Back to Archived Journals » Advances in Genomics and Genetics » Volume 5
Designing metabolic engineering strategies with genome-scale metabolic flux modeling
Authors Yen J, Tanniche I, Fisher A, Gillaspy G, Bevan D, Senger R
Received 2 September 2014
Accepted for publication 18 November 2014
Published 30 January 2015 Volume 2015:5 Pages 93—105
DOI https://doi.org/10.2147/AGG.S58494
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 4
Editor who approved publication: Dr John Martignetti
Jiun Y Yen,1,2 Imen Tanniche,1 Amanda K Fisher,1–3 Glenda E Gillaspy,2 David R Bevan,2,3 Ryan S Senger1
1Department of Biological Systems Engineering, 2Department of Biochemistry, 3Genomics, Bioinformatics, and Computational Biology Interdisciplinary Program, Virginia Tech, Blacksburg, VA, USA
Abstract: New in silico tools that make use of genome-scale metabolic flux modeling are improving the design of metabolic engineering strategies. This review highlights the latest developments in this area, explains the interface between these in silico tools and the experimental implementation tools of metabolic engineers, and provides a way forward so that in silico predictions can better mimic reality and more experimental methods can be considered in simulation studies. The several methodologies for solving genome-scale models (eg, flux balance analysis [FBA], parsimonious FBA, flux variability analysis, and minimization of metabolic adjustment) all have unique advantages and applications. There are two basic approaches to designing metabolic engineering strategies in silico, and both have demonstrated success in the literature. The first involves: 1) making a genetic manipulation in a model; 2) testing for improved performance through simulation; and 3) iterating the process. The second approach has been used in more recently designed in silico tools and involves: 1) comparing metabolic flux profiles of a wild-type and ideally engineered state and 2) designing engineering strategies based on the differences in these flux profiles. Improvements in genome-scale modeling are anticipated in areas such as the inclusion of all relevant cellular machinery, the ability to understand and anticipate the results of combinatorial enrichment experiments, and constructing dynamic and flexible biomass equations that can respond to environmental and genetic manipulations.
Keywords: genome-scale modeling, genome-scale modeling, flux balance analysis, flux variability analysis, minimization of metabolic adjustment, metabolic bottleneck, pathway optimization
A brief introduction to genome-scale metabolic flux modeling
A “genome-scale” metabolic flux model (GEM) consists of a network of biochemical reactions that is reconstructed based on the genomic sequence and annotation of a cell. Assuming a “steady-state” metabolism (ie, a snapshot of metabolism at one time point) is reached on a short time-scale, these reactions can be represented by a linear system of equations. Then, problems such as maximizing specific chemical production or growth can be solved efficiently by linear programming. GEMs and their uses have been reviewed thoroughly, and they are most basically used to predict reaction flux, which is the overall rate of metabolite conversion.1,2 Often, laboratory measurements including the rates of substrate consumption, product formation, and growth are used as model constraints so calculations coincide with observations. Other model constraints can be derived from reaction thermodynamics,3 cellular regulatory networks,4 and -omics datasets.5 GEMs have been constructed and utilized for intensively studied model organisms with well-annotated genomes (eg, Escherichia coli. MG1655 [bacteria],6 Saccharomyces cerevisiae. [yeast],7 Mus musculus. [mouse],8 and Arabidopsis thaliana. [plant]9). In addition, homology algorithms have enabled GEM construction of the less-studied organisms. For example, the European Bioinformatics Institute has constructed draft GEMs for 2,630 organisms across phylogenetic domains using automated model-building methods,10 and the Model SEED also contains several GEMs and has the ability to custom-build GEMs for annotated genomes submitted by the user.11 The construction of high-quality models often requires expert-informed manual curation,12 but automated reconstruction provides foundations for further improvement. Although GEMs have been built for species of all domains, microbes still dominate GEM reconstructions and studies due to their relative genomic simplicity, usefulness in biotechnology, and the pathogenicity of some species.
GEMs have extraordinary utility for biological discovery, and novel computational tools have been developed to predict metabolic engineering strategies, which are then validated in the laboratory. Much research in metabolic engineering is focusing on the synthesis of valuable chemicals, biofuels, and pharmaceuticals. Model-guided metabolic engineering presents significant advantages, notably the minimization of laboratory resource use and time required to develop productive strains. Using GEM predictions to design strains enables researchers to engineer product yield/selectivity, substrate utilization, and growth rate. Future developments are anticipated to allow engineering of toxicity responses, cellular differentiation, culture density, and cellular interactions with other cells and materials. Some of the computational tools for predicting gene targets in GEMs for metabolic engineering have been reviewed.1 The focuses of this review are: 1) how predictions from different tools have been translated into experimental metabolic engineering strategies and 2) which of the experimental methods available are (or are not) represented in the computational (in silico) tools. Since the experimental toolset for metabolic engineering is expanding, this review also addresses how new tools can be incorporated in the in silico design strategies.
In silico metabolic engineering tools
It has been long believed that cells (especially microbes) maintain optimal growth as their primary objective. It has been shown that an additional objective of a minimal adjustment between initial and engineered states also exists.13 Imposing the goal of chemical overproduction by metabolic engineering often conflicts with the optimal growth objective. Thus, genome-scale modeling serves to establish the relationship between target chemical production and growth. In silico metabolic engineering tools seek to identify genetic manipulations to alter this relationship so that stable strains with high chemical production and growth can be achieved. This section describes the various methods available for solving GEMs, and it highlights those used when metabolism has been engineered. In addition, this section presents the recent advances in in silico tools used with GEMs to generate metabolic engineering strategies for the overproduction of a targeted chemical. In this review, in silico metabolic engineering tools are classified as “top-down” or “bottom-up”. The top-down algorithms generate/apply metabolic modifications in silico and then simulate their effects on the dual objectives (ie, productivity and growth) through genome-scale metabolic flux modeling. The procedure is repeated until optimal metabolic modifications are identified. On the other hand, bottom-up algorithms generate separate flux solutions where: 1) growth is maximized and 2) product formation of interest is maximized. Differences between the two flux distributions are identified as targets to design metabolic engineering strategies. These approaches are reviewed in detail in the following section; however, first the methods for generating metabolic flux solutions of GEMs are summarized.
Flux balance analysis and its variants
The fundamental approaches of constraint-based modeling have been reviewed,1,2 and a subset of these applicable to metabolic engineering are described here. The essential base of almost all predictive tools is flux balance analysis (FBA), which solves the linear system of biological reactions given the “pseudo-” steady-state assumption and an objective function (eg, maximize growth or chemical production rate) using linear programming. The flux balance equation is now commonly written as S · v=0, where S is an m-by-n matrix containing stoichiometric coefficients for each biochemical reaction. Each compound is represented by a row of the matrix, and each reaction is represented in a column. The vector v contains flux values for all n reactions of the system. The system also contains a “biomass equation” that describes cell growth. This is often composed of stoichiometric amounts of macromolecules (eg, protein, DNA, RNA, lipids, cell wall), small molecules, and adenosine triphosphate (ATP) hydrolysis required for growth “maintenance”.14 FBA solves the system of equations given an objective function and constraints (upper and lower) for each flux contained in v. Flux constraints are imposed from laboratory measurements, thermodynamic predictions, and regulatory rules; many reaction fluxes are left unconstrained. Two other useful approaches are parsimonious FBA (pFBA)15 and flux variability analysis (FVA).16 Since the number of reactions is typically greater than the number of compounds in GEMs, multiple FBA solutions exist, and techniques that explore this solution space have been reviewed. The pFBA algorithm was developed to provide the FBA solution that meets optimality with a minimized total flux in the system. In addition, FVA serves the purpose of calculating the possible flux distributions of all reactions.
As mentioned earlier, cellular metabolism changes when a genetic manipulation is introduced in vivo. However, dramatic shifts in metabolism, on a global level, toward optimality are not immediate.17,18 Thus, a flux distribution predicted in silico that captures this initial response of a cell, instead of one that describes massive flux reorganization toward optimality, provides a better description of the cellular response to genetic changes. For this reason, the minimization of metabolic adjustment (MOMA) algorithm was developed to predict the optimal flux distribution of altered metabolism that would require the smallest change from that of wild-type metabolism.17 This concept has since been validated by 13C-isotope tracing studies.13 Similar to MOMA, the regulatory on/off minimization (ROOM) tool hypothesizes that a cell attempts to compensate for genetic manipulations through the fewest number of enzymatic reactions by gene regulation.18 Additional studies have shown that, in time, cells will evolve from this minimized flux redistribution state to the FBA solution.19 This concept, introduced over a decade ago,20 is shown in Figure 1. The goal of metabolic engineering is to alter the metabolic network of a cell so that optimal growth and target chemical production are coupled (meaning a product must be formed as the cell reaches an optimum growth rate). This approach leads to stable strains capable of industrial production. As a cell is engineered, MOMA/ROOM can predict the immediate outcome of genetic manipulations, and FBA (or pFBA) predicts the long-term evolved state of the cell. In the following sections, the top-down and bottom-up in silico metabolic engineering tools are discussed, and a summary of these tools is given in Table 1. However, it is important to note that not all tools are designed to consider evolution and long-term strain stability, which are critically important if an industrial process is going to consider chemostat cultivation over batch processing in which the microbe is replaced frequently.
 | Figure 1 The relationship between the target chemical production flux and the growth rate for wild-type (solid line) and an engineered strain (dash line). The initial wild-type optima determined by FBA (bottom right) can be engineered and the resulting state predicted with MOMA/ROOM. Evolution will eventually optimize growth, which can be predicted by FBA/pFBA. Combinatorial addition of metabolic capabilities can expand the solution space beyond the wild-type potential. |
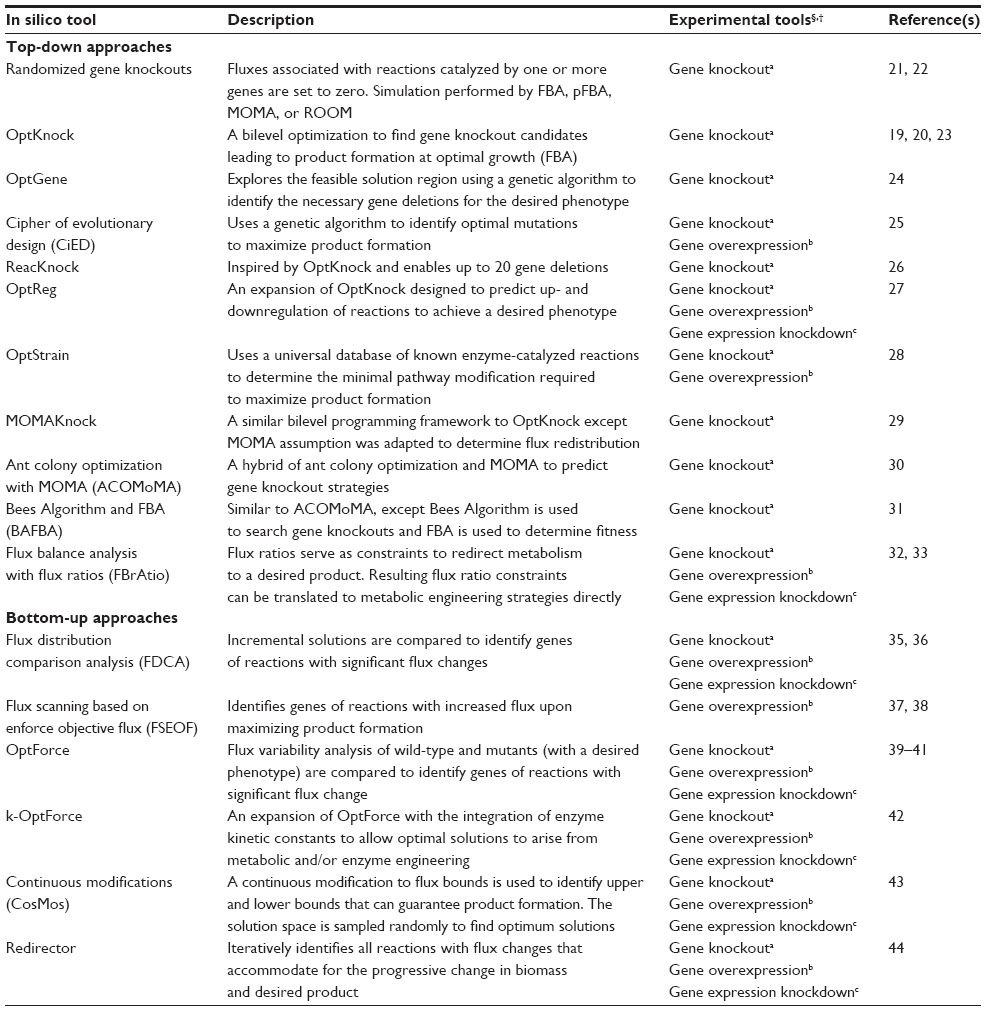 | Table 1 Summary of in silico tools for generating metabolic engineering strategies and the experimental tools that can be used for implementation |
Top-down in silico tools for designing metabolic engineering strategies
As mentioned previously, a top-down approach is defined here as one in which genetic manipulations are made in silico, and then genome-scale modeling is used to determine whether the strategy is beneficial. The concept is shown in Figure 2. The simplest strategy to employ is creating single-gene knockouts. This is done in silico by constraining all reactions associated with a gene of interest to zero and performing FBA or MOMA/ROOM to look for knockouts that enhance target chemical production without compromising growth. This method was used in a well-known study to identify gene knockouts in E. coli, resulting in the overproduction of L-valine.21 Here, single-, double-, and triple-gene knockouts were investigated in silico using MOMA to predict resulting phenotypes. Using a multifaceted approach that included the in silico gene deletion study, an industrially relevant strain capable of producing over 7.5 g/L of L-valine (2.27-fold improvement over wild-type) was engineered. This method was also used to generate all single- and double-gene knockout combinations in S. cerevisiae. in an effort to overproduce succinate.22 FBA was used in calculations, and three single knockouts (Δmdh, Δoac1, and Δdic1) were selected for experimental validation. The Δdic1. strategy was successful, yet non-intuitive for succinate production, and this study demonstrated an important proof-of-concept for designing strains by in silico predictions followed by experimental validations. OptKnock was one of the first in silico metabolic engineering tools, and it guides the selection of gene knockouts in order to couple maximized product formation to growth.20 It uses FBA and identifies a limited number of gene knockouts, which serve to reshape the growth and product formation relationship as shown in Figure 1. Successful identification of gene knockout targets, followed by adaptive evolution to achieve FBA predictions, have led to industrially relevant strains capable of producing lactic acid,19 1,4-butanediol,23 and others. Since its introduction, other inspired approaches have attempted to extend its capabilities (ie, increase the potential number of gene candidates for knockout) by reconsidering the bilevel optimization framework. Approaches such as OptGene24 and the cipher of evolutionary design (CiED)25 relied on an evolutionary algorithm to select gene targets, and improved functionality was noted. ReacKnock has emerged recently with a new approach to the mixed integer bilevel optimization problem and enables up to 20 gene deletion predictions in a short amount of computational time.26 In their publication, the authors provide ReacKnock- and OptKnock-designed gene knockout strategies to produce succinate, ethanol, acetate, hydrogen, formate, glycolate, D-lactate, fumarate, and threonine from E. coli.26 OptReg extended OptKnock to include gene overexpressions,27 and OptStrain allowed incorporation of non-native metabolic pathways for the production of new chemicals.28 Other approaches, such as MOMAKnock29 have focused on the limitations of FBA and have sought to implement MOMA in the automated design of gene knockout strategies. Further modifications have combined ant colony optimization (ACO) methods with MOMA in an algorithm called ACOMoMA. The ACOMoMA approach was applied to produce an improved gene knockout strategy for succinate production from E. coli.30 Another development achieved significant results using a hybrid of Bees Algorithm and FBA (BAFBA; a metaheuristic procedure) to design gene knockouts for succinate and lactate production.31
 | Figure 2 Example workflow to design metabolic engineering strategies using “top-down” and “bottom-up” approaches. Several different in silico tools apply these strategies in different forms. In all cases, the objective is to maximize production of a target chemical (shown here as vtarget). The following metabolic engineering strategies are shown: (KO) gene knockout, (OX) gene overexpression, and (KD) gene expression knockdown. |
While most in silico designs rely on gene knockouts, others infer gene overexpression and partial gene knockdowns as metabolic engineering strategies. In general, the flux change of a reaction may be the result of: 1) directly engineering genes of the catalyzing enzymes; 2) engineering the availability of reaction precursors and substrates upstream; or 3) eliminating bottlenecks downstream. Thus, these strategies are all major contributors to a metabolic adjustment. A recent approach called FBA with flux ratios (FBrAtio) considers strategies of gene overexpression, knockout, and partial knockdown for designing metabolic engineering strategies.32,33 FBrAtio examines how multiple enzymes compete for the same substrate and allow the distribution of this substrate to be modified and included as a flux ratio constraint in a GEM. Flux ratio constraints can be modified, and pFBA is used to predict global flux distributions. This procedure has been used to design metabolic engineering strategies for several chemicals by different organisms. The concept of the flux ratio constraint was first introduced for two enzymes that compete for the same compound.32 However, this was later expanded to include all enzymes competing for the same compound.33 FBrAtio has been used to model the metabolic shift in Clostridium acetobutylicum. from acids to solvents production as well as predict a high-ethanol-producing phenotype.32 In addition, it has been used to examine metabolic engineering strategies for: 1) cellulose overproduction by A. thaliana; 2) isobutanol production by yeast; 3) acetone production by Synechocystis; 4) hydrogen production by E. coli; and 5) mixed solvents production by C. acetobutylicum.33 The purpose of this study was to demonstrate further improvements of experimental implementations where possible with “fine-tuned” metabolic engineering strategies derived by FBrAtio. With Arabidopsis, it was shown experimentally that the overexpression of a heterologous uridine diphosphate (UDP)–glucose pyrophosphorylase (UGPase) increased cellulose production by approximately 25%.34 The FBrAtio approach predicted that further increased uridine triphosphate (UTP) consumption by the UGPase could continue to increase cellulose production up to 30%–50% (compared to wild-type) before UTP depletion impacted the growth of the plants negatively.
Bottom-up in silico tools for designing metabolic engineering strategies
The tools classified as bottom-up approaches rely on multiple objective functions in genome-scale modeling to design metabolic engineering strategies. The flux distribution comparison analysis (FDCA) provides a good example of this. First, a GEM is solved by FBA to maximize growth. Then, the GEM is solved (by linear MOMA [lMOMA]) to maximize the production of a chemical of interest. The differences between the flux distributions are considered, and rules for up- or downregulation of genes are determined based on significant changes between the flux distributions.35 FDCA has been used to improve lycopene production by 174% in an E. coli strain already capable of high lycopene production,36 and it identified 51 potential gene targets, including five novel gene knockout targets and four novel gene overexpression targets. The flux scanning based on enforced objective flux (FSEOF) approach was also developed to enhance lycopene production.37 This approach also begins with maximizing biomass formation of a GEM with FBA, but the flux of product formation is constrained to be equal to the experimentally observed flux in the wild-type organism. Then, the theoretical maximum product formation rate is calculated in a new simulation by setting this as the objective function. FSEOF works by maximizing the cell growth rate while the target product formation rate is increased gradually from its initial value toward its theoretical maximum. Targets for gene overexpression are identified as fluxes that increase throughout simulations without changing direction. This method identified 35 gene overexpression targets for lycopene production by E. coli. FVA was then employed to narrow these potential targets by selecting those showing increases outside of the ranges due to flux variability.37 This approach can also be used with an altered biomass equation to accommodate intracellular target (eg, protein) accumulation. For example, the human superoxide dismutase (hSOD) enzyme was overproduced in Pichia pastoris using predicted gene knockout and overexpression strategies from MOMA and FSEOF, respectively.38
OptForce is another bottom-up approach that has enabled the incorporation of gene knockouts, overexpressions, and knockdowns as metabolic engineering strategies.39 OptForce also allows (and encourages) the incorporation of experimentally measured metabolic flux data of the wild-type and a strain engineered to overproduce a target chemical. In general, flux variability is calculated for both wild-type and engineered strains, and the flux ranges are compared for each reaction. Candidates for metabolic engineering are identified as those reactions where there is no overlap between possible flux ranges. OptForce then performs a secondary optimization (a top-down procedure) where the minimal set of metabolic interventions is identified to achieve a desired goal. OptForce has been used in several applications, including the overexpression of succinate39 and fatty acids of specified chain length in E. coli.40 In addition, OptForce was used to design a metabolic engineering strategy leading to a four-fold increase in intracellular malonyl-CoA concentration in E. coli, which was then utilized for the production of naringenin (a valuable plant secondary metabolite).41 The recent extension k-OptForce has enabled the incorporation of enzyme kinetic constants, where possible, and returns metabolic engineering strategies (ie, gene knockout, overexpression, or knockdown) along with kinetic parameters that could be altered by enzyme engineering.42 This approach can consider relevant phenomena, such as substrate inhibition, that cannot be modeled using flux-based approaches alone. The continuous modifications (CosMos) approach significantly differs from OptForce in that changes to flux bounds are modified continuously, rather than by FVA results. CosMos then minimizes product formation given a constrained non-zero growth rate, and looks for modified flux constraints that still yield product formation under these conditions.43
Finally, the Redirector approach is different in that it relies on an artificial objective function consisting of contributions from growth and metabolic flux redirected into a product-forming pathway and does not rely on manipulating flux bounds.44 Redirector can also design gene knockout, overexpression, or knockdown metabolic engineering strategies, and the manipulation of algorithm parameters can alter the number of manipulations returned by the algorithm. The production of fatty acids by E. coli MG1655 was chosen as a test case of the algorithm. The algorithm designed strategies capable of reaching 80% of the theoretical yield for myristoyl-CoA while maintaining 20% biomass yield.44 The global implementation of FBrAtio (currently in press) is also classified as an approach that does not manipulate flux bounds to derive metabolic engineering strategies. The global FBrAtio uses flux distribution maps of maximized growth and product formation using pFBA and designs flux ratio constraints that enable product formation and growth.
Experimental metabolic engineering tools
The available in silico metabolic engineering tools return strategies consisting of gene knockout, overexpression, and/or knockdown (and enzyme engineering for k-OptForce). There are several ways in which these strategies can be implemented, but current in silico tools do not consider this level of detail. In this section, many common (but certainly not all) experimental implementation methods are reviewed along with their relationship to in silico predictions. For example, returning a gene overexpression strategy does not explain how it should be implemented. If it must be encoded on a plasmid, what type and strength of promoter/ribosomal binding site (RBS) combination should be used? What copy number of plasmid should be used? Since plasmid copy number per cell is heterogeneous, what impacts will this have? Will plasmid replication demand cellular resources that influence metabolic flux predictions? What are the impacts of antibiotic resistance genes? Should one or multiple copies of the gene of interest be knocked into the genome? Or, should a native promoter/RBS be tuned instead? If so, to what levels? Finally, what impact will this genetic manipulation have on the resulting phenotype? Will this significantly impact cell composition, metabolic flux distribution, and predictions? These and more questions will be addressed by in silico tools that returned “fine-tuned” metabolic engineering strategies (eg, overexpress a target gene by 70% relative to wild-type) and take into account changing cell phenotype by updating the GEM biomass equation.
Manipulating gene expression
Here, basic experimental strategies for gene expression manipulations are reviewed in the context of genome-scale modeling. Clearly, not all tools and approaches can be discussed here, but the basics are identified. Manipulations can occur at the transcriptional, translational, and posttranslational levels, with emphasis on the first two in microbes. Several experimental methods exist for generating gene knockouts that involve chromosomal integration for gene disruption. Of course, chromosomal integration can also be used to knock-in useful genes/regulatory elements. One particularly popular method for single-gene targeting is the polymerase chain reaction (PCR)-based version of λ red recombineering.45 It has also been used for the introduction of site-directed mutations, promoter tuning/replacement, and reporter genes for promoter tagging experiments.46 The knock-in/knockout (KIKO) vectors facilitate the chromosomal integration of large DNA segments (including multigene cassettes and entire pathways) at specific well-characterized loci using λ red recombination.47 Other means of gene knockout involve the use of transposons or homologous recombination mediated by phage-derived elements, and more advanced genetic systems are required for other microbes, such as the clostridia.48,49 In higher plant species, such as A. thaliana, genomic integration is accomplished using an Agrobacterium-mediated method that makes use of its ability to transfer DNA from its tumor-inducing plasmid into the plant host genome.50,51 This technology has been used for both gene disruption and knock-in in Arabidopsis. Gene knockouts (and knock-ins) appear to be the most benign to genome-scale modeling predictions, as long as plasmids and antibiotic resistance markers are removed. Indeed, the presence of plasmids and antibiotics (even with effective antibiotic resistance genes) has been shown to alter cell phenotypes.52 The GEM biomass equation describes the cell phenotype, and how this equation should be altered by the presence of plasmids, antibiotics, or other genetic or environmental manipulations remains a subject for research. This makes clustered regularly interspaced short palindromic repeats (CRISPR)-Cas systems53 attractive for genome editing from a genome-scale modeling standpoint. While the mechanisms of plasmid replication are understood, this cellular machinery is not yet encoded in GEMs, creating a divergence between the in silico and experimental systems. In addition, gene knockouts, knock-ins, and genome editing are designed to alter metabolism. When successful, this alters the cellular phenotype; thus, the biomass equation must be updated accordingly. However, this will require predictions or a simplified method of measurement, both of which are discussed later.
With this knowledge, it is easy to see why gene overexpression methods may lead to greater metabolic burden and uncertainty with genome-scale modeling, especially when a gene is overexpressed from a plasmid. Techniques that minimize the ATP maintenance requirements of a cell are preferred and are more effectively modeled. With gene overexpression, promoter and RBS engineering have enabled significant progress. Controllable gene expression has launched the field of synthetic biology and led to the quest to design genetic circuits.54 Furthermore, promoter tuning55 with RBS optimization can improve metabolic pathway function.56 Tools, such as the RBS calculator,57 are enabling RBS design based on thermodynamic principles. In these cases, it becomes clear that synthetic designs are enabling pathway overexpression by orders of magnitude, and at some point, cellular resources are depleted (eg, transfer RNA [tRNA] pools), creating competition between cell growth and pathway expression. This is not yet accounted for by genome-scale modeling and presents a unique opportunity to integrate metabolic pathway tuning with genome-wide metabolic activity.
Gene expression tuning can also be engineered at the posttranslational level, where interactions with mRNA are the major focus. Small RNA (sRNA) bind targeted mRNA (through complementary base-pairing) and modulate its translation.58 The majority of sRNAs have been identified as translation repressors, and binding generally occurs at or near the RBS.59 Thermodynamic-based design has enabled “fine-tuned” gene expression knockdowns,60 and these have proven advantageous in a metabolic engineering strategy to produce phenol from glucose.61 Similarly, artificial small interfering RNA and microRNA have been widely used in plant systems to reduce gene expression.62 Achieving stable gene integration in higher plants can be problematic. An alternative is to employ viral-induced gene silencing approaches.63 While these technologies enable gene knockdown, they are generally operated from plasmid-based systems, which provide the same challenges to genome-scale modeling as mentioned previously.
Combinatorial approaches
Using genome-scale modeling to predict the outcomes of combinatorial metabolic engineering experiments is an area for many future advances. As shown in Figure 1, the addition of new genetic material (either synthetic or from other organisms) can expand the product-forming capabilities of an organism. Combinatorial approaches can involve induced chromosomal mutations, random insertion of transposons, genome shuffling, transcription factor engineering,64 or even randomized chromosomal insertion of synthetic DNA.65 Lycopene production has been engineered successfully through: 1) a combination of model-driven and transposon-based combinatorial knockouts66 and 2) the multiplex automated genome engineering (MAGE) platform, which relies on synthetic DNA insertion.65 In addition, gene overexpression libraries offer the opportunity to insert the genomic capabilities of a single organism or a metagenome. This strategy has proven successful in locating genomic sequences to confer tolerance to furfural,67 among many others. The simultaneous expression of dual libraries on a plasmid and fosmid led to a unique combination of gene enrichment that increased acid tolerance in E. coli by 9,000-fold.68 Expanding the genome to confer resistance to toxins or new/improved metabolic capabilities has the potential to redefine the relationship between product formation and culture growth, as shown in Figure 1. In the case of conferring resistance to toxins, often uncharacterized or non-obvious library fragments are selected during enrichment.69 This is often because toxicity mechanisms, as well as many cellular interactions, are multigenic and still not understood fully. While genome-scale modeling cannot provide these types of predictions, where the interaction mechanisms are uncharacterized, the metabolic potentials through the completion and addition of new pathways and enzymes are predictable. It is likely that the theoretical limits of metabolic enhancement due to library enrichment can be found through genome-scale modeling, and the emergence of metagenomic GEMs will likely contain the metabolic potentials.
Phenotyping
Phenotyping refers to the monitoring of cell chemical composition and differentiation. This is critical because the GEM biomass equation contains the cell chemical composition and is representative of the cellular phenotype, which is known to change with genetic and environmental perturbations. The role of the biomass equation has been shown to be crucial in genome-scale modeling,14,70 creating the need for accurate and near real-time monitoring techniques to interface with GEMs. In silico optimization methods have shown promising results,70 but it is likely that an experimental approach will be needed as a supplement. Traditional methods of biomass equation generation are laborious and involve offline analytical methods. Analysis of heterogeneous populations of differentiating (eg, sporulating) microbes is now possible using flow cytometry.71 In addition, Raman spectroscopy has recently proven useful for near real-time phenotyping of E. coli Raman spectroscopy also does not require the use of chemical labels and is nondestructive to the sample. In one application, Raman spectroscopy was used to resolve fatty acids (saturated, unsaturated, and cyclopropane), cell membrane fluidity, amino acids, and total protein content of cultures exposed to toxic 1.2% volume per volume 1-butanol (and control cultures) over a 180-minute time course.72 In another approach, “chemometric fingerprinting”, a multivariate statistical analysis involving principal component analysis and linear discriminate analysis, was used to classify the E. coli phenotypes resulting from exposure to different classes of antibiotics.73 Chemometric fingerprinting is unique in that it uses the entire Raman spectrum to characterize a phenotype, whereas most approaches focus on only a few well-defined characteristic bands of the spectrum. With these types of near real-time analyses, GEM biomass equations can become dynamic and responsive to environmental and genetic changes. With current offline methods of phenotype characterization, this level of detail is not possible. However, with easily accessible phenotyping capabilities, biomass equations can be updated easily, leading to improved genome-scale modeling performance.
The path forward
New metabolic engineering targets and opportunities with plants
Deriving metabolic engineering strategies with genome-scale modeling is proving to be efficient and informative. As research continues to derive de novo metabolic pathways to synthesize valuable chemicals, optimization of product yield to meet industrial demands will be inevitable. Still, the variety of potential products from microbes remains limited and may be expanded in the near term by looking into complex eukaryotic species, such as plants. There are many valuable compounds made by plants that are not available elsewhere. For example, oil seed crops (eg, soybeans) produce edible vegetable oil that is used throughout the world. Although the pathways for lipid biosynthesis in higher plants have been studied for years, understanding of the crucial regulatory mechanisms of these pathways remains limited. Thus, engineering plants to accumulate high levels of healthy omega-3 long-chain polyunsaturated fatty acids,74 or modified non-native fatty acids as replacements for petroleum-derived chemicals in industrial processes75 is desirable. Similarly, central carbon metabolism is a target for understanding the relationship between the regulation of carbon partitioning and biomass production in plants. Identifying metabolic bottlenecks in the production of cellulose, the energy-rich polymer that is targeted for consolidated bioprocessing,76 could enhance efforts to produce more cellulose per plant in the field. Likewise, modeling is being applied to the goal of reducing plant lignin, a phenolic polymer in the secondary wall that limits our use of cellulosic biomass during industrial processing.77 One caveat of reducing lignin is that optimal plant growth must also be preserved, and GEMs may be uniquely positioned to tackle this issue because they can theoretically integrate metabolic behavior with plant growth.78 Enhancing the vitamin content of edible plants is another active area of research.79 For some vitamin synthesis pathways, enough information exists to begin the application of genome-scale modeling to increase the concentration of vitamins to meet minimal requirements for humans.80 In the future, it may be possible to use genome-scale modeling to tackle issues such as optimizing plant growth under stressful or poor nutrient growth conditions. In these cases, genome-scale models would have to account for complex interactions between stress, hormone, and other signaling pathways that impact biomass synthesis and composition.81 In addition, a related application is to understand how to limit plant yield loss due to pests, by engineering known, disease-resistance pathways.81 All of these approaches will require flexible biomass equations that can respond to manipulations, and a complex multicellular plant will likely require tissue-specific GEMs that will integrate to form an overall plant phenotype.
Enzyme engineering for pathway redirection
The k-OptForce in silico tool is among the first to incorporate the concept of enzyme engineering to redirect metabolic flux for the production of target chemicals. Kinetics-based approaches to genome-scale metabolic modeling are emerging,82,83 and soon enzyme redesign will be a valid metabolic engineering strategy. Direct genome editing, which is preferred over insertion of plasmids and markers that consume cellular resources, will enable easy implementation. Enzyme engineering is a complex field itself and beyond the scope of this review, but effective in silico methods are emerging and are expected to play a role in enzyme redesign. Improvements in hardware and software performance will continue to expand the range and size of enzyme engineering problems and systems that can be studied. Current computational approaches can be divided into bioinformatics, molecular modeling, and de novo design.84 Bioinformatics approaches are typically based on analysis of evolutionary data and can be used to change activity, selectivity, and stability within a family of enzymes. Molecular modeling approaches (eg, molecular dynamics, quantum mechanics/molecular mechanics simulations) have considerable potential to address challenges in computational enzyme design and redesign. In particular, advances in these methods may enable improved calculation of binding affinities and energy barriers, which will enhance understanding of enzyme specificity.85 De novo design is also showing increasing promise in designing enzymes, including those that catalyze reactions for which nature has not designed a catalyst. Notably, these novel methods may be enhanced by the application of molecular modeling approaches.86
Increasing modeling accuracy
Finally, the path forward must focus on methods that increase the accuracy of genome-scale metabolic flux modeling and improve agreements with 13C-isotopomer tracing studies. In our experience, there are four areas for immediate improvement. The first area includes the incorporation of a more detailed account of cellular machinery in GEMs. As mentioned previously, the ATP maintenance approximation of the GEM biomass equation should be replaced by mechanistic accounts. This must also allow for the identification of metabolic burdens of plasmids and altered metabolic states as a result of genome editing. There are current ongoing efforts of “whole cell modeling” that aim to include cellular machinery in modeling efforts. These models are showing promise of being able to predict phenotypes as well as better integrate and explain -omics datasets.87 Second, more accurate biomass equations are needed. Whether these will be derived computationally or experimentally remains to be seen, and there are good arguments for both approaches. Third, a more accurate representation of flux branching at critical metabolic nodes is needed. This occurs when multiple enzymes can consume the same metabolite. Ultimately, the laws of thermodynamics (including enzyme availability) determine how that metabolite is distributed among the competing enzymes. Current methods of FBA, pFBA, FVA, ROOM (etc) do not consider this level of detail. FBrAtio provides this capability, though significant strides are needed to first translate the biophysical constraints into flux ratio constraints. Finally, the roles of redox states in product secretion profiles and the influx/efflux of protons across the cell membrane need to be included as constraints in GEMs. In addition, efforts in these areas will supplement the many useful emerging tools that are focusing on genomic regulation and -omics dataset integrations. All of these will improve genome-scale modeling accuracy, which is needed for deriving effective metabolic engineering strategies.
Acknowledgments
Funding was provided by the National Science Foundation (NSF1243988 and NSF1254242), the US Department of Agriculture (2010-65504-20346 and the HATCH program), and the Institute for Critical Technologies and Applied Science at Virginia Tech.
Disclosure
The authors report no conflicts of interest in this work.
References
Tomar N, De RK. Comparing methods for metabolic network analysis and an application to metabolic engineering. Gene. 2013;521(1):1–14. | |
Oberhardt MA, Palsson BØ, Papin JA. Applications of genome-scale metabolic reconstructions. Mol Syst Biol. 2009;5:320. | |
Henry CS, Broadbelt LJ, Hatzimanikatis V. Thermodynamics-based metabolic flux analysis. Biophys J. 2007;92(5):1792–1805. | |
Chandrasekaran S, Price ND. Probabilistic integrative modeling of genome-scale metabolic and regulatory networks in Escherichia coli and Mycobacterium tuberculosis. Proc Natl Acad Sci U S A. 2010;107(41):17845–17850. | |
Hyduke DR, Lewis NE, Palsson BØ. Analysis of omics data with genome-scale models of metabolism. Mol Biosyst. 2013;9(2):167–174. | |
Orth JD, Conrad TM, Na J, et al. A comprehensive genome-scale reconstruction of Escherichia coli metabolism – 2011. Mol Syst Biol. 2011;7:535. | |
Heavner BD, Smallbone K, Price ND, Walker LP. Version 6 of the consensus yeast metabolic network refines biochemical coverage and improves model performance. Database (Oxford). 2013;2013:bat059. | |
Selvarasu S, Karimi IA, Ghim GH, Lee DY. Genome-scale modeling and in silico analysis of mouse cell metabolic network. Mol Biosyst. 2010;6(1):152–161. | |
de Oliveira Dal’Molin CG, Quek LE, Palfreyman RW, Brumbley SM, Nielsen LK. AraGEM, a genome-scale reconstruction of the primary metabolic network in Arabidopsis. Plant Physiol. 2010;152(2):579–589. | |
Büchel F, Rodriguez N, Swainston N, et al. Path2Models: large-scale generation of computational models from biochemical pathway maps. BMC Syst Biol. 2013;7:116. | |
Aziz RK, Devoid S, Disz T, et al. SEED servers: high-performance access to the SEED genomes, annotations, and metabolic models. PLoS One. 2012;7(10):e48053. | |
Thiele I, Palsson BØ. A protocol for generating a high-quality genome-scale metabolic reconstruction. Nat Protoc. 2010;5(1):93–121. | |
Schuetz R, Zamboni N, Zampieri M, Heinemann M, Sauer U. Multidimensional optimality of microbial metabolism. Science. 2012;336(6081):601–604. | |
Senger RS. Biofuel production improvement with genome-scale models: the role of cell composition. Biotechnol J. 2010;5(7):671–685. | |
Lewis NE, Hixson KK, Conrad TM, et al. Omic data from evolved E. coli are consistent with computed optimal growth from genome-scale models. Mol Syst Biol. 2010;6:390. | |
Gudmundsson S, Thiele I. Computationally efficient flux variability analysis. BMC Bioinformatics. 2010;11:489. | |
Segre D, Vitkup D, Church GM. Analysis of optimality in natural and perturbed metabolic networks. Proc Natl Acad Sci U S A. 2002;99(23):15112–15117. | |
Shlomi T, Berkman O, Ruppin E. Regulatory on/off minimization of metabolic flux changes after genetic perturbations. Proc Natl Acad Sci U S A. 2005;102(21):7695–7700. | |
Fong SS, Burgard AP, Herring CD, et al. In silico design and adaptive evolution of Escherichia coli for production of lactic acid. Biotechnol Bioeng. 2005;91(5):643–648. | |
Burgard AP, Pharkya P, Maranas CD. Optknock: a bilevel programming framework for identifying gene knockout strategies for microbial strain optimization. Biotechnol Bioeng. 2003;84(6):647–657. | |
Park JH, Lee KH, Kim TY, Lee SY. Metabolic engineering of Escherichia coli for the production of L-valine based on transcriptome analysis and in silico gene knockout simulation. Proc Natl Acad Sci U S A. 2007;104(19):7797–7802. | |
Agren R, Otero JM, Nielsen J. Genome-scale modeling enables metabolic engineering of Saccharomyces cerevisiae for succinic acid production. J Ind Microbiol Biotechnol. 2013;40(7):735–747. | |
Yim H, Haselbeck R, Niu W, et al. Metabolic engineering of Escherichia coli for direct production of 1,4-butanediol. Nat Chem Biol. 2011;7(7):445–452. | |
Patil KR, Rocha I, Forster J, Nielsen J. Evolutionary programming as a platform for in silico metabolic engineering. BMC Bioinformatics. 2005;6:308. | |
Fowler ZL, Gikandi WW, Koffas MA. Increased malonyl coenzyme A biosynthesis by tuning the Escherichia coli metabolic network and its application to flavanone production. Appl Environ Microbiol. 2009;75(18):5831–5839. | |
Xu Z, Zheng P, Sun J, Ma Y. ReacKnock: identifying reaction deletion strategies for microbial strain optimization based on genome-scale metabolic network. PLoS One. 2013;8(12):e72150. | |
Pharkya P, Maranas CD. An optimization framework for identifying reaction activation/inhibition or elimination candidates for overproduction in microbial systems. Metab Eng. 2006;8(1):1–13. | |
Pharkya P, Burgard AP, Maranas CD. OptStrain: a computational framework for redesign of microbial production systems. Genome Res. 2004;14(11):2367–2376. | |
Ren S, Zeng B, Qian X. Adaptive bi-level programming for optimal gene knockouts for targeted overproduction under phenotypic constraints. BMC Bioinformatics. 2013;14 Suppl 2:S17. | |
Chong SK, Mohamad MS, Mohamed Salleh AH, Choon YW, Chong CK, Deris S. A hybrid of ant colony optimization and minimization of metabolic adjustment to improve the production of succinic acid in Escherichia coli. Comput Biol Med. 2014;49:74–82. | |
Choon Y, Mohamad M, Deris S, et al. Identifying gene knockout strategies using a hybrid of Bees Algorithm and flux balance analysis for in silico optimization of microbial strains. In: Omatu S, De Paz Santana JF, González SR, Molina JM, Bernardos AM, Rodríguez JMC, editors. Distributed Computing and Artificial Intelligence. Vol 151. Springer Berlin Heidelberg; 2012:371–378. | |
McAnulty MJ, Yen JY, Freedman BG, Senger RS. Genome-scale modeling using flux ratio constraints to enable metabolic engineering of clostridial metabolism in silico. BMC Syst Biol. 2012;6(1):42. | |
Yen JY, Nazem-Bokaee H, Freedman BG, Athamneh AI, Senger RS. Deriving metabolic engineering strategies from genome-scale modeling with flux ratio constraints. Biotechnol J. 2013;8(5):581–594. | |
Wang Q, Zhang X, Li F, Hou Y, Liu X, Zhang X. Identification of a UDP-glucose pyrophosphorylase from cotton (Gossypium hirsutum L.) involved in cellulose biosynthesis in Arabidopsis thaliana. Plant Cell Rep. 2011;30(7):1303–1312. | |
Meng H, Lu Z, Wang Y, Wang X, Zhang S. In silico improvement of heterologous biosynthesis of erythromycin precursor 6-deoxyerythronolide B in Escherichia coli. Biotechnol Bioproc Eng. 2011;16(3):445–456. | |
Wang JF, Meng HL, Xiong ZQ, Zhang SL, Wang Y. Identification of novel knockout and up-regulated targets for improving isoprenoid production in E. coli. Biotechnol Lett. 2014;36(5):1021–1027. | |
Choi HS, Lee SY, Kim TY, Woo HM. In silico identification of gene amplification targets for improvement of lycopene production. Appl Environ Microbiol. 2010;76(10):3097–3105. | |
Nocon J, Steiger MG, Pfeffer M, et al. Model based engineering of Pichia pastoris central metabolism enhances recombinant protein production. Metab Eng. 2014;24:129–138. | |
Ranganathan S, Suthers PF, Maranas CD. OptForce: an optimization procedure for identifying all genetic manipulations leading to targeted overproductions. PLoS Comput Biol. 2010;6(4):e1000744. | |
Ranganathan S, Tee TW, Chowdhury A, et al. An integrated computational and experimental study for overproducing fatty acids in Escherichia coli. Metab Eng. 2012;14(6):687–704. | |
Xu P, Ranganathan S, Fowler ZL, Maranas CD, Koffas MA. Genome-scale metabolic network modeling results in minimal interventions that cooperatively force carbon flux towards malonyl-CoA. Metab Eng. 2011;13(5):578–587. | |
Chowdhury A, Zomorrodi AR, Maranas CD. k-OptForce: integrating kinetics with flux balance analysis for strain design. PLoS Comput Biol. 2014;10(2):e1003487. | |
Cotten C, Reed JL. Constraint-based strain design using continuous modifications (CosMos) of flux bounds finds new strategies for metabolic engineering. Biotechnol J. 2013;8(5):595–604. | |
Rockwell G, Guido NJ, Church GM. Redirector: designing cell factories by reconstructing the metabolic objective. PLoS Comput Biol. 2013;9(1):e1002882. | |
Datsenko KA, Wanner BL. One-step inactivation of chromosomal genes in Escherichia coli K-12 using PCR products. Proc Natl Acad Sci U S A. 2000;97(12):6640–6645. | |
Thomason LC, Court D, Bubunenko M, et al. Recombineering: genetic engineering in bacteria using homologous recombination. Curr Protoc Mol Biol. 2007;Chapter 1:Unit 1.16. | |
Sabri S, Steen JA, Bongers M, Nielsen LK, Vickers CE. Knock-in/Knock-out (KIKO) vectors for rapid integration of large DNA sequences, including whole metabolic pathways, onto the Escherichia coli chromosome at well-characterised loci. Microb Cell Fact. 2013;12:60. | |
Heap JT, Pennington OJ, Cartman ST, Carter GP, Minton NP. The ClosTron: a universal gene knock-out system for the genus Clostridium. J Microbiol Methods. 2007;70(3):452–464. | |
Tracy BP, Jones SW, Papoutsakis ET. Inactivation of σE and σG in Clostridium acetobutylicum illuminates their roles in clostridial-cell-form biogenesis, granulose synthesis, solventogenesis, and spore morphogenesis. J Bacteriol. 2011;193(6):1414–1426. | |
Tsuda K, Qi Y, Nguyen le V, et al. An efficient Agrobacterium-mediated transient transformation of Arabidopsis. Plant J. 2012;69(4):713–719. | |
Alonso JM, Stepanova AN, Leisse TJ, et al. Genome-wide insertional mutagenesis of Arabidopsis thaliana. Science. 2003;301(5633):653–657. | |
Walter A, Reinicke M, Bocklitz T, et al. Raman spectroscopic detection of physiology changes in plasmid-bearing Escherichia coli with and without antibiotic treatment. Anal Bioanal Chem. 2011;400(9):2763–2773. | |
Jiang W, Bikard D, Cox D, Zhang F, Marraffini LA. RNA-guided editing of bacterial genomes using CRISPR-Cas systems. Nat Biotechnol. 2013;31(3):233–239. | |
Alper H, Fischer C, Nevoigt E, Stephanopoulos G. Tuning genetic control through promoter engineering. Proc Natl Acad Sci U S A. 2005;102(36):12678–12683. | |
Hammer K, Mijakovic I, Jensen PR. Synthetic promoter libraries – tuning of gene expression. Trends Biotechnol. 2006;24(2):53–55. | |
Pfleger BF, Pitera DJ, Smolke CD, Keasling JD. Combinatorial engineering of intergenic regions in operons tunes expression of multiple genes. Nat Biotechnol. 2006;24(8):1027–1032. | |
Salis MH, Mirsky EA, Voigt CA. Automated design of synthetic ribosome binding sites to control protein expression. Nat Biotechnol. 2009;27(10):946–950. | |
Waters LS, Storz G. Regulatory RNAs in bacteria. Cell. 2009;136(4):615–628. | |
Aiba H. Mechanism of RNA silencing by Hfq-binding small RNAs. Curr Opin Microbiol. 2007;10(2):134–139. | |
Yoo SM, Na D, Lee SY. Design and use of synthetic regulatory small RNAs to control gene expression in Escherichia coli. Nat Protoc. 2013;8(9):1694–1707. | |
Kim B, Park H, Na D, Lee SY. Metabolic engineering of Escherichia coli for the production of phenol from glucose. Biotechnol J. 2014;9(5):621–629. | |
Tiwari M, Sharma D, Trivedi PK. Artificial microRNA mediated gene silencing in plants: progress and perspectives. Plant Mol Biol. 2014;86(1–2):1–18. | |
Lange M, Yellina AL, Orashakova S, Becker A. Virus-induced gene silencing (VIGS) in plants: an overview of target species and the virus-derived vector systems. Methods Mol Biol. 2013;975:1–14. | |
Santos CN, Stephanopoulos G. Combinatorial engineering of microbes for optimizing cellular phenotype. Curr Opin Chem Biol. 2008;12(2):168–176. | |
Wang HH, Isaacs FJ, Carr PA, et al. Programming cells by multiplex genome engineering and accelerated evolution. Nature. 2009;460(7257):894–898. | |
Alper H, Miyaoku K, Stephanopoulos G. Construction of lycopene-overproducing E. coli strains by combining systematic and combinatorial gene knockout targets. Nat Biotechnol. 2005;23(5):612–616. | |
Glebes TY, Sandoval NR, Reeder PJ, Schilling KD, Zhang M, Gill RT. Genome-wide mapping of furfural tolerance genes in Escherichia coli. PLoS One. 2014;9(1):e87540. | |
Nicolaou SA, Gaida SM, Papoutsakis ET. Coexisting/Coexpressing Genomic Libraries (CoGeL) identify interactions among distantly located genetic loci for developing complex microbial phenotypes. Nucleic Acids Res. 2011;39(22):e152. | |
Borden JR, Jones SW, Indurthi D, Chen Y, Papoutsakis ET. A genomic-library based discovery of a novel, possibly synthetic, acid-tolerance mechanism in Clostridium acetobutylicum involving non-coding RNAs and ribosomal RNA processing. Metab Eng. 2010;12(3):268–281. | |
Senger RS, Nazem-Bokaee H. Resolving cell composition through simple measurements, genome-scale modeling, and a genetic algorithm. Methods Mol Biol. 2013;985:85–101. | |
Tracy BP, Gaida SM, Papoutsakis ET. Development and application of flow-cytometric techniques for analyzing and sorting endospore-forming clostridia. Appl Environ Microbiol. 2008;74(24):7497–7506. | |
Zu TN, Athamneh AI, Wallace RS, Collakova E, Senger RS. Near real-time analysis of the phenotypic responses of Escherichia coli to 1-butanol exposure using Raman spectroscopy. J Bacteriol. 2014;196(23):3983–3991. | |
Athamneh AI, Alajlouni RA, Wallace RS, Seleem MN, Senger RS. Phenotypic profiling of antibiotic response signatures in Escherichia coli using Raman spectroscopy. Antimicrob Agents Chemother. 2014;58(3):1302–1314. | |
Napier JA, Haslam RP, Beaudoin F, Cahoon EB. Understanding and manipulating plant lipid composition: metabolic engineering leads the way. Curr Opin Plant Biol. 2014;19:68–75. | |
Yu XH, Prakash RR, Sweet M, Shanklin J. Coexpressing Escherichia coli cyclopropane synthase with Sterculia foetida Lysophosphatidic acid acyltransferase enhances cyclopropane fatty acid accumulation. Plant Physiol. 2014;164(1):455–465. | |
Youngs H, Somerville C. Development of feedstocks for cellulosic biofuels. F1000 Biol Rep. 2012;4:10. | |
Chen HC, Song J, Wang JP, et al. Systems biology of lignin biosynthesis in Populus trichocarpa: heteromeric 4-coumaric acid:coenzyme a ligase protein complex formation, regulation, and numerical modeling. Plant Cell. 2014;26(3):876–893. | |
Collakova E, Yen JY, Senger RS. Are we ready for genome-scale modeling in plants? Plant Sci. 2012;191–192:53–70. | |
Wilson SA, Roberts SC. Metabolic engineering approaches for production of biochemicals in food and medicinal plants. Curr Opin Biotechnol. 2014;26:174–182. | |
Mintz-Oron S, Meir S, Malitsky S, Ruppin E, Aharoni A, Shlomi T. Reconstruction of Arabidopsis metabolic network models accounting for subcellular compartmentalization and tissue-specificity. Proc Natl Acad Sci U S A. 2012;109(1):339–344. | |
Suzuki N, Rivero RM, Shulaev V, Blumwald E, Mittler R. Abiotic and biotic stress combinations. New Phytol. 2014;203(1):32–43. | |
Chakrabarti A, Miskovic L, Soh KC, Hatzimanikatis V. Towards kinetic modeling of genome-scale metabolic networks without sacrificing stoichiometric, thermodynamic and physiological constraints. Biotechnol J. 2013;8(9):1043–1057. | |
Jamshidi N, Palsson BO. Formulating genome-scale kinetic models in the post-genome era. Mol Syst Biol. 2008;4:171. | |
Damborsky J, Brezovsky J. Computational tools for designing and engineering enzymes. Curr Opin Chem Biol. 2014;19:8–16. | |
Kiss G, Çelebi-Ölçüm N, Moretti R, Baker D, Houk KN. Computational enzyme design. Angew Chem Int Ed Engl. 2013;52(22):5700–5725. | |
Privett HK, Kiss G, Lee TM, et al. Iterative approach to computational enzyme design. Proc Natl Acad Sci U S A. 2012;109(10):3790–3795. | |
Karr JR, Sanghvi JC, Macklin DN, et al. A whole-cell computational model predicts phenotype from genotype. Cell. 2012;150(2):389–401. |
 © 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.