Back to Journals » International Journal of Nanomedicine » Volume 13
Design of pH-sensitive methotrexate prodrug-targeted curcumin nanoparticles for efficient dual-drug delivery and combination cancer therapy
Authors Xie JJ, Fan ZX, Li Y, Zhang YY, Yu F, Su GH, Xie LY, Hou ZQ
Received 22 September 2017
Accepted for publication 11 January 2018
Published 9 March 2018 Volume 2018:13 Pages 1381—1398
DOI https://doi.org/10.2147/IJN.S152312
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Professor Lei Yang
Jiajiang Xie,1,2,* Zhongxiong Fan,2,* Yang Li,2,* Yinying Zhang,2 Fei Yu,3 Guanghao Su,4 Liya Xie,5 Zhenqing Hou2
1Xiamen Xianyue Hospital, Xiamen, China; 2Research Center of Biomedical Engineering of Xiamen, Key Laboratory of Biomedical Engineering of Fujian Province, Fujian Provincial Key Laboratory for Soft Functional Materials Research, Department of Biomaterials, College of Materials, Xiamen University, Xiamen, China; 3College of Medicals, Xiamen University, Xiamen, China; 4Children’s Hospital of Soochow University, Suzhou, China; 5The First Affiliated Hospital of Xiamen University, Xiamen, China
*These authors contributed equally to this work
Aim: We designed acid-labile methotrexate (MTX) targeting prodrug self-assembling nanoparticles loaded with curcumin (CUR) drug for simultaneous delivery of multi-chemotherapeutic drugs and combination cancer therapy.
Methods: A dual-acting MTX, acting as both an anticancer drug and as a tumor-targeting ligand, was coupled to 1,2-distearoyl-sn-glycero-3-phosphoethanolamine-N-[aldehyde(polyethylene glycol)-2000] via Schiff’s base reaction. The synthesized prodrug conjugate (DSPE-PEG-Imine-MTX) could be self-assembled into micellar nanoparticles (MTX-Imine-M) in aqueous solution, which encapsulated CUR into their core by hydrophobic interactions (MTX-Imine-M-CUR).
Results: The prepared MTX-Imine-M-CUR nanoparticles were composed of an inner hydrophobic DSPE/CUR core and an outside hydrophilic bishydroxyl poly (ethyleneglycol) (PEG) shell with a self-targeting MTX prodrug corona. The imine linker between 1,2-distearoyl-sn-glycero-3-phosphoethanolamine-N-[aldehyde(polyethyleneglycol)-2000] and MTX, as a dynamic covalent bond, was strong enough to remain intact in physiological pH, even though it is rapidly cleaved in acidic pH. The MTX-Imine-M-CUR could codeliver MTX and CUR selectively and efficiently into the cancer cells via folate receptor-mediated endocytosis followed by the rapid intracellular release of CUR and the active form of MTX via the acidity of endosomes/lysosomes. Moreover, the MTX-Imine-M-CUR resulted in significantly higher in vitro and in vivo anticancer activity than pH-insensitive DSPE-PEGAmide-MTX assembling nanoparticles loaded with CUR (MTX-Amide-M-CUR), MTX unconjugated DSPE-PEG assembling micellar nanoparticles loaded with CUR (M-CUR), combination of both free drugs, and individual free drugs.
Conclusion: The smart system provided a simple, yet feasible, drug delivery strategy for targeted combination chemotherapy.
Keywords: pH-sensitive prodrug, self-assembly, targeting, combination therapy, nanoparticles
Introduction
Over the last 30 years, there has been a sharp increase in the occurrence of human cancers.1 Although it could be treated with chemotherapy, the clinical outcome is limited due to the serious side effects, low therapeutic efficiency, and multidrug resistance.2,3 Considering the above obstacles, combination therapy has been conceived as one of the primary options to treat cancer.3,4 The significant advantage is that it could not only damage or kill cancer cells with multiple drugs to achieve the synergism via different cell cycle stages and mechanisms of action but also suppress the drug resistance.5–7
Methotrexate (MTX) is one of the effective wide-spectrum chemotherapeutic drugs used for treating a wide range of human cancers, such as cervical, breast, lung, head, neck, skin, and other malignant tumors.8–10 MTX could inhibit the activity of dihydrofolate reductase enzyme participating in folate metabolism.9,11 However, its clinical application is limited due to the poor pharmacokinetics and severe systemic toxicity following systemic administration.12–14
Curcumin (CUR), a natural polyphenol extracted from turmeric, has attracted increasing attention on account of its anticancer, antioxidant, anti-inflammatory, and hepatoprotective properties.15–18 As a potent anticancer and antioxidant agent, CUR can inhibit cell proliferation of a large variety of tumors through topoisomerase II-mediated DNA cleavage.19,20 Unfortunately, its clinical application has been severely restricted due to the poor stability, limited water solubility, and inadequate accumulation at the tumor sites.21 Thus, the application of CUR and MTX in combination therapy suffers from some drawbacks.
Nanoparticles are regarded as an innovative platform for drug delivery because of their ability to improve the pharmacokinetics and biodistribution of the drug while reducing the drug toxicity.22 Specifically, compared with the conventional combination drug therapy, nanoparticle-based multidrug delivery system could target the tumor region or cells via the enhanced permeability and retention (EPR) effect, release the drug in a sustained manner, and even provide various synergistic effects.22–25 Stimuli-responsive nanoparticles have been rapidly developed on the basis of subtle differences between the physiological microenvironments of tumor and normal tissues.26 For instance, the pH at extracellular microenvironment of the tumor is lower (~6.8) due to more active aerobic glycolysis, and the acidity in intracellular endo/lysosomes is even increased (pH 4.5–6.5).26 The pH-responsive polymer prodrug-based nanoparticles attract considerable research interest owing to controlled drug release.27,28 The most widely used intracellular acid-responsive covalent linkers in polymer prodrug include hydrazone, imine, acetal, boronate linker, etc.27,29,30 In addition, tumor-specific targeting strategies by nanoparticles have been extensively studied in chemotherapy for minimizing systemic toxicity and augmenting therapeutic efficacy.31–33 Therefore, the structural design of the nanoparticles integrating the functions of selective tumor cell targeting, specific intracellular drug release, and drug combination is crucial for anticancer drug delivery.
1,2-distearoyl-sn-glycero-3-phosphoethanolamine-N-[(polyethylene glycol)-2000] (DSPE-PEG), a kind of PEGylated lipid, is a US Food and Drug Administration-approved pharmaceutical additive. Nanoparticles self-assembled by the amphiphilic DSPE-PEG lipopolymer integrate both the polymer- and lipid-based nanoparticles and are attractive therapeutic drug carriers for anticancer drug delivery.34,35 DSPE-PEG lipopolymer self-assembling nanoparticles possess nanoscale particle size (<100 nm), fine in vivo biocompatibility/nontoxicity/non-cytotoxicity, low critical micelle concentration (10−6 M), high colloidal stability, long-circulating property, and passive tumor-targeting ability via EPR effect.36 Moreover, these nanoparticles could efficiently encapsulate a variety of poorly water-soluble anticancer drugs within their hydrophobic DSPE core.
Our group and certain other groups reported that MTX has some affinity for folate receptors because of the structural similarity with folic acid (FA); therefore, MTX could not only serve as an anticancer drug to exert its anticancer effect but also act as a targeting moiety that binds with the folate receptor to exert its targeting function.8,37,38 In this study, combining the advantages of anticancer drug MTX for active tumor targeting, DSPE-PEG for drug carriers, and imine linkage for acid-triggered intracellular release, a novel targeting DSPE-PEG-Imine-MTX amphiphilic lipopolymer prodrug was synthesized by conjugating the aldehyde group of DSPE-PEG with the aromatic amino group in MTX via Schiff’s base reaction (Figure 1A). Due to the amphiphilicity, DSPE-PEG-Imine-MTX and DSPE-PEG-methoxy (DSPE-MPEG) could be self-assembled into self-targeting and pH-responsive micellar nanoparticles, in which CUR was encapsulated within their hydrophobic core (designated as MTX-Imine-M-CUR; Figure 1B). Herein, the in vitro dual-drug release profile, cellular uptake, subcellular location, and intracellular drug delivery of MTX-Imine-M-CUR were investigated. Besides, the in vitro anticancer activity in folate receptor-overexpressing human cervical carcinoma cell line HeLa cells and human breast carcinoma cell line MCF-7 cells, and the in vivo tumor inhibition effect in HeLa tumor-bearing nude mice was evaluated. Our results revealed that the smart MTX-Imine-M-CUR had potential as a self-targeting and pH-responsive multidrug delivery system for synergistic cancer therapy (Figure 1C).
 | Figure 1 (A) Synthetic route of DSPE-PEG-Imine-MTX conjugate via imine reaction between the aldehyde group of DSPE-PEG-CHO and the aromatic amino group of MTX. (B) Schematic representation of preparation of MTX unconjugated DSPE-PEG assembling micellar nanoparticles loaded with CUR (M-CUR), DSPE-PEG-Amide-MTX nanoparticles (MTX-Amide-M-CUR), and DSPE-PEG-Imine-MTX nanoparticles (MTX-Imine-M-CUR). (C) Schematic representation of active selective cellular uptake via folate receptor-mediated endocytosis, pH-controlled intracellular dual-drug release, and combination therapy of MTX-Imine-M-CUR nanoparticles after passive tumor accumulation by EPR effect. |
Materials and methods
Materials
CUR was purchased from Aladdin Industrial Corporation (Shanghai, China). 1,2-distearoyl-sn-glycero-3-phosphoethanolamine (DSPE), 1,2-distearoyl-sn-glycero-3-phosphoethanolamine-N-[carboxyl(polyethylene glycol)-2000] (DSPE-PEG-COOH), and DSPE-MPEG were provided by Avanti Polar Lipids. Cy5.5 was from Molecular Probes Inc. (Eugene, OR, USA). DSPE-PEG-Cy5.5 was prepared according to our previous literature.43 Bis-hydroxyl poly (ethyleneglycol) (PEG, molecular weight of ~2,000) was provided by Sinopeg Biotech Co., Ltd. (Xiamen, China). MTX and FA were purchased from Bio Basic Inc. (Markham, Ontario, Canada). N, N′-Dicyclohexylcarbodiimide, N-hydroxysuccinimide (NHS), and N, N′-disuccinimidyl carbonate were purchased from Sigma-Aldrich Co., Ltd. (St Louis, MO, USA). DAPI was obtained from Molecular Probes Inc. (Eugene, OR, USA). MTT was purchased from Amresco (Solon, OH, USA). RPMI-1640, trypsin, and penicillin–streptomycin were ordered from Sigma Chemical Corp. (St Louis, MO, USA). Fetal bovine serum was purchased from Thermo Fisher Scientific (Waltham, MA, USA). All other chemicals and reagents were purchased from Sigma-Aldrich Co., Ltd., unless otherwise noted.
Synthesis of DSPE-PEG-Imine-MTX
First, DSPE-PEG-hydroxyl (DSPE-PEG-OH) was synthesized according to the reported literature.46 Second, DSPE-PEG-aldehyde (DSPE-PEG-CHO) was synthesized by the oxidation of DSPE-PEG-OH with acetic anhydride in dimethyl sulfoxide (DMSO)/chloroform according to the reported procedure.47 Finally, DSPE-PEG-Imine-MTX was synthesized starting from DSPE-PEG-CHO lipopolymer and MTX molecule by reductive amination via Schiff’s base formation.39 DSPE-PEG-CHO (56.2 mg, 20 μmol) and MTX (13.6 mg, 30 μmol) were dissolved in 20 mL of DMSO/chloroform in a round-bottom flask, and then a few drops of acetic acid were added to the flask with molecular sieves. The mixture was degassed through three freeze–pump–thaw procedures and made to react at 40°C for 48 h. The resultant product was evaporated and precipitated into excess dry, ice-cold ethyl ether and acetone (10:1) mixture. The crude precipitate was separated by decantation and redissolved in dichloromethane. The unreacted or excess MTX was removed from the conjugated one via filtration. The filtrate was concentrated and reprecipitated into a mixture of ice-cold ethyl ether and acetone (10:1). The reprecipitate was separated by decantation, washed three times with ice-cold ethyl ether to remove traces of DMSO, dried in vacuum overnight, and stored at −20°C.
In addition, DSPE-PEG-Amide-MTX was synthesized starting from DSPE-PEG-COOH and MTX molecule via an amide bond according to our previous reports with some modification.41,48–50 In a typical reaction, DSPE-PEG-COOH (84.8 mg, 30 μmol), N, N′-dicyclohexylcarbodiimide (7.5 mg, 36 μmol), and NHS (4.2 mg, 36 μmol) were dissolved in DMSO with continuous stirring under a nitrogen atmosphere. After 30 min, MTX (20.4 mg, 45 μmol) was added to the DMSO solution containing activated DSPE-PEG-COOH. The mixture was allowed to react for 72 h with continuous stirring under a nitrogen atmosphere. The resulting mixture was filtered to remove the white precipitate of dicylcohexyl urea and precipitated in an ice-cold mixture of ethyl ether and acetone (10:1). The crude precipitate was separated by decantation and redissolved in dichloromethane. The unreacted or excess MTX was removed from the conjugated one via filtration. The filtrate was concentrated and reprecipitated into a mixture of ice-cold ethyl ether and acetone (10:1). The reprecipitate was separated by decantation, washed three times with ice-cold ethyl ether to remove traces of DMSO, dried in vacuum overnight, and stored at −20°C.
Characterization of DSPE-PEG-Imine-MTX
The 1H nuclear magnetic resonance (1H NMR) spectrum was obtained on a Bruker AV400 MHz NMR spectrometer (Bruker, Billerica, MA, USA). The ultraviolet–visible (UV–vis) absorption spectrum was recorded with a Perkin Elmer Lambda 750 UV–vis–near-infrared spectrophotometer (Perkin-Elmer, Norwalk, CT, USA). The XRD spectrum was recorded with an X-ray diffractometer (Phillips X’pert Pro Super; Panalytical, Almelo, the Netherlands). The MALDI-TOF-MS spectrum was obtained using a Bruker Reflex III MALDI-TOF mass spectrometer (Bruker Daltonics, Bremen, Germany). The molecular weight was determined by GPC using Waters analytical columns Styragel HT2 (7.8×300 mm). Tetrahydrofuran was used as the mobile phase at a flow rate of 1.0 mL/min under 30°C.
Preparation of MTX-Imine-M-CUR
MTX-Imine-M nanoparticles loaded with CUR were prepared by “one-pot ultrasonic emulsification”. In brief, 10 mg of DSPE-PEG-Imine-MTX and 15 mg of DSPE-MPEG powder were placed into a round-bottom flask. Then, certain amount of CUR in chloroform was added to the flask and mixed thoroughly until the mixture was completely dissolved. Afterward, 30 mL of deionized water was added to the mixture and the flask was placed in an ultrasonic bath cleaner for ultrasonic emulsification for about 10 min at 100 W power. The organic solvent was then removed by evaporation while stirring overnight. After that, the unincorporated drug (precipitate) was removed through polycarbonate membranes with 0.2 and 0.1 μm pores using a Mini-extruder (Avanti, Alabaster, AL, USA) by 10 extrusion cycles.
The MTX unconjugated DSPE-PEG assembling micellar nanoparticles loaded with CUR (M-CUR) nanoparticles were prepared using an identical procedure except that DSPE-PEG-Imine-MTX was replaced by DSPE-MPEG at an equivalent molar for addressing the specificity of MTX-Imine-M nanoparticles toward folate receptors. The MTX-Amide-M-CUR nanoparticles were prepared using an identical procedure except that DSPE-PEG-Imine-MTX was replaced by DSPE-PEG-Amide-MTX (discussed above) at an equivalent molar for addressing the target release of MTX-Imine-M nanoparticles toward a significantly acidic environment.
Characterization of MTX-Imine-M-CUR
The hydrodynamic particle size, PDI, and zeta potential were measured by DLS and electrophoretic light scattering using a Malvern Zetasizer 2000 (Malvern Instruments, Malvern, UK). The morphology was observed through TEM (JEM2100; JEOL, Tokyo, Japan) operated at an accelerating voltage of 200 kV. The XRD spectrum was recorded by an X-ray diffractometer.
Drug loading content
To measure the amount of CUR and MTX loaded (encapsulated or conjugated) within MTX-Imine-M-CUR, the lyophilized MTX-Imine-M-CUR was dissolved in anhydrous DMSO by ultrasonication. After filtration by a 0.22 μm filter membrane, a part of the filtrate was analyzed for the determination of drug loading content by a high-performance liquid chromatography method as described in our previous studies.17,41 The drug loading content of MTX-Imine-M-CUR was calculated as (amount of drug in MTX-Imine-M-CUR)/(weight of MTX-Imine-M-CUR) ×100%.
In vitro drug release
In vitro release profiles of both MTX and CUR from MTX-Imine-M-CUR were determined in phosphate buffer saline (PBS) or acetate buffered saline at 37°C at different pH values of 7.4 and 5.0. Typically, 2 mL of MTX-Imine-M-CUR was transferred into a dialysis bag (molecular weight cut-off =1,000 Da), which was then immersed into 58 mL of buffer saline medium with constant shaking at 100 rpm at 37°C in darkness. Then, an equivalent volume of the external buffer was periodically collected at predetermined time points. Subsequently, 2 mL of fresh medium was immediately replenished. The amount of released drug was determined by high-performance liquid chromatography method as described earlier in this paper.
Cell culture
HeLa human cervical carcinoma cells and MCF-7 human breast carcinoma cells were cultured in FA-deficient RPMI-1640. All cell lines were obtained from American Type Culture Collection and grown in culture media supplemented with 10% fetal bovine serum and 1% penicillin–streptomycin (100 U/mL). The cells were cultured in an incubator (Thermo Fisher Scientific) at 37°C under a humidified atmosphere containing 5% CO2.
In vitro cellular uptake
Both CLSM and flow cytometry were performed to determine the cellular uptake and subcellular distribution of MTX-Imine-M-CUR toward folate receptor-overexpressing cancer cells.
Qualitative cellular uptake of MTX-Imine-M-CUR was performed by CLSM. HeLa or MCF-7 cells (high expression of the folate receptors) were seeded into six-well plates at a density of 1.0×104 cells per well, incubated at 37°C for 24 h, and then treated with MTX-Imine-M-CUR for different treatment time periods (0.5 and 2 h) at 37°C. After treatment, the cells were washed three times and then fixed by 4% paraformaldehyde for 15 min. Then, the cells were washed with PBS and the nuclei were stained by DAPI for 10 min. Finally, the cells were imaged using a Leica TCS SP5 CLSM with excitation at 405 nm for DAPI and 488 nm for CUR. Free CUR, M-CUR, and MTX-Amide-M-CUR were used as controls. In addition, to investigate the folate receptor-mediated internalization of MTX-Imine-M-CUR, excess of free FA was incubated with cells for 1 h prior to the addition of MTX-Imine-M-CUR.
Quantitative cellular uptake of MTX-Imine-M-CUR was measured by flow cytometry analysis. HeLa cells (high expression of the folate receptors) were seeded at a density of 2.0×105 cells per well into six-well plates with 2 mL of cell culture medium and cultured for 24 h. After that, the cell culture medium was replaced with fresh RPMI-1640 followed by addition of free CUR, M-CUR, MTX-Amide-M-CUR, and MTX-Imine-M-CUR at an equivalent CUR concentration. After coincubation for 2 h, the cell culture medium was removed and the cells were washed with PBS. After that, the cells were detached with trypsin/EDTA, harvested by centrifugation at 1,500 rpm for 5 min at 4°C, and resuspended in PBS. Data were obtained on a FACSCalibur flow cytometer (Becton Dickinson, San Jose, CA, USA) and analyzed using Cell Quest software.
Subcellular location
HeLa cells were seeded, cultured for 24 h at 37°C and 5% CO2, and then incubated with MTX-Imine-M-CUR for 1 h. Subsequently, the cells were stained by 200 nM of LysoTracker Red (Thermo Fisher Scientific) at 37°C for 20 min and fixed with 4% paraformaldehyde for 15 min. Finally, the cells were washed by cold PBS twice, stained with DAPI for 10 min, and imaged using a Leica TCS SP5 CLSM.
Intracellular drug/nanocarriers delivery
To investigate the intracellular drug/nanocarrier delivery of MTX-Imine-M-CUR, Cy5.5-labeled DSPE-PEG was synthesized for use as a near-infrared fluorescent probe (DSPE-PEG-Cy5.5 was conjugated between DSPE-PEG-NH2 and Cy5.5 NHS ester via amide linkage). HeLa cells were seeded into 24-well plates at a density of 1.0×104 cells per well. After culturing for 24 h, the cells were treated with MTX-Imine-MCy5.5-CUR at 37°C for predetermined incubation time periods. Afterward, the cells were washed three times with PBS and fixed with 4% (v/v) paraformaldehyde for 15 min. Finally, the cells were washed by cold PBS twice, stained with DAPI for 10 min, and observed using a Leica TCS SP5 CLSM.
Animals
Female BALB/c nude mice (20±2 g, 4–6 weeks) were purchased from Vital River Laboratory Animal Technology Co., Ltd (Beijing, China). All animals were acclimatized in a laminar flow room at a controlled temperature of 25°C±2°C, relative humidity of 50%–60%, and 12 h light–dark cycles with standard diet ad libitum for 1 week prior to experiment. All the experimental procedures were performed in accordance with the Principles of Care and Use of Laboratory Animals and were approved by the Xiamen University Laboratory Animal Management Ethics Committee for the Care and Use of Laboratory Animals.
In vivo antitumor effect
In vivo antitumor effect was evaluated on HeLa tumor-bearing BALB/c nude mice. In brief, 0.2 mL of cell suspension containing 2×106 HeLa cells was inoculated subcutaneously into the right armpit of mice (18–22 g, 4–6 weeks old). When the tumor reached around 100 mm3, the tumor-bearing mice were randomly divided into six groups. Mice were intravenously administrated with 0.2 mL of 0.9% NaCl, free CUR, free CUR/MTX, M-CUR, MTX-Amide-M-CUR, or MTX-Imine-M-CUR at an equivalent CUR concentration of 8.0 mg/kg and MTX concentration of 5.1 mg/kg via the tail vein every 3 days for four times. The tumor size and body weight of the mice were monitored during treatment. Tumor size was measured every 2 or 3 days by a caliper, and tumor volume (V) was calculated by the following formula: V (mm3) =0.5× L × W2, where L and W represent the largest diameter and the smallest diameter, respectively.
Histologic analyses
Histologic analyses were further performed on HeLa tumor-bearing BALB/c nude mice. According to the above in vivo antitumor studies, the mice were sacrificed on the 18th day. Afterward, the tumors were excised, weighed, fixed with 4% paraformaldehyde, and embedded in paraffin. Finally, 4–5 μm thick slices of tumor tissues were stained by H&E, and the stained tumor sections were observed using optical microscopy.
Statistical analysis
Data were described as mean ± SD value. The differences among different groups were calculated using independent samples t-test of SPSS. P<0.05 was indicated by a single asterisk (*) and was considered statistically significant. P<0.01 was indicated by a double asterisk (**) and was considered highly statistically significant.
Results and discussion
Synthesis and characterization of DSPE-PEG-Imine-MTX
We proposed a novel approach, which was fundamentally different from the traditional strategy, using a pair of two types of orthogonal molecules (an anticancer drug and a targeting ligand). It was based on the use of a dual-acting single molecule (MTX) that functions as both a targeting ligand (to bind with cancer cell-overexpressing receptors) and an anticancer drug (to induce cytotoxicity following cellular uptake).
To endow the tumor-selective targeting capacity of small molecular anticancer drug MTX and seek its biomedical applications, an amphiphilic conjugate of DSPE-PEG-Imine-MTX was synthesized via the reductive amination reaction of Schiff’s base between the aromatic amino group of MTX molecule and the aldehyde group of DSPE-PEG-CHO polymer.39 The synthetic route of DSPE-PEG-Imine-MTX amphiphilic polymer prodrug is shown in Figure 1A. The chemical structure of DSPE-PEG-Imine-MTX was characterized by 1H NMR spectroscopy, UV-vis spectrophotometer, X-ray diffractometer (XRD), matrix-assisted laser desorption/ionization time-of-flight mass spectrometry (MALDI-TOF-MS), and gel permeation chromatography (GPC), as shown in Figure S1. As shown in Figure S1A, the 1H NMR spectrum of DSPE-PEG-Imine-MTX exhibited the proton peaks of methylene (-CH2-) and methyl (-CH3) groups of DSPE (δ=1.2 and 0.8 ppm), the sharp proton peaks of repeated units (-CH2-CH2-O-) of PEG (δ=3.5 ppm), and the characteristic proton peaks (δ=8.6, 7.7, and 6.8 ppm) belonging to the p-phenyl ring and the pteridine ring of MTX, respectively. Moreover, after conjugation of DSPE-PEG-CHO with MTX, the proton peak of aldehyde group derived from DSPE-PEG-CHO (δ=9.8 ppm) disappeared significantly and the new proton peak of imine group (δ=8.2 ppm) emerged in the 1H NMR spectrum of DSPE-PEG-Imine-MTX conjugate. As shown in Figure S1B, compared to the UV–vis absorption peak of MTX at 299 nm, we observed a 3 nm red shift in the UV–vis absorption spectrum of DSPE-PEG-Imine-MTX at 302 nm. This was possibly due to the successful linkage between DSPE-PEG-CHO and MTX. In addition, as shown in XRD spectra, DSPE-PEG-Imine-MTX conjugate showed no sharp diffraction peaks compared to the small molecular MTX and DSPE-PEG-CHO/MTX mixture (Figure S1C). The result demonstrated that the crystalline structure of MTX was significantly disrupted in the polymer prodrug, indicating that MTX was coupled to DSPE-PEG-CHO.
The MALDI-TOF-MS spectra revealed that the peak of molecular mass of DSPE-PEG-CHO that appeared at 2,400–3,200 Da was rightshifted to 2,900–3,700 Da after conjugation with MTX (Figure S1D). The result implied that DSPE-PEG-CHO was successfully coupled to MTX possessing a molecular mass of ~500 Da. Furthermore, an obvious increase in experimental molecular mass was found in the GPC spectra after conjugation of MTX to DSPE-PEG-CHO (Figure S1E), as the molecular mass values of DSPE-PEG-CHO, MTX, and DSPE-PEG-Imine-MTX were 2,810, 454, and 3,250, respectively. The above results of 1H NMR, UV–vis, XRD, MALDI-TOF-MS, and GPC analysis confirmed the successful synthesis of DSPE-PEG-Imine-MTX polymer prodrug.
Preparation and characterization of pH-sensitive DSPE-PEG-Imide-MTX assembling micellar nanoparticles loaded with CUR (MTX-Imine-M-CUR)
Because of inherent amphiphilicity, DSPE-PEG-Imine-MTX and DSPE-MPEG could spontaneously self-assemble into micellar nanoparticles (MTX-Imine-M) composed of an inner hydrophobic DSPE core and an outside hydrophilic PEG shell with a self-targeting prodrug corona when exposed to the aqueous solution. The CUR drug could be encapsulated within the hydrophobic core of MTX-Imine-M (MTX-Imine-M-CUR) through hydrophobic interactions. The MTX-Imine-M-CUR nanoparticles were characterized by dynamic light scattering (DLS) and transmission electron microscope (TEM). In addition, the M-CUR (nanocarriers were self-assembled by DSPE-MPEG) and the MTX-Amide-M-CUR (nanocarriers were self-assembled by DSPE-PEG-Amide-MTX and DSPE-MPEG) were fabricated as comparative formulations (Figure 1B). The TEM observation of all drug formulations (Figure 2A–C) confirmed the monodispersed spherical nanoparticles. With DLS measurement, all nanoparticles showed a mean hydrodynamic particle size of 20–30 nm with negative surface charge (Figure 2A–C), which might facilitate preventing potential aggregations via electrostatic repulsion and decrease the nonspecific interaction between nanoparticles and plasma protein/red blood cells. Furthermore, the M-CUR, MTX-Amide-M-CUR, and MTX-Imine-M-CUR nanoparticles exhibited a CUR-loading capacity of ~8.4%, ~8.0%, and ~7.9% (weight ratio), respectively. In addition, the MTX-Amide-M-CUR and the MTX-Imine-M-CUR nanoparticles exhibit an MTX-loading capacity of ~5.1% and ~5.0% (weight ratio), respectively.
 | Figure 2 Hydrodynamic size distribution, zeta potential, and TEM images of (A) M-CUR, (B) MTX-Amide-M-CUR, and (C) MTX-Imine-M-CUR. (D) XRD spectra of CUR, MTX-Amide-M nanocarriers, MTX-Amide-M nanocarriers/CUR mixture, MTX-Amide-M-CUR nanosystems, MTX-Imine-M nanocarriers, MTX-Imine-M nanocarriers/CUR mixture, and MTX-Imine-M-CUR nanosystems. (E) In vitro release profiles of CUR from M-CUR, MTX-Amide-M-CUR, and MTX-Imine-M-CUR in PBS buffer (pH 7.4 and 5.0) at 37°C. (F) In vitro release profiles of MTX from MTX-Amide-M-CUR and MTX-Imine-M-CUR in PBS buffer (pH 7.4 and 5.0) at 37°C. |
XRD analysis was used to confirm the existing state of CUR in the nanoparticles (Figure 2D). The typical crystal peaks of CUR were still found in the physical mixture of CUR and MTX-Imine-M (or MTX-Amide-M) nanocarriers, while they disappeared in the MTX-Imine-M-CUR (or MTX-Amide-M-CUR) nanoparticles, suggesting the CUR was uniformly dispersed in the nanoparticles.
In vitro colloidal stability
The colloidal stability of MTX-Imine-M-CUR was also investigated by incubating the nanoparticles in water and PBS. The hydrodynamic particle size and polydispersity index (PDI) of the nanoparticles were continuously monitored up to 72 h. As shown in Figure S2, no remarkable change and no obvious aggregation were observed throughout the test. In addition, the PDI values were smaller than 0.200, indicating good dispersity of the nanoparticles. It might be vital for the nanoparticles to keep these characteristics constant before arriving at the tumor sites.22,40 Thus, MTX-Imine-M-CUR could be utilized as a potential drug delivery formulation for intravenous administration.
In vitro drug release
The in vitro dual-drug release profiles from MTX-Imine-M-CUR were determined in buffers at different environmental pH values (7.4 and 5.0). The M-CUR and MTX-Amide-M-CUR were used as controls. As depicted in Figure 2E, the CUR release rate from M-CUR, MTX-Amide-M-CUR, and MTX-Imine-M-CUR was promoted by a decrease in environmental pH. However, the release profiles of the three drug formulations at the same pH values were similar, indicating the surface functionalization of MTX did not significantly modify the release of CUR.37
A pH-dependent release of MTX was also clearly observed from MTX-Imine-M-CUR instead of MTX-Amide-M-CUR (Figure 2F). Specifically, at pH 5.0, only 20% of the total MTX was released from MTX-Imine-M-CUR, whereas the accumulative release amount increased to ~60% at pH 5.0 under the same conditions. On the basis of this result, it could be reasonably concluded that DSPE-PEG-Imine-MTX could remain relatively stable in a neutral environment (ie, normal physiological fluid). Afterward, the release rate of MTX from DSPE-PEG-Imine-MTX was dramatically accelerated via the rapid cleavage of imine bond when the nanoparticles were exposed to an acidic environment (ie, endo/lysosomes). Thus, the pH-responsive dual-drug release pattern of MTX-Imine-M-CUR might be facilitated to reduce the burst release and leakage of the drug.17,41
In vitro cellular uptake
Confocal laser scanning microscopy (CLSM) was used to study the cellular uptake behavior of nanoparticles, and CUR was used as an intrinsic fluorescence probe. HeLa cells (high overexpression of folate receptors) were incubated with free CUR, M-CUR, and MTX-Imine-M-CUR for 0.5 and 2 h at 37°C. The cell nuclei were stained by 4′,6-diamidino-2-phenylindole dihydrochloride (DAPI) to identify the cell nuclei. Figure 3A shows that in terms of all kinds of drug formulations, the green fluorescence signal gradually became stronger with an increase of incubation time. Moreover, a stronger green fluorescence intensity was clearly observed in HeLa cells treated with MTX-Imine-M-CUR (or MTX-Amide-M-CUR) compared with free CUR and M-CUR for the same incubation time (Figures 3A and 4A). This result indicates the manner of time-dependent cellular uptake of CUR molecules and CUR-loaded nanoparticles. Similar results were also demonstrated in MCF-7 cells because of high overexpression of folate receptors on the cell membrane (Figure 3B). The result indicates that regardless of the linkage type, surface functionalization of MTX on the nanoparticles could achieve much more efficient cellular uptake due to the much higher multivalent affinity of multiple MTX ligands from the nanosystems to multiple folate receptors overexpressed on HeLa or MCF-7 cells.
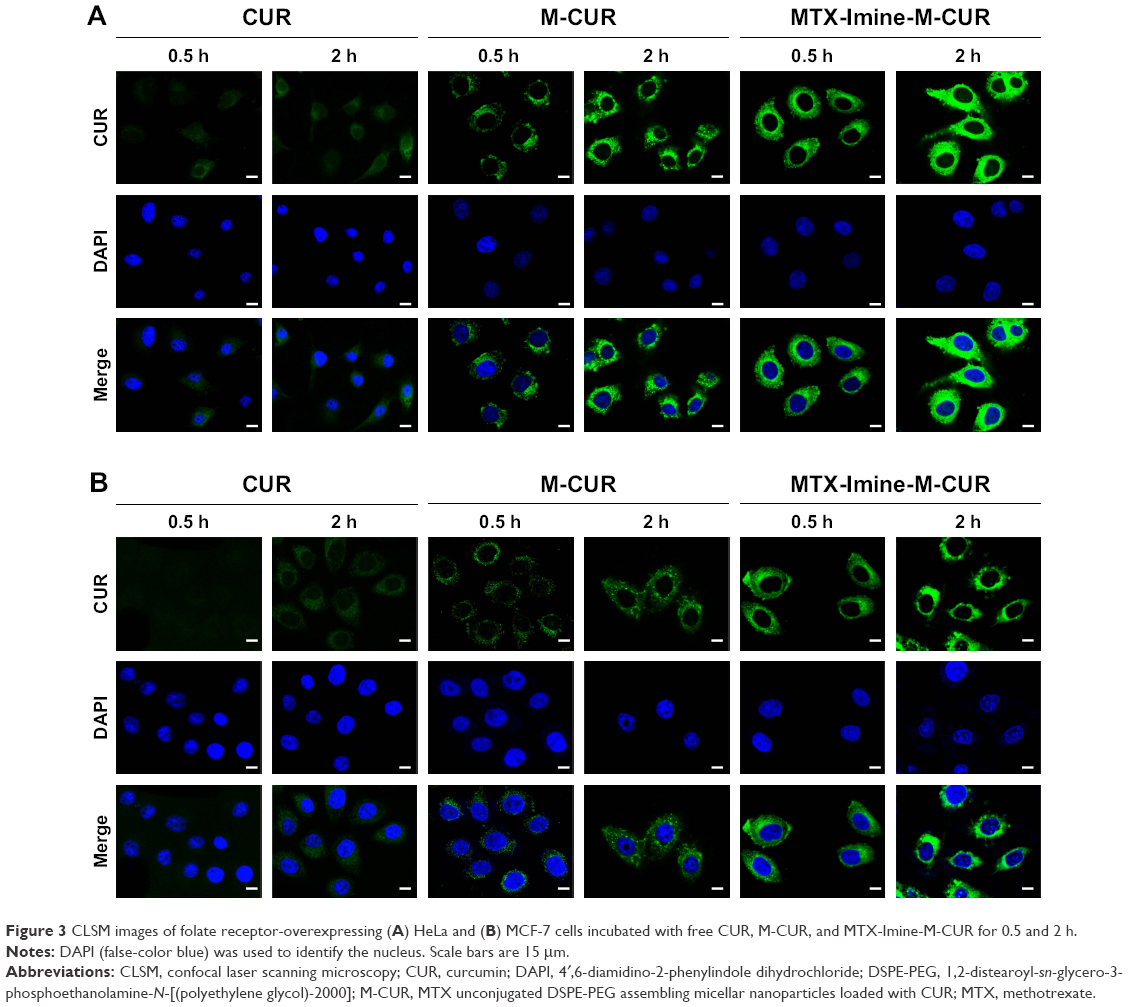 | Figure 3 CLSM images of folate receptor-overexpressing (A) HeLa and (B) MCF-7 cells incubated with free CUR, M-CUR, and MTX-Imine-M-CUR for 0.5 and 2 h. |
 | Figure 4 (A) CLSM images, (B) flow cytometry profiles, and (C) mean fluorescence intensity of HeLa cells incubated with free CUR, M-CUR, MTX-Amide-M-CUR (without/with FA pretreatment), or MTX-Imine-M-CUR (without/with FA pretreatment) for 2 h. Error bars indicate SD (n=4). *P<0.05. (D) Subcellular location of MTX-Imine-M-CUR in HeLa cells after incubation for 1 h. |
Flow cytometry
Flow cytometry was also performed to determine the cellular uptake efficacy of the nanoparticles. HeLa cells were incubated with free CUR, M-CUR, and MTX-Imine-M-CUR at 37°C for 2 h. Cells cultured with culture medium were used as control to eliminate interference by autofluorescence. As shown in Figure 4B and C, the flow cytometric histogram of MTX-Imine-M-CUR group (or MTX-Amide-M-CUR group) exhibited a significant shift to higher fluorescence intensity region compared to that of M-CUR or free CUR group. It is well known that free CUR enters into the cells through the cellular membrane via simple diffusion. Besides, M-CUR nanoparticles were possibly internalized by the cancer cells by adsorptive endocytosis due to the effect of their nanosize. Both quantitative and qualitative results indicate that the MTX-Imine-M-CUR (or MTX-Amide-M-CUR) could be specifically internalized by the HeLa cells via folate receptor-mediated endocytosis.
Competitive inhibition test
To investigate the specificity of MTX-Imine-M-CUR toward folate receptors, we conducted a competitive inhibition test on HeLa and MCF-7 cells. Cells were pretreated with excess of free FA (inhibitor of folate receptor-mediated endocytosis) for 1 h and subsequently treated with MTX-Imine-M-CUR and MTX-Amide-M-CUR. When cancer cells were pretreated with an excess of free FA followed by incubation with the MTX-Imine-M-CUR and MTX-Amide-M-CUR, their enhanced intracellular internalization was drastically suppressed (Figure 4A–C). The result demonstrates that MTX-functionalized micellar nanosystems (MTX-Imine-M-CUR and MTX-Amide-M-CUR) could selectively bind to folate receptors.
On the basis of the above discussions, the comprehensive results of cellular uptake, internalization, and competitive inhibition test confirmed the effectiveness of self-assembled MTX-Imine-M and MTX-Amide-M as active targeting drug carriers based on the folate receptor-mediated endocytosis.
Subcellular location
To examine the subcellular localization of MTX-Imine-M-CUR, LysoTracker Red was used to identify the acidic lysosomes. After incubation of HeLa cells with MTX-Imine-M-CUR for 1 h (Figure 4D), the majority of red fluorescence of MTX-Imine-M-CUR was localized within the lysosomes. This result indicates that MTX-Imine-M-CUR nanoparticles were internalized through the endocytosis pathway.
Besides, to directly image the dynamic distribution of CUR drug/MTX-Imine-M nanocarriers of MTX-Imine-M-CUR nanosystems in the cancer cells, 1,2-distearoyl-sn-glycero-3-phosphoethanolamine-N-[Cy5.5(polyethylene glycol)-2000] (DSPE-PEG-Cy5.5) was used as a near-infrared fluorescent dye to label MTX-Imine-M nanocarriers (MTX-Imine-MCy5.5). As shown in Figure 5A, the green and red fluorescence signals were mainly located in the cytoplasm of HeLa cells after incubation with MTX-Imine-MCy5.5-CUR for 1 h. Interestingly, after 6 h of incubation, the green fluorescence signal was accumulated in both the cytoplasm and nucleus, while the majority of red fluorescence signal was still distributed in the cytoplasm. The result powerfully implies that MTX-Imine-M-CUR could effectively transport CUR to both cell cytoplasm and nucleus by integration of enhanced cellular uptake (via folate receptor-mediated endocytosis; Figures 3 and 4A–C) and accelerated intracellular drug release (induced by acidic pH; Figures 2E and 4D).
 | Figure 5 (A) Intracellular drug/nanocarriers distribution of HeLa cells treated with MTX-Imine-M-CUR for 1 and 6 h. DSPE-PEG-Cy5.5 (false-color red) was used to label nanocarriers. (B, C) In vitro cell viability of (B) HeLa cells and (C) MCF-7 cells incubated with free CUR, free CUR/MTX mixture, M-CUR nanosystems, MTX-Amide-M-CUR nanosystems, or MTX-Imine-M-CUR nanosystems for 24 h. (D, E) In vitro cell viability of (D) HeLa and (E) MCF-7 cells incubated with MTX-Imine-M-CUR nanosystems without/with FA pretreatment for 24 h. |
In vitro cytotoxicity
We further investigated the cytotoxicity of MTX-Imine-M-CUR in HeLa and MCF-7 cells. The cell viability was investigated by a standard MTT assay after exposure of MTX-Imine-M-CUR to various CUR concentrations for 24 h and the IC50 values were also calculated. In addition, free CUR, free CUR/MTX, M-CUR, and MTX-Amide-M-CUR were used as controls. As shown in Figure 5B and C, it was clear that after incubation of HeLa or MCF-7 cells for 24 h, the cell inhibition effect of MTX-Imine-M-CUR was significantly higher than that of MTX-Amide-M-CUR, M-CUR, free CUR/MTX, and free CUR. Specifically, the IC50 of MTX-Imine-M-CUR was calculated as 16.6 μg/mL toward the HeLa cells. In contrast, the IC50 of MTX-Amide-M-CUR, M-CUR, free CUR/MTX, and free CUR was calculated as 27.4, 40.8, 52.6, and 55.9 μg/mL (corresponding to the concentration of CUR), respectively. To further confirm the synergy in the MTX-Imine-M-CUR, the combination index method was carried out.42 The combination index value of MTX-Imine-M-CUR toward HeLa cells was calculated as 0.52 (0.61 for MCF-7 cells), which was much smaller than 1, indicating a synergisitic effect. Furthermore, the cell inhibition effect of MTX-Imine-M-CUR and MTX-Amide-M-CUR with pretreatment of free FA was lower than that of unpretreated group (Figure 5D and E). Specifically, the IC50 of MTX-Imine-M-CUR without and with pretreatment of free FA was calculated as 16.6 and 27.2 μg/mL toward the HeLa cells, respectively. The result was possibly due to the inhibition of folate receptor-mediated endocytosis of MTX surface-functionalized nanosystems, which caused a decrease in intracellular drug concentration and anticancer activity.
On the basis of the above results, it is clear that MTX-Imine-M-CUR could be specifically taken up by the tumor cells through folate receptor-mediated endocytosis pathway. Simultaneously, as MTX-Imine-M-CUR was exposed to the significantly acidic endo/lysosomal microenvironment of the tumor cells, the imine linkers of DSPE-PEG-Imine-MTX prodrug rather than the amine ones of DSPE-PEG-Amide-MTX prodrug were rapidly cleaved, thus accelerating the release of the active form of MTX and exerting a combinational anticancer effect with the released CUR. Therefore, MTX-Imine-M-CUR might have potential as an effective multidrug delivery system due to its high tumor specificity and pH-responsive drug release behavior.
In vivo antitumor effect
For in vivo therapeutic efficacy studies, the xenografted HeLa tumor-bearing nude mice were treated with MTX-Imine-M-CUR nanosystems at a CUR concentration of 8 mg/kg via intravenous administration. Also, 0.9% NaCl, free CUR, free CUR/MTX mixture, M-CUR nanosystems, and MTX-Amide-M-CUR nanosystems were used as controls. At the end of the antitumor study, free CUR treatment produced no significant tumor growth inhibition compared to 0.9% NaCl group. Treatment with a combination of free CUR and MTX also inhibited tumor growth slightly compared to 0.9% NaCl group. In addition, the reduction in tumor growth of M-CUR group was significantly higher than that of free CUR group. Moreover, the tumor volume of mice treated with MTX-Amide-M-CUR or MTX-Imine-M-CUR was obviously smaller than that of mice treated with MTX/CUR or M-CUR (Figure 6A). Furthermore, a statistically significant difference (P<0.05) in tumor volume change was observed between MTX-Amide-M-CUR and MTX-Imine-M-CUR groups. These results could be explained by the fact that both CUR and MTX were simultaneously delivered into the tumor cells by prolonged blood circulation (Figure S3), the EPR effect mediated by the nanosystems (passive targeting), the MTX prodrug-mediated endocytosis via folate receptor (active targeting), pH-triggered dual-drug release from the nanosystems (controlled release), and combining the anticancer activity of dual-drug (MTX surface-functionalized nanosystems could inhibit the activity of dihydrofolate reductase enzyme; Figure S4).26,32,43
 | Figure 6 In vivo antitumor efficacy of HeLa tumor-bearing nude mice after intravenous injection of 0.9% NaCl, free CUR, free CUR/MTX, M-CUR, MTX-Amide-M-CUR, or MTX-Imine-M-CUR at an equivalent dose of CUR (8 mg/kg). |
The body weight was monitored throughout the study. As shown in Figure 6B, all groups showed no obvious loss in body weight at the given concentration during treatment. The result implies that MTX-Imine-M-CUR along with other treatments did not produce significant in vivo systemic toxicity on mice. At the end of the in vivo studies, the tumors were excised and weighed. The tumor weights of MTX-Amide-M-CUR and MTX-Imine-M-CUR groups were significantly lower than those of free CUR and free CUR/MTX mixture groups (Figure 6C). Besides, the MTX-Imine-M-CUR group exhibited the highest tumor inhibition efficiency in all treatment groups.
Histologic analyses
To further evaluate the antitumor effect of HeLa tumor-bearing BALB/c nude mice treated with different formulations, histologic analyses were performed on H&E-stained tumor sections. As shown in Figure 6D, much larger necrotic regions with irregular cell morphology were obviously observed in the MTX-Imine-M-CUR group compared with the MTX-Amide-M-CUR, M-CUR, MTX/CUR, and CUR groups, which was well consistent with the result of tumor growth inhibition. The H&E staining results further confirmed the remarkable in vivo antitumor efficacy and sufficient cellular shrinkage/nuclear fragmentation by MTX-Imine-M-CUR treatment. Therefore, the smart MTX-Imine-M-CUR nanosystems could mediate tumor-selective cellular uptake and drug release, resulting in efficiently improved therapeutic effect. Additionally, animal models were still the basic translational model in the preclinical studies and clinical trials. However, animal models are limited in their ability to faithfully mirror the extremely complex process of human carcinogenesis, physiology, and progression, which may lead to translational limitations.44,45
Conclusion
We have developed a prodrug-based nanosystem for the codelivery of MTX and CUR. A type of DSPE-PEG-Imine-MTX as a self-targeting and pH-responsive prodrug was successfully synthesized via Schiff’s base reaction between MTX and DSPE-PEG. When exposed to an aqueous solution, the DSPE-PEG-Imine-MTX prodrug could spontaneously self-assemble into MTX-Imine-M nanoparticles encapsulating CUR drug within their hydrophobic DSPE core. The MTX-Imine-M-CUR nanoparticles could release the active form of MTX and CUR more efficiently in acidic media at pH 5.0 than at pH 7.4 due to the pH-sensitive imine linkage between DSPE-PEG and MTX. In vitro cellular uptake and internalization studies proved that MTX-Imine-M-CUR and MTX-Amide-M-CUR nanoparticles could be selectively taken up by the cancer cells via folate receptor-mediated endocytosis, which showed the much more efficient cellular uptake efficiency compared with MTX unfunctionalized M-CUR nanoparticles and the free drug. Furthermore, MTX-Imine-M-CUR exhibited significantly stronger in vitro cytotoxicity and in vivo tumor inhibition efficiency than MTX-Amide-M-CUR nanoparticles, M-CUR nanoparticles, combination of both free drugs, and individual free drugs toward HeLa/MCF-7 cells and HeLa tumor-bearing nude mice. Our work demonstrates that MTX-Imine-M-CUR nanosystems have a great potential as effective chemotherapeutic formulations in targeting and treating cancer.
Acknowledgments
The work is supported by the National Natural Science Foundation of China (31271071), the China Postdoctoral Science Foundation (2016M602074), the Special Funding of China Postdoctoral Science Foundation (2017T100472), the Natural Science Foundation of Fujian Province of China (2016J01406), the Clinical Medicine Science and Technology Project of Jiangsu Province (BL2013015), and the Xiamen Science and Technology Project (3502Z20174071).
Disclosure
The authors report no conflicts of interest in this work.
References
Siegel RL, Miller KD, Jemal A. Cancer statistics, 2016. CA Cancer J Clin. 2016;66:7–30. | ||
Brigger I, Dubernet C, Couvreur P. Nanoparticles in cancer therapy and diagnosis. Adv Drug Deliv Rev. 2002;54:631–651. | ||
Wu Q, Yang Z, Nie Y, Shi Y, Fan D. Multi-drug resistance in cancer chemotherapeutics: mechanisms and lab approaches. Cancer Lett. 2014;347:159–166. | ||
Lehar J, Krueger AS, Avery W, et al. Synergistic drug combinations tend to improve therapeutically relevant selectivity. Nat Biotechnol. 2009;27:659–666. | ||
Liu Z, Jiao Y, Wang Y, Zhou C, Zhang Z. Polysaccharides-based nanoparticles as drug delivery systems. Adv Drug Deliv Rev. 2008;60:1650–1662. | ||
Tian G, Zheng X, Zhang X, et al. Tpgs-stabilized Naybf4:Er upconversion nanoparticles for dual-modal fluorescent/Ct imaging and anticancer drug delivery to overcome multi-drug resistance. Biomaterials. 2015;40:107–116. | ||
Song L, Pan Z, Zhang H, et al. Dually folate/Cd44 receptor-targeted self-assembled hyaluronic acid nanoparticles for dual-drug delivery and combination cancer therapy. J Mater Chem B. 2017;5:6835–6846. | ||
Thomas TP, Huang B, Choi SK, et al. Polyvalent dendrimer-methotrexate as a folate receptor-targeted cancer therapeutic. Mol Pharm. 2012;9:2669–2676. | ||
Roberts GC, Feeney J, Birdsall B, Charlton P, Young D. Methotrexate binding to dihydrofolate reductase. Nature. 1980;286:309. | ||
Zeb A, Qureshi OS, Kim HS, Cha JH, Kim HS, Kim JK. Improved skin permeation of methotrexate via nanosized ultradeformable liposomes. Int J Nanomedicine. 2016;11:3813–3824. | ||
Boechat AL, De Oliveira CP, Tarrago AM, et al. Methotrexate-loaded lipid-core nanocapsules are highly effective in the control of inflammation in synovial cells and a chronic arthritis model. Int J Nanomedicine. 2015;10:6603–6614. | ||
Mount C, Featherstone J. Rheumatoid arthritis market. Nat Rev Drug Discov. 2005;4:11–12. | ||
Cronstein BN. Low-dose methotrexate: a mainstay in the treatment of rheumatoid arthritis. Pharmacol Rev. 2005;57:163–172. | ||
Shao Z, Shao J, Tan B, et al. Targeted lung cancer therapy: preparation and optimization of transferrin-decorated nanostructured lipid carriers as novel nanomedicine for co-delivery of anticancer drugs and DNA. Int J Nanomedicine. 2015;10:1223–1233. | ||
Aggarwal BB, Kumar A, Bharti AC. Anticancer potential of curcumin: preclinical and clinical studies. Anticancer Res. 2003;23:363–398. | ||
Banerjee S, Chakravarty AR. Metal complexes of curcumin for cellular imaging, targeting, and photoinduced anticancer activity. Acc Chem Res. 2015;48:2075–2083. | ||
Xie J, Li Y, Song L, Pan Z, Ye S, Hou Z. Design of a novel curcumin-soybean phosphatidylcholine complex-based targeted drug delivery systems. Drug Deliv. 2017;24:707–719. | ||
Zhao X, Chen Q, Liu W, et al. Codelivery of doxorubicin and curcumin with lipid nanoparticles results in improved efficacy of chemotherapy in liver cancer. Int J Nanomedicine. 2015;10:257–270. | ||
Ketron AC, Gordon ON, Schneider C, Osheroff N. Oxidative metabolites of curcumin poison human type II topoisomerases. Biochemistry. 2013;52:221–227. | ||
Zhang Y, Yang C, Wang W, et al. Co-delivery of doxorubicin and curcumin by ph-sensitive prodrug nanoparticle for combination therapy of cancer. Sci Rep. 2016;6:21225. | ||
He S, Qi Y, Kuang, G, et al. Single-stimulus dual-drug sensitive nanoplatform for enhanced photoactivated therapy. Biomacromolecules. 2016;17:2120–2127. | ||
Peer D, Karp JM, Hong S, Farokhzad OC, Margalit R, Langer R. Nanocarriers as an emerging platform for cancer therapy. Nat Nanotechnol. 2007;2:751–760. | ||
Wang K, Huang Q, Qiu F, Sui M. Non-viral delivery systems for the application in P53 cancer gene therapy. Curr Med Chem. 2015;22:4118–4136. | ||
Wen R, Banik B, Pathak RK, Kumar A, Kolishetti N, Dhar S. Nanotechnology inspired tools for mitochondrial dysfunction related diseases. Adv Drug Deliv Rev. 2016;99:52–69. | ||
Zhang B, Wang K, Si J, Sui M, Shen Y. Charge-reversal polymers for biodelivery. In: Gu Z, editor. Bioinspired and Biomimetic Polymer Systems for Drug and Gene Delivery. Darmstadt: Wiley-VCH Verlag GmbH Co., KGaA; 2014:223–242. | ||
Shi J, Kantoff PW, Wooster R, Farokhzad OC. Cancer nanomedicine: progress, challenges and opportunities. Nat Rev Cancer. 2017;17:20–37. | ||
Pang X, Jiang Y, Xiao Q, Leung AW, Hua H, Xu C. pH-responsive polymer-drug conjugates: design and progress. J Control Release. 2016;222:116–129. | ||
Cai Y, Sun Z, Fang X, et al. Synthesis, characterization and anti-cancer activity of pluronic F68-curcumin conjugate micelles. Drug Deliv. 2016;23:2587–2595. | ||
Zhao Y, Ren W, Zhong T, et al. Tumor-specific ph-responsive peptide-modified ph-sensitive liposomes containing doxorubicin for enhancing glioma targeting and anti-tumor activity. J Control Release. 2016;222:56–66. | ||
Liang W, Chow MY, Lau PN, et al. Inhalable dry powder formulations of sirna and ph-responsive peptides with antiviral activity against H1n1 influenza virus. Mol Pharm. 2015;12:910–921. | ||
Maeda H, Wu J, Sawa T, Matsumura Y, Hori K. Tumor vascular permeability and the Epr effect in macromolecular therapeutics: a review. J Control Release. 2000;65:271–284. | ||
Bertrand N, Wu J, Xu X, Kamaly N, Farokhzad OC. Cancer nanotechnology: the impact of passive and active targeting in the era of modern cancer biology. Adv Drug Delivey Rev. 2014;66:2–25. | ||
Yan J, Wang Y, Zhang X, Liu S, Tian C, Wang H. Targeted nanomedicine for prostate cancer therapy: docetaxel and curcumin co-encapsulated lipid-polymer hybrid nanoparticles for the enhanced anti-tumor activity in vitro and in vivo. Drug Deliv. 2016;23:1757–1762. | ||
Duncan R. Polymer conjugates as anticancer nanomedicines. Nat Rev Cancer. 2006;6:688–701. | ||
Wagner V, Dullaart A, Bock AK, Zweck A. The emerging nanomedicine landscape. Nat Biotechnol. 2006;24:1211–1217. | ||
Lukyanov AN, Torchilin VP. Micelles from lipid derivatives of water-soluble polymers as delivery systems for poorly soluble drugs. Adv Drug Deliv Rev. 2004;56:1273–1289. | ||
Li Y, Lin J, Wu H, et al. Orthogonally functionalized nanoscale micelles for active targeted codelivery of methotrexate and mitomycin C with synergistic anticancer effect. Mol Pharm. 2015;12:769–782. | ||
Chen Y, Zhang W, Huang Y, et al. The therapeutic effect of methotrexate-conjugated pluronic-based polymeric micelles on the folate receptor-rich tumors treatment. Int J Nanomedicine. 2015;10:4043–4057. | ||
Li Y, Song L, Lin J, et al. Programmed nanococktail based on pH-responsive function switch for self-synergistic tumor-targeting therapy. ACS Appl Mater Interfaces. 2017;9:39127–39142. | ||
Torchilin V. Tumor delivery of macromolecular drugs based on the epr effect. Adv Drug Delivey Rev. 2011;63:131–135. | ||
Li Y, Lin J, Huang Y, et al. Self-targeted, shape-assisted, and controlled-release self-delivery nanodrug for synergistic targeting/anticancer effect of cytoplasm and nucleus of cancer cells. ACS Appl Mater Interfaces. 2015;7:25553–25559. | ||
Hu M, Huang P, Wang Y, Su Y, Zhou L, Zhu X, Yan D. Synergistic combination chemotherapy of camptothecin and floxuridine through self-assembly of amphiphilic drug-drug conjugate. Bioconjug Chem. 2015;26:2497–2506. | ||
Li Y, Lin J, Yang X, et al. Self-assembled nanoparticles based on amphiphilic anticancer drug-phospholipid complex for targeted drug delivery and intracellular dual-controlled release. ACS Appl MaterInterfaces. 2015;7:17573–17581. | ||
Beura LK, Hamilton SE, Bi K, et al. Normalizing the environment recapitulates adult human immune traits in laboratory mice. Nature. 2016;532:512. | ||
Breschi A, Gingeras TR, Guigo R. Comparative transcriptomics in human and mouse. Nat Rev Genet. 2017;18:425–440. | ||
Zalipsky S, Brandeis E, Newman MS, Woodle MC. Long circulating, cationic liposomes containing amino-peg-phosphatidylethanolamine. FEBS Lett. 1994;353:71–74. | ||
Zheng JN, Xie HG, Yu WT, et al. Chitosan-G-Mpeg-modified alginate/chitosan hydrogel microcapsules: a quantitative study of the effect of polymer architecture on the resistance to protein adsorption. Langmuir. 2010;26:17156–17164. | ||
Li Y, Lin J, Wu H, et al. Novel methotrexate prodrug-targeted drug delivery system based on peg-lipid-PLA hybrid nanoparticles for enhanced anticancer efficacy and reduced toxicity of mitomycin C. J Mater Chem B. 2014;2:6534–6548. | ||
Guo Y, Zhang Y, Ma J, et al. Light/magnetic hyperthermia triggered drug released from multi-functional thermo-sensitive magnetoliposomes for precise cancer synergetic theranostics. J Control Release. Epub 2017 Apr 22. | ||
Liu G, Ma J, Li Y, et al. Core-interlayer-shell Fe3o4@Msio2@lipid-PEG-methotrexate nanoparticle for multimodal imaging and multistage targeted chemo-photodynamic therapy. Int J Pharm. 2017;521:19–32. |
Supplementary materials
 | Figure S1 (A) 1H NMR, (B) UV–vis absorption, (C) XRD, (D) MALDI-TOF-MS, and (E) GPC spectra of DSPE-PEG-CHO, MTX, and DSPE-PEG-Imine-MTX amphiphilic polymer prodrug. |
 | Figure S2 In vitro stability of hydrodynamic size distribution of MTX-Imine-M-CUR nanosystems in (A) water and (B) PBS over 72 h. Error bars indicate SD (n=3). |
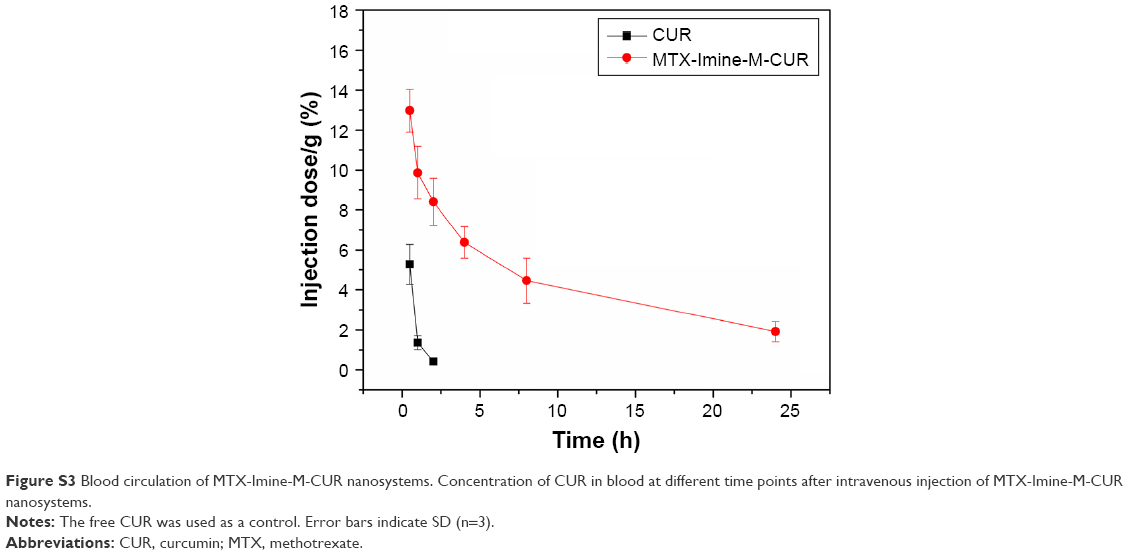 | Figure S3 Blood circulation of MTX-Imine-M-CUR nanosystems. Concentration of CUR in blood at different time points after intravenous injection of MTX-Imine-M-CUR nanosystems. |
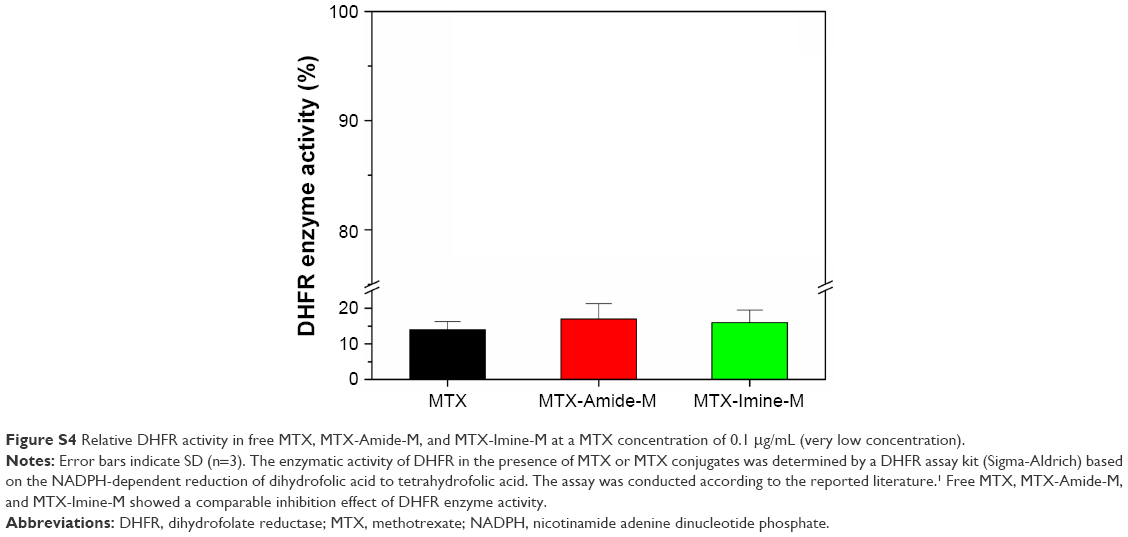 | Figure S4 Relative DHFR activity in free MTX, MTX-Amide-M, and MTX-Imine-M at a MTX concentration of 0.1 μg/mL (very low concentration). |
Reference
Lachelt U, Wittmann V, Muller K, et al. Synthetic polyglutamylation of dual-functional mtx ligands for enhanced combined cytotoxicity of poly(I:C) nanoplexes. Mol Pharmaceutics. 2014;11:2631–2639. |
 © 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.