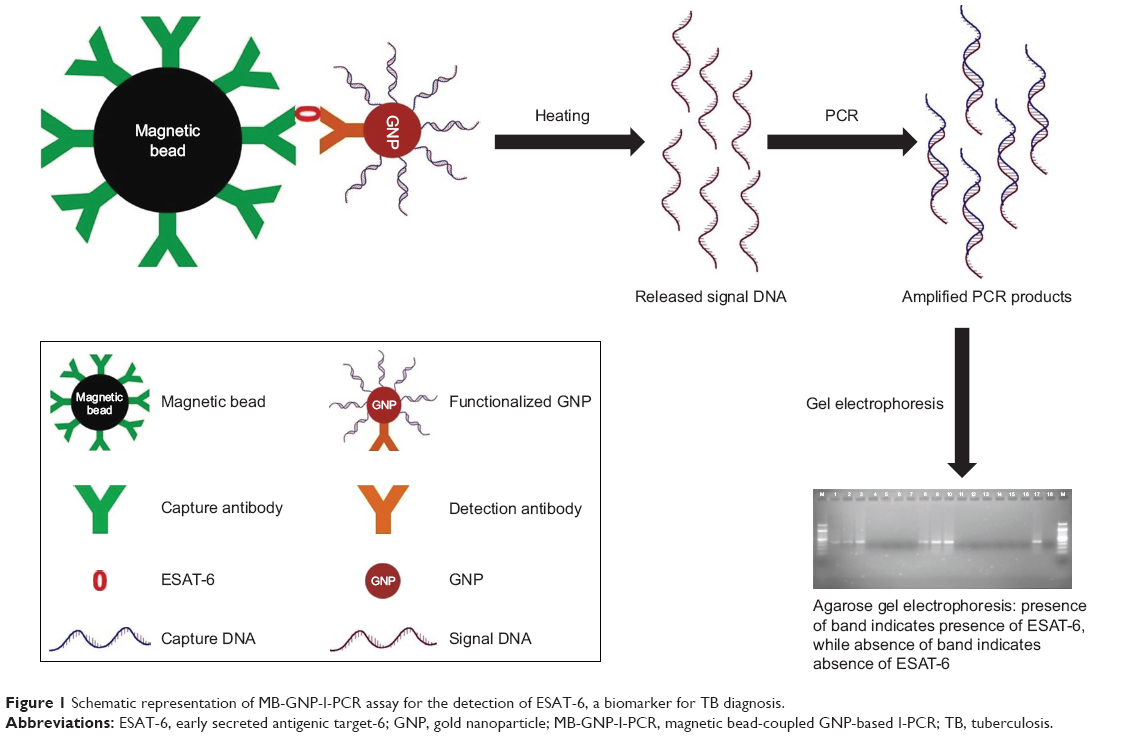 | Figure 1 Schematic representation of MB-GNP-I-PCR assay for the detection of ESAT-6, a biomarker for TB diagnosis. |
Statistical analysis
Experiments were performed in triplicates for the ELISA and magneto-ELISA, and the average of three independent experiments along with SD was calculated for each assay. The Student’s t-test and P-value were calculated to show the significance and reproducibility of experiments. P-value <0.05 was considered significant. All analyses were carried out using SPSS for Windows statistical software (SPSS Inc., Chicago, IL, USA).
Results and discussion
The nonspecific binding occurring with the use of classical solid format in I-PCR can be circumvented by the use of NPs-based assay, which could also lead to automated one-step assay.10,24 The large surface area of GNPs allows a higher and faster interaction between the capture antibody (coupled with MBs) and the respective antigen in comparison with robostrip wells used in conventional I-PCR.9,24 In this study, we characterized the functionalized GNPs and designed a modified BCA assay, ie, MB-GNP-I-PCR assay for the detection of M. tuberculosis purified ESAT-6 protein.
The formation of GNPs was confirmed by UV-vis absorption spectroscopy, where the presence of a single peak at 524 and 539 nm was related to the characteristic surface plasmon resonance (SPR) band of GNPs, which indicated that the GNPs were spherical and of different sizes, ie, ~20 and ~60 nm, respectively (Figure 2A and B). This is in agreement with their previous study by Baptista et al,31 where red color was observed for non-aggregated GNP solution because of SPR at an absorbance peak of ~526 nm. Also, the solution remained red after the addition of thiolated oligonucleotides to GNPs.31 TEM images revealed synthesis of spherical, slightly polydispersed GNPs of average size of ~20 and ~60 nm, respectively (Figures 2D and 4A). SEM images also exhibited spherical, slightly polydispersed and aggregated forms of GNPs with smooth surfaces (Figure 2E), while EDS spectrum showed the presence of strong peak at 2.12 keV, typical for crystalline GNPs, and no other significant peak belonging to impurities was observed throughout the EDS spectrum (Figure 2F), thus indicating the formation of impurity free GNPs. Similar EDS peaks were also observed earlier by Anuradha et al32 for GNPs synthesized using Pistia stratiotes. SEM analysis of commercially obtained MBs showed irregular shapes varying from spherical to rods. TEM micrograph of MBs also confirmed their irregular shapes with sizes varying between 1 and 6 μm (Figure 2G and H).
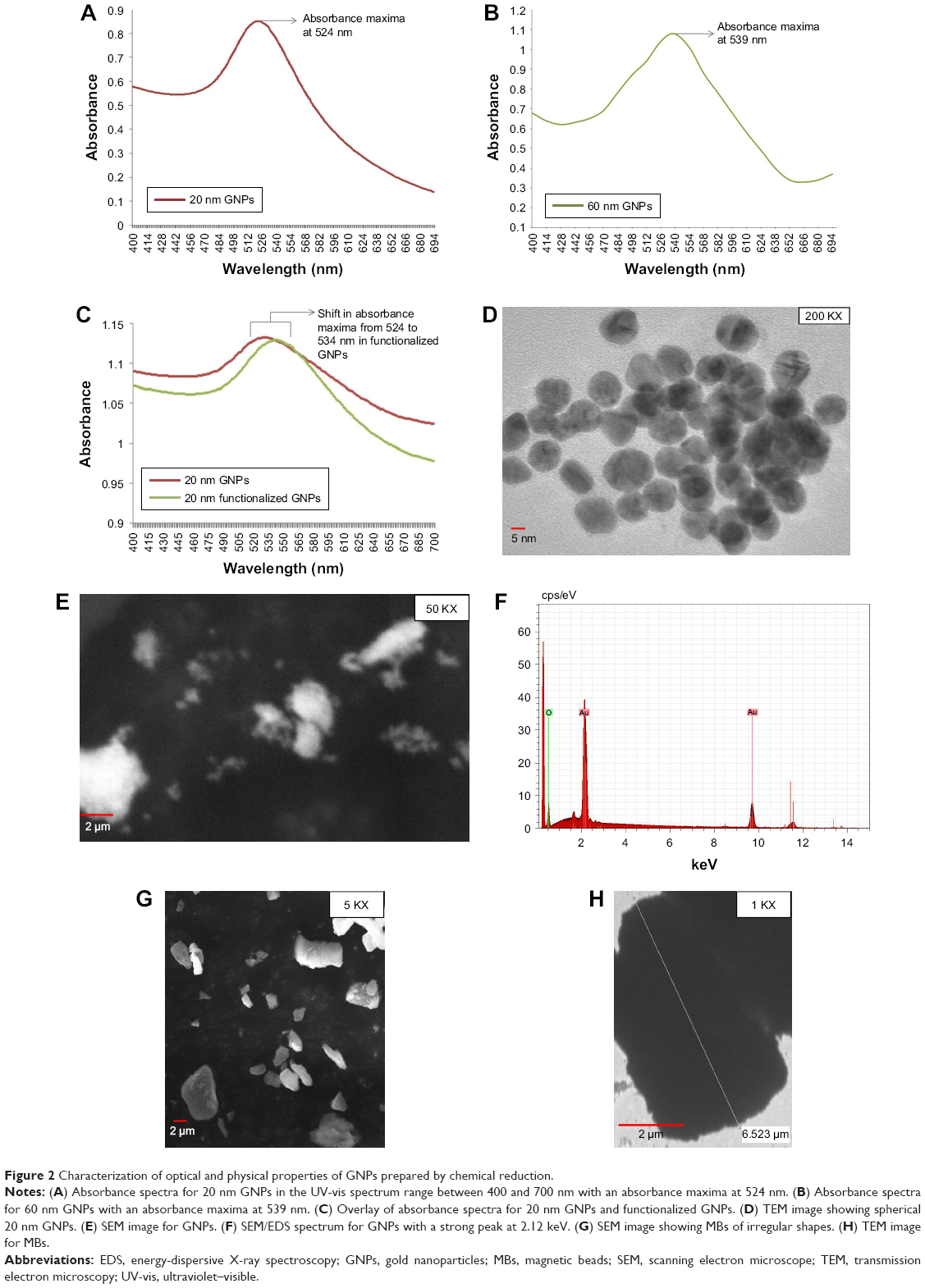 | Figure 2 Characterization of optical and physical properties of GNPs prepared by chemical reduction. |
Any change in size and shape of NPs could yield change and displacement of SPR, which in turn might change their appearance, color and absorption properties.33 In this study, unconjugated GNPs exhibited the absorption peak at 524 nm, whereas functionalized GNPs revealed a shift in the absorption maxima from 524 to 534 nm (red shift), thus confirming the attachment of antibodies with the GNPs (Figure 2C). In a similar manner, a change to higher wavelength values by 5–10 nm was also observed by Potůčková et al28 after functionalization of GNPs with antibodies and oligonucleotides. Both functionalized GNPs and MBs coupled with antibodies showed no significant change in their shape by TEM analysis (Figure 3). To further validate the attachment of antibodies with GNPs, 20 nm GNPs were conjugated with primary rabbit anti-ESAT-6 pAb followed by incubation with secondary goat anti-rabbit IgG conjugated with 60 nm GNPs and examined by TEM. Notably, 20 and 60 nm GNPs were found to be in very close proximity appearing partially like a rosette, (Figure 4A–C) probably due to binding of primary antibodies with the secondary antibodies, thus confirming the attachment of detection antibodies with GNPs. However, no attachment was observed between 20 and 60 nm GNPs in controls (Figure 4D–F), thus indicating that the attachment was due to the specific binding of primary antibodies with the secondary antibodies and not due to aggregation of GNPs or other nonspecific binding. Similar binding of primary antibodies (coupled to 30 nm GNPs) with the secondary antibodies (coupled to 5 nm GNPs) for the detection of IL-3 and stem cell factor was also demonstrated by TEM analysis.28 In fact, such binding resulted in the formation of proper rosettes comprising 30 nm GNPs surrounded by few 5 nm GNPs, thus validating the coupling of antibodies with GNPs.
 | Figure 3 Validation of functionalized GNPs and MBs coupled with antibodies. |
 | Figure 4 (A) TEM visualization of 60 nm GNPs; (B, C) 20 nm GNPs with rabbit anti-ESAT-6 pAb were incubated with 60 nm GNPs coupled with secondary goat anti-rabbit IgG for 1 hour at RT. After washing, GNPs were observed by TEM; (D) control, 20 nm GNPs were conjugated with 1% BSA, followed by conjugation with 60 nm GNPs coupled with secondary goat anti-rabbit IgG; (E) control, 20 nm GNPs (with no rabbit anti-ESAT-6 pAb) and 60 nm GNPs (with no secondary goat anti-rabbit IgG); (F) control, 20 nm GNPs (with rabbit anti-ESAT-6 pAb) and 60 nm GNPs (with no secondary goat anti-rabbit IgG). Red arrows indicate large GNPs (60 nm) and blue arrows indicate small GNPs (20 nm). |
In this study, coupling of antibodies and thiolated oligonucleotides to functionalized GNPs was also confirmed by ELISA and PCR. In comparison with unbound GNPs, functionalized GNPs showed a visible color reaction with ELISA (Figure 5A). The mean OD of functionalized GNPs (0.817±0.0021, P<0.001) coupled with anti-ESAT-6 pAb was found to be significantly higher than the OD of the GNPs with no coupled anti-ESAT-6 pAb (0.0743±0.0006; Figure 5B), which confirmed the specific attachment of antibodies to the functionalized GNPs. In addition, PCR also validated the conjugation of signal DNA and revealed the presence of a specific 76 bp product in the functionalized GNPs, while no such product was observed in controls (Figure 5C). Notably, the conjugation of capture antibodies with the MBs was confirmed by a color reaction and OD measurement by magneto-ELISA (Figure 6A and B). The antibody conjugated MBs showed a significantly higher mean OD (0.791±0.021, P<0.001) than the MBs without capture antibodies (mean OD 0.057±0.0004; Figure 6B). Use of magneto-ELISA has also been previously reported for confirming the coupling of MBs with the antibodies for detection of hepatitis B surface antigen.30
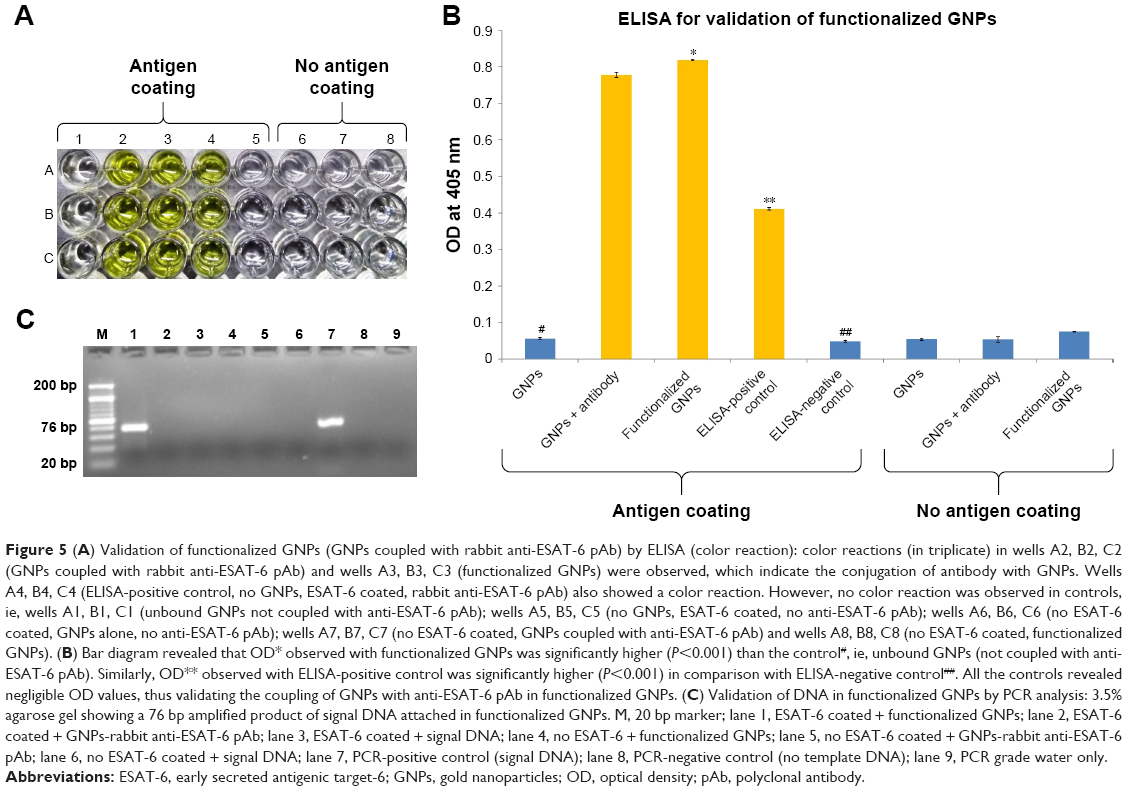 | Figure 5 (A) Validation of functionalized GNPs (GNPs coupled with rabbit anti-ESAT-6 pAb) by ELISA (color reaction): color reactions (in triplicate) in wells A2, B2, C2 (GNPs coupled with rabbit anti-ESAT-6 pAb) and wells A3, B3, C3 (functionalized GNPs) were observed, which indicate the conjugation of antibody with GNPs. Wells A4, B4, C4 (ELISA-positive control, no GNPs, ESAT-6 coated, rabbit anti-ESAT-6 pAb) also showed a color reaction. However, no color reaction was observed in controls, ie, wells A1, B1, C1 (unbound GNPs not coupled with anti-ESAT-6 pAb); wells A5, B5, C5 (no GNPs, ESAT-6 coated, no anti-ESAT-6 pAb); wells A6, B6, C6 (no ESAT-6 coated, GNPs alone, no anti-ESAT-6 pAb); wells A7, B7, C7 (no ESAT-6 coated, GNPs coupled with anti-ESAT-6 pAb) and wells A8, B8, C8 (no ESAT-6 coated, functionalized GNPs). (B) Bar diagram revealed that OD* observed with functionalized GNPs was significantly higher (P<0.001) than the control#, ie, unbound GNPs (not coupled with anti-ESAT-6 pAb). Similarly, OD** observed with ELISA-positive control was significantly higher (P<0.001) in comparison with ELISA-negative control##. All the controls revealed negligible OD values, thus validating the coupling of GNPs with anti-ESAT-6 pAb in functionalized GNPs. (C) Validation of DNA in functionalized GNPs by PCR analysis: 3.5% agarose gel showing a 76 bp amplified product of signal DNA attached in functionalized GNPs. M, 20 bp marker; lane 1, ESAT-6 coated + functionalized GNPs; lane 2, ESAT-6 coated + GNPs-rabbit anti-ESAT-6 pAb; lane 3, ESAT-6 coated + signal DNA; lane 4, no ESAT-6 + functionalized GNPs; lane 5, no ESAT-6 coated + GNPs-rabbit anti-ESAT-6 pAb; lane 6, no ESAT-6 coated + signal DNA; lane 7, PCR-positive control (signal DNA); lane 8, PCR-negative control (no template DNA); lane 9, PCR grade water only. |
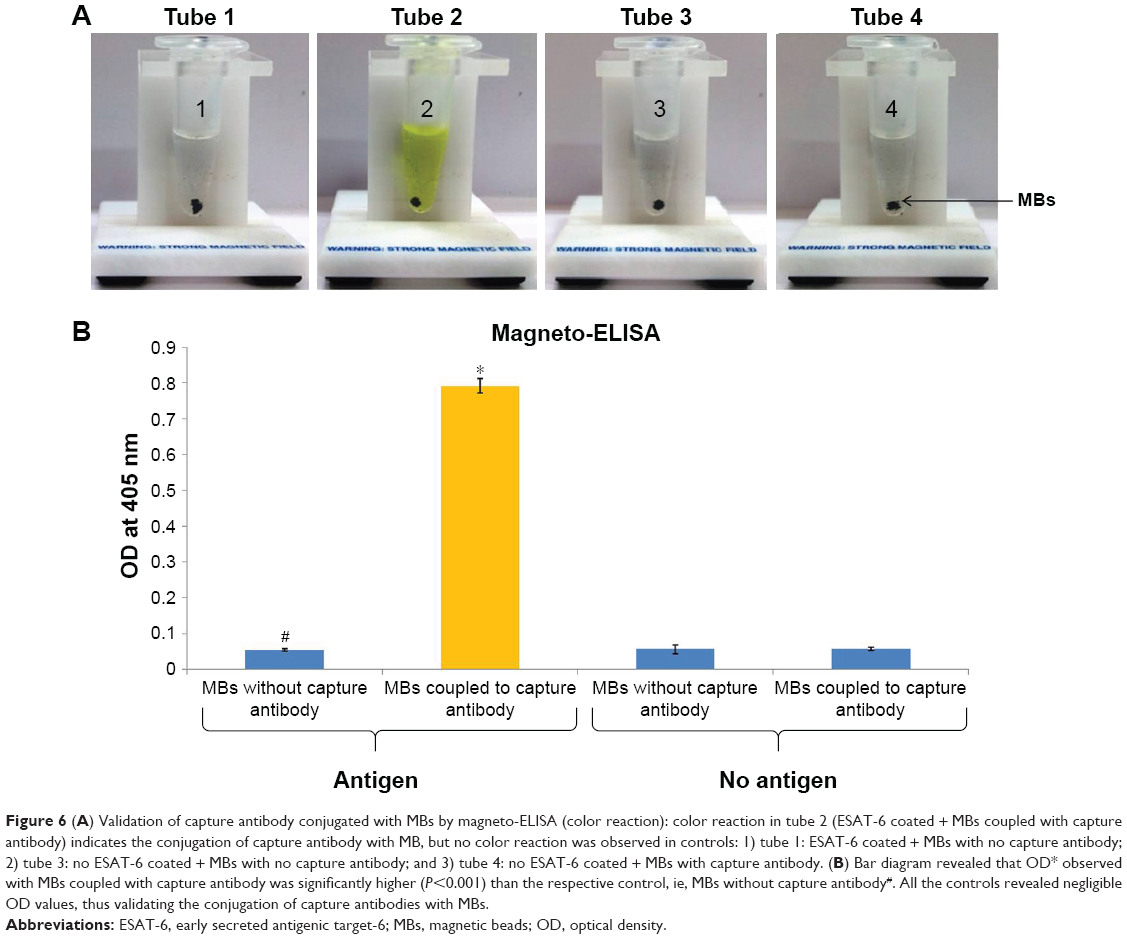 | Figure 6 (A) Validation of capture antibody conjugated with MBs by magneto-ELISA (color reaction): color reaction in tube 2 (ESAT-6 coated + MBs coupled with capture antibody) indicates the conjugation of capture antibody with MB, but no color reaction was observed in controls: 1) tube 1: ESAT-6 coated + MBs with no capture antibody; 2) tube 3: no ESAT-6 coated + MBs with no capture antibody; and 3) tube 4: no ESAT-6 coated + MBs with capture antibody. (B) Bar diagram revealed that OD* observed with MBs coupled with capture antibody was significantly higher (P<0.001) than the respective control, ie, MBs without capture antibody#. All the controls revealed negligible OD values, thus validating the conjugation of capture antibodies with MBs. |
Our MB-GNP-I-PCR assay revealed limit of detection (LOD) of 10 fg/mL for M. tuberculosis purified ESAT-6 (Figure 7A), while ELISA could detect up to 1 ng/mL for the same, thus demonstrating that MB-GNP-I-PCR revealed 105-fold higher sensitivity than ELISA. These results are in agreement with the previous reports for the detection of prostate-specific antigen (LOD of 10 fg/mL) by BCA assay using NPs.34 In a similar manner, the LOD of 1 fg/mL for hepatitis C virus core antigen by MB-GNP-I-PCR and 10 fg/mL for Hantaan virus nucleocapsid protein in serum samples as well as foot and mouth disease viruses particles in BHK-21 cells by GNP-I-PCR assay have been reported.21,22,35 Furthermore, functionalized GNPs coupled with anti-ESAT-6 pAb employed in the assay did not cross-react with other RD antigens such as purified CFP-10, CFP-21 (Rv1984c) and MPT64 as well as Ag85B (Figure 7B), which demonstrated the specificity of ESAT-6 detection with MB-GNP-I-PCR. However, we could not calculate the recovery of the MB-GNP-I-PCR as it is the only qualitative and affirmative test indicating either the presence or the absence of ESAT-6 marker, but the theoretical recovery appears to be 100% since the LOD of ESAT-6 remained the same (10 fg/mL) in both PBS and spiked saliva samples (Figure 7A and C). It is intriguing that the LOD of purified ESAT-6 by MB-GNP-I-PCR was ten-fold lesser than the conventional I-PCR performed in robostrip wells3 (solid format). This could be due to aggregation of NPs in aqueous system resulting from nonspecific binding of functionalized GNPs to the solid phase or the salts present in reagents might be altering the optical, electrical and mechanical properties of NPs.14,36,37 The finding that the saliva samples of healthy individuals spiked with purified ESAT-6 displayed the same LOD by MB-GNP-I-PCR as that observed in PBS suggests that the reduced background signals were associated with MB-GNP-I-PCR, thus showing no sample matrix effect. This could be explained by the liquid format displayed by MB-GNP-I-PCR, which permits a thorough washing of the captured antigen and MBs/GNPs, thus leading to removal of unbound antigens/antibodies, which in turn decreases nonspecific binding and leads to diminished background signals.12,24 However, an LOD of 10 ng/mL was observed by ELISA in spiked saliva samples, thereby showing a sample matrix effect. Similarly, the conventional I-PCR in robostrip wells (solid format) also revealed a sample matrix effect as higher LOD (100-fold) of 100 fg/mL was observed for Ag85B detection in spiked saliva samples.2
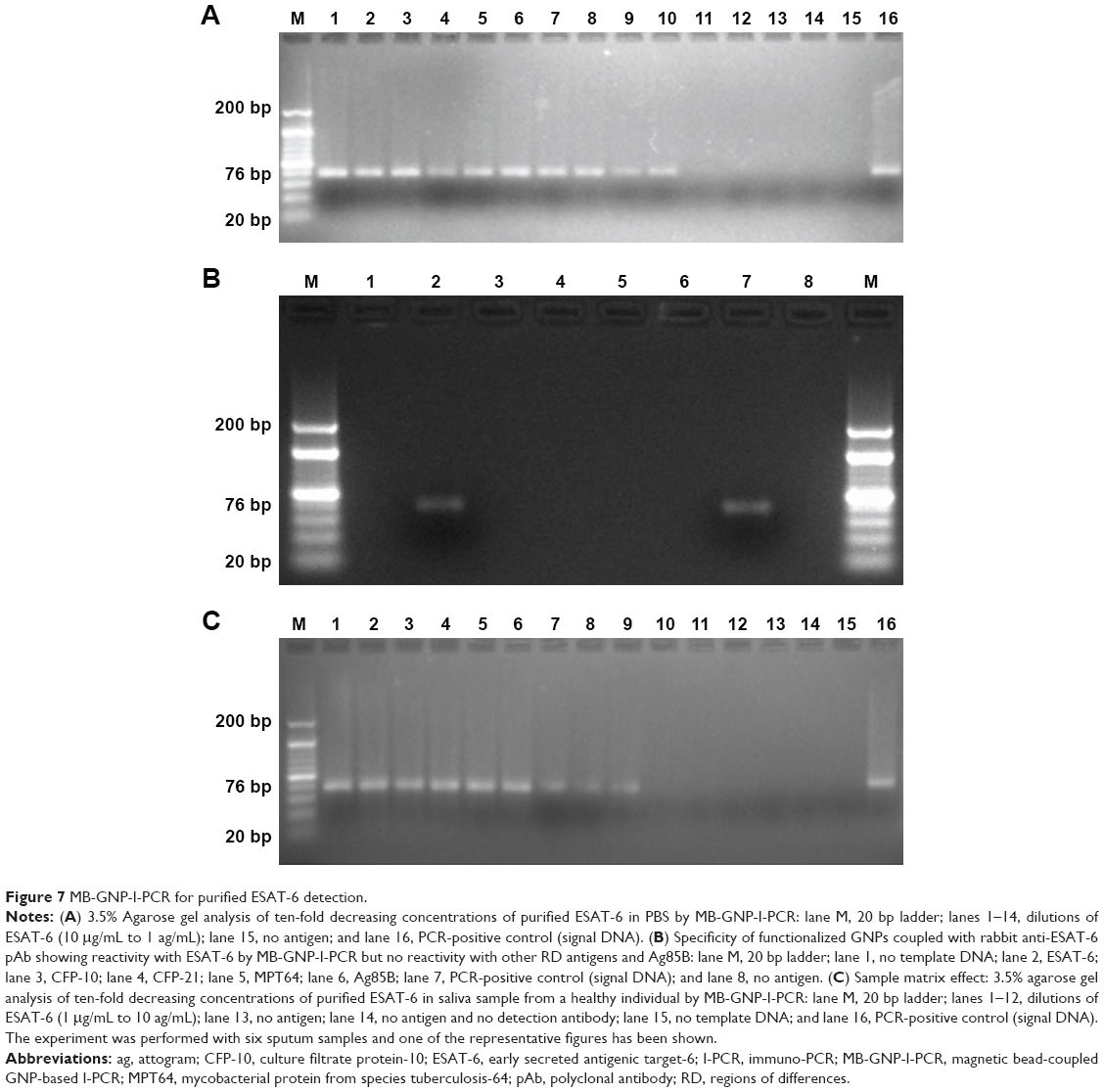 | Figure 7 MB-GNP-I-PCR for purified ESAT-6 detection. |
In contrast to synthetic MBs, Wacker et al30 employed biogenic magnetosome particles isolated from Magnetospirillum gryphiswaldense for designing magneto I-PCR for the detection of recombinant HBsAg in human serum with an LOD of 320 pg/mL, whereas we observed an LOD of 10 fg/mL in human saliva for the detection of recombinant ESAT-6. The use of biogenic magnetosomes has been reported to possess some advantages over the synthetic MBs as better signal-to-noise ratios and lesser SDs are observed owing to their smaller size, monodispersity and higher magnetization.30,38 However, the use of biogenic magnetosomes also has some disadvantages due to limited stability of their biological membranes and limited tolerance toward the detergents, high salt concentrations and repeated centrifugation.30,38 Similar to our study, Barletta et al39 employed synthetic MBs to design immuno-MB real-time I-PCR assay for the detection of HIV p24 protein and could detect <1 HIV-1 virion in plasma samples, which may have important implications to study HIV pathogenesis and anti-retroviral therapy. HIV-1 p24 Gag protein has also been detected in plasma samples of HIV-1-infected subjects with high accuracy by MB-GNP-I-PCR.40 Interestingly, magnetic gold particles have been used to design I-PCR for the detection of Giardia lamblia cysts with the LOD of approximately five cysts,23 thus proposing its utility for the fast screening of samples from drinking water, lakes, swimming pools, etc.
Similar to this study, Perez et al12 designed MB-GNP real-time I-PCR assay, known as NP-amplified I-PCR assay with dual amplification for the detection of respiratory syncytial virus surface protein in infected Hep-2 cell extracts with the LOD of 4.1 PFU/mL, an improvement of >1,000-fold and 4-fold over ELISA and real-time PCR, respectively. By using the biotinylated DNA-streptavidin nanostructures, magneto I-PCR was found to enhance the valency of the antibody binding, which in turn increased the sensitivity of assay30,41 but such studies used an average DNA/complex ratio of only 5:1 and further increased DNA ratios revealed an instability of the streptavidin conjugates. However, several-fold higher NP valence amplification could be achieved prior to tag amplification by MB-GNP-I-PCR assay.21 The first step of signal amplification is achieved by using multivalent GNPs to release multiple signal DNA molecules/antibody binding events.12 After signal DNA is released, PCR provides a second level of amplification, which shows several-fold enhanced sensitivity for the detection of antigen (eg, ESAT-6), in comparison with ELISA.
Conclusion
MB-GNP-I-PCR (liquid format) has displayed improved technology as the conjugation of GNPs with the antibodies and DNA in functionalized GNPs is relatively simple than the conventional I-PCR (solid format) based on streptavidin–biotin or SMCC system. Moreover, the assay has lesser incubation/wash steps, which reduces the overall duration. Hence, the assay leads to diminished background signals with no sample matrix effect and also the reliable detection of purified ESAT-6 up to 10 fg/mL in spiked saliva samples. Further work is in progress to extend these findings for the detection of M. tuberculosis antigens by MB-GNP-I-PCR in body fluids of TB patients.
Acknowledgments
The financial support by the Department of Biotechnology (DBT), Government of India, New Delhi (BT/PR8054/MED/29/710/2013), University Grant Commission (UGC) (JNU-UPOEII; Government of India, New Delhi) and Department of Science and Technology (DST), Government of India, New Delhi (JNU-DST-PURSE) is gratefully acknowledged. B Dahiya acknowledges DBT (DBT/JRF/14/AL/243) for providing Junior/Senior Research Fellowship. The authors thank Suman Sharma for critically reading the manuscript. Part of the work of this study was incorporated in the abstract (available online)42 for the International Biotechnology and Pharmaceutical Industry Forum, organized by Clyto Conferences; however, the conference was cancelled and the research findings were not presented.
Disclosure
The authors report no conflicts of interest in this work.
References
WHO. Global tuberculosis control. WHO Report. Geneva: World Health Organization; 2017. | ||
Singh N, Sreenivas V, Gupta KB, et al. Diagnosis of pulmonary and extrapulmonary tuberculosis based on detection of mycobacterial antigen 85B by immuno-PCR. Diagn Microbiol Infect Dis. 2015;83(4):359–364. | ||
Mehta PK, Singh N, Dharra R, et al. Diagnosis of tuberculosis based on the detection of a cocktail of mycobacterial antigen 85B, ESAT-6 and cord factor by immuno-PCR. J Microbiol Methods. 2016;127:24–27. | ||
Sharma S, Raj A, Singh N, et al. Development of real-time immuno-PCR for the quantitative detection of mycobacterial PstS1 in tuberculosis patients. J Microbiol Methods. 2017;132:134–138. | ||
Kalra M, Khuller GK, Grover A, Behera D, Wanchu A, Verma I. Utility of a combination of RD1 and RD2 antigens as a diagnostic marker for tuberculosis. Diagn Microbiol Infect Dis. 2010;66(2):153–161. | ||
Mehta PK, Kalra M, Khuller GK, Behera D, Verma I. Development of an ultrasensitive polymerase chain reaction-amplified immunoassay based on mycobacterial RD antigens: implications for the serodiagnosis of tuberculosis. Diagn Microbiol Infect Dis. 2012;72(2):166–174. | ||
Hasan Z, Jamil B, Ashraf M, et al. Differential live Mycobacterium tuberculosis-, M. bovis BCG-, recombinant ESAT6-, and culture filtrate protein 10-induced immunity in tuberculosis. Clin Vaccine Immunol. 2009;16(7):991–998. | ||
Mehta PK, Dahiya B, Sharma S, et al. Immuno-PCR, a new technique for the serodiagnosis of tuberculosis. J Microbiol Methods. 2017;139:218–229. | ||
Mehta PK, Raj A, Singh NP, Khuller GK. Detection of potential microbial antigens by immuno-PCR (PCR-amplified immunoassay). J Med Microbiol. 2014;63(Pt 5):627–641. | ||
Assumpção AL, da Silva RC. Immuno-PCR in cancer and non-cancer related diseases: a review. Vet Q. 2016;36(2):63–70. | ||
Torres-Sangiao E, Holban AM, Gestal MC. Advanced nanobiomaterials, vaccines, diagnosis and treatment of infectious diseases. Molecules. 2016;21(7):1–21. | ||
Perez JW, Vargis EA, Russ PK, Haselton FR, Wright DW. Detection of respiratory syncytial virus using nanoparticle amplified immuno-polymerase chain reaction. Anal Biochem. 2011;410(1):141–148. | ||
Perez JW, Adams NM, Zimmerman GR, Haselton FR, Wright DW. Detecting respiratory syncytial virus using nanoparticle-amplified immuno-PCR. Methods Mol Biol. 2013;1026:93–110. | ||
Nam JM, Park SJ, Mirkin CA. Bio-barcodes based on oligonucleotide-modified nanoparticles. J Am Chem Soc. 2002;124(15):3820–3821. | ||
Nam JM, Thaxton CS, Mirkin CA. Nanoparticle-based bio-bar codes for the ultrasensitive detection of proteins. Science. 2003;301(5641):1884–1886. | ||
Nam JM, Stoeva SI, Mirkin CA. Bio-bar-code-based DNA detection with PCR-like sensitivity. J Am Chem Soc. 2004;126(19):5932–5933. | ||
Nam JM, Wise AR, Groves JT. Colorimetric bio-barcode amplification assay for cytokines. Anal Chem. 2005;77(21):6985–6988. | ||
Stoeva SI, Lee JS, Smith JE, Rosen ST, Mirkin CA. Multiplexed detection of protein cancer markers with biobarcoded nanoparticle probes. J Am Chem Soc. 2006;128(26):8378–8379. | ||
Tang S, Zhao J, Storhoff JJ, et al. Nanoparticle-based biobarcode amplification assay (BCA) for sensitive and early detection of human immunodeficiency type 1 capsid (p24) antigen. J Acquir Immune Defic Syndr. 2007;46(2):231–237. | ||
Oh BK, Nam JM, Lee SW, Mirkin CA. A fluorophore-based bio-barcode amplification assay for proteins. Small. 2006;2(1):103–108. | ||
Chen L, Wei H, Guo Y, Cui Z, Zhang Z, Zhang XE. Gold nanoparticle enhanced immuno-PCR for ultrasensitive detection of Hantaan virus nucleocapsid protein. J Immunol Methods. 2009;346(1–2):64–70. | ||
Ding YZ, Liu YS, Zhou JH, et al. A highly sensitive detection for foot-and-mouth disease virus by gold nanopariticle improved immuno-PCR. Virol J. 2011;8(1):148. | ||
Deng MJ, Ji XC, Xiao XZ, et al. Immuno-PCR for detection of Giardia lamblia cysts in water. J AOAC Int. 2014;97(2):561–566. | ||
Malou N, Raoult D. Immuno-PCR: a promising ultrasensitive diagnostic method to detect antigens and antibodies. Trends Microbiol. 2011;19(6):295–302. | ||
Turkevich J, Stevenson PC, Hillier J. A study of the nucleation and growth processes in the synthesis of colloidal gold. Discuss Faraday Soc. 1951;11:55–75. | ||
Liu J, Lu Y. Preparation of aptamer-linked gold nanoparticle purple aggregates for colorimetric sensing of analytes. Nat Protoc. 2006;1(1):246–252. | ||
Kumar D, Mutreja I, Sykes P. Seed mediated synthesis of highly mono-dispersed gold nanoparticles in the presence of hydroquinone. Nanotechnology. 2016;27(35):355601–355615. | ||
Potůčková L, Franko F, Bambousková M, Dráber P. Rapid and sensitive detection of cytokines using functionalized gold nanoparticle-based immuno-PCR, comparison with immuno-PCR and ELISA. J Immunol Methods. 2011;371(1–2):38–47. | ||
Singh N, Sreenivas V, Sheoran A, et al. Serodiagnostic potential of immuno-PCR using a cocktail of mycobacterial antigen 85B, ESAT-6 and cord factor in tuberculosis patients. J Microbiol Methods. 2016;120:56–64. | ||
Wacker R, Ceyhan B, Alhorn P, Schueler D, Lang C, Niemeyer CM. Magneto immuno-PCR: a novel immunoassay based on biogenic magnetosome nanoparticles. Biochem Biophys Res Commun. 2007;357(2):391–396. | ||
Baptista PV, Koziol-Montewka M, Paluch-Oles J, Doria G, Franco R. Gold-nanoparticle-probe-based assay for rapid and direct detection of Mycobacterium tuberculosis DNA in clinical samples. Clin Chem. 2006;52(7):1433–1434. | ||
Anuradha J, Abbasi T, Abbasi SA. An eco-friendly method of synthesizing gold nanoparticles using an otherwise worthless weed pistia (Pistia stratiotes L.). J Adv Res. 2015;6(5):711–720. | ||
Shevchenko E, Talapin D, Kornowski A, et al. Colloidal crystals of monodisperse FePt nanoparticles grown by a three-layer technique of controlled oversaturation. Adv Mater. 2002;14(4):287–290. | ||
Bao YP, Wei TF, Lefebvre PA, et al. Detection of protein analytes via nanoparticle-based bio bar code technology. Anal Chem. 2006;78(6):2055–2059. | ||
Yin HQ, Ji CF, Yang XQ, et al. An improved gold nanoparticle probe-based assay for HCV core antigen ultrasensitive detection. J Virol Methods. 2017;243:142–145. | ||
Lind K, Kubista M. Development and evaluation of three real-time immuno-PCR assemblages for quantification of PSA. J Immunol Methods. 2005;304(1–2):107–116. | ||
Niemeyer CM, Adler M, Wacker R. Detecting antigens by quantitative immuno-PCR. Nat Protoc. 2007;2(8):1918–1930. | ||
Ceyhan B, Alhorn P, Lang C, Schüler D, Niemeyer CM. Semisynthetic biogenic magnetosome nanoparticles for the detection of proteins and nucleic acids. Small. 2006;2(11):1251–1255. | ||
Barletta J, Bartolome A, Constantine NT. Immunomagnetic quantitative immuno-PCR for detection of less than one HIV-1 virion. J Virol Methods. 2009;157(2):122–132. | ||
Kim EY, Stanton J, Korber BT, et al. Detection of HIV-1 p24 Gag in plasma by a nanoparticle-based bio-barcode-amplification method. Nanomedicine. 2008;3(3):293–303. | ||
Niemeyer CM, Adler M, Pignataro B, et al. Self-assembly of DNA-streptavidin nanostructures and their use as reagents in immuno-PCR. Nucleic Acids Res. 1999;27(23):4553–4561. | ||
Mehta P. Immuno-PCR, a novel tool for an early diagnosis of tuberculosis [abstract]; 2017. Available from: https://www.clytoconferences.com/dr-promod-mehta. Accessed July 26 2017. |
 © 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.