")
Back to Journals » International Journal of Nanomedicine » Volume 12
Design of theranostic nanomedicine (II): synthesis and physicochemical properties of a biocompatible polyphosphazene–docetaxel conjugate
Authors Jun YJ, Park JH, Avaji PG, Park KS, Lee KE, Lee HJ, Sohn YS
Received 20 April 2017
Accepted for publication 13 June 2017
Published 27 July 2017 Volume 2017:12 Pages 5373—5386
DOI https://doi.org/10.2147/IJN.S140073
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 4
Editor who approved publication: Dr Thomas Webster
Yong Joo Jun,1,* Jung Hyun Park,2,* Prakash G Avaji,1 Kyung Su Park,3 Kyung Eun Lee,3 Hwa Jeong Lee,2 Youn Soo Sohn1
1C & Pharm, Ewha Womans University, Seodaemun-gu, Seoul, Republic of Korea; 2Graduate School of Pharmaceutical Sciences, Ewha Womans University, Seodaemun-gu, Seoul, Republic of Korea; 3Advanced Analysis Center, Korea Institute of Science and Technology, Seongbuk-gu, Seoul, Republic of Korea
*These authors contributed equally to this work
Abstract: To prepare an efficient theranostic polyphosphazene–docetaxel (DTX) conjugate, a new drug delivery system was designed by grafting a multifunctional lysine ethylester (LysOEt) as a spacer group along with methoxy poly(ethylene glycol) (MPEG) to the polyphosphazene backbone ([NP]n), and then DTX was conjugated to the carrier polymer using acid-cleavable cis-aconitic acid (AA) as a linker. The resultant polyphosphazene–DTX conjugate, formulated as [NP(MPEG550)3(Lys-OEt)(AA)(DTX)]n and named “Polytaxel”, exhibited high water solubility and stability by forming stable polymeric micelles as shown in its transmission electron microscopy image and dynamic light scattering measurements. Another important aspect of Polytaxel is that it can easily be labeled with various imaging agents using the lysine amino group, enabling studies on various aspects, such as its organ distribution, tumor-targeting properties, pharmacokinetics, toxicity, and excretion. The pharmacokinetics of Polytaxel was remarkably improved, with prolonged elimination half-life and enhanced area under the curve. Ex vivo imaging study of cyanine dye-labeled Polytaxel showed that intravenously injected Polytaxel is long circulating in the blood stream and selectively accumulates in tumor tissues. Polytaxel distributed in other organs was cleared from all major organs at ~6 weeks after injection. The in vitro study of DTX release from the carrier polymer showed that >95% of conjugated DTX was released at pH 5.4 over a period of 7 days. Xenograft trials of Polytaxel using nude mice against the human gastric tumor cell line MKN-28 showed complete tumor regression, with low systemic toxicity. Polytaxel is currently in preclinical study.
Keywords: docetaxel, polyphosphazene, anticancer drug, nanomedicine, theranostics
Introduction
DTX is a semisynthetic analog of PTX, belonging to the taxane family of antimitotic agents, which are very effective anticancer agents against a wide spectrum of malignancies, such as breast, ovarian, prostate, non-small cell lung, and gastric cancers.1–4 The cytotoxic action of the taxane family is attributed to interference with function of microtubules in the cell by inhibiting their depolymerization, inducing cell death.5,6 DTX is known to be twice as potent as PTX in efficacy, but its photoinstability7,8 causes inconvenience in drug synthesis and patient treatment. Furthermore, similar to PTX, DTX should be solubilized using a surfactant solution of Polysorbate 80 (Tween® 80) and 13% ethanol due to its low water solubility (7 μg/mL).9 Such a formulated DTX, named “Taxotere®”, exhibits several adverse effects, including neurotoxicity and neutropenia, due to the agent itself and hypersensitivity to the solvent system.1,10,11
Therefore, tremendous efforts using a variety of nanotechnologies have been made over the past decades to overcome such severe toxicity and solubility issues associated with small molecular drugs. Among them, one of the most promising approaches is “polymer therapy,” 12,13 which can be classified into two chemically different methods. One is to physically encapsulate an anticancer drug using polymeric micelles or nanoparticles,14–18 and the other is to chemically conjugate the drug molecules by covalent bonding to a carrier polymer directly or by means of a biodegradable linker.1 Physical encapsulation of DTX using polymeric micelles is technically easy but has been reported to show critical instability when entrapped in micelles, resulting in precipitation of DTX from the hydrophobic micelle core in aqueous solution or causing toxicities due to the released DTX in blood circulation.
On the other hand, chemical conjugation of DTX molecules to a carrier polymer requires a rational design based on the DTX release kinetics at the tumor site and very careful manipulation during synthesis of the polymer–DTX conjugate to protect the intact molecular structure of DTX. There are not many research reports on polymer–DTX conjugate drugs. In 2012, Ernsting et al19,20 reported their first synthesis of a carboxymethylcellulose–DTX conjugate named “Cellax” in Canada and continued to study its biodistribution, pharmacokinetics, efficacy, and safety. However, according to their most recent report, Cellax could not enter preclinical study due to safety concerns.21 In the same year, a Japanese group reported a new DTX conjugate with an amphiphilic block copolymer composed of PEG-poly(aspartate).22 There are also a few reports on DTX-entrapped core-cross-linked polymeric micelles23 and DTX-binding peptide-modified polymeric micelles.24
These polymer–DTX conjugates are more difficult to prepare and characterize, compared with physically encapsulated polymeric micelles of DTX, but the polymer–DTX conjugates form more stable polymeric micelles due to the highly hydrophobic DTX molecules oriented in the hydrophobic micelle core. Most recently, it has been pointed out that, in order to translate nanomedicine into clinical use, drug safety and pharmacology are the crucial factors.25 In particular, in the case of polymeric drugs, clearance of the injected polymer conjugate from the major organs is a critical safety factor, as is the design of an appropriate linker to optimize pharmacology. In fact, it is extremely difficult to design a polymeric drug conjugate using the conventional drug delivery systems composed of organic polymers, since most natural or synthetic organic polymers are not biodegradable, except for polyesters and poly(amino acids), and it is difficult to modify the functional groups bonded to the carbon–carbon rigid organic backbone.
On the other hand, polyphosphazenes are a new class of hybrid polymers composed of an inorganic backbone called “phosphazene” grafted with two organic side groups (R, R′) and are represented by the formula [N = P(R)(R′)]n. Depending on the structure and properties of the side groups R and R′, a variety of new materials with a wide range of physicochemical properties can be designed and synthesized.26,27 Furthermore, the polyphosphazene backbone is known to be hydrolytically degradable in weak acidic media similar to the microenvironment of tumor cells.25
In this study, a new biocompatible drug delivery system was successfully designed for conjugation with a hydrophobic anticancer drug DTX, satisfying various physicochemical properties. A hydrophilic methoxy-PEG with an average molecular weight of 550 (MPEG550) was introduced into the polyphosphazene backbone as a side group R for prolonged blood circulation, along with a multifunctional amino acid lysine as another side group R′ for direct conjugation with DTX molecules, yielding an amphiphilic polyphosphazene–DTX conjugate named “Polytaxel”. Various studies were performed to characterize the morphology and physicochemical properties of Polytaxel and to evaluate its organ distribution, pharmacokinetics, in vivo efficacy, clearance, and toxicity.
Materials and methods
Materials
DTX and PTX were obtained from Samyang Genex (Daejeon, Republic of Korea). All other chemicals that are used for synthesis and animal studies, including MPEG550, THF, formic acid, dimethyl sulfoxide, and anhydrous ethanol, were supplied by Sigma-Aldrich (St Louis, MO, USA). Human alveolar basal epithelial carcinoma cell line (A549) and gastric tumor cell line (MKN-28) were obtained from Korean Cell Line Bank (Seoul, Republic of Korea). Male Sprague Dawley rats and BALB/C nude mice were supplied by Orient Bio Inc (Gyeonggi-do, Republic of Korea).
Instruments and measurements
1H-NMR spectra were recorded using a Varian NMR spectrophotometer operating at 500 MHz in Fourier transform mode. Proton decoupled 31P-NMR spectra were measured with a Varian NMR spectrophotometer operating at 500 MHz using phosphoric acid as an internal standard. The TEM image of Polytaxel in aqueous solution (20 mg/mL) was measured using 200 kV Tecnai F20 (FEI, Eindhoven, the Netherlands). The IR spectra were measured using an FT/IR-6100 Fourier transform IR spectrophotometer. The particle size, distribution, and the zeta potentials of the polyphosphazene carrier polymer and Polytaxel were measured in 0.5% aqueous solution by DLS using a Malvern zetasizer instrument (Nano-ZS: Malvern Instruments, Malvern, UK). We measured the LCST by the cloud point method for 5% polymer conjugate in distilled water and PBS contained in a glass capillary immersed in an oil bath. We estimated the solution stability of Polytaxel in saline by measuring its CMC by the pyrene fluorescence method.28 The fluorescence intensities of plasma and various organs, including liver, lung, kidney, spleen, muscle, and tumor, were measured using a Kodak Image Station 4000MM instrument (Kodak, New Haven, CT, USA). The plasma concentration of DTX was determined using an Agilent 1260 Infinity series HPLC system (Agilent Technologies, Santa Clara, CA, USA), and the amount of DTX in the major organs and tumor was estimated using an Agilent 1260 infinity HPLC system coupled with an Agilent 6460 triple quadrupole mass spectrometer detector.
Synthesis of the polyphosphazene carrier polymer [NP(MPEG550)3(Lys-OEt)]n
Poly(dichlorophosphazene) was prepared from hexachlorocyclotriphosphazene [(NPCl2)3], (2.0 g, 5.72 mmol) in the presence of AlCl3 (7.5 wt%) as a catalyst according to a published method.29 A typical synthetic procedure was as follows. The sodium salt of MPEG550 was prepared by reaction of MPEG550 (14.22 g, 25.85 mmol) with an excess amount of sodium metal (1.2 g, 52.17 mmol) in dry toluene at refluxing temperature for 12 h. After the resultant solution was filtered to remove excess sodium metal, the filtrate was dropped slowly into a solution of poly(dichlorophosphazene) (2 g, 17.3 mmol) dissolved in dry THF (100 mL). The reaction mixture was stirred for 5 h at −50°C and further stirred at room temperature for 12 h to yield PEGylated polyphosphazene. Meanwhile, Boc-lysine ethyl ester (Nα-Boc-Lys-OEt, 3.7 g, 13.5 mmol) was dissolved in dry chloroform (100 mL) and neutralized with dry triethylamine (Et3N, 13.6 mL, 134.4 mmol). This solution was added slowly to the PEGylated polymer solution and allowed to react at room temperature for 24 h. After the reaction mixture was filtered to remove the by-products (Et3N·HCl or NaCl salts), the filtrate was concentrated under reduced pressure and then dissolved in a small amount of ethanol, after which it was filtered again to remove insoluble salts and concentrated under reduced pressure.
To remove the protecting group (t-Boc) from the polymeric polyphosphazene derivative, the product was dissolved in a mixed solution of methylene chloride (20 mL) and TFA (20 mL) (1:1 ratio MC/TFA). The reaction mixture was stirred at room temperature for 6 h, and the solvent was evaporated under reduced pressure. The product was neutralized with NaHCO3 solution and dialyzed in water using regenerated cellulose membranes (MWCO: 3 kDa) for 1 d. The dialyzed solution was freeze-dried to obtain the pure carrier polymer [NP(MPEG550)3(Lys-OEt)]n, which was fractionated in distilled water using cellulose membranes (MWCO: 30 and 100 kDa) to obtain the final product in the molecular weight range of 30–100 kDa. All reactions were monitored using 31P-NMR spectroscopy. The total yield of the polymer material was ~75%. Unit molecular weight was 1942. 1H-NMR (CDCl3, ppm): δ 1.26 (bs, 3H, methyl protons of ethyl ester), 1.27–1.62 [brm, 4H, lysine–(–CH2–CH2–) protons], 1.71–1.91 [brm, 4H, lysine–(HN–CH2–CH2–) protons], 2.85 [brs, 1H, lysine–(–NH2–CH–) protons], 3.37 [s, 9H, terminal methoxy protons of MPEG–(–OCH3)], 3.46–3.76 [brm, 138H, ethylene protons of MPEG–(–CH2–CH2–)], 3.92–4.15 [brm, 6H, MPEG–(–P–O–CH2–) protons], 4.22 [brs, 2H, lysine ethyl ester–(–CH2–) protons]. 31P-NMR (CDCl3, ppm): δ −5.49 (–O–P–O–), 1.41 (–N–P–O–). LCST: >100°C.
Synthesis of the DTX-AA precursor
To prepare a DTX-AA precursor, cis-AA anhydride (0.145 g, 0.93 mmol) was dissolved in dry THF and activated by adding N, N′-DIC (0.117 g, 0.93 mmol) and DPTS (0.273 g, 0.93 mmol) at 0°C and further stirring for 2 h at the same temperature. DTX (0.3 g, 0.37 mmol) dissolved in dry THF was slowly added to this solution, and the reaction mixture was stirred continuously at room temperature for 40 h. Finally, NHS (0.107 g, 0.93 mmol) and DIPEA (0.24 g, 1.85 mmol) were added to the reaction mixture, and stirring was continued for another 5 h. The solvent was evaporated under reduced pressure, and the product was purified by column chromatography (methylene chloride: methanol; 90:10%, TLC; Rf =0.14). Yield was 73%. Molecular weight was 1061. 1H-NMR (CDCl3, ppm): δ 1.07–1.38 (brm, 15H, C16, C17, and C7′–9′ protons of DTX), 1.58–1.97 (brm, 8H, C14, C19, and C18 protons of DTX), 2.17–2.62 (brm, 5H, C6, and C22 protons of DTX), 2.69–2.82 (brm, 4H, C43, and C44 protons of succinate), 3.18–3.50 (m, 2H, C37 protons of AA), 3.65–3.71 (m, 1H, C3 proton of DTX), 4.15–4.26 (brm, 3H, C20, and C7 protons of DTX), 4.63 (brs, 1H, C2′ protons of DTX), 4.92 (brm, 2H, C5 and C10 protons of DTX), 5.25–5.33 (brm, 2H, C3′ and NH protons of DTX), 5.34–5.80 (brm, 2H, C2 protons of DTX and C40 protons of AA), 6.17 (brm, 1H, C13 protons of DTX), 7.27–7.59 (brm, 8H, C31-C35, and C26-C28 phenyl protons of DTX), 8.11 (brs, 2H, C25, and C29 phenyl protons of DTX).
Synthesis of the polyphosphazene–DTX conjugate, [NP(MPEG550)3(Lys-OEt)(AA)(DTX)]n
A solution of DTX-AA precursor (0.3 g, 0.28 mmol) in THF (10 mL) was added to a solution of carrier polymer [NP(MPEG550)3(Lys-OEt)]n (1 g, 0.51 mmol) dissolved in THF (10 mL). To this reaction mixture, DIPEA (0.2 g, 1.55 mmol) was added and stirred at 0°C for 12 h and then held at room temperature for 5 h. The reaction mixture was evaporated under reduced pressure, and the residue was dissolved in 30% ethanol–water mixture (50 mL). The solution was filtered with a 0.2 μm syringe filter, and the filtrate was dialyzed for 12 h in ethanol using a regenerated cellulose membrane (MWCO: 2000). The dialyzed solution was further fractionated to obtain the final product in the molecular weight range of 50–100 kDa using membranes with different MWCOs (50 and 100 kDa), and the final solution was freeze-dried to obtain the final conjugate product, Polytaxel. Yield was 82% and molecular weight was 2904. 1H-NMR (CDCl3, ppm): δ 1.24–1.48 (brm, 15H, C16, C17, and C7′-9′ protons of DTX, CH3 protons of ethyl ester, and –(CH2)2– protons of lysine), 1.60–1.82 (brm, 7H, C14, C19, and C18 protons of DTX, and lysine –(HN–CH2–CH2–) protons), 2.17–2.62 (brm, 2H, C6, and C22 protons of DTX), 2.65–3.22 (brm, 5H, C43, and C44 protons of succinate and lysine –(–CH–N–) protons), 3.36 (s, 9H, –OCH3– protons of lysine), 3.50–3.77 (brm, 141H, ethylene protons of MPEG–(–CH2–CH2–), C37 protons of AA, and C3 protons of DTX), 3.90–4.10 (brm, 6H, MPEG–(P–O–CH2–) protons), 4.22–4.29 (brm, 5H, lysine ethyl ester –(–CH2–CH3) protons, C20 and C7 protons of DTX), 4.52–6.80 (8H, C2′, C3′, C2, C5, C10, C13, and NH protons of DTX and C40 protons of AA), 7.3–7.59 (brm, 2H, C31–C35 and C26–C28 phenyl protons of DTX), 8.08 (brs, 2H, C25, and C29 phenyl protons of DTX). 31P-NMR (CDCl3, ppm): δ −6.01 (–O–P–O–), 0.96 (–N–P–O–). LCST: >100°C.
Labeling Polytaxel with the fluorescent dye Cy5.5® for imaging study
Polytaxel (60 mg, 4.0×10−5 mmol) in anhydrous DMF (5 mL), the coupling agent HBTU (7 mg, 1.8×10−3 mmol), and triethyl amine (0.5 mL) were added to a suspension of Cy5.5 monoacid (1 mg, 9.69×10−4 mmol), and the reaction mixture was stirred for 18 h at room temperature. Then, the reaction mixture was diluted with ethanol (50 mL) and centrifuged in Vivaspin® polyethersulfone ultrafiltration spin columns (MWCO: 50000) with capacity of 6 mL at 4,000 rpm. The final Cy5.5-labeled Polytaxel product was washed several times with water and saline.
Biodistribution and clearance studies of Cy5.5-labeled Polytaxel by ex vivo imaging
BALB/C nude mice (5 weeks old), weighing between 20 and 22 g, were housed in a specific pathogen-free room kept at a temperature of 23°C±3°C and relative humidity of 50%±5% with free access to a sterilized standard chow diet and water. The mice were acclimatized under this condition for 1 week prior to the experiments. All animal study protocols were reviewed and approved by the Institutional Animal Care and Use Committee (IACUC number 14-048) of Ewha Womans University, Seoul, Republic of Korea. The committee abides by the Institute of Laboratory Animal Resources (ILAR) guide of Korea.
Human alveolar basal epithelial carcinoma cell line (A549) was grown in RPMI 1640 media containing 10% fetal bovine serum at 37°C in a humidified 5% CO2 atmosphere. The A549 cells were suspended in serum-free RPMI 1640 media and inoculated subcutaneously at a density of 1×107 cells per 150 μL into the right flank region of each mouse. Tumors were measured using a caliper (CD-AX/APX; Mitutoyo America Corporation, Waltham, MA, USA) and tumor volumes were calculated using the following equation:30
|
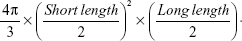
After 3 weeks, when the tumor was grown to 250–300 mm3, the Cy5.5-labeled Polytaxel was injected intravenously into the tail vein of the tumor-bearing mice. The animals were sacrificed at 12, 24, 48, 72, and 96 h after injection, and their liver, heart, lung, kidney, spleen, tumor, and muscle (from the left flank region) were harvested and weighed. Whole blood samples were collected by cardiac puncture with a syringe and centrifuged at 12,000 rpm for 15 min to separate plasma fractions. The fluorescence intensity of each organ was measured immediately. The fluorescence images of harvested organs and tumors were obtained using a 12-bit CCD camera (Kodak Image Station 4000MM instrument) equipped with a special C-mount lens and Cy5.5 bandpass excitation and emission filters (625–700 nm; Omega Optical, Brattleboro, VT, USA). After fluorescence intensity measurements, all organ and plasma samples were stored at −80°C for LC-MS/MS analysis.
For the study of clearance of Polytaxel from the major organs, BALB/C nude mice (5 weeks old, 20–22 g) were used after adaptation in a specific pathogen-free room for 1 week. Human alveolar basal epithelial carcinoma cell line (A549) was inoculated at a density of 1×107 cells per 150 μL to each mouse. When the tumor volume reached 200–250 mm3, Cy5.5-labeled Polytaxel was injected intravenously at the optimal dose based on DTX (30 mg/kg) into the tail vein of each of 16 mice. The mice were sacrificed according to the predetermined time schedules after injection at short intervals during the first week and then once every 2 weeks thereafter. The fluorescence images of harvested organs and tumors were obtained using the same equipment as mentioned in the section on organ distribution study.
Pharmacokinetic study
In order to compare the pharmacokinetic behavior of DTX conjugated to polyphosphazene carrier polymer with that of the monomeric DTX simply formulated with surfactant micelles, a pharmacokinetic study of Polytaxel was carried out following our previous protocol1 using healthy male Sprague-Dawley rats (7–8 weeks old), weighing between 235 and 265 g and purchased from Orient Bio Seongnam, Republic of Korea. All animal experiments using rats were approved by the IACUC of Ewha Womans University, Republic of Korea (IACUC number 2012-01-019).
In vitro release study of DTX from Polytaxel
Polytaxel (10 mg) was dissolved in distilled water (1 mL) in a colored vial, to which the same volume of a buffer solution at pH 5.4 or 7.4 was added. Ten sample solutions of each pH were prepared and fixed in a shaking incubator at 37°C. The vials were opened after predetermined time intervals, and the released DTX was extracted with ethyl acetate and subjected to HPLC analysis.
In vivo antitumor efficacy study
The in vivo antitumor efficacy of Polytaxel and Taxotere (as a reference) was evaluated against a gastric tumor cell line (MKN-28) using BALB/C nude mice (5 weeks old, 20–22 g). Animals were adapted under controlled temperature and humidity for 1 week prior to the experiments. MKN-28 cells cultured in RPMI 1640 media containing 10% fetal bovine serum and 1% antibiotic–antimycotic agent (cell culture media) at 37°C in a humidified 5% CO2 atmosphere were treated with 0.25% trypsin–EDTA solution. The incubated cells were centrifuged in cell culture media for 5 min. The concentration of cells in media was calculated from the number of cells counted using a hemocytometer (catalog number 0650030; Paul Marienfeld, Lauda-Königshofen, Germany). The tumor cells were suspended in serum-free RPMI 1640 media at a density of 5×106 cells in 150 μL of media and inoculated subcutaneously into the right flank region of each mouse. When the tumor volume reached about 100–150 mm3, the test drugs or normal saline as a control were injected intravenously via the tail vein of the mice according to a triple injection regimen on days 1, 5, and 9. In order to compare the antitumor efficacy of Polytaxel and Taxotere, tumor size and weights of the mice were measured every 2 or 3 d. The tumor volume was calculated using the equation provided earlier. All mice were sacrificed on the 40th day from the first injection.
Results and discussion
Synthesis and characterization of the polyphosphazene carrier polymer and its conjugate Polytaxel
The reaction scheme for synthesis of the polyphosphazene–DTX conjugate Polytaxel (7) is displayed in Figure 1.
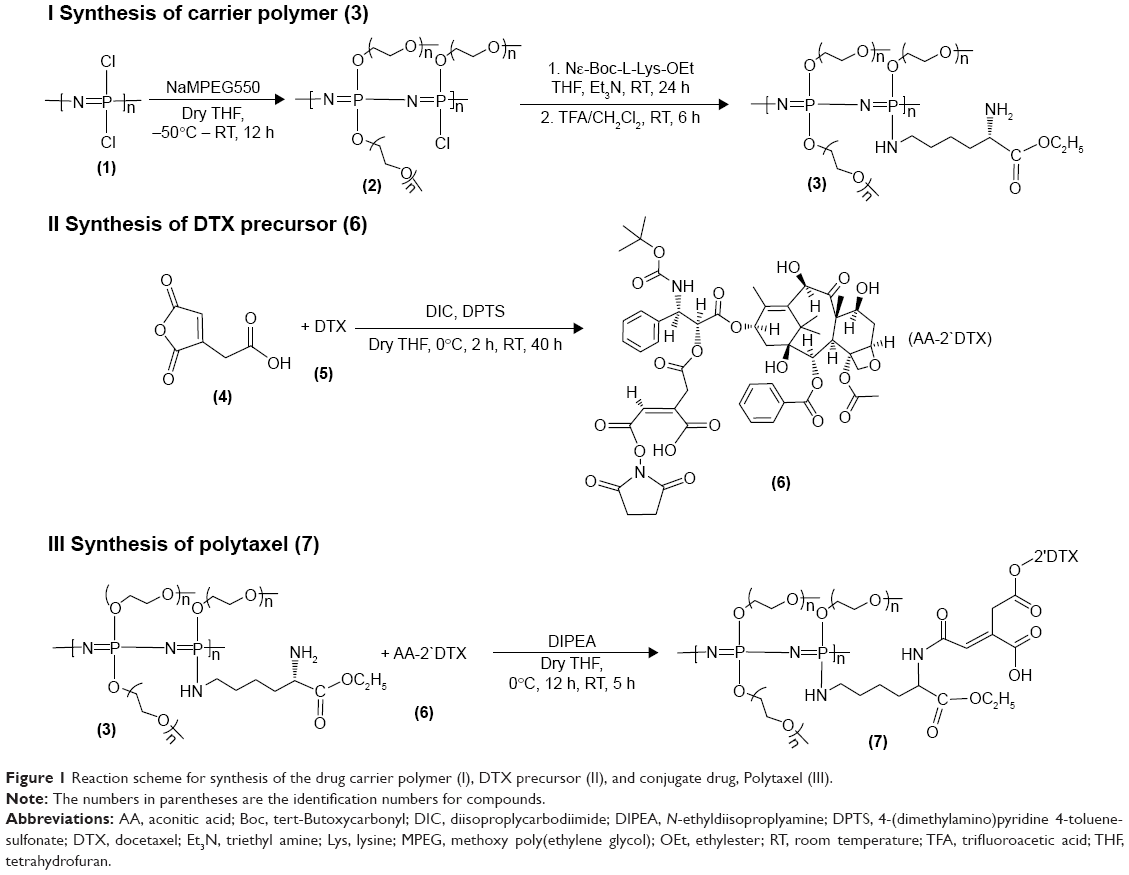 | Figure 1 Reaction scheme for synthesis of the drug carrier polymer (I), DTX precursor (II), and conjugate drug, Polytaxel (III). |
Synthesis of Polytaxel was performed in three steps, as shown in the reaction scheme. First, the carrier polymer [NP(MPEG550)3(LysEt)]n (3) was prepared by stepwise nucleophilic substitution of chloropolyphosphazene (1) with 3 mol of MPEG550 for prolonged blood circulation and 1 mol of a multifunctional amino acid, lysine ethyl ester, for conjugation with DTX. The conjugation of DTX to this carrier polymer can be performed in many different ways since there are many functional groups on both the carrier polymer and the DTX molecule.
However, the polymer conjugate drug was designed rationally based on the stability of the conjugate drug, morphology related with the tumor-targeting properties, and drug release kinetics relevant to drug efficacy. It is well known that the DTX molecule can be efficiently conjugated to carrier polymers containing carboxylic acids via esterification using the 2′-hydroxyl group of DTX. Therefore, DTX molecules can be conjugated to the lysine carboxylate group after hydrolysis; however, in this study, the release kinetics of DTX were critically considered, leading to the conclusion that AA should be used as the acid-cleavable linker for DTX conjugation. Therefore, instead of direct esterification of DTX with the lysine carboxylic acid, DTX was reacted with excess AA (4) to obtain an intermediate precursor (DTX-AA) (6), which was conjugated to the carrier polymer (3) to obtain the final DTX conjugate product (7). The final product, Polytaxel (7), was obtained as a dark purple solid that was soluble in water and polar organic solvents and found to self-assemble into very stable polymeric micelles in aqueous solution. All reaction intermediates and the final product were characterized by 1H- and 31P-NMR spectroscopy (Figures S1 and S2).
Morphology and physicochemical properties of Polytaxel
The morphology of polymer conjugate drugs is critically important because it is directly related with the passive tumor targeting properties of the polymer conjugates by the EPR effect.31 It is well known that, even though the particle size of polymers is not an absolute measure of tumor targeting, the average diameter of particles should be in the range of 20–100 nm for appreciable EPR effect. Therefore, we measured the particle sizes of the carrier polymer and Polytaxel using DLS, and the results are displayed in Figure 2A and B.
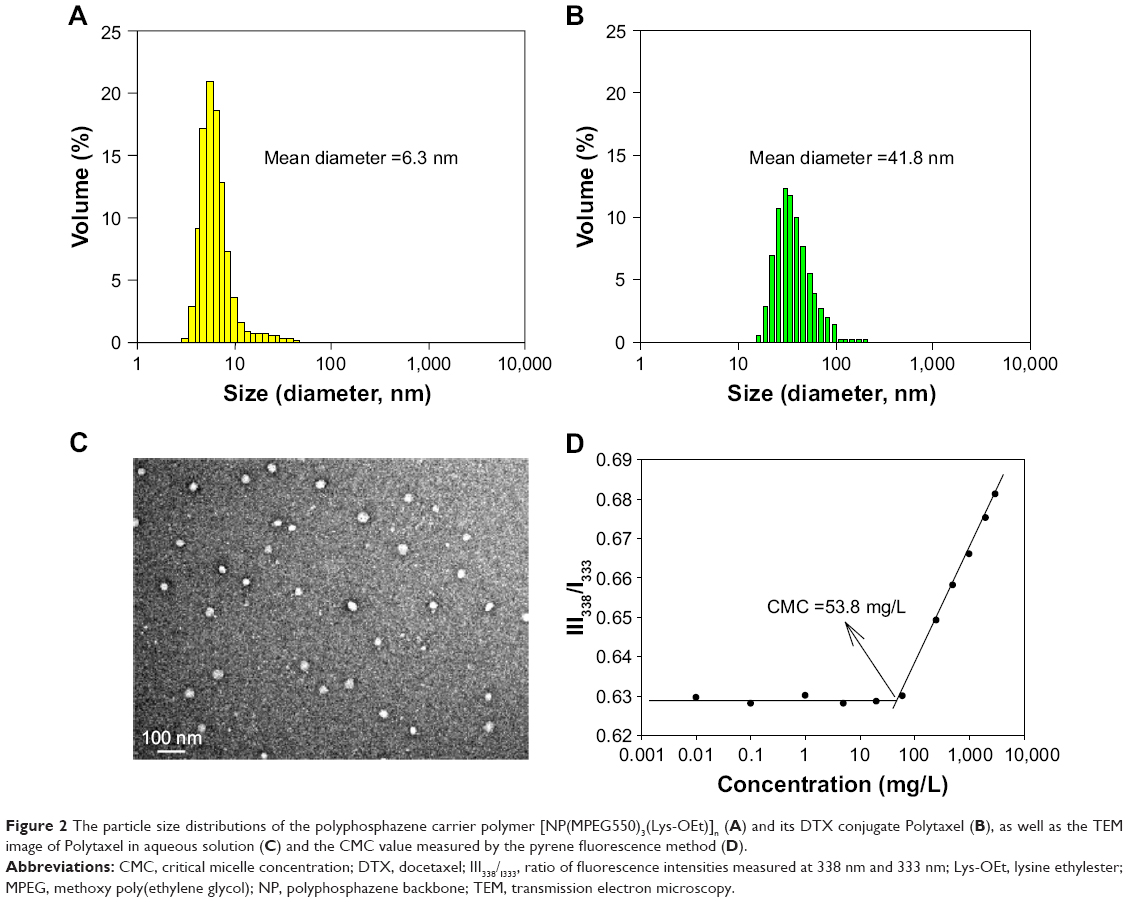 | Figure 2 The particle size distributions of the polyphosphazene carrier polymer [NP(MPEG550)3(Lys-OEt)]n (A) and its DTX conjugate Polytaxel (B), as well as the TEM image of Polytaxel in aqueous solution (C) and the CMC value measured by the pyrene fluorescence method (D). |
As seen in the figure, the average particle size of the carrier polymer was ~6.3 nm, indicating a moderate free dynamic volume, but the polymer conjugate Polytaxel exhibited a much larger particle size of 41.8 nm, which indicates that Polytaxel molecules are subjected to self-assembly to form larger polymeric micelles suitable for tumor targeting by the EPR effect.31 In order to confirm the exact morphology of Polytaxel, we measured its TEM image in aqueous solution, as displayed in Figure 2C. The spherical particle shape in the figure clearly indicates that Polytaxel forms large spherical micelles, as expected from its DLS measurement (Figure S3), and the CMC measured by the pyrene fluorescence method28 was 53.8 mg/L, as shown in Figure 2D.
Another important aspect of amphiphilic polymer conjugate drugs such as Polytaxel is that their LCST should be much higher than body temperature, since it is generally known that amphiphilic polymers with an LCST near the body temperature can be precipitated in aqueous solution.32 However, the LCST of Polytaxel was determined to be higher than 100°C, which is safe.
Pharmacokinetics of Polytaxel
In order to investigate the pharmacokinetic profiles of the DTX released from the newly developed polyphosphazene –DTX conjugate Polytaxel, compared with Taxotere as a reference, we performed a pharmacokinetic study in accordance with a slight modification of our previously reported method.1 The mean plasma concentration–time profiles of DTX obtained after intravenous injection of Taxotere and Polytaxel are shown in Figure 3.
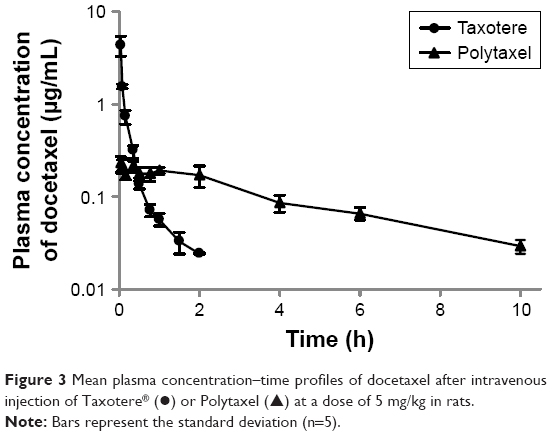 | Figure 3 Mean plasma concentration–time profiles of docetaxel after intravenous injection of Taxotere® (●) or Polytaxel (▲) at a dose of 5 mg/kg in rats. |
The pharmacokinetic parameters of DTX estimated from the plasma concentration–time data are presented in Table 1. In the reference Taxotere group, DTX was rapidly exposed from the core of the micelles to blood circulation and quickly cleared from the body after intravenous injection. The elimination half-life (t1/2) of DTX was very short (0.651±0.093 h). On the other hand, in the polymer conjugate group, the initial plasma concentration of released DTX was remarkably lower than that of Taxotere (0.244±0.053 μg/mL vs 8.76±3.22 μg/mL), even though both groups were administered the same dose. The t1/2 and the AUC for plasma concentration vs time of Polytaxel were increased approximately fivefold and 1.5-fold, respectively, compared with those of the reference. Based on the results of the improved pharmacokinetic parameters, the polyphosphazene–DTX conjugate was degraded very slowly in blood and long circulated in the body after injection. This might be due to the ester linkage properties of the AA spacer, which is known to be relatively stable in general physicochemical conditions, although very rapid cleavage occurs in the acidic tumoral pH condition.33
 | Table 1 PK parameters of docetaxel after intravenous injection of Polytaxel and Taxotere® at a dose of 5 mg/kg in rats |
Ex vivo imaging study of Cy-Polytaxel
To confirm the selective tumor targeting of Polytaxel, the time-dependent organ distribution of Cy5.5-labeled Polytaxel in the A549-tumor bearing mice was monitored by measuring the time-dependent fluorescence intensities of major organs and plasma. The fluorescence images of the organs, including liver, heart, lung, kidney, spleen, muscle, and tumor, as well as the plasma, harvested from mice at 12, 24, 48, 72, and 96 h after injection are displayed in Figure 4.
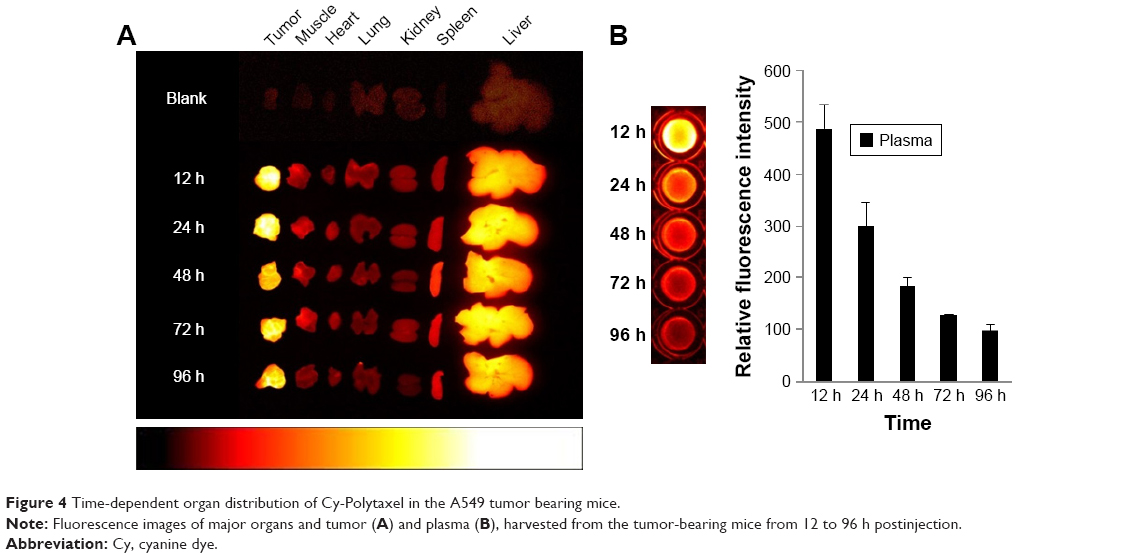 | Figure 4 Time-dependent organ distribution of Cy-Polytaxel in the A549 tumor bearing mice. |
As shown in Figure 4A, Cy-Polytaxel was mainly distributed in the tumor and reached a maximum at 24 h postinjection. The plasma concentration of Cy-Polytaxel exhibited a time-dependent decrease, as shown in Figure 4B; however, strong fluorescence intensity was still detected at the last time point. A quantitative diagram of the fluorescence intensity detected in each organ as well as the TTR representing the tumor selectivity are displayed in Figure 5A and B, respectively. The TTR was >4 at every time point, and the maximum value was 5.3 at 24 h postinjection. These results indicate the prolonged circulation of Polytaxel in the body due to the positively charged primary amine group in lysine and the stealth effect by the PEG surface of the polyphosphazene polymer.34,35
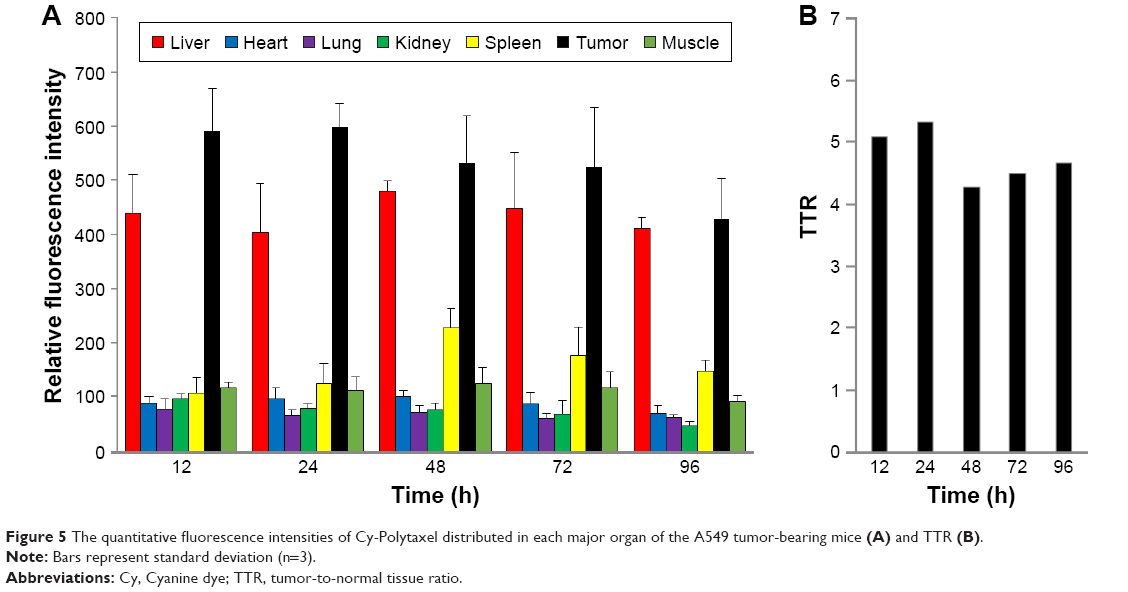 | Figure 5 The quantitative fluorescence intensities of Cy-Polytaxel distributed in each major organ of the A549 tumor-bearing mice (A) and TTR (B). |
Quantitative analysis of time-dependent DTX concentration in each organ using HPLC tandem MS
Each organ and tumor harvested from the sacrificed mice injected with Cy-labeled Polytaxel was homogenized in 20 μL of water and then extracted with acetonitrile for quantification of free DTX using HPLC tandem MS (LC-MS/MS) (Method section of Supplementary materials). Even though the amount of DTX in each organ and tumor does not exactly correspond to the result of ex vivo organ distribution of Polytaxel, a large amount of DTX was detected steadily up to 96 h in tumor tissue, as shown in Figure 6. The trend of time-dependent amounts of detectable DTX in each organ is not exactly proportional to the organ distributions of Polytaxel; however, Figure 6 shows that liver, spleen, and tumor are the major organs of Polytaxel accumulation, as was observed in the ex vivo organ distributions. The lower content of free DTX in tumor compared with that in liver could be due to lower extraction of DTX from the tumor tissue compared with liver under our extraction conditions.
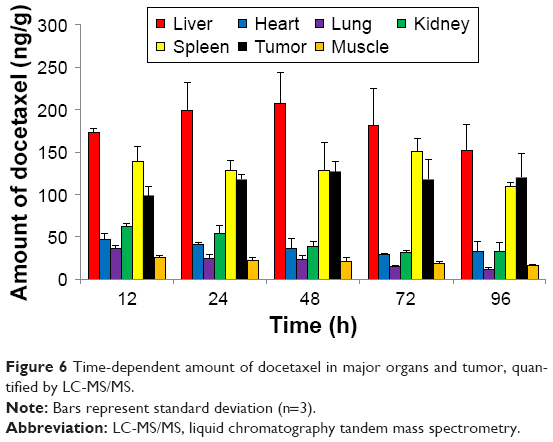 | Figure 6 Time-dependent amount of docetaxel in major organs and tumor, quantified by LC-MS/MS. |
Clearance of Cy-Polytaxel from the major organs
To confirm disappearance of the polymer-conjugated drug from the major organs, a clearance study of Cy5.5-labeled Polytaxel was carried out using tumor-bearing nude mice. The mice were sacrificed at scheduled time points after injection. Plasma and various organs, including liver, lung, kidney, spleen, tumor, and muscle, were harvested from the mice, and the fluorescence intensities of major organs and plasma were measured. All time-dependent clearance rates of Polytaxel from the major organs and tumor and the fluorescence images of organs and plasma harvested from the mice are displayed in Figure 7.
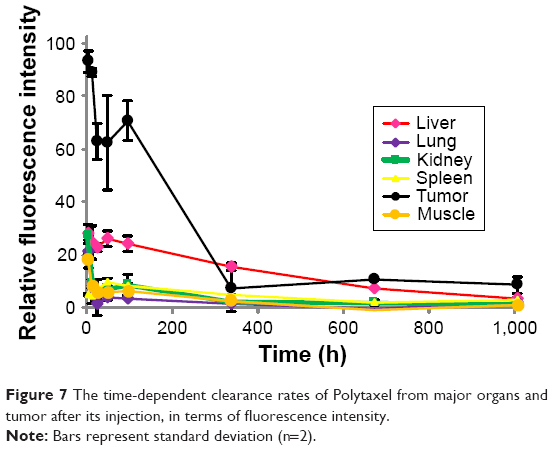 | Figure 7 The time-dependent clearance rates of Polytaxel from major organs and tumor after its injection, in terms of fluorescence intensity. |
One of the most critical safety factors of polymeric nanomedicine is clearance from human organs. As shown in Figure 7, Polytaxel was distributed dominantly in the tumor and accumulated in liver and spleen for 2 d after injection, after which it was excreted from the organs and tumor. Cy-Polytaxel was cleared from all major organs at ~6 weeks after injection.
In vitro release rate of DTX from Polytaxel
In order to investigate the pH-dependent drug release pattern of DTX from Polytaxel in physiological conditions, in vitro drug release experiments were performed in acidic (pH 5.4) and neutral (pH 7.4) buffer solutions at 37°C. The time-dependent DTX release profile from Polytaxel was determined using HPLC. The accumulated amounts of DTX released from the polymer conjugate Polytaxel in acidic and neutral buffer solutions are displayed in Figure 8.
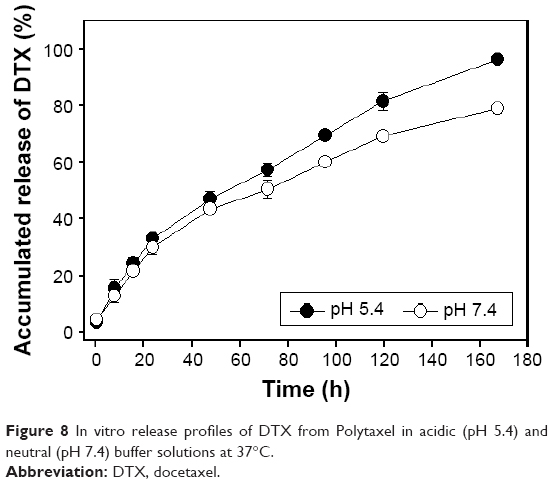 | Figure 8 In vitro release profiles of DTX from Polytaxel in acidic (pH 5.4) and neutral (pH 7.4) buffer solutions at 37°C. |
At acidic pH, >95% of the total DTX content of Polytaxel was released in 7 d, while ~80% DTX was released from Polytaxel in neutral solution. Such results are in accord with the biodegradability of polyphosphazenes,36 suggesting that AA linkers are appropriate for this use.37
In vivo antitumor efficacy study
In order to examine the antitumor efficacy of Polytaxel, compared with Taxotere as a positive control, we performed nude mouse xenograft trials against the gastric tumor cell line MKN-28. In this study, Taxotere was administered at a dose of 10 mg/kg and Polytaxel was given at two different doses of 10 and 20 mg/kg based on DTX content three times on days 1, 5, and 9. Saline solution was also injected into another group of mice as a negative control to confirm the xenograft model. The tumor size and weights of mice were measured every 2 or 3 d.
The results of the antitumor efficacy study and the changes in body weight are shown in Figures 9 and S4, respectively. Tumor growth inhibition was observed in all treated groups, but the antitumor efficacy of Polytaxel 10 mg/kg was lower than that of the same dose of Taxotere or Polytaxel 20 mg/kg. However, the higher dose of Polytaxel (20 mg/kg) exhibited complete tumor regression, with disappearance of the tumors after 30 d. Body weight changes were also observed during the experiments. When Taxotere was injected into mice, the mean body weight was decreased by 10%, while body weights of mice in Polytaxel groups were increased similarly to the saline group (Figure S3). The excellent antitumor efficacy and reduced systemic toxicity of Polytaxel seem to be due to the prolonged circulation in blood and the sustained but rapid release of DTX from the polymer conjugate.
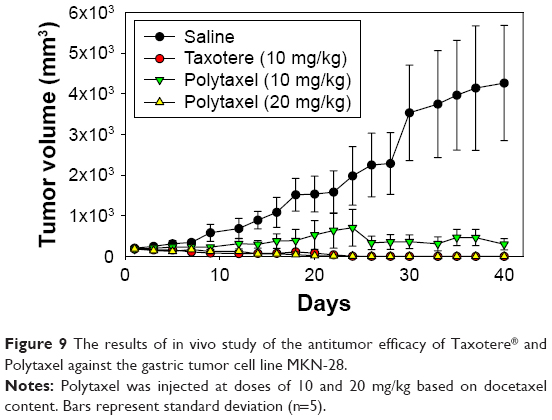 | Figure 9 The results of in vivo study of the antitumor efficacy of Taxotere® and Polytaxel against the gastric tumor cell line MKN-28. |
Conclusion
A new biocompatible theranostic polyphosphazene drug carrier polymer was designed by grafting a hydrophilic MPEG550 moiety for prolonged blood circulation and a multifunctional lysine into the polyphosphazene backbone as a spacer group. The highly potent anticancer agent DTX was conjugated to the carrier polymer using acid-cleavable cis-AA as a linker. The resultant polyphosphazene–DTX conjugate named “Polytaxel” could easily be labeled with the imaging agent Cy5.5, and various theranostic studies could be performed using animals. These amphiphilic conjugate drug molecules were found to self-assemble into stable polymeric micelles with a mean particle size of 41.3 nm and a diameter suitable for passive tumor targeting by the EPR effect. The pharmacokinetics of Polytaxel was remarkably improved, with prolonged elimination half-life and enhanced AUC. Polytaxel was long circulated in the bloodstream and accumulated in tumor tissue, rather than in normal organs, with high TTR but was cleared from all major organs after approximately 6 weeks postinjection. Furthermore, Polytaxel exhibited excellent antitumor efficacy by showing complete tumor regression in a nude mouse xenograft model against the human gastric cancer MKN-28 cell line, with low systemic toxicity. Polytaxel has entered preclinical studies.
Abbreviations
AA, aconitic acid; AUC, area under the curve; CCD, charge coupled device; CMC, critical micelle concentration; Cy, cyanine dye; Cy5.5, cyanine 5.5; DIC, diisoproplycarbodiimide; DIPEA, N-ethyldiisoproplyamine; DLS, dynamic light scattering; DMF, dimethylformamide; DPTS, 4-(dimethylamino)pyridine 4-toluenesulfonate; DTX, docetaxel; EPR, enhanced permeability and retention; HBTU, N,N,N′,N′-tetramethyl-O-(1H-benzotriazol-1-yl)uronium hexafluorophosphate; HPLC, high-performance liquid chromatography; IR, infrared; LC, liquid chromatography; LCST, lower critical solution temperature; Lys, lysine; MC, methylene chloride; MPEG, methoxy poly(ethylene glycol); MS, mass spectrometry; MWCO, molecular weight cutoff; NHC, N-hydroxysuccinimide; NMR, nuclear magnetic resonance; NP, polyphosphazene backbone; OEt, ethylester; PBS, phosphate-buffered saline; PEG, poly(ethylene glycol); PTX, paclitaxel; Rf, retention factor; TEM, transmission electron microscopy; TFA, trifluoroacetic acid; THF, tetrahydrofuran; TLC, thin layer chromatography; TTR, tumor-to-normal tissue ratio.
Acknowledgments
This study was supported by grants from the Korean Health Technology R&D Project, Ministry of Health and Welfare, Republic of Korea (HI11C0532) and the National Research Foundation of Korea (NRF) funded by the Korean government (MEST) (2017R1A2B4007869 and NRF-2014R1A1A2055876) as well as by C & Pharm.
Disclosure
YJJ, PGA, and YSS are employed by C & Pharm, a venture company at Ewha Womans University. The authors report no conflicts of interest in this work.
References
Jun YJ, Jadhav VB, Min JH, et al. Stable and efficient delivery of docetaxel by micelle-encapsulation using a tripodal cyclotriphosphazene amphiphile. Int J Pharm. 2012;422(1–2):374–380. | ||
Hennenfent KL, Govindan R. Novel formulations of taxanes: a review. Old wine in a new bottle? Ann Oncol. 2006;17(5):735–749. | ||
Bissery MC, Nohynek G, Sanderink GJ, Lavelle F. Docetaxel (Taxotere): a review of preclinical and clinical experience. Part I: preclinical experience. Anticancer Drugs. 1995;6(3):339–355. | ||
van Oosterom AT, Schrijvers D. Docetaxel (Taxotere): a review of preclinical and clinical experience. Part II: clinical experience. Anticancer Drugs. 1995;6(3):356–368. | ||
He L, Orr GA, Horwitz SB. Novel molecules that interact with microtubules and have functional activity similar to Taxol. Drug Discov Today. 2001;6(22):1153–1164. | ||
Herbst RS, Khuri FR. Mode of action of docetaxel – a basis for combination with novel anticancer agents. Cancer Treat Rev. 2003;29(5):407–415. | ||
Vasu DR, Moses BJ, Vyas K, et al. Isolation and characterization of impurities in docetaxel. J Pharm Biomed Anal. 2006;40(3):614–622. | ||
Kumar D, Tomar RS, Deolia SK, Mitra M, Mukherjee R, Burman AC. Isolation and characterization of degradation impurities in docetaxel drug substance and its formulation. J Pharm Biomed Anal. 2007;43(4):1228–1235. | ||
Nuijen B, Bouma M, Schellens JH, Beijnen JH. Progress in the development of alternative pharmaceutical formulations of taxanes. Invest New Drugs. 2001;19(2):143–153. | ||
van Zuylen L, Verweij J, Sparreboom A. Role of formulation vehicles in taxane pharmacology. Invest New Drugs. 2001;19(2):125–141. | ||
Persohn E, Canta A, Schoepfer S, et al. Morphological and morphometric analysis of paclitaxel and docetaxel-induced peripheral neuropathy in rats. Eur J Cancer. 2005;41(10):1460–1466. | ||
Haag R, Kratz F. Polymer therapeutics: concepts and applications. Angew Chem Int Ed Engl. 2006;45:1198–1215. | ||
Kabanov AV, Okano T. Challenges in polymer therapeutics: state of the art and prospects of polymer drugs. Adv Exp Med Biol. 2003;519:1–27. | ||
Shin HC, Alani AW, Rao DA, Rockich NC, Kwon GS. Multi-drug loaded polymeric micelles for simultaneous delivery of poorly soluble anticancer drugs. J Control Release. 2009;140(3):294–300. | ||
Gaucher G, Marchessault RH, Leroux JC. Polyester-based micelles for the parenteral delivery of taxanes. J Control Release. 2010;143:2–12. | ||
Carstens MG, de Jong PH, van Nostrum CF, et al. The effect of core composition in biodegradable oligomeric micelles as taxane formulations. Eur J Pharm Biopharm. 2008;68(3):596–606. | ||
Elsabahy M, Perron ME, Bertrand N, Yu GE, Leroux JC. Solubilization of docetaxel in poly(ethylene oxide)-block-poly(butylene/styrene oxide) micelles. Biomacromolecules. 2007;8(7):2250–2257. | ||
Lee SW, Yun MH, Jeong SW. Development of docetaxel-loaded intravenous formulation, Nanoxel-PM™ using polymer-based delivery system. J Control Release. 2011;155:262–271. | ||
Ernsting MJ, Tang WL, MacCallum NW, Li SD. Preclinical pharmacokinetic, biodistribution, and anti-cancer efficacy studies of a docetaxel-carboxymethylcellulose nanoparticle in mouse models. Biomaterials. 2012;33(5):1445–1454. | ||
Ernsting MJ, Foltz WD, Undzys E, Tagami T, Li SD. Tumor-targeted drug delivery using MR-contrasted docetaxel-carboxymethylcellulose nanoparticles. Biomaterials. 2012;33:3931–3941. | ||
Hoang B, Ernsting MJ, Roy A, Murakami M, Undzys E, Li SD. Docetaxel-carboxymethylcellulose nanoparticles target cells via a SPARC and albumin dependent mechanism. Biomaterials. 2015;59:66–76. | ||
Harada M, Iwata C, Saito H, et al. NC-6301, polymeric micelle rationally optimized for effective release of docetaxel is potent but is less toxic than native docetaxel in vivo. Int J Nanomedicine. 2012;7:2713–2727. | ||
Hu Q, Rijcken CJ, Bansal R, Hennink WE, Storm G, Prakash J. Complete regression of breast tumor with a single dose of docetaxel-entrapped core-cross-linked polymeric micelles. Biomaterials. 2015;53:370–378. | ||
Logie J, Ganesh AN, Aman AM, Al-awar RS, Shoichet MS. Preclinical evaluation of taxane-binding peptide-modified polymeric micelles loaded with docetaxel in an orthotopic breast cancer mouse model. Biomaterials. 2017;123:30–47. | ||
Wicki A, Witzigmann D, Balasubramanian V, Huwyler J. Nanomedicine in cancer therapy: challenge, opportunities, and clinical applications. J Control Release. 2015;200:138–157. | ||
Sohn YS, Jun YJ. Poly- and cyclotriphosphazenes as drug carriers for anticancer therapy. In: Andrianov AK, editor. Polyphosphazenes for Biomedical Applications. Hoboken, NJ: Wiley; 2009:249–275. | ||
Allcock HR, Pucher SR, Scopelianos AG. Poly[(amino acid ester)phosphazenes] as substrates for the controlled release of small molecules. Biomaterials. 1994;15(8):563–569. | ||
Inoue T, Chen G, Nakamae K, Hoffman AS. An AB block copolymer of oligo(methyl methacrylate) and poly(acrylic acid) for micellar delivery of hydrophobic drugs. J Control Release. 1998;51(2–3):221–229. | ||
Sohn YS, Cho YH, Beak H, Jung OS. Synthesis and properties of low molecular weight polyphosphazenes. Macromolecules. 1995;28:7566–7568. | ||
Lee HJ, Pardridge WM. Monoclonal antibody radiopharmaceuticals: cationization, pegylation, radimetal chelation, pharmacokinetics, and tumor imaging. Bioconjugate Chem. 2003;14:546–553. | ||
Maeda H, Wu J, Sawa T, Matsumura Y, Hori K. Tumor vascular permeability and the EPR effect in macromolecular therapeutics: a review. J Control Release. 2000;65(1–2):271–284. | ||
Zu JZ, Moon SH, Jeong B, Sohn YS. Thermosensitive micelles from PEGylated oligopeptides. Polymer. 2007;48:3673–3678. | ||
Song CW, Griffin R, Park HJ. Influence of tumor pH on therapeutic response. In: Teicher BA, editor. Cancer Drug Resistance. (Chap. 2). Totowa, NJ: Humana Press; 2006:21–42. | ||
Salmaso S, Caliceti P. Stealth properties to improve therapeutic efficacy of drug nanocarriers. J Drug Deliv. 2013;2013:19. | ||
Blanco E, Shen H, Ferrari M. Principles of nanoparticle design for overcoming biological barriers to drug delivery. Nat Biotechnol. 2015;33(9):941–951. | ||
Allcock HR, Fuller TJ, Matsumura K. Hydrolysis pathways for aminophosphazenes. Inorg Chem. 1982;21:515–521. | ||
Yoo HS, Lee EA, Park TG. Doxorubicin-conjugated biodegradable polymeric micelles having acid-cleavable linkages. J Control Release. 2002;82(1):17–27. |
Supplementary materials
Methods
Analytical method to estimate the time-dependent amounts of docetaxel in major organs by liquid chromatography tandem mass spectrometry (LC-MS/MS)
For quantification of the time-dependent concentrations of docetaxel in various organs and plasma, an Agilent 1260 infinity high-performance liquid chromatography (HPLC) system coupled with an Agilent 6460 triple quadrupole mass spectrometer detector was used in this study. An Agilent Poroshell® 120 EC–C18 (3.0×50 mm, particle size 2.7 μm) column thermostated at 30°C was used for separation. The mobile phase was a mixture of water containing 0.1% formic acid (A) and acetonitrile containing 0.1% formic acid (B). The flow rate was 0.3 mL/min. The gradient elution was performed as follows. The initial condition, 90% A and 10% B, was maintained to equilibrate the column. From 0.5 min to 4 min, the proportion of B was gradually increased to 90% and maintained thus for the next 1 min. Then, the mobile phases were recovered to the initial condition for reequilibrium of the system prior to the next injection. The total running time was 8.5 min. All samples were maintained at 10°C during the analysis.
The MS detector was operated in positive electrospray ionization (ESI) mode in multiple reaction monitoring (MRM) mode. The ion source parameters were as follows. The drying gas temperature was maintained at 300°C, and the drying gas flow rate was 11 L/min. The nebulizer pressure and the capillary voltage were set to 45 psi and 3,500 V, respectively. The sheath gas temperature was maintained at 400°C and the sheath gas flow rate was 11 L/min. The first quadrupole was set to select sodium adducts as the precursor ion of the analyte, ie, docetaxel (m/z 830.3), and the internal standard, paclitaxel (m/z 876.3). The product ion of each analyte (m/z 549.3 for docetaxel and m/z 591.3 for paclitaxel) was selected by the third quadrupole. The fragmentor voltage and collision energy were 175 and 22 for docetaxel, and 185 and 20 for paclitaxel, respectively. The LC-MS/MS system control and data analysis were operated with Agilent MassHunter QQQ Qualitative and Quantitative analysis software programs (version B.04.00).
For plasma sample preparation, 5 μL of the internal standard, paclitaxel, was added to 50 μL of plasma, and the analytes were extracted with 500 μL of acetonitrile. The mixture was vortexed vigorously for 3 min and centrifuged at 12,000 rpm for 15 min. Then, 400 μL of supernatant was transferred to a new microtube and evaporated to dryness under a gentle stream of nitrogen gas at room temperature. The residue was reconstituted in 50 μL of 70% acetonitrile by vortexing for 1.5 min. The clear solution was transferred to a clean insert in a vial and 5 μL of the solution was injected into the LC-MS/MS system for analysis. For sample preparation of organ tissue, each organ and tumor was homogenized in 20 μL of water per 1 mg of tissue using a homogenizer (T10, IKA, Staufen, Germany). Next, 50 μL of tissue homogenate was assayed similar to the method for plasma.
 | Figure S1 1H-NMR spectra of the polyphosphazene carrier polymer [NP(MPEG550)3(Lys-OEt)]n (A) and its conjugate, Polytaxel [NP(MPEG550)3(Lys-OEt)(AA)(DTX)]n (B). |
 | Figure S2 31P-NMR spectrum of polyphosphazene carrier polymer [NP(MPEG550)3(Lys-OEt)]n (A) and its Polytaxel conjugate [NP(MPEG550)3(Lys-OEt)(AA)(DTX)]n (B). |
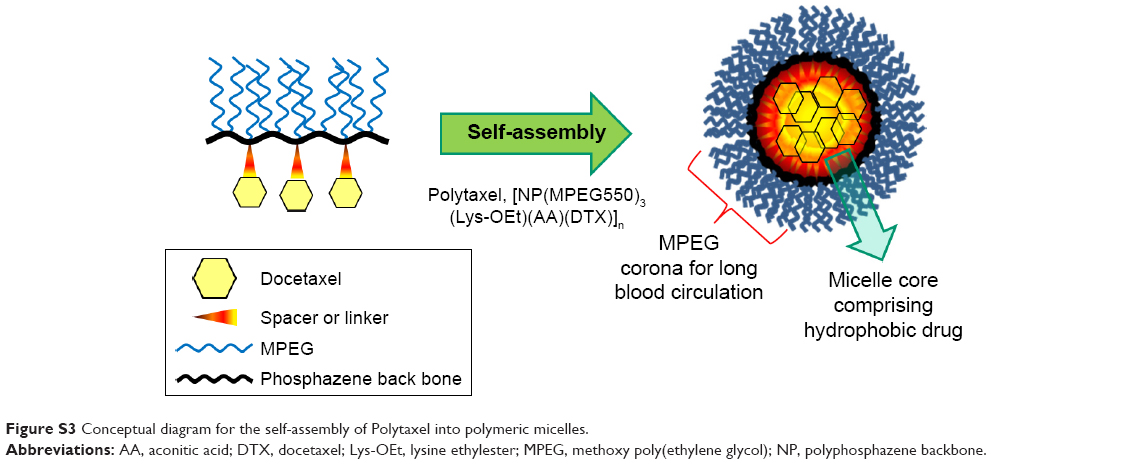 | Figure S3 Conceptual diagram for the self-assembly of Polytaxel into polymeric micelles. |
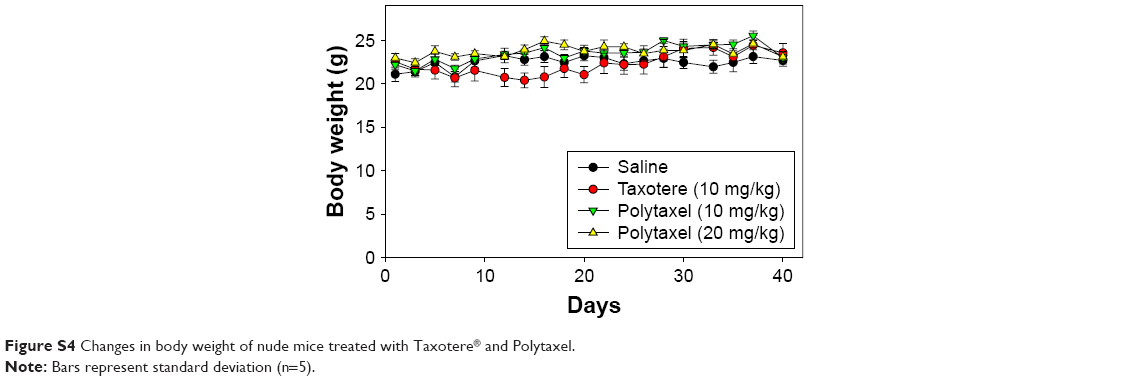 | Figure S4 Changes in body weight of nude mice treated with Taxotere® and Polytaxel. |
 © 2017 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2017 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.