Back to Journals » Drug Design, Development and Therapy » Volume 10
Design, synthesis, and antihypertensive activity of curcumin-inspired compounds via ACE inhibition and vasodilation, along with a bioavailability study for possible benefit in cardiovascular diseases
Authors Zhuang X, Liao L, Dong X, Hu X, Guo Y, Du Z, Liao X, Wang L
Received 14 September 2015
Accepted for publication 9 October 2015
Published 5 January 2016 Volume 2016:10 Pages 129—139
DOI https://doi.org/10.2147/DDDT.S96315
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Prof. Dr. Wei Duan
Xiao-dong Zhuang,1,* Li-zhen Liao,2,* Xiao-bian Dong,1 Xun Hu,1 Yue Guo,1 Zhi-min Du,1 Xin-xue Liao,1 Li-chun Wang1
1Department of Cardiology, First Affiliated Hospital, Sun Yat-sen University, Guangzhou, People’s Republic of China; 2Department of Health, Guangdong Pharmaceutical University, Guangzhou Higher Education Mega Center, Guangzhou, People’s Republic of China
*These authors contributed equally to this work
Abstract: This study describes the synthesis of a novel series of curcumin-inspired compounds via a facile synthetic route. The structures of these derivatives were ascertained using various spectroscopic and analytic techniques. The pharmacological effects of the target analogs were assessed by assaying their inhibition of angiotensin-converting enzyme (ACE). All of the synthesized derivatives exhibited considerable inhibition of ACE, with half-maximal inhibitory concentrations ranging from 1.23 to 120.32 µM. In a docking analysis with testicular ACE (tACE), the most promising inhibitor (4j) was efficiently accommodated in the deep cleft of the protein cavity, making close interatomic contacts with Glu162, His353, and Ala356, comparable with lisinopril. Compounds 4i, 4j, 4k, and 4l were further selected for determination of their vasodilator activity (cardiac output and stroke volume) on isolated rat hearts using the Langendorff technique. The bioavailability of compound 4j was determined in experimental mice.
Keywords: curcumin, synthesis, ACE inhibition, antihypertensive, vasodilation, bioavailability
Introduction
Hypertension is deemed to be a major contributing factor to several cardiovascular illnesses, including coronary heart disease, ischemia, and hemorrhagic stroke.1 According to recent estimates from the World Health Organization, hypertension is responsible for the premature death of 7.5 million people each year, accounting for approximately 13% of total deaths.2 This disorder is not confined to poor countries, often affecting also those in developed economies; for instance, approximately 77.9 million adults in the US are affected by hypertension.3 Despite various therapeutic interventions to control or treat hypertension, no individual clinical agent entirely serves the purpose. Patients often have to take multiple drugs, each of which targets various mediators responsible for high blood pressure. Such agents might include angiotensin-converting enzyme (ACE) inhibitors/angiotensin-receptor blockers, β-blockers, and diuretics.
Drugs belonging to the ACE inhibitor family suppress the endogenous conversion of angiotensin I to angiotensin II. Under the influence of angiotensin II, the blood vessels are constricted, which results in elevated blood pressure.4 Consequently, the heart has to use greater force to propel the blood into the arterioles, which exacerbates heart failure together with weak cardiac muscles. It has been shown that levels of angiotensin II are elevated in various cardiovascular illnesses. Thus, its inhibition lowers blood pressure and the amount of energy required for the heart to maintain normal blood circulation.5
Despite substantial progress toward an effective understanding of acute heart failure in recent decades, its management remains challenging. Major therapeutic interventions are based on diuretics supplemented by vasodilators. Vasodilators have been found to significantly improve cardiac performance by decreasing arteriolar resistance. In particular, these agents appreciably increase venous capacitance, which results in enlargement of the relative size of the venous blood pool along with decrease in preload and associated pressures.6 Thus, it can be suggested that vasodilators indirectly act on the heart, and reduce the cardiac burden of both acute and chronic heart failure.
Together with antihypertensive drugs, vasodilators specifically correct major hemodynamic irregularities that arise as a result of primary hypertension and can act in an additive manner to treat hypertension (Figure 1).7 This approach offers several advantages and exerts beneficial effects on associated cardiac illnesses, and it improves the risk factors associated with the disease.
 | Figure 1 Dual mode of action to treat hypertension. |
Curcumin (diferuloylmethane), an active principal constituent of Curcuma longa (family: Zingiberaceae), is known for its diverse medicinal properties. It was first isolated in 1815, and its chemical structure was established by J Milobedzka and V Lampe (Germany) approximately 100 years later.8 The importance of curcumin in drug discovery is easily seen by the fact that, within a span of approximately two decades, more than 2,000 publications on the subject have been indexed in the PubMed database. These studies have concluded that curcumin acts as a multifaceted agent, exhibiting antioxidant, antibacterial, antifungal, antiviral, anti-inflammatory, antiproliferative, and proapoptotic effects by targeting various key pathological functions.9
However, because of poor bioavailability, the clinical significance of curcumin is greatly compromised and various efforts have concentrated on improving its bioavailability.10 The basic approach to this has adopted two strategies: the first strategy focuses on the development of delivery options for curcumin (eg, nanoparticle, liposome, micelles, emulsion, and microparticles), while the second strategy is concerned with the structural modification of curcumin. The latter approach has been widely accepted due to its versatility and the ease of introducing chemical diversity.11–15
This report describes the development of novel synthetic analogs inspired by curcumin and their subsequent pharmacologic evaluation. In addition, metabolic and bioavailability studies were conducted in mice.
Experimental
The chemicals were procured from Sigma-Aldrich (St Louis, MO, USA) and used without further purification. Established spectroscopic and analytic methods were used to ascertain the purity and integrity of the synthesized derivatives, including Fourier transform infrared (FTIR) spectroscopy, 1H-nuclear magnetic resonance (NMR), 13C NMR, and mass spectroscopy. The completion of the reaction was monitored by thin-layer chromatography using silica gel G-coated Al plates (0.5 mm thickness; Merck, Darmstadt, Germany). After spotting, the plates were evaluated in an iodine chamber. The uncorrected melting points of products were determined using a melting point apparatus (Electrothermic model, MP-1). FTIR (in KBr) spectra were determined using a PerkinElmer Spectrum RX-I spectroscope (Perkin Elmer Instruments, Buckinghamshire, UK). Proton and carbon NMR spectra (1H NMR and 13C NMR) were recorded using a Bruker Avance II 400 Fourier-Transform-NMR spectrometer (Bruker Corporation, Billerica, MA, USA) in Dimethyl sulfoxide-d6 (DMSO-d6), with tetramethylsilane (TMS) as the internal standard. The chemical shifts are expressed as δ values (ppm), downfield from TMS as the internal standard. Liquid chromatography–mass spectroscopy on a Waters ZQ-4000 using the electrospray ionization technique was used to record the mass spectra of the synthesized derivatives. The elemental analysis was conducted using the Vario Elemental Analyzer (Elementar, Hanau, Germany).
General procedure for synthesis of substituted 3,5-bisarylidene-piperidin-4-ones, 3 (a–p)
4-Piperidone hydrate hydrochloride (0.10 mol) was taken along with desired aromatic aldehydes (0.20 mol) and acetic acid (150 mL) in a 250 mL round bottom flask. Additionally, through this reaction mixture, HCl (g) was flushed for 30 minutes with continuous stirring to afford solid pale-orange-colored precipitate. After that, HCl was removed and the mixture was stirred constantly overnight. The resultant precipitate was then filtered to furnish the corresponding analogs 3 (a–p).
Synthesis of title analogs 4 (a–p)
To the above solution of respective compound 3, appropriate acyl chloride (0.01 mol) was added in the presence of dry pyridine at room temperature for 2 hours. The resultant mixture was then diluted with ethyl acetate and extracted with 3 N HCl, saturated solution of NaHCO3 (aq.), and brine. After rigorous extraction, the pooled solution was dried over magnesium sulfate and the solvent was removed. The resulting solid was dissolved in methanol and cooled saturated solution of ammonia was added to it. This mixture was kept in a sealed container overnight at room temperature with constant stirring. On the next day, before opening the vessel, it was cooled, and evaporated to dryness. The residue was further purified with the help of column chromatography with the assumed polarity of the solvent to afford the pure crystalline 4 (a–p).
(3E,5E)-3,5-dibenzylidene-1-ethylpiperidin-4-one (4a)
Yield: 83%; mp: 168°C–169°C; molecular weight (MW): 303.16; Rf: 0.59; FTIR (νmax; cm−1 KBr): 3,128 (C–Hstr aromatic), 2,982 (C–Hstr alkanes), 1,727 (C=Ostr), 1,523 (C=Cstr), 1,476 (C=Nstr), 985, 882; 1H NMR (400 MHz, DMSO, TMS) δ ppm: 7.64 (s, 2H, CH), 7.48 (m, 4H, Ar–H), 7.41 (m, 4H, Ar–H), 7.34 (s, 2H, Ar–H), 3.41 (m, 4H, methylene), 2.61 (s, 2H, methylene), 1.09 (s, 3H, CH3); 13C NMR (100 MHz, DMSO) δ ppm: 186.4, 145.8, 140.5, 135.3, 128.8, 128.2, 127.9, 53.5, 52.5, 13.5; Mass: 304.18 (M + H)+; elemental analysis for C21H21NO: calculated: C, 82.13; H, 6.98; N, 4.62. Found: C, 82.09; H, 6.99; N, 4.63.
(3E,5E)-3,5-dibenzylidene-1-propylpiperidin-4-one (4b)
Yield: 75%; mp: 182°C–183°C; MW: 317.42; Rf: 0.52; FTIR (νmax; cm−1 KBr): 3,136 (C–Hstr aromatic), 2,989 (C–Hstr alkanes), 1,721 (C=Ostr), 1,529 (C=Cstr), 1,471 (C=Nstr), 989, 886; 1H NMR (400 MHz, DMSO, TMS) δ ppm: 7.67 (s, 2H, CH), 7.58 (m, 4H, Ar–H), 7.45 (m, 4H, Ar–H), 7.31 (s, 2H, Ar–H), 3.45 (m, 4H, methylene), 2.46 (s, 2H, methylene), 1.43 (s, 2H, methylene), 0.92 (s, 3H, CH3); 13C NMR (100 MHz, DMSO) δ ppm: 186.3, 145.9, 140.7, 135.3, 128.7, 128.3, 127.2, 59.6, 53.8, 21.6, 11.8; Mass: 318.46 (M + H)+; elemental analysis for C22H23NO: calculated: C, 83.24; H, 7.30; N, 4.41. Found: C, 83.27; H, 7.31; N, 4.40.
(3E,5E)-3,5-dibenzylidene-1-butylpiperidin-4-one (4c)
Yield: 68%; mp: 187°C–188°C; MW: 331.45; Rf: 0.69; FTIR (νmax; cm−1 KBr): 3,128 (C–Hstr aromatic), 2,991 (C–Hstr Alkanes), 1,716 (C=Ostr), 1,522 (C=Cstr), 1,476 (C=Nstr), 982, 884; 1H NMR (400 MHz, DMSO, TMS) δ ppm: 7.64 (s, 2H, CH), 7.57 (m, 4H, Ar–H), 7.43 (m, 4H, Ar–H), 7.32 (s, 2H, Ar–H), 3.41 (m, 4H, methylene), 2.45 (s, 2H, methylene), 1.36 (s, 2H, methylene), 1.31 (s, 2H, methylene), 0.98 (s, 3H, CH3); 13C NMR (100 MHz, DMSO) δ ppm: 186.2, 145.8, 140.4, 135.3, 128.7, 128.2, 127.9, 57.1, 53.8, 30.2, 20.4, 13.7; Mass: 333.48 (M+H)+; elemental analysis for C23H25NO: calculated: C, 83.34; H, 7.60; N, 4.23. Found: C, 83.36; H, 7.58; N, 4.21.
(3E,5E)-3,5-dibenzylidene-1-pentylpiperidin-4-one (4d)
Yield: 62%; mp: 180°C–181°C; MW: 345.48; Rf: 0.79; FTIR (νmax; cm−1 KBr): 3,138 (C–Hstr aromatic), 2,978 (C–Hstr alkanes), 1,719 (C=Ostr), 1,518 (C=Cstr), 1,468 (C=Nstr), 987, 881; 1H NMR (400 MHz, DMSO, TMS) δ ppm: 7.62 (s, 2H, CH), 7.59 (m, 4H, Ar–H), 7.41 (m, 4H, Ar–H), 7.34 (s, 2H, Ar–H), 3.38 (m, 4H, methylene), 2.43 (s, 2H, methylene), 1.35 (s, 2H, methylene), 1.32 (s, 2H, methylene), 1.28 (s, 2H, methylene), 1.02 (s, 3H, CH3); 13C NMR (100 MHz, DMSO) δ ppm: 186.4, 145.9, 140.2, 135.3, 128.7, 128.2, 127.6, 57.3, 53.2, 29.5, 28.2, 22.4, 14.1; Mass: 346.46 (M + H)+; elemental analysis for C24H27NO: calculated: C, 83.44; H, 7.88; N, 4.05. Found: C, 83.42; H, 7.88; N, 4.04.
(3E,5E)-3,5-bis(3,4-dihydroxybenzylidene)-1-ethylpiperidin-4-one (4e)
Yield: 59%; mp: 203°C–204°C; MW: 367.40; Rf: 0.71; FTIR (νmax; cm−1 KBr): 3,448 (O–Hstr), 3,142 (C–Hstr aromatic), 2,984 (C–Hstr alkanes), 1,714 (C=Ostr), 1,512 (C=Cstr), 1,458 (C=Nstr), 981, 879; 1H NMR (400 MHz, DMSO, TMS) δ ppm: 7.65 (s, 2H, CH), 7.17–6.93 (m, 6H, Ar–H), 5.42 (s, 4H, Ar–OH), 3.39 (m, 4H, methylene), 2.68 (s, 2H, methylene), 1.04 (s, 3H, CH3); 13C NMR (100 MHz, DMSO) δ ppm: 186.5, 146.7, 145.9, 145.2, 140.8, 127.9, 126.4, 117.3, 115.2, 53.5, 52.4, 13.5; Mass: 368.42 (M + H)+; elemental analysis for C21H21NO5: calculated: C, 68.65; H, 5.76; N, 3.81. Found: C, 68.67; H, 5.75; N, 3.81.
(3E,5E)-3,5-bis(3,4-dihydroxybenzylidene)-1-propylpiperidin-4-one (4f)
Yield: 81%; mp: 208°C–209°C; MW: 381.42; Rf: 0.78; FTIR (νmax; cm−1 KBr): 3,452 (O–Hstr), 3,146 (C–Hstr aromatic), 2,987 (C–Hstr alkanes), 1,718 (C=Ostr), 1,513 (C=Cstr), 1,464 (C=Nstr), 986, 872; 1H NMR (400 MHz, DMSO, TMS) δ ppm: 7.61 (s, 2H, CH), 7.19–6.93 (m, 6H, Ar–H), 5.41 (s, 4H, Ar–OH), 3.41 (m, 4H, methylene), 2.48 (s, 2H, methylene), 1.42 (s, 2H, methylene), 1.02 (s, 3H, CH3); 13C NMR (100 MHz, DMSO) δ ppm : 186.4, 146.5, 145.8, 140.8, 127.8, 126.4, 117.2, 115.4, 59.8, 53.9, 21.8, 11.8; Mass: 382.46 (M + H)+; elemental analysis for C22H23NO5: calculated: C, 69.28; H, 6.08; N, 3.67. Found: C, 69.30; H, 6.07; N, 3.67.
(3E,5E)-1-butyl-3,5-bis(3,4-dihydroxybenzylidene)piperidin-4-one (4g)
Yield: 73%; mp: 212°C–213°C; MW: 395.45; Rf: 0.67; FTIR (νmax; cm−1 KBr): 3,459 (O–Hstr), 3,142 (C–Hstr aromatic), 2,989 (C–Hstr alkanes), 1,724 (C=Ostr), 1,529 (C=Cstr), 1,478 (C=Nstr), 981, 879; 1H NMR (400 MHz, DMSO, TMS) δ ppm: 7.59 (s, 2H, CH), 7.19–6.91 (m, 6H, Ar–H), 5.39 (s, 4H, Ar–OH), 3.42 (m, 4H, methylene), 2.47 (s, 2H, methylene), 1.38 (s, 2H, methylene), 1.29 (s, 2H, methylene), 1.04 (s, 3H, CH3); 13C NMR (100 MHz, DMSO) δ ppm: 186.5, 146.8, 145.7, 140.6, 127.9, 126.3, 117.2, 115.4, 57.2, 53.9, 30.8, 20.5, 13.8; Mass: 396.48 (M + H)+; elemental analysis for C23H25NO5: calculated: C, 69.86; H, 6.37; N, 3.54. Found: C, 69.89; H, 6.39; N, 3.54.
(3E,5E)-3,5-bis(3,4-dihydroxybenzylidene)-1-pentylpiperidin-4-one (4h)
Yield: 77%; mp: 219°C–221°C; MW: 409.47; Rf: 0.56; FTIR (νmax; cm−1 KBr): 3,462 (O–Hstr), 3,147 (C–Hstr aromatic), 2,993 (C–Hstr alkanes), 1,727 (C=Ostr), 1,534 (C=Cstr), 1,472 (C=Nstr), 989, 871; 1H NMR (400 MHz, DMSO, TMS) δ ppm: 7.62 (s, 2H, CH), 7.21–6.93 (m, 6H, Ar–H), 5.37 (s, 4H, Ar–OH), 3.39 (m, 4H, methylene), 2.46 (s, 2H, methylene), 1.37 (s, 2H, methylene), 1.31 (s, 2H, methylene), 1.27 (s, 2H, methylene), 1.02 (s, 3H, CH3); 13C NMR (100 MHz, DMSO) δ ppm: 186.5, 146.7, 145.8, 140.4, 127.9, 126.4, 117.3, 115.2, 57.5, 53.9, 29.5, 28.2, 22.6, 14.2; Mass: 410.49 (M + H)+; elemental analysis for C24H27NO5: calculated: C, 70.40; H, 6.65; N, 3.42. Found: C, 70.42; H, 6.67; N, 3.43.
(3E,5E)-1-ethyl-3,5-bis(3,4,5-trimethoxybenzylidene)piperidin-4-one (4i)
Yield: 65%; mp: 232°C–233°C; MW: 483.55; Rf: 0.67; FTIR (νmax; cm−1 KBr): 3,156 (C–Hstr aromatic), 2,983 (C–Hstr alkanes), 2,857 (O–CH3str), 1,721 (C=Ostr), 1,539 (C=Cstr), 1,475 (C=Nstr), 982, 876; 1H NMR (400 MHz, DMSO, TMS) δ ppm: 7.58 (s, 2H, CH), 6.73– (m, 4H, Ar–H), 3.92 (s, 18H, Ar–OCH3), 3.37 (m, 4H, methylene), 2.68 (s, 2H, methylene), 1.04 (s, 3H, CH3); 13C NMR (100 MHz, DMSO) δ ppm: 186.3, 153.2, 145.4, 140.2, 138.6, 125.3, 107.2, 60.8, 56.3, 53.8, 52.9, 13.6; Mass: 484.58 (M + H)+; elemental analysis for C27H33NO7: calculated: C, 67.06; H, 6.88; N, 2.90. Found: C, 67.09; H, 6.89; N, 2.89.
(3E,5E)-1-propyl-3,5-bis(3,4,5-trimethoxybenzylidene)piperidin-4-one (4j)
Yield: 69%; mp: 238°C–239°C; MW: 497.58; Rf: 0.72; FTIR (νmax; cm−1 KBr): 3,159 (C–Hstr aromatic), 2,987 (C–Hstr alkanes), 2,859 (O–CH3str), 1,723 (C=Ostr), 1,545 (C=Cstr), 1,478 (C=Nstr), 987, 872; 1H NMR (400 MHz, DMSO, TMS) δ ppm: 7.63 (s, 2H, CH), 6.79– (m, 4H, Ar–H), 3.91 (s, 18H, Ar–OCH3), 3.39 (m, 4H, methylene), 2.47 (s, 2H, methylene), 1.43 (s, 2H, methylene), 0.92 (s, 3H, CH3); 13C NMR (100 MHz, DMSO) δ ppm: 186.4, 153.2, 145.9, 140.7, 138.5, 125.2, 107.5, 60.9, 59.7, 56.2, 53.9, 21.7, 11.8; Mass: 498.61 (M + H)+; elemental analysis for C28H35NO7: calculated: C, 67.59; H, 7.09; N, 2.81. Found: C, 67.61; H, 7.08; N, 2.81.
(3E,5E)-1-butyl-3,5-bis(3,4,5-trimethoxybenzylidene)piperidin-4-one (4k)
Yield: 74%; mp: 252°C–253°C; MW: 511.61; Rf: 0.57; FTIR (νmax; cm−1 KBr): 3,163 (C–Hstr aromatic), 2,992 (C–Hstr alkanes), 2,861 (O–CH3str), 1,714 (C=Ostr), 1,548 (C=Cstr), 1,474 (C=Nstr), 989, 877; 1H NMR (400 MHz, DMSO, TMS) δ ppm: 7.65 (s, 2H, CH), 6.82– (m, 4H, Ar–H), 3.89 (s, 18H, Ar–OCH3), 3.37 (m, 4H, methylene), 2.48 (s, 2H, methylene), 1.37 (s, 2H, methylene), 1.31 (s, 2H, methylene), 0.96 (s, 3H, CH3); 13C NMR (100 MHz, DMSO) δ ppm: 186.4, 153.2, 145.9, 140.8, 138.4, 125.4, 107.4, 60.9, 57.2, 56.6, 53.8, 30.8, 20.5, 13.9; Mass: 512.63 (M + H)+; elemental analysis for C29H37NO7: calculated: C, 68.08; H, 7.29; N, 2.74. Found: C, 68.09; H, 7.30; N, 2.74.
(3E,5E)-1-pentyl-3,5-bis(3,4,5-trimethoxybenzylidene)piperidin-4-one (4l)
Yield: 63%; mp: 267°C–268°C; MW: 525.63; Rf: 0.65; FTIR (νmax; cm−1 KBr): 3,167 (C–Hstr aromatic), 2,994 (C–Hstr alkanes), 2,865 (O–CH3str), 1,716 (C=Ostr), 1,543 (C=Cstr), 1,476 (C=Nstr), 981, 879; 1H NMR (400 MHz, DMSO, TMS) δ ppm: 7.63 (s, 2H, CH), 6.79– (m, 4H, Ar–H), 3.87 (s, 18H, Ar–OCH3), 3.39 (m, 4H, methylene), 2.49 (s, 2H, methylene), 1.36 (s, 2H, methylene), 1.32 (s, 2H, methylene), 1.28 (s, 2H, methylene), 0.92 (s, 3H, CH3); 13C NMR (100 MHz, DMSO) δ ppm: 186.4, 153.9, 145.8, 140.6, 138.5, 125.6, 107.8, 60.9, 57.4, 56.2, 53.9, 29.4, 28.5, 22.1, 14.2; Mass: 526.67 (M + H)+; elemental analysis for C30H39NO7: calculated: C, 68.55; H, 7.48; N, 2.66. Found: C, 68.57; H, 7.47; N, 2.66.
(3E,5E)-1-ethyl-3,5-bis(4-nitrobenzylidene)piperidin-4-one (4m)
Yield: 76%; mp: 236°C–237°C; MW: 393.39; Rf: 0.68; FTIR (νmax; cm−1 KBr): 3,169 (C–Hstr aromatic), 2,997 (C–Hstr alkanes), 1,726 (C=Ostr), 1,549 (C=Cstr), 1,537 (NO2str), 1,479 (C=Nstr), 984, 892; 1H NMR (400 MHz, DMSO, TMS) δ ppm: 8.28–8.06 (m, 4H, Ar–H), 7.74– (s, 2H, C–H), 3.42 (m, 4H, methylene), 2.68 (s, 2H, methylene), 1.03 (s, 3H, CH3); 13C NMR (100 MHz, DMSO) δ ppm: 186.4, 147.2, 145.9, 141.3, 140.8, 129.6, 123.9, 53.6, 52.4, 13.5; Mass: 394.41 (M + H)+; elemental analysis for C21H19N3O5: calculated: C, 64.12; H, 4.87; N, 10.68. Found: C, 64.14; H, 4.89; N, 10.69.
(3E,5E)-3,5-bis(4-nitrobenzylidene)-1-propylpiperidin-4-one (4n)
Yield: 86%; mp: 242°C–243°C; MW: 407.42; Rf: 0.75; FTIR (νmax; cm−1 KBr): 3,172 (C–Hstr aromatic), 2,985 (C–Hstr alkanes), 1,729 (C=Ostr), 1,552 (C=Cstr), 1,534 (NO2str), 1,473 (C=Nstr), 987, 895; 1H NMR (400 MHz, DMSO, TMS) δ ppm: 8.27–8.04 (m, 4H, Ar–H), 7.68– (s, 2H, C–H), 3.41 (m, 4H, methylene), 2.45 (s, 2H, methylene), 1.42 (s, 2H, methylene), 0.96 (s, 3H, CH3); 13C NMR (100 MHz, DMSO) δ ppm: 186.5, 147.3, 145.8, 141.4, 140.8, 129.3, 123.9, 59.6, 53.9, 21.7, 11.8; Mass: 408.43 (M + H)+; elemental analysis for C22H21N3O5: calculated: C, 64.86; H, 5.20; N, 10.31. Found: C, 64.88; H, 5.21; N, 10.32.
(3E,5E)-1-butyl-3,5-bis(4-nitrobenzylidene)piperidin-4-one (4o)
Yield: 79%; mp: 254°C–255°C; MW: 421.45; Rf: 0.70; FTIR (νmax; cm−1 KBr): 3,177 (C–Hstr aromatic), 2,989 (C–Hstr alkanes), 1,723 (C=Ostr), 1,559 (C=Cstr), 1,537 (NO2str), 1,478 (C=Nstr), 983, 892; 1H NMR (400 MHz, DMSO, TMS) δ ppm: 8.24–8.03 (m, 4H, Ar–H), 7.78– (s, 2H, C–H), 3.39 (m, 4H, methylene), 2.47 (s, 2H, methylene), 1.36 (s, 2H, methylene), 1.31 (s, 2H, methylene), 0.99 (s, 3H, CH3); 13C NMR (100 MHz, DMSO) δ ppm: 186.4, 147.2, 145.9, 141.4, 140.8, 129.3, 123.9, 57.3, 53.6, 30.8, 20.6, 13.8; Mass: 422.46 (M + H)+; elemental analysis for C23H23N3O5: calculated: C, 65.55; H, 5.50; N, 9.97. Found: C, 65.58; H, 5.51; N, 9.98.
(3E,5E)-3,5-bis(4-nitrobenzylidene)-1-pentylpiperidin-4-one (4p)
Yield: 74%; mp: 263°C–264°C; MW: 435.47; Rf: 0.59; FTIR (νmax; cm−1 KBr): 3,182 (C–Hstr aromatic), 2,985 (C–Hstr alkanes), 1,728 (C=Ostr), 1,551 (C=Cstr), 1,539 (NO2str), 1,474 (C=Nstr), 987, 896; 1H NMR (400 MHz, DMSO, TMS) δ ppm: 8.27–8.05 (m, 4H, Ar–H), 7.74– (s, 2H, C–H), 3.42 (m, 4H, methylene), 2.48 (s, 2H, methylene), 1.38 (s, 2H, methylene), 1.32 (s, 2H, methylene), 1.27 (s, 2H, methylene), 0.94 (s, 3H, CH3); 13C NMR (100 MHz, DMSO) δ ppm: 186.3, 147.3, 145.8, 141.4, 140.8, 129.3, 123.9, 57.6, 53.9, 29.6, 28.3, 22.6, 14.2; Mass: 436.49 (M + H)+; elemental analysis for C24H25N3O5: calculated: C, 66.19; H, 5.79; N, 9.65. Found: C, 66.21; H, 5.80; N, 9.65.
Pharmacologic activity
In vitro ACE inhibition assay
As described earlier, a colorimetric method was used to determine the in vitro ACE inhibitory activity of the synthesized derivatives. The determination began with the preparation of a suspension of 1 g rabbit lung acetone powder (Sigma-Aldrich) in 10 mL of 0.05 M sodium borate buffer (pH 8.2) containing 0.3 M NaCl and 0.5% Triton X-100. This suspension was then centrifuged at 15,000 rpm for 60 minutes at 4°C. The resulting supernatant served as the source of ACE. This assay was based on determination of the level of hippuric acid generated after hydrolysis of the substrate, hippuryl histidyl leucine (HHL; Sigma-Aldrich); the level of hydrolysis was directly related to the inhibitory ability of the compound. A 1 mM solution of both test and standard drug (lisinopril) was preincubated with 7 mL ACE for 10 minutes at 37°C. To obtain a similar concentration for avoiding the interference, the final volume was adjusted using 0.05 M sodium borate buffer (pH 8.2) containing 0.3 M NaCl. A total of 50 mL of 5 mM substrate (HHL) was used to start the enzyme reaction, and the mixture was incubated at 37°C. After 30 minutes, the reaction was stopped by adding 0.1 mL of 1 M HCl. The hippuric acid thus produced was then allowed to react with 0.2 mL of pyridine and 0.1 mL of benzene sulfonyl chloride (Sigma-Aldrich), to form a yellow solution. ACE inhibition was expressed as percentage inhibition and calculated using the following equation:
|
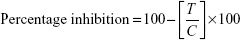
where T is the absorbance of the test reaction and C the absorbance of the control reaction.
The IC50 was calculated using nonlinear regression. A dose–response curve was obtained by plotting the percentage inhibition versus the concentration.
Vasodilator activity
For determination of the vasodilatory effect, Wistar albino rats of either sex, weighing 175–200 g, were acquired. The experiment was approved by the Institutional Animal Ethical Committee. The rats were fed a standard laboratory diet and had ad libitum access to water. The hearts of the experimental rats were excised and rapidly transferred to Krebs–Henseleit buffer solution at 37°C. The pericardial and lung tissues were removed and the aorta was cut just below the point of division. After insertion of a glass cannula, perfusion with oxygenated Krebs–Henseleit buffer solution was started. Each heart was then transferred to a glass perfusion apparatus and the heart rate was measured through a chronometer connected to a physiograph. The solution of test compounds (10 μg) along with a standard solution (10 μg sodium nitroprusside) was administered by injection into the perfusion medium just above the aortic cannula, and the cardiac output was measured using a drop counter. Before recording the parameters induced by BaCl2, normal heart activity was recorded.16
NO donor activity
Solutions of test compounds were dissolved in DMSO, maintaining the concentration at 100 μmol. The resulting solution was added to phosphate buffer (pH 7.4, 50 mM) containing 5 mM L-cysteine hydrochloride and made up to the final volume with the required quantity of buffer. The reaction mixture was kept for 3 hours at 37°C and 500 μL of Griess reagent was added. After 20 minutes, the absorbance of the solution was determined at 550 nm. Sodium nitrite solution (5–50 μM) was used as the standard for the calibration curve.
Bioavailability study of compound 4j
The plasma concentration of the test compound was analyzed in 20- to 25-week-old Swiss Webster mice. The experiment was duly approved by the Institutional Animal Ethical Committee. Mice were divided into control and test groups (n=5), with the test group receiving compound 4j (3 mg per 30 g body weight) by oral administration using a dosing needle. Whole blood was collected and 4j was isolated from the plasma. For this, the samples were first precipitated with methanol (40%) and centrifuged (14,000 rpm) for 10 minutes. The resulting supernatant was used for high performance liquid chromatography analysis. Ascentis RP-Amide was used as the stationary phase for determining 4j, using gradient elution (buffer A [50 mmol/L NaH2PO4, pH 4.3] for 0–5 minutes, a linear change from buffer A to buffer B [70% acetonitrile] for 5–20 minutes, and buffer B for 5 minutes) at a flow rate of 0.7 mL/min. The level of analyte was determined using an electrochemical detector.
Results and discussion
Chemistry
As depicted in Figure 2, the target compounds were developed using a short and cost-effective route. The synthesis was started using commercially available diverse aldehydes (2 eq.), which were allowed to react with 4-piperidone hydrate hydrochloride (1 eq.) in acetic acid. This reaction was carried out in an atmosphere of dry HCl gas to afford salts of 3,5-diaryl-methylene-piperidone acid (3). This salt served as a template for the next reaction, in which it was acylated with numerous acyl chlorides to produce the target derivatives, 4 (a–p).
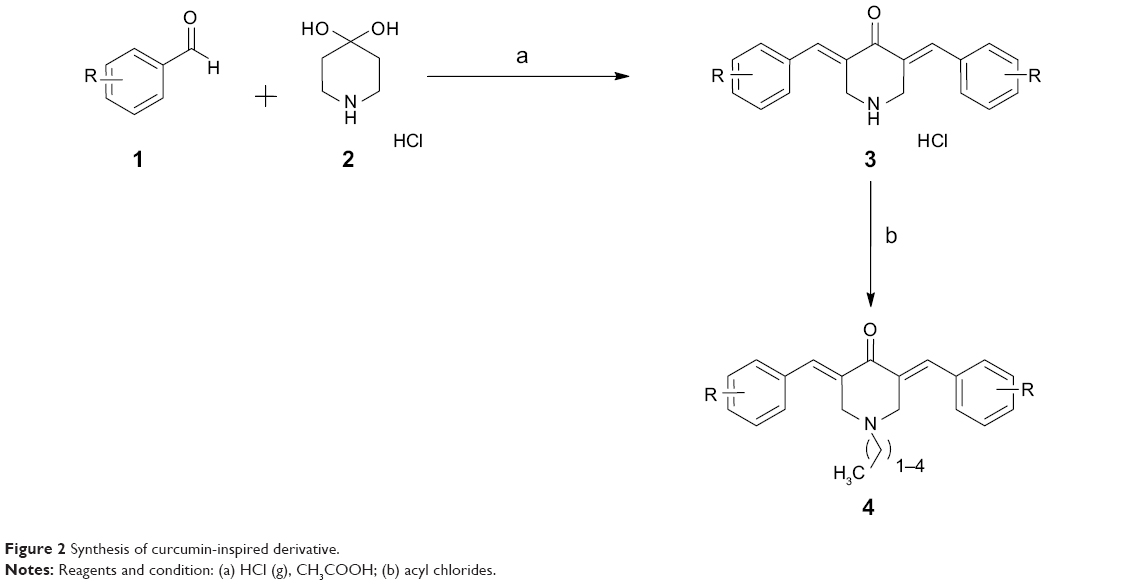 | Figure 2 Synthesis of curcumin-inspired derivative. |
Pharmacological activity
ACE inhibition assay
The inhibitory activity of target analogs against ACE was determined in an enzyme inhibition assay using a colorimetric method (Table 1). The experiment was carried out at 1.0 μM concentration of test compounds and using lisinopril as a standard. The target compounds were designed in such a way as to create diversity around the core skeleton using the easiest possible synthetic steps. Therefore, aldehydes were chosen based upon their chemical character (ie, electron-withdrawing and -donating groups). Later, different chain lengths of acyl chlorides were used to assess their effects on the bioactivity of the compounds. A comparative inhibition analysis found that all of the synthesized derivatives exhibited considerable inhibition of ACE, with half-maximal inhibitory concentrations (IC50) ranging from 1.23 to 120.32 μM. Among the tested derivatives, compound 4j was identified as the most promising inhibitor; the lowest level of activity was exhibited by compound 4o. The results showed that nonsubstituted derivatives (R=H) exhibited moderate activity against ACE. Similar inhibitory potency (≈50 μM) was demonstrated for compounds 4a and 4b, while lengthening the N-acyl chain (compounds 4c and 4d) led to loss of the activity.
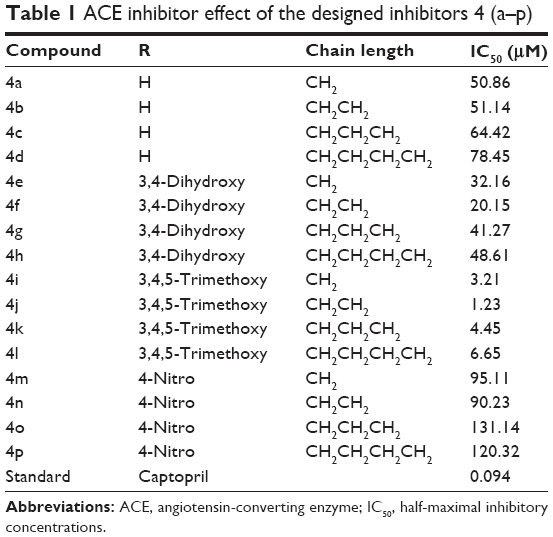 | Table 1 ACE inhibitor effect of the designed inhibitors 4 (a–p) |
The introduction of substituents resulted in marked improvements in inhibitory potency. For example, compound 4e showed an IC50 of 32.16 μM against ACE. Furthermore, significant improvements in activity were observed for the next compound (4f), which contained an ethyl group as a linker. However, a more than twofold decline in activity was observed upon increasing the N-acyl linker length (4g and 4h). The most prominent activity was shown by compounds that contained a trimethoxy substituent (4i–4l). Compound 4j demonstrated prominent inhibition of ACE, with an IC50 of 1.23 μM. However, this activity was reduced on further extension of the chain length, with minimal activity in the case of compound 4l. The rest of the compounds (4m–4p) showed less activity against ACE.
Structure–activity relationship studies suggested that structural variations in the compounds had a marked influence on activity. It has previously been found that substituents play a critical role in generating activity. Compounds containing an electron-donating group proved to be more efficient inhibitors than their electron-withdrawing congeners, while nonsubstituted analogs had a mild effect on activity. The length of the acyl side chain has its own influence on the activity. Particularly, compounds containing a 2-N-acyl chain were found to be more efficient inhibitors than those with different chain lengths. A detailed structure–activity relationship analysis is shown in Figure 3.
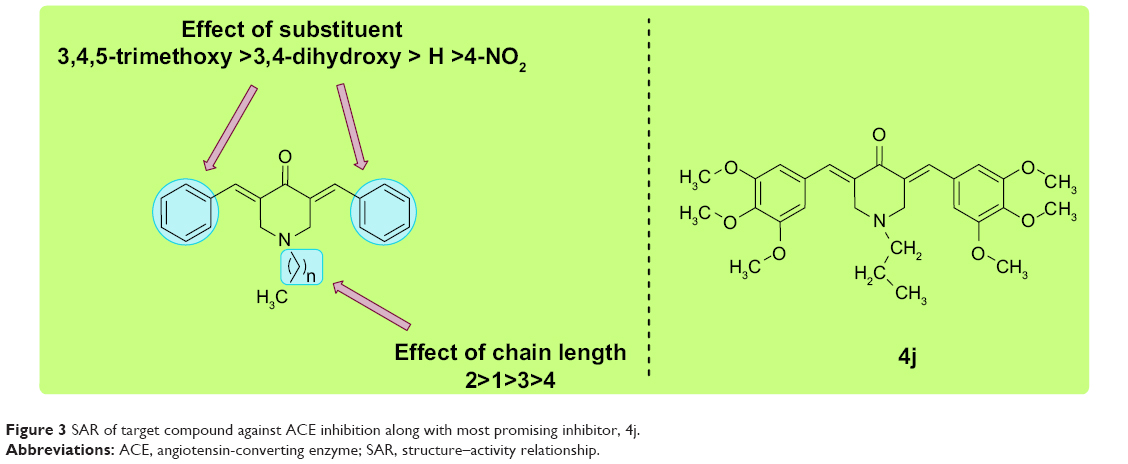 | Figure 3 SAR of target compound against ACE inhibition along with most promising inhibitor, 4j. |
Docking study
Docking is considered to be a powerful simulation of the molecular recognition process. It is used to illustrate the probable molecular interaction of a designed ligand with the protein of interest, predict the affinity and activity of the ligand, and identify the energy of the interaction between the ligand and protein.17 To provide efficient results, it uses algorithms such as molecular dynamics, Monte Carlo stimulation, and fragment-based search methods. Thus, on the basis of the in vitro results, a docking study was performed of compound 4j with tACE.
The crystal structure of human ACE (Protein Data Bank [PDB]: 1O86) was used for this study. The cocrystallized ligand of the protein (ie, lisinopril) was extracted, and heteroatoms and water molecules were removed to “clean” the protein for further analysis. The analysis was carried out using the CDOCKER program within Discovery Studio 2.5 (Accelrys, San Diego, CA, USA). Compound 4j was found to be efficiently accommodated in the deep cleft of the protein cavity (Figure 4), making close interatomic contacts through the formation of H-bonds with Glu162, His353, and Ala356. The trimethoxy fragment of the compound was found to be buried inside the major cleft of the active site, surrounded by the key catalytic residues (ie, Tyr523, Tyr520, Glu152, Asp453, Val380, Ala354, His353, Ser355, Lys454, Ala356, His387, and Glu384). Moreover, the aromatic fragment of the ligand (ie, trimethoxy phenyl) was found to be oriented toward the aromatic residues of the active site (Tyr523 and Tyr520), and was identified as the main determinant of its stabilization in the active site. Comparing the molecular interactions of 4j versus lisinopril against tACE revealed a high degree of resemblance.18 Thus, it appeared that compound 4j might have a similar mechanism of action.
 | Figure 4 Docking study of compound 4j in the active site of human ACE. |
Vasodilator activity
Animals have long been used to understand human biology. In particular, in cardiovascular research, animal models have allowed the study of associated diseases in the early stages, as well as mechanisms of therapeutic interventions. In this study, we used the Langendorff technique to generate a hypoxic–ischemic injury in a rat model. This technique has increased the ease with which arterial and venous perfusion samples can be obtained and allows direct or indirect monitoring of such parameters as arteriovenous differences in pH, PO2, ion content, substrate uptake, metabolite clearance, and enzyme leakage. Moreover, biopsies can be retrieved for biochemical or morphologic studies, or the whole heart can be used. The Langendorff preparation is stable and can be maintained for many hours, with minimal cardiac output and oxygen requirements.19
Among the designed analogs, those compounds showing most promise with respect to ACE inhibition (ie, 4i, 4j, 4k, and 4l) were further selected for determination of their vasodilator activity. For this, an in vitro experiment was conducted using 10 μg of the test compounds on isolated rat hearts using the Langendorff technique. The ability of the compound to influence vasodilatation was determined by quantifying cardiac output and stroke volume in the experimental subjects. The effect of the compounds was also determined in the presence of barium chloride (BaCl2)-induced vasoconstriction (via inhibition of the K+ ion channels), which leads to reductions in cardiac output and stroke volume.
As shown in Figure 5, none of the tested derivatives was associated with improvements in the heart rate. However, the reduced cardiac output and stroke volume under the influence of BaCl2 were significantly elevated on treatment with the test compounds (Figures 6 and 7). Therefore, in experimental rats, the tested analogs were associated with improvements in cardiac blood flow without influencing the contraction rate. This suggests that the target site of these compounds might be the coronary blood vessels, rather than the cardiac muscles.
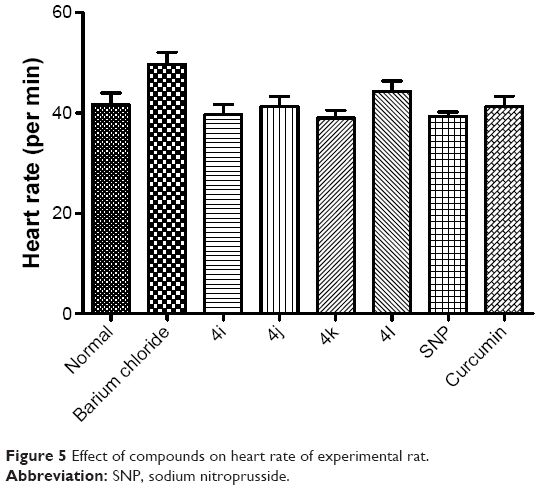 | Figure 5 Effect of compounds on heart rate of experimental rat. |
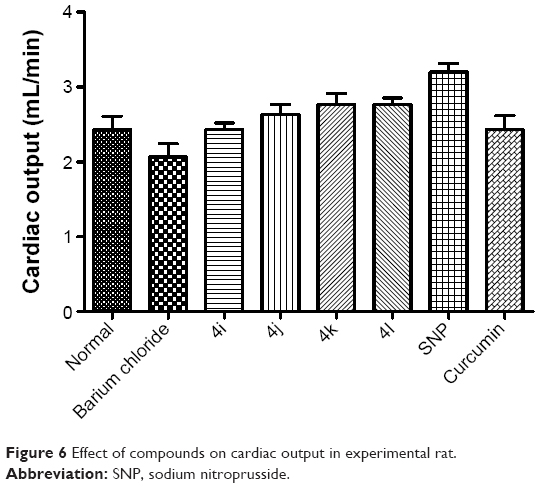 | Figure 6 Effect of compounds on cardiac output in experimental rat. |
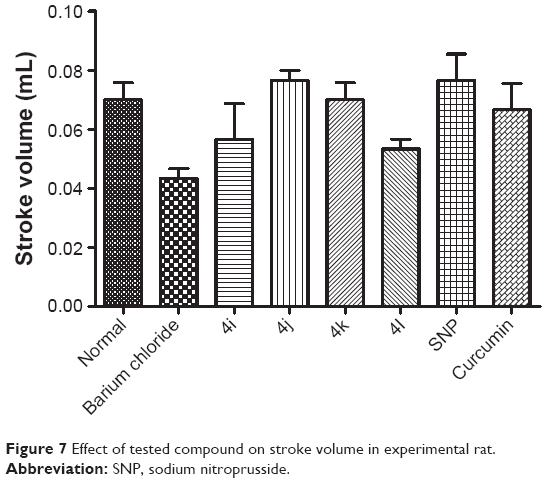 | Figure 7 Effect of tested compound on stroke volume in experimental rat. |
To explore the probable mechanism of action of these derivatives, they were further tested for their ability to donate nitric oxide (NO) by the Griess reaction. NO is a radical gas that is also known as endothelium-derived relaxing factor. It is used by the endothelium of blood vessels to signal to the surrounding smooth muscle, via a cascade of biological events, resulting in the relaxation of arteries and veins. This relaxation leads to vasodilatation and increased blood flow. Therefore, the ability to release NO˙ was calculated as the percentage of NO2˙ (ie, as the ratio between the number of NO2˙ mol detected and the number of mol of test derivatives, multiplied by 100). At the tested dose, the entire set of compounds showed considerable NO donor activity (Figure 8). Moreover, it was surprising to find that an increasing number of N-acyl side chains was associated with improvements in donor capability, with the highest donor activity being seen for compound 4l. Thus, chain length might have a prominent influence on activity.
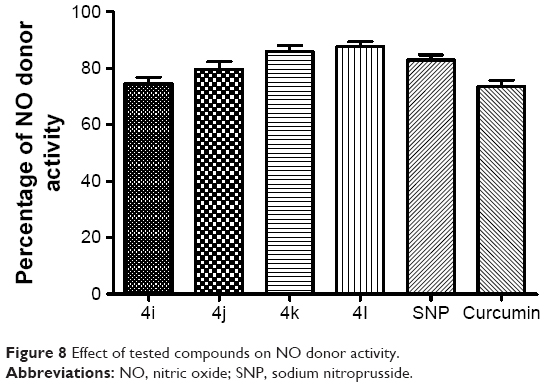 | Figure 8 Effect of tested compounds on NO donor activity. |
Because these compounds are derived from curcumin, it is important to compare the effects of the most promising of these compounds with those of curcumin. As can be seen in Figures 5–8, the administration of curcumin did not result in any significant changes in the parameters discussed above, when compared with the normal control group.
Bioavailability of compound 4j after oral administration in mice
The poor bioavailability of curcumin seriously hampers its clinical efficacy and utility. A study have indicated that the oral bioavailability of curcumin is very poor in experimental rats because of its erratic absorption and ease of metabolism via glucuronidation.15 Because we developed the compounds in this study with the aim of improving the bioavailability of curcumin, it was important to gain insight regarding their bioavailability. Therefore, the plasma concentrations of the most promising inhibitor, 4j, were determined using high-performance liquid chromatography after oral administration of 3 mg per 30 g body weight to experimental mice, along with parallel administration of curcumin in other group 2 g/kg.
In this bioavailability determination experiment, compound 4j was not detected until 10 minutes. As time went on, however, its concentration rose sharply (to 35 μmol/L) and the plasma concentration of the drug was elevated until 40 minutes, which corresponded to the maximal concentration (Figure 9). Thereafter, the concentration of 4j declined rapidly due to metabolism. The same pattern was not seen with curcumin. Because of its low solubility, it took nearly 80 minutes before curcumin was first observed in the blood. The concentration gradually increased over time, reaching a maximum at 120 minutes before declining. These results suggest that compound 4j is not prone to metabolic degradation and was able to persist for a long period of time in the subject.
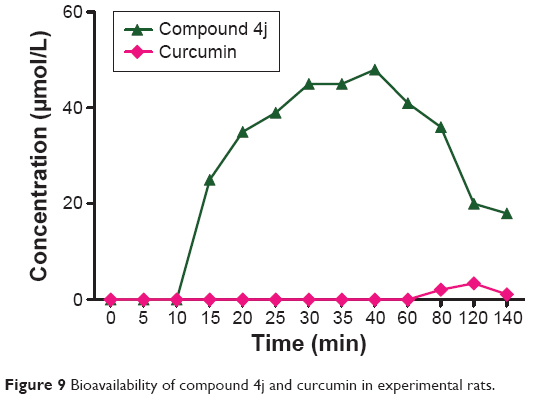 | Figure 9 Bioavailability of compound 4j and curcumin in experimental rats. |
Conclusion
In this study, we created a series of antihypertensive compounds based upon curcumin in excellent yields via short synthetic steps. These derivatives were able to reduce hypertension in vitro and in experimental animal models via ACE inhibition and vasodilation of the arterioles. The potentiation of NO donor activity was identified as a probable mechanism for the vasodilatory activity. Moreover, the most promising inhibitor, 4j, was found to resist metabolic deactivation in mice, resulting in better in vivo bioavailability compared with curcumin. With its excellent pharmacological activity and bioavailability, compound 4j might serve as a template for the future discovery of novel agents and hence it warrants further research. Further studies are underway and will be communicated in due course.
Disclosure
The authors report no conflicts of interest in this work.
References
Franklin SS, Wong ND. Hypertension and cardiovascular disease: contributions of the Framingham heart study. Global Heart J. 2013;8(1):49–57. | ||
Burden: mortality, morbidity and risk factors. Available from: www.who.int/nmh/publications/ncd_report_chapter1.pdf. Accessed on July 28, 2015. | ||
Chobanian AV, Bakris GL, Black HR, et al. The Seventh Report of the Joint National Committee on Prevention, Detection, Evaluation, and Treatment of High Blood Pressure: the JNC 7 report. JAMA. 2003;289(19):2560–2572. | ||
Borghi C, Rossi F. Role of the Renin-Angiotensin-Aldosterone system and its pharmacological inhibitors in cardiovascular diseases: complex and critical issues. High Blood Press Cardiovasc Prev. Epub September 24, 2015. | ||
Sennesael J, Verbeelen D. Therapeutic efficacy and tolerance of two orally active angiotensin converting enzyme inhibitors in patients undergoing regular hemodialysis. Kidney Int. 1985;28:A252. | ||
Davies MK, Gibbs CR, Lip GYH. Management: diuretics, ACE inhibitors, and nitrates. Br Med J. 2000;320(7232):428–431. | ||
Osterziel KJ, Julius S. Vasodilators in the treatment of hypertension. Compr Ther. 1982;8(11):43–52. | ||
Basnet P, Skalko-Basnet N. Curcumin: an anti-inflammatory molecule from a curry spice on the path to cancer treatment. Molecules. 2011;16:4567–4598. | ||
Aggarwal BB, Sung B. Pharmacological basis for the role of curcumin in chronic diseases: an age-old spice with modern targets. Trends Pharmacol Sci. 2009;30(2):85–94. | ||
Anand P, Kunnumakkara AB, Newman RA, Aggarwal BB. Bioavailability of curcumin: problems and promises. Mol Pharm. 2007;4(6):807–818. | ||
Košťálováa D, Bezákováb L, Račkovác L, Mošovskáa S, Šturdík E. Therapeutic potential of curcumin in medicinal chemistry. Acta Chim Slovaca. 2013;6(1):89–99. | ||
Meng X-Y, Zhang H-X, Mezei M, Cui M. Molecular docking: a powerful approach for structure-based drug discovery. Curr Comput Aided Drug Des. 2011;7(2):146–157. | ||
Ahmad W, Kumolosasi E, Jantan I, Bukhari SN, Jasamai M. Effects of novel diarylpentanoid analogues of curcumin on secretory phospholipase A2, cyclooxygenases, lipo-oxygenase, and microsomal prostaglandin E synthase-1. Chem Biol Drug Des. 2014;83(6):670–681. | ||
Bukhari SN, Lauro G, Jantan I, Bifulco G, Amjad MW. Pharmacological evaluation and docking studies of α, β-unsaturated carbonyl based synthetic compounds as inhibitors of secretory phospholipase A2, cyclooxygenases, lipoxygenase and proinflammatory cytokines. Bioorg Med Chem. 2014;22(15):4151–4161. | ||
Bukhari SN, Jantan I, Unsal Tan O, Sher M, Naeem-Ul-Hassan M, Qin HL. Biological activity and molecular docking studies of curcumin-related α, β-unsaturated carbonyl-based synthetic compounds as anticancer agents and mushroom tyrosinase inhibitors. J Agric Food Chem. 2014;62(24):5538–5547. | ||
Ringman JM, Frautschy SA, Cole GM, Masterman DL, Cummings JL. A potential role of the curry spice curcumin in Alzheimer’s disease. Curr Alzheimer Res. 2005;2(2):131–136. | ||
Bukhari SN, Butt AM, Amjad MW, Ahmad W, Shah VH, Trivedi AR. Synthesis and evaluation of chalcone analogues based pyrimidines as angiotensin converting enzyme inhibitors. Pak J Biol Sci. 2013;16(21):1368–1372. | ||
Addla D, Jallapally A, Kanwal A, Sridhar B, Banerjee SK, Kantevari S. Design, synthesis and evaluation of novel 2-hydroxypyrrolobenzodiazepine-5,11-dione analogues as potent angiotensin converting enzyme (ACE) inhibitors. Bioorg Med Chem. 2013;21(15):4485–4493. | ||
Skrzypiec-Spring M, Grotthus B, Szeląg A, Schulz R. Isolated heart perfusion according to Langendorff – still viable in the new millennium. J Pharmacal Toxicol Meth. 2007;55:113–126. |
 © 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.