Back to Journals » International Journal of Nanomedicine » Volume 9 » Issue 1
Design of multifunctional magnetic iron oxide nanoparticles/mitoxantrone-loaded liposomes for both magnetic resonance imaging and targeted cancer therapy
Authors He Y, Zhang L, Zhu D ![]() , Song C, Sun H
, Song C, Sun H
Received 13 February 2014
Accepted for publication 3 May 2014
Published 22 August 2014 Volume 2014:9(1) Pages 4055—4066
DOI https://doi.org/10.2147/IJN.S61880
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Yingna He,1 Linhua Zhang,2 Dunwan Zhu,2 Cunxian Song2
1Laboratory of Chinese Medicine Pharmacology, College of Pharmacy, Hebei University of Chinese Medicine, Shijiazhuang, Hebei, People’s Republic of China; 2Key Laboratory of Biomedical Material of Tianjin, Institute of Biomedical Engineering, Peking Union Medical College and Chinese Academy of Medical Sciences, Tianjin, People’s Republic of China
Abstract: Tumor-targeting multifunctional liposomes simultaneously loaded with magnetic iron oxide nanoparticles (MIONs) as a magnetic resonance imaging (MRI) contrast agent and anticancer drug, mitoxantrone (Mit), were developed for targeted cancer therapy and ultrasensitive MRI. The gonadorelin-functionalized MION/Mit-loaded liposome (Mit-GML) showed significantly increased uptake in luteinizing hormone–releasing hormone (LHRH) receptor overexpressing MCF-7 (Michigan Cancer Foundation-7) breast cancer cells over a gonadorelin-free MION/Mit-loaded liposome (Mit-ML) control, as well as in an LHRH receptor low-expressing Sloan-Kettering HER2 3+ Ovarian Cancer (SK-OV-3) cell control, thereby leading to high cytotoxicity against the MCF-7 human breast tumor cell line. The Mit-GML formulation was more effective and less toxic than equimolar doses of free Mit or Mit-ML in the treatment of LHRH receptors overexpressing MCF-7 breast cancer xenografts in mice. Furthermore, the Mit-GML demonstrated much higher T2 enhancement than did Mit-ML controls in vivo. Collectively, the study indicates that the integrated diagnostic and therapeutic design of Mit-GML nanomedicine potentially allows for the image-guided, target-specific treatment of cancer.
Keywords: multifunctional liposome, magnetic resonance imaging, theranostic nanomedicine, mitoxantrone, gonadorelin
Introduction
Cancer is one of the most challenging problems facing modern medicine. Despite rapid advances in diagnostic procedures and treatments, the overall survival rate from cancer has not improved substantially over the past 30 years.1 The limitations of chemotherapy, radiotherapy, and surgery are apparent, and have been the impetus for the development of novel approaches that are used for the accurate detection of early-stage cancer and targeted therapies.
In recent years, nanomedicine platforms that combine therapeutic agents, molecular targeting, and diagnostic imaging capabilities, termed as theranostics, are emerging as the next generation of multifunctional nanomedicine to improve the therapeutic outcome of cancer therapy.2–4 Unlike traditional small molecular contrast agents or drugs, theranostic nanomedicine has the potential to provide simultaneous molecular diagnosis and therapeutic treatment in one system.5 Such integrated diagnostic and therapeutic designs allow for the timely tailoring of nanomedicine modules to address the challenges of tumor heterogeneity and adaptive resistance, which can ultimately help achieve the goal of personalized therapy for cancer.6,7
Magnetic iron oxide nanoparticles (MIONs) have been shown to be suitable for use as theranostic agents by employing their intrinsic diagnostic capabilities in the context of magnetic resonance imaging (MRI) applications.8 Surface modifications may be easily introduced through conjugation with targeting moieties (for example, antibodies, peptides, small molecules, or aptamers), fluorescence dyes, genes, or drugs to provide multimodal functionalities.9,10 MIONs are promising, however, shortcomings such as low dispersion in solvents, wide particle size distribution, toxicity, and lack of tissue-specific targeting in vivo have limited their use to the diagnosis and treatment of cancer. These challenges become more outstanding as MIONs are further scaled down in size. The high surface area-to-volume ratio of these nanoparticles results in a tendency to aggregate and absorb plasma proteins upon intravenous injection, leading to rapid clearance by the reticuloendothelial system.11 Additionally, they are limited in their capacity for drug loading and rapid drug clearance after intravenous administration.12 Thus, MIONs are commonly protected with a polymer coating to improve their dispersity and stability.13
Liposomes have been intensively investigated for the sustained and controlled delivery of imaging and therapeutic agents for cancer diagnosis and cancer treatment, which can result in high diagnostic and therapeutic efficiency and low side effects.14 Coating MIONs with liposomes can prevent them from aggregation and opsonization, while evading nanoparticle uptake by the reticuloendothelial system, increasing colloidal stability in physiological solutions, and increasing its blood circulation time.8 Moreover, liposomes can be easily conjugated with ligands that target disease-specific receptors or other molecules.15
Gonadorelin is a peptide analogue of luteinizing hormone–releasing hormone (LHRH), and it has high affinity for LHRH membrane receptors, which are characteristically overexpressed in many tumors, including in ovarian and endometrial (~80%), prostate (~90%), and breast (~50%) cancers, while its expression is scarce in healthy tissues.16–18 Studies employing LHRH receptor-targeted dendrimers,19 nanoparticles,20 liposomes,21,22 and so forth were able to substantially enhance intratumoral accumulation and anticancer efficacy and, hence, indicate that LHRH-receptor mediated targeting is effective despite the differences in the size, molecular mass of the components, composition and architecture of the carriers.
In this study, we present a simple yet efficient technique to prepare multifunctional liposomes carrying both MIONs and a conventional chemotherapeutic agent, mitoxantrone (Mit), using gonadorelin as a model targeting agent conjugated on the liposomes. We systematically examined the efficacy of these gonadorelin-functionalized multifunctional liposomes to serve as a therapeutic agent and as an MRI contrast agent. The gonadorelin-functionalized multifunctional liposomes demonstrated more sensitive targeted detection by MRI and better therapeutic efficacy in vitro and in vivo. This integrated liposomal nanomedicine that combines cancer targeting, MRI visibility, and therapeutic delivery should contribute to the growing arsenal for the targeted therapy of cancer.
Materials and methods
Materials
Gonadorelin was purchased from ProSpec-Tany TechnoGene Ltd. (Rehovot, Israel). The sequence of gonadorelin was Pyr-His-Trp-Ser-Tyr-Gly-Leu-Arg-Pro-Gly-NH2 (molecular weight: 1,182.3). Mit hydrochloride was obtained from Chongqing Kailin Pharmaceutical Co., Ltd. (Chongqing, People’s Republic of China). Hydrogenated soybean phosphatidylcholine (HSPC) and (N-[carbonyl-methoxy polyethylene glycol 2000]-1,2-distearoyl-sn-glycero-3-phosphor-ethan-olamine sodium salt) (PEG2000-DSPE) were purchased from Lipoid GmbH (Ludwigshafen am Rhein, Germany). Cholesterol, 2-iminothiolane (Traut’s reagent), 3-(4,5-Dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT), and Sephadex® G-10 were purchased from Sigma-Aldrich Co. (St Louis, MO, USA). Maleimide–PEG2000-DSPE (Mal-PEG2000-DSPE) was purchased from Avanti Polar Lipids, Inc. (Alabaster, AL, USA). Phosphate buffered saline (PBS) (pH 7.4; 290 mOsm/L) was purchased from Tianjin Haoyang Biologicals Technology Co., Ltd. (Tianjin, People’s Republic of China). MIONs (10 mg/mL for Fe3O4 in PBS; mean diameter: 10 nm) were from Tianjin Baseline Chromtech Co. (Tianjin, People’s Republic of China). The bicinchoninic acid (BCA) kit was purchased from Shanghai Shenergy Biocolor Bioscience & Technology Company, Ltd. (Shanghai, People’s Republic of China). Ammonium sulfate and all other chemicals used in this study were of analytical grade or of high-performance liquid chromatography grade.
Cell line
Human MCF-7 (Michigan Cancer Foundation-7) breast cancer cells (a cancer cell line overexpressing LHRH receptors) and human ovarian cancer Sloan-Kettering HER2 3+ Ovarian Cancer (SK-OV-3) cells (a cancer cell line with low expression of LHRH receptors) were purchased from the American Type Culture Collection (Manassas, VA, USA). Cells were cultured in Roswell Park Memorial Institute (RPMI) 1640 medium (Sigma-Aldrich Co.) supplemented with 10% fetal bovine serum (FBS) (Tianjin Haoyang Biologicals Technology Co., Ltd.). Cells were grown at 37°C in a humidified atmosphere of 5% CO2 (vol/vol) in air. All experiments were performed on cells during the exponential growth phase.
Preparation of gonadorelin-functionalized Mit and MION-loaded liposomes
MION-loaded liposomes (MLs) were prepared by a lipid film hydration method. A total of 100 μmol of a lipid mixture composed of HSPC, cholesterol, and PEG2000-DSPE (molar ratio, 90:10:0.4) was dissolved in chloroform. Subsequently, the solvent was removed by rotary evaporation, followed by additional drying under a vacuum overnight to remove residual solvent. After the addition of 2 mg of MIONs, the dried lipid film was hydrated with a solution containing 300 mM of ammonium sulfate at 60°C for 1 hour. The resulting large multilamellar vesicles were further homogenized to small unilamellar vesicles by sonication (VCX 130 PB; Sonics & Materials, Inc., Newtown, CT, USA) for 10 minutes at 4°C. Nonencapsulated MIONs were removed by centrifugation at 1,000 g for 15 minutes.
Mit was encapsulated into the preformed MLs using a transmembrane ammonium sulfate gradient-driven loading procedure. The procedure consisted of initially exchanging the buffer by dialysis against a sucrose (300 mM)–histidine (10 mM) buffer (pH 7.5) using dialysis tubing (Pierce Chemical Co, Rockford, IL, USA), with a 10–12 KDa molecular weight cutoff for 4 hours, and incubating the liposomes at 60°C for 5 minutes before adding Mit to achieve a final lipid-to-drug weight ratio of 10:1. The resulting mixture was incubated at 60°C for an additional 15 minutes.
To prepare the gonadorelin conjugates with PEG2000-DSPE, the dried Mal-PEG2000-DSPE lipids were hydrated in (4-[2-hydroxyethyl]-piperazine-1-ethanesulfonic acid) (HEPES) buffer (pH 8.0) at 60°C to form micelles. Simultaneously, gonadorelin was modified with 2-iminothiolane to introduce the sulfhydryl groups, as described previously.22 Briefly, fourfold molar excess of 2-iminothiolane dissolved in HEPES buffer (25 mM HEPES, 140 mM NaCl; pH 8.0) was added to the gonadorelin peptide solution (1:10 weight/volume) and incubated for 1 hour at room temperature. The unreacted 2-iminothiolane was removed by size exclusion chromatography using a Sephadex® G10 column (Sigma-Aldrich Co.) with degassed HEPES buffer (25 mM HEPES, 140 mM NaCl; pH 8.0). Thiolated gonadorelin was then added to the Mal-PEG2000-DSPE micelles solution at a 2:1 molar ratio. The mixture was incubated overnight under a nitrogen atmosphere at room temperature. Free maleimide groups were quenched with excess histidine for 30 minutes at room temperature. Gonadorelin–PEG2000-DSPE micelles were then concentrated in a Centriprep® Centrifugal Filter, using a size cutoff of 3,000 Da (EMD Millipore, Billerica, MA, USA).
To obtain gonadorelin-functionalized Mit-loaded MLs (Mit-GML), gonadorelin–PEG2000-DSPE micelles were added into the preformed Mit-loaded ML (Mit-ML) solution at 1:1,000 molar ratios and incubated at 60°C for 1 hour. The final suspension of liposomes was purified by exhaustive dialysis (size cutoff: 12–14 KDa) against a sucrose (300 mM)–histidine (10 mM) buffer (pH 7.5) at 4°C to remove free Mit and gonadorelin–PEG2000-DSPE. Liposomes mimicking Mit-GML, but containing no Mit (GML) were prepared similarly, except for the omission of the Mit loading procedure.
Physical characterization of Mit-GML
Liposomes were characterized by dynamic light scattering techniques (Zetasizer Nano ZS; Malvern Instruments, Malvern, UK) at 25°C and at a scattering angle of 90°. The liposomes were diluted appropriately in PBS (pH 7.4) before measurement. The morphology of Mit-GML was observed by transmission electron microscopy (Hitachi H7100; Hitachi Ltd., Tokyo, Japan).
Gonadorelin–PEG2000-DSPE conjugates were analyzed by ultraviolet (UV) absorbance using a spectral scanning multimode reader (Varioskan Flash; Thermo Fisher Scientific, Waltham, MA, USA): the UV spectra were recorded from 220–400 nm in PBS (pH 7.4; 0.5 mg polymer/mL). The peptide coupling efficiency of Mit-GML was determined by a protein assay method. In short, the coupled gonadorelin was separated from free gonadorelin using ultrafiltration (size cutoff: 50 KDa) at 10,000 rpm for 30 minutes. The liposomes in the supernatant were solubilized by 0.3% TritonTM-X100 (Sigma-Aldrich Co.), and then the amounts of free gonadorelin were determined by a micro BCA assay according to the manufacturer’s instructions. The coupling efficiency was calculated using the following formula:
|
|
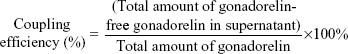
The encapsulation efficiency of Mit and MION was calculated as the percentage of the actual amount of loaded agent (Mit or MION) added in the liposomes over the theoretical amount of corresponding agent added in the liposomes. Mit loading was determined by a spectral scanning multimode reader. First, the liposome solutions were lysed with 3% Triton-X100. The suspended MIONs were removed by centrifugation, and the upper solution was collected to measure the Mit amount by UV absorbance at 650 nm. MION loading was determined based on ferrous ion by the o-phenanthroline method. Briefly, a liposomal or ferrofluid sample (0.1 mL) was mixed with 0.1 mL of Triton-X100 solution (5%, v/v) before ionizing the magnetite by adding 0.5 mL of concentrated HCl; 0.5 mL of a hydroxylamine hydrochloride solution (1.44 M) was then added to reduce ferric ion. After 15 minutes, 1 mL of o-phenanthroline solution (12.6 mM) was added, the mixture was neutralized with 0.25 mL of 12 M NaOH, and the pH was adjusted to about 4.0 by adding the necessary volume of 50 mM citrate buffer. Finally, the absorbance was read at 509 nm in a VarioskanTM Flash spectral scanning multimode reader. The calibration curve was performed with several amounts of a solution of Fe3O4 in 12 M of HCl.
Drug release was examined in isotonic glucose–histidine (10 mM) buffer (pH 7.5) containing 10% human plasma. Liposomes at a concentration of 0.5 mM HSPC, diluted with the release medium, were sealed into dialysis tubes with a cutoff size of 12–14 KDa. Then, liposome-loaded dialysis tubes were incubated in 100 mL of the release medium for 72 hours at 37°C with continuous stirring at medium speed. At specific time intervals, aliquots were withdrawn and replaced with an equal volume of the release medium. The samples were treated and analyzed using the method mentioned in the biodistribution studies. The amount of drug released was calculated based on the standard curve obtained for Mit in the buffers. The in vitro release experiments were performed in duplicate to confirm their reproducibility.
The in vitro stability of liposomes was also analyzed. Mit-GML and Mit-ML were incubated in PBS (which were centrifuged earlier at 1,000 g for 10 minutes to remove aggregates) at 4°C. At each time point, the particle size and polydispersity of liposomes were measured using dynamic laser light scattering (Zetasizer Nano ZS); this was performed in triplicate at room temperature and at a scattering angle of 90°.
In vitro cell uptake experiments
The cellular uptake and distribution of free Mit, Mit-GML, and Mit-ML were performed using confocal laser scanning microscopy. MCF-7 cells overexpressing LHRH receptors, or SK-OV-3 cells with low expression levels of LHRH receptors were seeded at densities of 2×105 cm−2 in 35 mm diameter culture dishes, and they were left to adhere and recover over 24 hours at 37°C in a humidified atmosphere of 5% CO2. Cells were treated with free Mit, Mit-GML, or Mit-ML (Mit concentration: 10 μg/mL) for 4 hours at 37°C. Then, the cells were washed three times with cold PBS (pH 7.4) and stained with Hoechst 33342 nucleic acid stain (Thermo Fisher Scientific) for nucleus. Cell samples were examined on a Zeiss LSM 510 confocal laser scanning microscope (Carl Zeiss Meditec AG, Jena, Germany). Hoechst 33342 and Mit were excited at 346 nm and 514 nm with emissions at 460 nm and 680 nm, respectively. Negative control cells were treated identically except for the omission of Mit formulations.
Cell proliferation assay
The cytotoxicity of Mit, Mit-ML, and Mit-GML against MCF-7 cells was studied using an MTT viability assay. MCF-7 cells were seeded in a 96-well plate at a density of 2×105 cells per well in 200 μL of RPMI 1640 medium with 10% FBS and incubated overnight. Culture medium containing Mit, Mit-ML, and Mit-GML at Mit concentrations of 0.3 ng/mL, 3 ng/mL, 30 ng/mL, and 300 ng/mL were then added in sextuple, and incubation continued for an additional 120 hours. For control wells, the same volume of RPMI 1640 was included in each group. At designated time points, the medium was discarded and fresh medium containing 200 μL of MTT was added to each well. The cells were incubated for 4 hours at 37°C, after which 200 μL of dimethyl sulfoxide was added to each well for 5 minutes, and the absorbance was measured at 570 nm using a VarioskanTM Flash spectral scanning multimode reader. Cell viability was calculated as follows:
Viability (%) = Atreated/Acontrol ×100%. | (2) |
Where Atreated is the fluorescence absorbance of the cells incubated with Mit or Mit-loaded liposomes suspension, while Acontrol was the averaged absorbance of the cells without drug treatment.
Breast cancer animal models
Female athymic nude BALB/c mice weighing 12–14 g were obtained from the Institute of Laboratory Animal Science, Chinese Academy of Medical Sciences and Peking Union Medical College (Peking, People’s Republic of China), and they were allowed to acclimate for 1 week in the animal facility before any intervention was initiated. All experimental procedures were conducted with the approval of, and in accordance with, the guidelines of the Institutional Animal Care and Use Committee at the Peking Union Medical College. Under direct visualization, MCF-7 breast cancer cells (4×106) were implanted into the mammary fat pads of the mice. The time for the appearance of the tumor varies from one mouse to another, and it usually takes 10–14 days. Mice were constantly monitored, and they were allowed to have free access to food and water; tumor volumes were then calculated using the formula,
(π/6) × length × width2, | (3) |
by measuring the dimensions of the tumor at regular time intervals.
Biodistribution studies
In vivo biodistribution studies were done in MCF-7 tumor-bearing BALB/c mice. Mice with size-matched tumors (~300 mm3) were randomly assigned to different treatment groups and injected through the tail vein with various formulations of Mit-GML, Mit-ML, and free Mit at a Mit equivalent dose of 2.5 mg/kg. At time points of 1 hour, 4 hours, and 24 hours postinjection, mice were anesthetized and sacrificed. After perfusion, xenograft tumors and mouse organs were removed and homogenized.
The Mit concentrations in plasma and tissue homogenate samples were then assayed by high-performance liquid chromatography. Before analysis, the samples were extracted by protein precipitation. Mit was separated on a Zorbax SB C18 column (150 mm×4.6 mm, 5 μm; Agilent Technologies, Santa Clara, CA, USA) at 35°C, and quantified by UV absorbance at 650 nm. The mobile phase consisted of acetonitrile and solutions containing 30 mM of sodium 1-heptanesulfonate and 9.0 mL/L of glacial acetic acid (37:63, v/v) at a flow rate of 1 mL/minute.
In vivo antitumor efficacy study
The therapeutic treatment of MCF-7 tumor-bearing mice was started after the tumor reached a mean diameter of 5.0 (±0.7) mm with a Mit equivalent dose intensity of 2.5 mg/kg every 7 days (day 0, day 7, and day 14). Mice were randomly divided into one of four groups: 1) saline control; 2) free Mit; 3) Mit-ML; and 4) Mit-GML. Each treatment group consisted of 6–8 mice, and they were treated with the corresponding formulation via the tail vein for a total of three times. Tumor diameters were measured, and approximate tumor volume was calculated as
(π/6) × length × width2 | (4) |
every 3 days during the treatment period of 21 days to monitor tumor growth. Changes in the body weights were also monitored for 21 days after the first administration of each formula.
In vitro MRI
Mit-ML and Mit-GML were diluted to final concentrations of 0 μg Fe/mL, 0.25 μg Fe/mL, 2.5 μg Fe/mL, and 25 μg Fe/mL in saline, and imaged in a 24-well plate on a Signa excite 1.5 T MR scanner (GE Healthcare Bio-Sciences Corp., Piscastaway, NJ, USA). The MRI T2 signal intensities within the region of interest were measured. T2-weighted images were acquired using the following parameters: 1.5 T; fast spin echo sequence; and repetition time/echo time of 4,000/103.7 ms.
In vivo MRI
An MRI on tumor-bearing mice was performed after the tumor diameter reached 7–10 mm. Twelve animals were divided into one of three groups, according to the injected material. Animals were injected (10 mL/kg) intravenously via their tail vein with Mit-GML (0.15 mg Fe/mL), Mit-ML (0.15 mg Fe/mL), or saline (0.10 mL). An MRI was performed before and 2 hours after (2–2.5 hours postcontrast) contrast injection using a clinical MR scanner (GE Signa excite 1.5 T; GE Healthcare Bio-Sciences Corp.). Animals were anesthetized with 10% chloral hydrate (intraperitoneally, 20 g/0.1 mL) before MRI. An eight channel wrist joint coil was used to MRI when mice were placed in the imaging chamber. T2-weighted images were acquired using the following parameters: 1.5 T; fast spin echo sequence; repetition time ¼, 3,600 ms; and echo time ¼, 83.5 ms.
Statistical analysis
All data are expressed as the mean ± standard deviation. The statistical significance of differences in the tumor volume of each group was analyzed by repeated measures analysis of variance with Bonferroni’s multiple comparison test. All data were calculated using the commercially available software package, SPSS 10.0 software (IBM Corporation, Armonk, NY, USA), and statistical significance was established at P<0.05.
Results
Preparation and physical characterization of liposomes
The preparation of Mit-GML involved: 1) the conjugation of gonadorelin with PEG2000-DSPE; 2) the preparation of drug- and MION-loaded untargeted liposomes; and 3) the attachment of the gonadorelin–PEG2000-DSPE conjugate to the drug- and MION-loaded untargeted liposomes. The modification of gonadorelin with the DSPE-PEG2000 lipid, and with the formation of the stable thioether linkage, resulted in an active gonadorelin–PEG2000-DSPE conjugate. The conjugation reaction and purification were confirmed by UV spectrometry. Figure 1 shows the UV spectrometry of DSPE-PEG2000, gonadorelin–PEG2000-DSPE, and gonadorelin; the presence of the peak at 270-280 nm for the conjugates indicates the peptide’s incorporation to polyethylene glycol (PEG) chain.
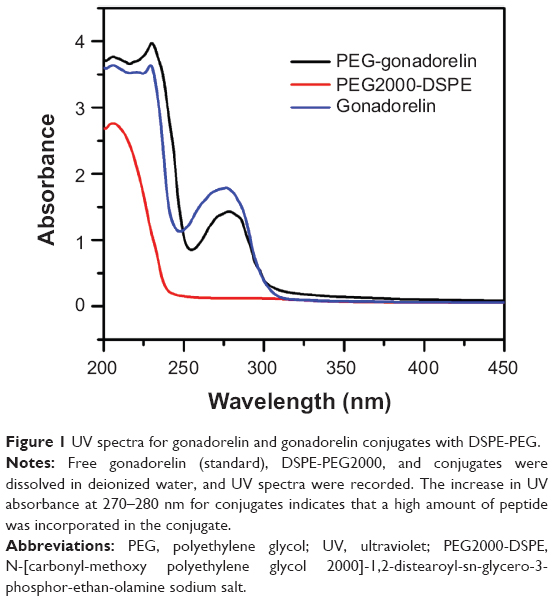 | Figure 1 UV spectra for gonadorelin and gonadorelin conjugates with DSPE-PEG. |
Then, via coincubation with the liposomes, a spontaneous process allowed for the transfer of the gonadorelin–PEG-DSPE conjugate from their loose micelles onto the liposome surface. This postinsertional liposome modification method yielded approximately 70% peptide coupling efficiency through the indirect quantification of unreacted peptides with the BCA protein assay. The peptide incorporation was not accompanied by any significant loss of the liposomal Mit.
The physicochemical characteristics of Mit-GML were quite similar to those of Mit-ML (Table 1), and both negatively-charged liposomes were characterized by a narrow size distribution (Figure 2A). Transmission electron microscopy studies suggested that both liposomes had a spherical morphology (Figure 2B). The two liposome solutions contained 1 mg/mL of Mit and 0.3 mg/mL of iron. The encapsulation efficiency of Mit and magnetite were about 95.7% and 93.2%, respectively.
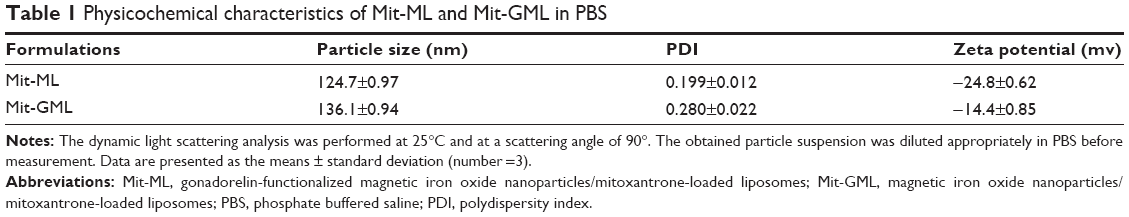 | Table 1 Physicochemical characteristics of Mit-ML and Mit-GML in PBS |
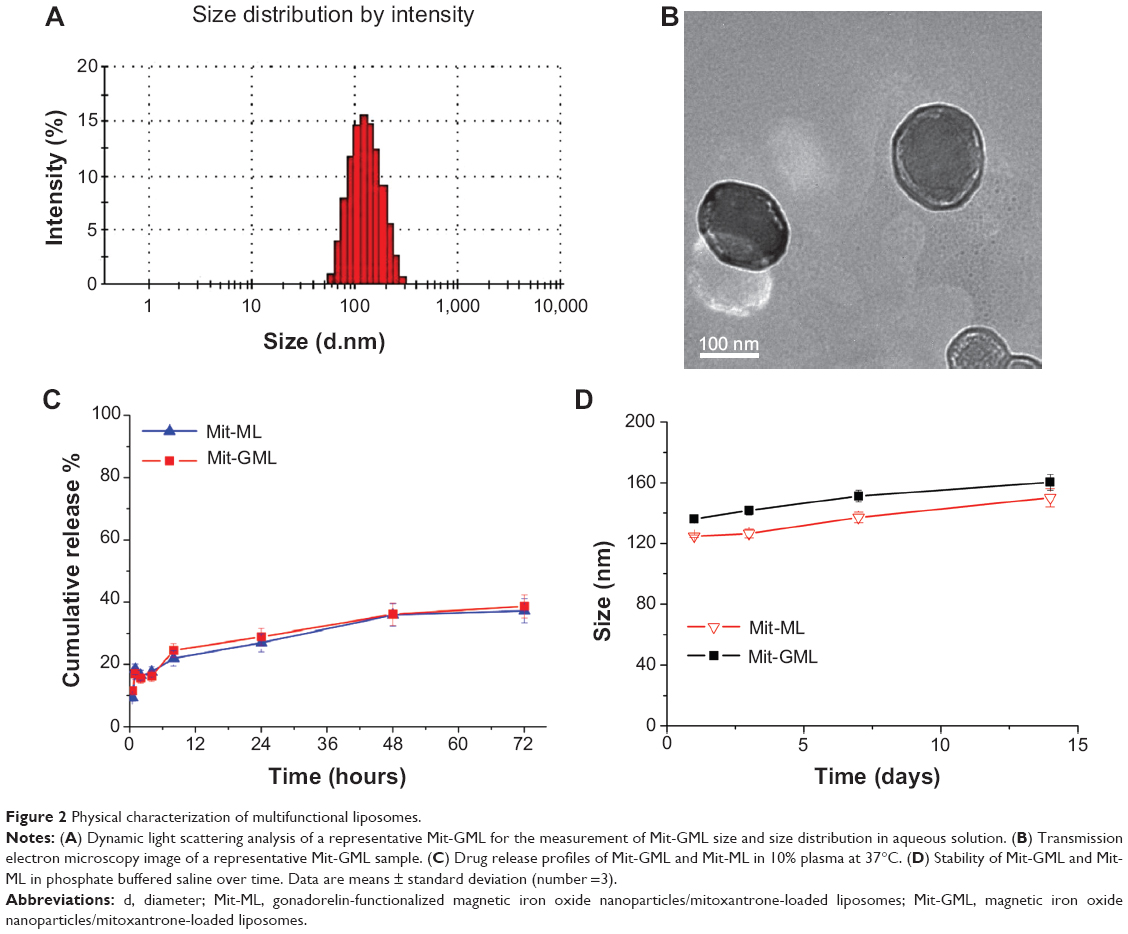 | Figure 2 Physical characterization of multifunctional liposomes. |
Drug release experiments were performed in 10% human plasma. Approximately 38% of Mit from Mit-GML was released within 72 hours, as compared to ~38% from Mit-ML (Figure 2C). Both formulations were capable of sustained drug release over a long period of time with no burst, which indicates that the modification of liposomes with the targeting groups had no influence on drug retention.
Stability experiments showed that the Mit-GML and Mit-ML were stable in PBS at 4°C for 2 weeks (Figure 2D). Combined with the drug release profile, the two liposomal formulations are expected to be stable and retain sufficient amounts of drug after systemic administration.
In vitro tumor-targeting activity of Mit-GML
Human breast carcinomas MCF-7 cells and human ovarian carcinomas SK-OV-3 cells known for their high and low levels of LHRH receptor expression, respectively,23,24 were used for these studies. As show in Figure 3, treatment of MCF-7 cells with free Mit for 4 hours led to strong Mit fluorescence in both the cytoplasm and the nucleus. Cells treated with Mit-GML showed a similar level of Mit fluorescence in the cytoplasm, but they showed somewhat lower levels of fluorescence in the nucleus. In contrast, cells treated with Mit-ML alone showed low-level cytoplasmic and nuclear Mit fluorescence.
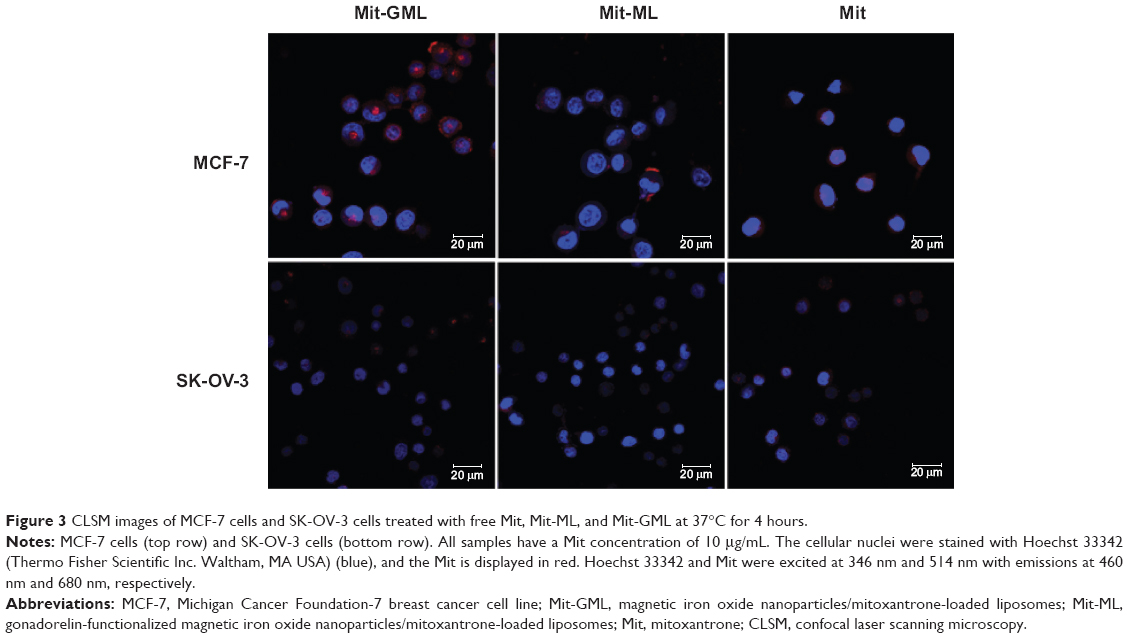 | Figure 3 CLSM images of MCF-7 cells and SK-OV-3 cells treated with free Mit, Mit-ML, and Mit-GML at 37°C for 4 hours. |
To further confirm the targeting effect of Mit-GML, the SK-OV-3 cells which low-expressing LHRH receptor incubated with free Mit, Mit-GML and Mit-ML were evaluated under the same conditions described above. The reduced uptake of Mit-GML by SK-OV-3 cells is clearly shown in Figure 3 (bottom row), when compared with MCF-7 cells under the same conditions. Collectively, these data indicated that Mit-GML is efficiently bound to the cells that overexpress LHRH receptors, and then internalized by receptor-mediated endocytosis.
In vitro anticancer effect of Mit-GML
The cytotoxicity of free Mit, Mit-ML, and Mit-GML against MCF-7 cells was carried out by a cell growth inhibition assay. MCF-7 cells were treated with free Mit and Mit-loaded liposomes at various Mit equivalent concentrations. Cell viability was calculated using the ratio of the number of MCF-7 cells of the treated group over the number of untreated controls. As shown in Figure 4, free Mit showed the highest cytotoxicity regardless of the Mit concentration. The cell viability of MCF-7 cells incubated with both Mit-GML and Mit-GML decreased with increasing Mit concentrations. However, at each Mit concentration, the cytotoxicity of Mit-GML was significantly higher than that of Mit-ML (P<0.05), which can be attributed to the higher cell uptake exhibited by Mit-GML due to LHRH receptor-mediated endocytosis.
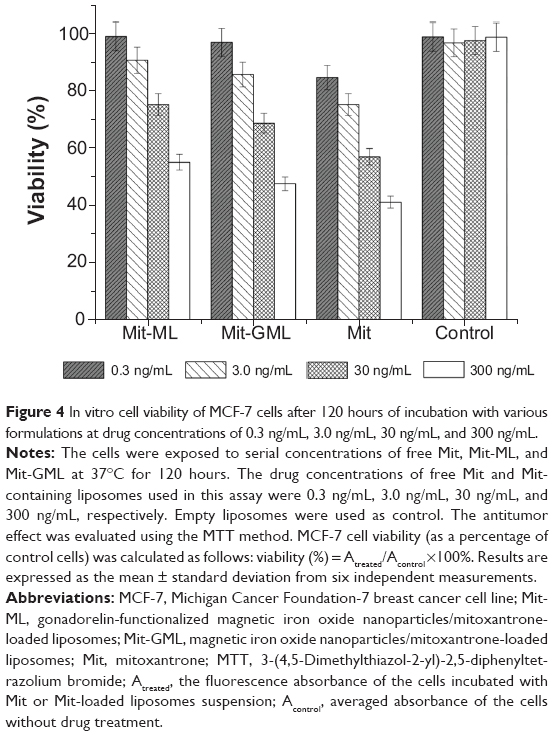 | Figure 4 In vitro cell viability of MCF-7 cells after 120 hours of incubation with various formulations at drug concentrations of 0.3 ng/mL, 3.0 ng/mL, 30 ng/mL, and 300 ng/mL. |
Tissue biodistribution of Mit-GML
The biodistribution of the liposomes in various organs over 24 hours (Figure 5) clearly demonstrated an enhanced tumor accumulation of Mit-GML and Mit-ML. In the studied tumor models, the tumor accumulation of Mit-GML was almost sevenfold higher than that of the free Mit after 24 hours postinjection. On the other hand, Mit-GML had a higher accumulation in the tumor at 24 hours postinjection compared to Mit-ML. We assume that modification of Mit-ML with gonadorelin have made them depend less on enhanced permeability and retention (EPR) effect, and this has resulted in an increase in the tumor accumulation of Mit-GML compared to that of the EPR dependent Mit-ML.
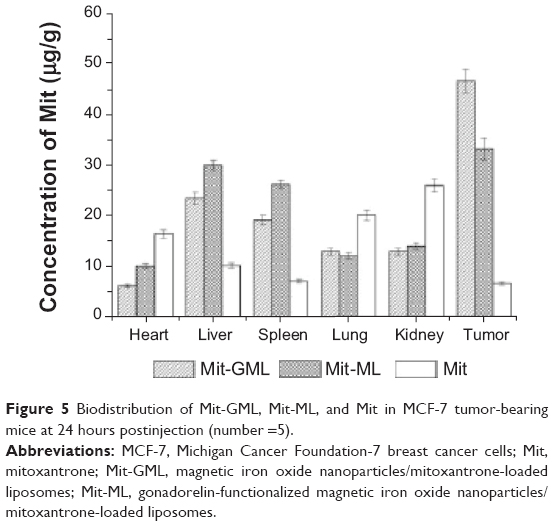 | Figure 5 Biodistribution of Mit-GML, Mit-ML, and Mit in MCF-7 tumor-bearing mice at 24 hours postinjection (number =5). |
Comparison of in vivo antitumor effects
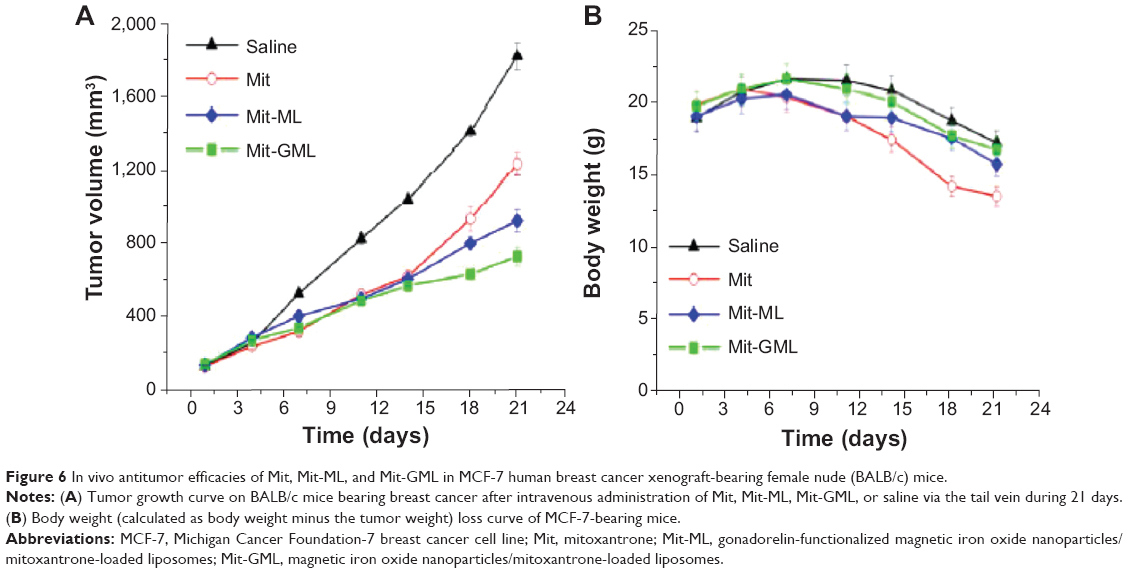
To address the utility of Mit-GML from the viewpoint of an in vivo antitumor effect, free Mit, Mit-GML, and Mit-ML were intravenously administered to the tumor-bearing mice (number =6), respectively. Figure 6A shows the time profiles of the tumor volume up to 21 days after the first administration of different formulations. In the animal models, all of the Mit-containing formulas suppressed tumor growth when compared to the saline-treated group (control). The Mit-GML-treated animals displayed markedly smaller tumor volumes and less body weight changes (Figure 6B) in comparison with the Mit- or Mit-ML-treated animals. These significant therapeutic results confirm the important role of tumor-specific binding and the internalization of the Mit-GML in achieving elevated local concentrations of the chemotherapeutic agent inside the tumor interstitium; this is in contrast to the in vitro data, where free Mit was more effective in terms of suppressing tumor growth than were Mit-GML or Mit-ML.
 | Figure 6 In vivo antitumor efficacies of Mit, Mit-ML, and Mit-GML in MCF-7 human breast cancer xenograft-bearing female nude (BALB/c) mice. |
In vitro and in vivo MRI
To evaluate the utility of Mit-GML as an MRI contrast agent, the signal intensity of Mit-ML and Mit-GML was first measured using a 1.5 T MRI system in vitro. The MRI signal intensity of all agents decreased according to the iron concentration in the T2-weighted images, and the signal intensity of Mit-ML and Mit-GML were similar at the same iron concentration (Figure 7A).
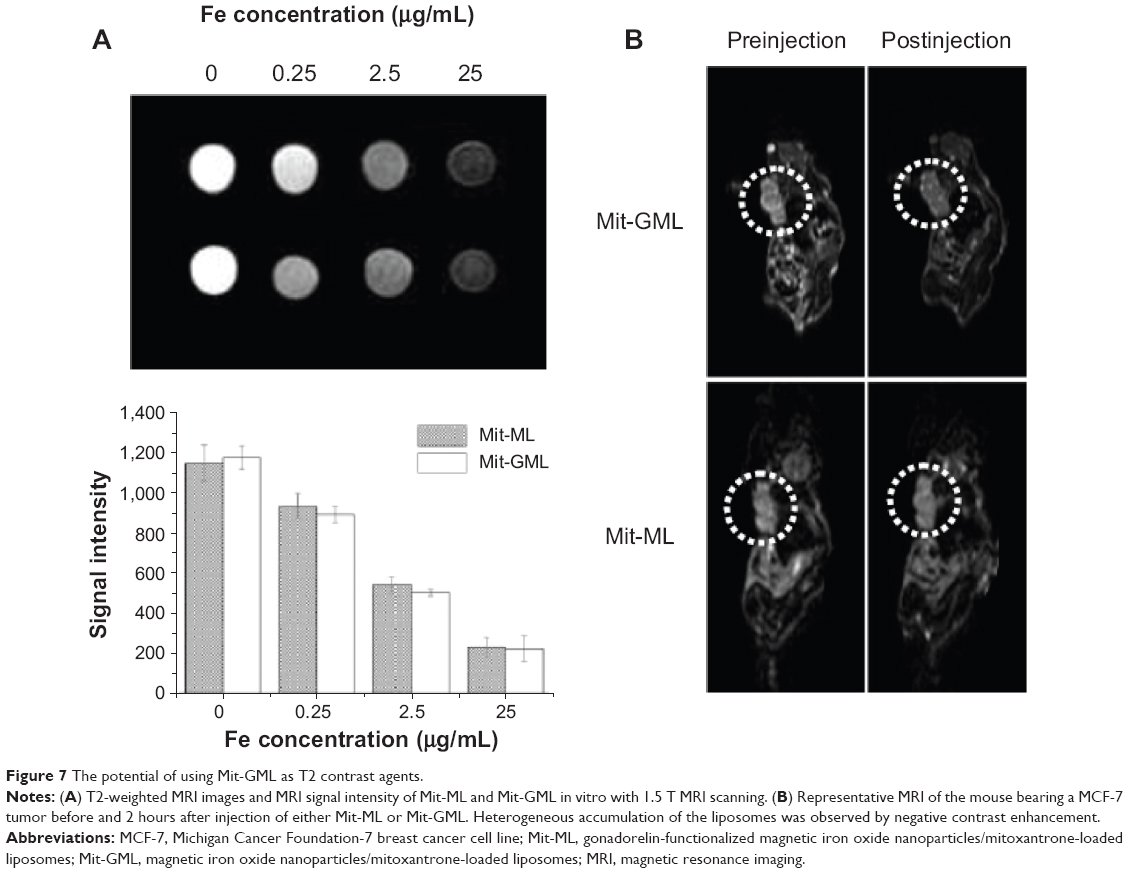 | Figure 7 The potential of using Mit-GML as T2 contrast agents. |
The imaging potential of Mit-ML and Mit-GML was also investigated in athymic mice with MCF-7 xenografts. Tumor-bearing mice were injected with either Mit-ML or Mit-GML. MR images were acquired with a fast spin echo MRI sequence prior to the administration of the liposome and at 2 hours postinjection. The images obtained showed a decrease in T2 in the tumors of the mice receiving either Mit-ML or Mit-GML (Figure 7B). However, at this time point, the tumor signal intensity value following an injection of Mit-GML decreased significantly when compared to Mit-ML, according to the T2-weighted images.
Discussion
The binding of the receptors and their ligands has shown high specificity and selectivity, strong affinity, and obvious biological effect.25 In the present research, we have designed, developed, and optimized multifunctional liposomes for codelivering MION and Mit, and we used a targeting ligand, gonadorelin, to functionalize the liposomes for the purpose of obtaining LHRH receptor-mediated endocytosis. Several analytical tools were employed to determine the physical characteristics of Mit-ML and Mit-GML, and the two liposomal formulations possess comparable sizes and loading contents for Mit and MION.
Gonadorelin was coupled to end-functionalized groups in PEG-DSPE micelles, and the gonadorelin–PEG-DSPE conjugates were then transferred, in a simple incubation step, from the micelles into the outer monolayer of the preformed liposomes. The attachment of gonadorelin to the Mit-ML did not significantly affect their drug-loading capacity and release profile. Both formulations were capable of sustained drug release over a long period of time with no burst.
Uptake of the Mit-GMLs and intracellular accumulation of its Mit cargo were superior to that of untargeted Mit-MLs in MCF-7 cells that overexpress LHRH receptors (Figure 3), but not in SK-OV-3 cells with low LHRH receptor expression, indicating the involvement of receptor-mediated endocytosis. In addition, the confocal laser scanning microscopy images also showed that free Mit was primarily localized in the nuclei of the MCF-7 cells; Mit delivered via Mit-loaded liposomes was largely accumulated in the cytoplasm of the MCF-7 cells after 4 hours of incubation. This is likely due to the slow release of the encapsulated drug from a nanocarrier (Figure 2C), and it may be the result of the different cellular uptake mechanisms between free Mit and Mit-loaded liposomes.26,27 Free Mit is known to be transported into cells via diffusion, while Mit-loaded liposomes are taken up by the MCF-7 cells via an endocytosis process. The intense Mit accumulation in the nucleus for free Mit occurred because the free Mit molecules that had diffused into the cytoplasm were rapidly transported into the nucleus and were avidly bound to the chromosomal deoxyribonucleic acid. In the case of Mit-loaded liposomes, they were taken into the cells via endocytosis and thus were initially accumulated in the endosomes where controlled Mit release began. Eventually, the released Mit would diffuse into the cytoplasm and then into the nucleus.
The cytotoxicity of free Mit, Mit-ML, and Mit-ML against MCF-7 cells was evaluated using an MTT assay. The cytotoxicity of Mit-GML was consistently higher than that of Mit-ML at all Mit concentrations tested due to the enhanced cellular uptake attributed to LHRH receptor-mediated endocytosis. Moreover, free Mit showed the highest cytotoxicity regardless of the Mit concentration. This is likely due to the slow release of the encapsulated drug from a nanocarrier and the slower accumulation of liposomes in the cells in comparison with the free drug. Nonetheless, free Mit does not have tumor-targeting abilities in vivo, which can lead to severe side effects.
To evaluate the influence of the LHRH-targeting group on the delivery of Mit to its targeted site, we used mice bearing xenografts of MCF-7 human breast cancer. Tumor growth for the different formulations exhibited the following trend: control > Mit-ML > Mit-GML. Tumor growth reduction in Mit-ML and Mit-GML groups could be distinguished only at the end of the treatment (second week). This could be due to one or more of the following reasons: 1) the increased tumor accumulation of the drug-loaded liposome was aided by passive targeting due to the EPR effect; and 2) circulating liposomes could also serve as a depot for a sustained release of Mit, which resulted in the increased overall exposure of the tumor to the drug. The LHRH-targeting groups further significantly improved the antitumor effect of the drug-loaded liposome.
It should be noted that numerous systemic toxicities such as cardiotoxicity and bone marrow suppression prevent the administration of higher Mit doses during therapy.27,28 Loading of Mit into the nanocarriers could allow for the use of higher drug doses.28–30 In support of this assumption, Figure 6B shows that both Mit-GML and Mit-ML did not induce obvious body weight loss, while the same dose of the free Mit produced considerable body weight loss (P<0.05), which indicated systemic toxicity of free Mit.
In this research, we embedded the MION into the hydrophilic cores of liposomes in the hope of obtaining desirable T2-negative contrast effects, and our ultimate goal was to develop a kind of MRI-visible nanodevice whose tumor targeting efficiency could be monitored noninvasively. MRI was chosen as our imaging modality due to its high spatial and temporal resolution and excellent soft tissue contrast, so it can provide cellular-level imaging and be used as a noninvasive strategy to monitor tumor targeting efficiency. One of the major limitations in MRI applications is the inadequate contrast sensitivity of the MR probes.31 For example, traditional T1-based contrast agents (for example, gadolinium–diethylene triamine pentaacetic acid) have only mM detection levels, which are not sensitive enough for imaging tumor markers at much lower concentrations.32 The use of target-specific contrast agents has the potential to greatly improve the ability of MRI to detect lesions, allowing for earlier detection and more accurate delineation of tumor boundaries.33,34
As shown in Figure 7A, the T2-weighted MR image of Mit-GML presented a dark MR contrast in a concentration-dependent manner in vitro, which is indicative of its strong negative contrast. We further performed in vivo MRI experiments in live animals to demonstrate the diagnostic ability of Mit-GML for targeted cancer detection. Images acquired 2 hours postinjection showed a decrease in T2 in the tumors of the mice receiving either Mit-ML or Mit-GML. However, at this time point, mice injected with the targeting Mit-GML displayed more significant contrast enhancement (darkening) heterogeneously throughout the tumor. These results suggest that Mit-GML is a good candidate as an MRI contrast agent.
Conclusion
In conclusion, we developed and characterized the theragnostic MION and Mit-loaded liposomes. Mit-GML significantly facilitated the specific delivery of Mit to tumor cells overexpressing LHRH receptors, and it efficiently inhibited tumor growth. Also, tumor accumulation of Mit-GML can be noninvasively measured by MRI. We anticipate that cancer theragnosis with Mit-GML presents a new strategy in cancer treatment, in which targeted imaging, drug delivery, and real-time noninvasive monitoring for therapeutic efficacy are carried out simultaneously.
Acknowledgments
The authors are grateful to the National Natural Science Foundation of China (no 51103180, and no 81100100), Hebei College Education Science Foundation of China (no QN20131101), Hebei Provincial Natural Science Foundation of China (no H2014206359), and Tianjin Natural Science Foundation of China (no 11JCYBJC02400) for funding this work.
Disclosure
The authors report no conflicts of interest in this work.
References
Jemal A, Siegel R, Xu J, Ward E. Cancer statistics, 2010. CA Cancer J Clin. 2010;60(5):277–300. | ||
Kelkar SS, Reineke TM. Theranostics: combining imaging and therapy. Bioconjug Chem. 2011;22(10):1879–1903. | ||
Bhojani MS, Van Dort M, Rehemtulla A, Ross BD. Targeted imaging and therapy of brain cancer using theranostic nanoparticles. Mol Pharm. 2010;7(6):1921–1929. | ||
Cabral H, Nishiyama N, Kataoka K. Supramolecular nanodevices: from design validation to theranostic nanomedicine. Acc Chem Res. 2011;44(10):999–1008. | ||
Fernandez-Fernandez A, Manchanda R, McGoron AJ. Theranostic applications of nanomaterials in cancer: drug delivery, image-guided therapy, and multifunctional platforms. Appl Biochem Biotechnol. 2011;165(7–8):1628–1651. | ||
Lammers T, Kiessling F, Hennink WE, Storm G. Nanotheranostics and image-guided drug delivery: current concepts and future directions. Mol Pharm. 2010;7(6):1899–1912. | ||
Kim K, Kim JH, Park H, et al. Tumor-homing multifunctional nanoparticles for cancer theragnosis: simultaneous diagnosis, drug delivery, and therapeutic monitoring. J Control Release. 2010;146(2):219–227. | ||
Yu MK, Park J, Jon S. Targeting strategies for multifunctional nanoparticles in cancer imaging and therapy. Theranostics. 2012;2(1):3–44. | ||
Soenen SJ, Hodenius M, De Cuyper M. Magnetoliposomes: versatile innovative nanocolloids for use in biotechnology and biomedicine. Nanomedicine (Lond). 2009;4(2):177–191. | ||
Bothun GD, Lelis A, Chen Y, Scully K, Anderson LE, Stoner MA. Multicomponent folate-targeted magnetoliposomes: design, characterization, and cellular uptake. Nanomedicine. 2011;7(6):797–805. | ||
Sun C, Du K, Fang C, et al. PEG-mediated synthesis of highly dispersive multifunctional superparamagnetic nanoparticles: their physicochemical properties and function in vivo. ACS Nano. 2010;4(4):2402–2410. | ||
Yallapu MM, Foy SP, Jain TK, Labhasetwar V. PEG-functionalized magnetic nanoparticles for drug delivery and magnetic resonance imaging applications. Pharm Res. 2010;27(11):2283–2295. | ||
Torchilin V. Multifunctional and stimuli-sensitive pharmaceutical nanocarriers. Eur J Pharm Biopharm. 2009;71(3):431–444. | ||
Taheri A, Dinarvand R, Ahadi F, Khorramizadeh MR, Atyabi F. The in vivo antitumor activity of LHRH targeted methotrexate-human serum albumin nanoparticles in 4T1 tumor-bearing Balb/c mice. Int J Pharm. 2012;431(1–2):183–189. | ||
Muthu MS, Leong DT, Mei L, Feng SS. Nanotheranostics – application and further development of nanomedicine strategies for advanced theranostics. Theranostics. 2014;4(6):660–677. | ||
Schally AV, Comaru-Schally AM, Nagy A, et al. Hypothalamic hormones and cancer. Front Neuroendocrinol. 2001;22(4):248–291. | ||
Schally AV, Engel JB, Emons G, Block NL, Pinski J. Use of analogs of peptide hormones conjugated to cytotoxic radicals for chemotherapy targeted to receptors on tumors. Curr Drug Deliv. 2011;8(1):11–25. | ||
Schally AV, Szepeshazi K, Nagy A, Comaru-Schally AM, Halmos G. New approaches to therapy of cancers of the stomach, colon and pancreas based on peptide analogs. Cell Mol Life Sci. 2004;61(9):1042–1068. | ||
Minko T, Patil ML, Zhang M, et al. LHRH-targeted nanoparticles for cancer therapeutics. Methods Mol Biol. 2010;624:281–294. | ||
Talelli M, Rijcken CJ, Lammers T, et al. Superparamagnetic iron oxide nanoparticles encapsulated in biodegradable thermosensitive polymeric micelles: toward a targeted nanomedicine suitable for image-guided drug delivery. Langmuir. 2009;25(4):2060–2067. | ||
Saad M, Garbuzenko OB, Ber E, et al. Receptor targeted polymers, dendrimers, liposomes: which nanocarrier is the most efficient for tumor-specific treatment and imaging? J Control Release. 2008;130(2): 107–114. | ||
He Y, Zhang L, Song C. Luteinizing hormone-releasing hormone receptor-mediated delivery of mitoxantrone using LHRH analogs modified with PEGylated liposomes. Int J Nanomedicine. 2010;5:697–705. | ||
Tang Q, Cao B, Wu H, Cheng G. Selective gene delivery to cancer cells using an integrated cationic amphiphilic peptide. Langmuir. 2012;28(46):16126–16132. | ||
Taratula O, Garbuzenko OB, Kirkpatrick P, et al. Surface-engineered targeted PPI dendrimer for efficient intracellular and intratumoral siRNA delivery. J Control Release. 2009;140(3):284–293. | ||
Mohanty C, Das M, Kanwar JR, Sahoo SK. Receptor mediated tumor targeting: an emerging approach for cancer therapy. Curr Drug Delivery. 2011;8(1):45–58. | ||
Zhang X, Guo S, Fan R, Yu M, Li F, Zhu C, Gan Y. Dual-functional liposome for tumor targeting and overcoming multidrug resistance in hepatocellular carcinoma cells. Biomaterials. 2012;33(29):7103–7114. | ||
Smith LA, Cornelius VR, Plummer CJ, Levitt G, Verrill M, Canney P, Jones A. Cardiotoxicity of anthracycline agents for the treatment of cancer: systematic review and meta-analysis of randomised controlled trials. BMC Cancer. 2010;10:337–350. | ||
Li C, Cui J, Li Y, et al. Copper ion-mediated liposomal encapsulation of mitoxantrone: the role of anions in drug loading, retention and release. Eur J Pharm Sci. 2008;34(4–5):333–344. | ||
Cui J, Li C, Wang L, et al. Ni2+-mediated mitoxantrone encapsulation: improved efficacy of fast release formulation. Int J Pharm. 2009; 368(1–2):24–30. | ||
Li C, Cui J, Wang C, et al. Lipid composition and grafted PEG affect in vivo activity of liposomal mitoxantrone. Int J Pharm. 2008;362(1–2): 60–66. | ||
Khemtong C, Kessinger CW, Gao J. Polymeric nanomedicine for cancer MR imaging and drug delivery. Chem Commun (Camb). 2009;(24):3497–3510. | ||
Guthi JS, Yang SG, Huang G, et al. MRI-visible micellar nanomedicine for targeted drug delivery to lung cancer cells. Mol Pharm. 2010; 7(1):32–40. | ||
Kohler N, Sun C, Fichtenholtz A, Gunn J, Fang C, Zhang M. Methotrexate-immobilized poly(ethylene glycol) magnetic nanoparticles for MR imaging and drug delivery. Small. 2006;2(6):785–792. | ||
Kohler N, Sun C, Wang J, Zhang M. Methotrexate-modified superparamagnetic nanoparticles and their intracellular uptake into human cancer cells. Langmuir. 2005;21(19):8858–8864. |
 © 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.