Back to Journals » Drug Design, Development and Therapy » Volume 16
Design and Optimization of Rivaroxaban-Cyclodextrin-Polymer Triple Complex Formulation with Improved Solubility
Authors Kang JH ![]() , Lee JE, Jeong SJ, Park CW
, Lee JE, Jeong SJ, Park CW ![]() , Kim DW, Weon KY
, Kim DW, Weon KY
Received 14 September 2022
Accepted for publication 8 December 2022
Published 16 December 2022 Volume 2022:16 Pages 4279—4289
DOI https://doi.org/10.2147/DDDT.S389884
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Professor Manfred Ogris
Ji-Hyun Kang,1,* Ji-Eun Lee,1,* So-Jeong Jeong,1 Chun-Woong Park,1 Dong-Wook Kim,2 Kwon-Yeon Weon3
1College of Pharmacy, Chungbuk National University, Cheongju, Korea; 2College of Pharmacy, Wonkwang University, Iksan, Korea; 3College of Pharmacy, Daegu Catholic University, Gyeongsan, Korea
*These authors contributed equally to this work
Correspondence: Dong-Wook Kim, College of Pharmacy, Wonkwang University, Iksan, 54538, Korea, Tel +82-63-229-7130, Fax +82-63-850-7309, Email [email protected] Kwon-Yeon Weon, College of Pharmacy, Daegu Catholic University, Gyeongsan, 38430, Korea, Tel +82-53-850-3616, Fax +82-53-850-3602, Email [email protected]
Purpose: This study aimed to ensure the convenience of administration and reproducibility of efficacy, regardless of the meal, by improving the solubility of rivaroxaban (RIV).
Methods: RIV is a non-vitamin K antagonist oral anticoagulants that exhibits a coagulation effect by directly inhibiting coagulation factor Xa. However, RIV has a very low solubility; therefore, it must be administered with a meal at high doses. We used a drug- hydroxypropyl-beta-cyclodextrin (CD)-water-soluble polymer triple complex (R-C-P complex) to solubilize RIV. Using Minitab, we evaluated the effect of each factor on RIV solubility and developed an optimal R-C-P complex formulation. The amount of CD, amount of polymer, and polymer type were set as the independent variables X1, X2, and X3, respectively. RIV solubility (Y1) and dissolution rate for 45 min in pH 4.5 medium (Y2) and pH 1.2 medium (Y3) were set as response variables.
Results: The most efficient RIV solubilization effect was obtained from the composition using CD and HPMC 2208, and physicochemical properties and dissolution parameters were analyzed. RIV in the R-C-P complex was present in an amorphous form and showed high solubility. Unlike commercial products, it showed a 100% dissolution rate. The R-C-P complex formulation secured high RIV solubility and 100% release regardless of pH.
Conclusion: The results imply that high-dose RIV can be administered regardless of the meal, reducing the risk of changing the drug effect due to the patient’s administration mistake.
Keywords: rivaroxaban, drug-cyclodextrin-polymer triple complex, solubilization, design of experiment
Introduction
Venous thromboembolism (VTE) is classified into deep vein thrombosis (DVT) and pulmonary embolism (PE).1 Vein blockage is caused by various factors promoting blood coagulation in the body, including hip/knee replacement, tumor removal, and obstetrics and gynecological surgery.2 Adequate early treatment is essential in PE, and anticoagulant therapy is paramount to reduce complications and prevent recurrence after acute treatment. After an initial 5–10 days of unfractionated heparin or low-molecular-weight heparin settlement, the patients are administered oral anticoagulant therapy. Non-vitamin K antagonist oral anticoagulants (NOACs), such as dabigatran, rivaroxaban, apixaban, and edoxaban, have been approved for use in Korea.3 NOACs with direct thrombin inhibition (dabigatran) and direct active factor Xa inhibition (rivaroxaban, apixaban, and edoxaban) pathways have a large therapeutic potential, a fixed dosing regimen, and few interactions with food or drugs. In addition, regular monitoring of international normalized ratio (INR) levels is not essential.1 NOACs have fewer side effects than warfarin; they exert different effects and do not require a dosage adjustment or monitoring.4
Rivaroxaban (RIV) is a NOAC that exhibits a coagulation effect by directly inhibiting coagulation factor Xa.5 It reduces the risk of DVT, PE, stroke, and systemic embolism in patients with nonvalvular atrial fibrillation. In addition, it is used to prevent VTE in adult patients who have undergone major orthopedic surgery of the lower extremities. The RIV dose is 10–20 mg once a day. RIV is a biopharmaceutics classification system class (BCS) II drug with low solubility (Approximately 5–7 μg/mL) and high permeability,6,7 and its bioavailability decreases by up to 66% when administered at high doses to an empty stomach.8–10 Therefore, RIV is administered with meals to ensure its bioavailability. Several studies have been conducted to improve the solubility of RIV and increase its fasting bioavailability. Research on self-nanoemulsifying drug delivery systems,11 solid dispersions,12 and sustained-release nanoparticles for RIV have been conducted.13 These studies showed that fasting bioavailability could be increased by improving the solubility of RIV.
There are various methods to improve solubility. Typically, the drug particle size reduction strategy is the most traditional method to improve the bioavailability of poorly soluble drugs to increase the solubility of substances without changing their chemical properties.14,15 Solid dispersions have long-term stability to maintain an amorphous form because the crystallization of the drug is inhibited by the polymer, aiding supersaturation.16–18 Using a self-microemulsifying drug delivery system, it is possible to solubilize poorly soluble drugs that are very stable and have high solubility by forming microdroplets.19–23 The hydroxypropyl-beta-cyclodextrin (CD) was used in this study. The CD is a cyclic oligosaccharide with a hydrophilic surface and a hydrophobic cavity in which D-(+) glucopyranose units form α-(1,4) glucosidic bonds. The hydrophobic region of the substrate was inserted into this hydrophobic cavity to form an inclusion complex, improving the solubility of poorly soluble drugs. In addition, drug-CD complexes can improve membrane permeability, stability, and taste.24–27
Previous studies on the RIV-CD complex had limitations in that solubility was increased, but the dissolution rate was not secured.28 Therefore, we intended to apply the drug-CD-polymer triple complex to RIV. The drug-CD-polymer triple complex can increase drug solubility compared to when cyclodextrin or water-soluble polymer is used separately. This can result from a synergistic effect between each component.29,30 In an aqueous solution, the water-soluble polymer helps the wettability of the particles, increasing the dissolution rate.31 In addition, the water-soluble polymer can reduce the mobility of CD and increase the solubility of the complex by changing the hydration properties of CD by stabilizing the aggregate in the aqueous solution.32–35
RIV, which is more convenient dose than other NOACs, does not affect meals when taking 10 mg doses. However, it must be administered with meals when taking 15 or 20 mg doses; this is due to reduced bioavailability resulting from low solubility. This study aimed to ensure the convenience of administration and reproducibility of efficacy, regardless of the meal, by improving the solubility of RIV. In this study, an RIV-CD-polymer triple complex (R-C-P complex) was used to improve the solubility of RIV. The R-C-P complex formulation was optimized by the design of experiments (DoEs).
Materials and Methods
Materials
RIV was a gift from the EOS Med CHEM Co., Ltd. (Jinan, China). CD was provided by Ashland, Inc. (Hydroxypropyl-beta-cyclodextrin, CAVASOL, Wilmington, DE, USA). Croscarmellose sodium was purchased from Roquette (Lesterem, Singapore). Microcrystalline cellulose was obtained from DuPont Nutrition (Newark, DE, USA). Hypromellose (HPMC) 2208 was purchased from Shin-Etsu (Tokyo, Japan). Lactose monohydrate was obtained from Meggle. (Wasserburg Inn, Germany). Povidone K 30 (PVP) was purchased from JRS Pharma (Polanco, Spain). Soluplus was supplied by BASF (Ludwigshafen, Germany). Magnesium stearate was purchased from FACI Asia Pacific Pte. Ltd. (Jurong Island, Singapore). A commercial product (Xarelto® tablet, 20 mg) was purchased from Bayer Korea, Ltd. (Seoul, Korea). Distilled water (DW) was purified by filtration in the laboratory. High-performance liquid chromatography (HPLC)-grade acetonitrile (ACN) and ethanol (EtOH) were used for the analysis. All other reagents were of reagent grade and used without further purification.
Preparation of the RIV-CD-Polymer Triple Complex (R-C-P Complex)
The R-C-P complex was prepared as follows. RIV was weighed and dissolved in 400 mL of acetone. The CD and the water-soluble polymer were weighed and completely dissolved in 200 mL of 50% (v/v) EtOH/DW solution (Table 1). The two solutions were then mixed and stirred for 30 min. The solution was evaporated using a rotary evaporator (N-N series; EYELA, Tokyo, Japan) to prepare the complex. The temperature was maintained at 65 °C, and the rotation speed was 50 rpm. The dry powder of the R-C-P complex was ground and passed through a sieve (#60 sieve, 250 μm) to remove any aggregates.
 |
Table 1 Composition of R-C-P Complex with Different Water-Soluble Polymers |
Screening of Water-Soluble Polymers
The R-C-P complex formulation for screening of water-soluble polymer was prepared using the same method as described above. The solubility of the R-C-P complex formulation with different polymers was evaluated as follows. The solubility evaluation standard was 80 μg/mL, the level at which 20 mg of RIV (the highest dose) can be dissolved in 250 mL of water, which is the solubility standard of BCS classification. Five milliliters of DW was placed in a 15 mL conical tube. The R-C-P complex with a 20 mg dose of RIV was added to the conical tube and mixed using a vortex mixer for 5 min. The conical tube was agitated for 72 h at 25 °C and 120 rpm in a shaking incubator (SI-600R, JEIO TECH, Daejeon, Korea). The suspension in the conical tube was centrifuged at 9425 ×g (10,000 rpm) for 5 min using a centrifuge (739R, Labogene, Seoul, KOREA), and the supernatant was collected using a 0.45 μm PVDF syringe filter (Whatman). The filtrate was diluted within the range of the calibration curve using a diluent (ACN:water = 80:20) and quantitatively analyzed using HPLC.
The release profile of the R-C-P complex with 20 mg of RIV was determined using the dissolution methods database of RIV in the US Food and Drug Administration (FDA) with minor modifications. In the original dissolution method of RIV in the FDA database, 0.4% sodium dodecyl sulfate (SDS) was added when a 20 mg dose of RIV was evaluated. However, in this study, SDS was excluded from the dissolution media to confirm its effect on improving solubility in the R-C-P complex. Therefore, the dissolution method we used was as follows: USP dissolution test apparatus II was used with a paddle speed of 75 rpm (Vision 8 elite, Hanson Co. Ltd., Los Angeles, CA, USA). The dissolution media used were 900 mL of acetate buffer solution (pH 4.5) or 0.1 N HCl (pH 1.2) at 37 °C. The pH 4.5 medium was derived from the dissolution method of RIV provided by the FDA, and the pH 1.2 medium was used to represent the fasting state. Samples (3 mL each) were collected from each dissolution vessel and filtered through a 0.45 μm PVDF syringe filter (Whatman, GE Healthcare, Chicago, IL, USA). An HPLC system (Agilent 1100 series, Agilent, Santa Clara, CA, USA) was used to identify and analyze RIV quantitatively. The column used was Kromasil 100-5-C (250 mm × 4.6 mm, 5 μm, Kromasil, Göteborg, Sweden). The mobile phase consisted of 550 mL of acetonitrile and 450 mL of DW in a 1000 mL volumetric flask, filtered through a 0.2 μm nylon membrane filter (Millipore, Billerica, MA, USA) and further degassed before use. The mobile phase was pumped through the column at a 1.2 mL/min flow rate. The column temperature was set to 40 °C, and the detection wavelength was 249 nm. The HPLC method was validated for selectivity, sensitivity, linearity, accuracy, precision, and recovery following the FDA industry guidelines.
The phase solubility test for RIV was performed as previously described.33,36 CD so-lutions of 0, 2, 5, 7, 10, 15, and 20% (w/v) were prepared using 0.5% (w/v) Soluplus or HPMC 2208 solution. Excess RIV was added to each solution and mixed using a vortex mixer for 5 min. The conical tube was agitated, centrifuged, and analyzed using the abovementioned method. The phase solubility profile was presented by plotting the molarity of dissolved RIV (mmol/L) against the molarity of CD (mmol/L). Further, the results were evaluated in terms of complexation constant and efficiency. The solubilization capacity of CD is mainly evaluated by the complex formation equilibrium constant “K” value. However, in poorly soluble drugs with a solubility of 1 mg/mL or less, there is a large error due to the inaccuracy of the initial concentration (S0) of the equation.37 Therefore, we compared the complexation efficiencies (CE), representing the ratio of complexed CD to free CD (Equation 1).
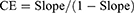
Full Factorial Design and Response Optimization of the R-C-P Complex
A 23 full factorial design was built for 12 different R-C-P complex formulations, containing two repetitions at the central point. Each independent variable was investigated at two levels (Table 2). The Minitab 19 software (Minitab Inc., Philadelphia, PA, USA) was used for the experimental design and statistical evaluation. The three independent variables were the amount of CD (X1, Low (−1): 64.7 mg as 1 equivalent with RIV; High (1): 194.1 mg as 3 equivalents with RIV), the amount of water-soluble polymer (X2, Low (−1): 10 mg; High (1): 40 mg), and the type of water-soluble polymer (X3, Low (−1): Soluplus; High (1): HPMC 2208). Solubility (Y1), amount released at 45 min in pH 4.5, dissolution media (Y2), and the amount released at 45 min in pH 1.2 dissolution media (Y3) were taken as DoE responses. When setting the models, a stepwise elimination criterion (α = 0.05) was used to eliminate the variables that were not significant for a given response. After the models were set, the response optimizer in Minitab was used to determine the optimal formulation, depending on the desired characteristics of the R-C-P complex. The goals were set to the minimum amount of the R-C-P complex/dose, maximum Y1, maximum Y2, and maximum Y3.
 |
Table 2 Variables, Levels of Variables, and Composition of the R-C-P Complex in Each Experiment Designed Using 23 Full Factorial Design |
The R-C-P complex formulations were prepared using the same methods as in Preparation of the RIV-CD-Polymer Triple Complex (R-C-P Complex). In addition, the water solubility and in vitro dissolution profile of the R-C-P complex formulation was obtained using the same method as in Screening of Water-Soluble Polymers.
Evaluation of an Optimal R-C-P Complex Formulation
The optimal R-C-P complex formulation was prepared using the composition obtained through response optimization. The preparation method is the same as in Preparation of the RIV-CD-Polymer Triple Complex (R-C-P Complex). In addition, the water solubility and in vitro dissolution profile of the optimal R-C-P complex formulation was obtained using the same method as in Screening of Water-Soluble Polymers.
X-ray diffraction (XRD) patterns of the RIV raw material, excipients, physical mixture, and R-C-P complex were analyzed using an EMPYREAN (Malvern Panalytical, Worcester-shire, UK) with Cu Kα radiation (wavelength of 1.54) generated at 30 mA and 40 kV. The samples were then placed on a silicon plate at room temperature. The 2 theta scans were conducted between 2° and 50° with a step size of 0.0001°/2 theta.
Infrared spectroscopy was performed using a Fourier-transform infrared (FT-IR) spec-trophotometer (Frontier, Perkin Elmer, Waltham, MA, USA) with the potassium bromide technique and deuterated triglycine sulfate (DTGS) detector. For each spectrum, 16 transient spectra were collected over 400–4000 cm−1 for the RIV raw material, excipients, and R-C-P complex.
The thermal properties of the RIV raw material, excipients, and R-C-P complex were analyzed using a DSC Q2000 thermal analyzer system (TA Instruments, New Castle, DE, USA). The samples were accurately weighed, loaded into an aluminum pan, and analyzed at a heating rate of 20 °C/min over a temperature range of 50–300 °C. The thermal response of the prepared sample was calculated using the TA Advantage/Universal Analysis software (v5.2.6, TA Instruments, New Castle, DE, USA).
Results
Screening of Water-Soluble Polymers
The water solubility of R-C-P complex formulations prepared using different water-soluble polymers was evaluated for polymer screening (Figure 1). We aimed to dissolve 20 mg or more of RIV in 250 mL of DW according to the criteria of the BCS class. The R-C-P complex was prepared using Soluplus and HPMC 2208 as S-1 and S-3 formulations, respectively, which met these criteria. The solubility of S-1 and S-3 was 189.87 ± 1.24 and 168.32 ± 1.72 μg/mL, respectively, and the solubility was increased by 5.5- and 4.9-folds, respectively, compared to the formulation consisting of RIV and CD. In contrast, the solubility of S-2, an R-C-P complex formulation using PVP K30, did not increase. These results suggested that Soluplus and HPMC 2208 work synergistically with CD in solubilizing RIV. Although many previous studies have confirmed the advantage of increasing the CD complexing efficiency by adding a hydrophilic polymer, the exact mechanism of this phenomenon remains unclear. Since different polymers have opposite effects in different drug-CD systems, their effects on CD complexing should be interpreted on a case-by-case basis.32,38 Therefore, specific interactions between RIV and polymers are important for explaining this phenomenon. The hydrophilic polymer used in our study induced micellar solubilization of drugs; however, this effect was not remarkable because only free drug molecules can form complexes with CD. It is of particular importance that these polymers stabilize CD and/or RIV-CD complex’s self-assembled structure and further enhance CD’s solubilization potential.39,40
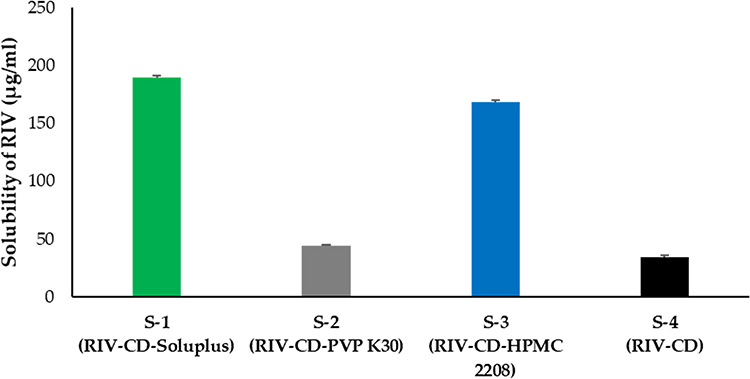 |
Figure 1 RIV solubility in the R-C-P complex with different polymers. |
For polymer selection, dissolution tests of R-C-P complex formulations using different polymers were performed (Figure 2). The dissolution test conditions for RIV listed in the FDA require that sodium lauryl sulfate (SLS) be added to the dissolution medium to secure the sink condition when a high-dose RIV tablet is tested. We proceeded in the absence of SLS to confirm the solubilizing effect of the R-C-P complex. The dissolution results of the medium using acetate buffer pH 4.5 simulating post-meal pH (Figure 2A) and HCl buffer pH 1.2 simulating pre-meal pH (Figure 2B) were the highest in the S-3 formulation using HPMC 2208. In addition, the dissolution rate of S-1 was significantly higher than that of S-4. Further, S-3 prepared using PVP showed a dissolution pattern similar to that of S-4. As shown in Figure 1, only approximately 20% of RIV was released from S-2 and S-4 at 45 min in the dissolution test because the solubilizing effect of RIV was low. However, drug release from S-1 and S-3 reached approximately 55% and 85% at 45 min, respectively, regardless of pH. Although S-1 had a higher solubility than S-3, S-3 exhibited a 1.5-times greater dissolution rate than S-1. In the case of S-1, the dissolution rate continued to rise, and if more time was given, it was thought to reach 100%. Solubilization is known to occur by binding of CD and RIV. Therefore, the S-1 formulation, which has superior solubilization effect than that of S-3, is estimated to have stronger CD and RIV binding, so the dissolution rate of S-1 is slower than that of S-3. Therefore, we proceeded using Soluplus and HPMC 2208 to confirm the synergy of the solubilization effect of CD and the polymer using a phase solubility test.
 |
Figure 2 The release profile of the R-C-P complex with different polymers in (A) pH 4.5 medium and (B) pH 1.2 medium (mean ± standard deviation, n = 3). |
Consequently, in the phase solubility test, the solubility of the R-C-P complex was higher in the solution containing Soluplus. In the phase solubility test, as the concentration of CD increased, a pattern close to a second-order curve was observed (Figure 3A). This is due to the solubilization effect in addition to CD inclusion, such as non-complex formation and micelle formation, as the ratio of CD increases.41 Therefore, to calculate the exact CE, the slope was calculated in the section where CD formed a straight line at a low concentration (Figure 3B). S0 obtained using HPLC analysis was 0.014, 0.051, and 0.021 mM of RIV in CD-only, 0.5% Soluplus, and 0.5% HPMC 2208 solutions, respectively. The slope calculated from the solubility plot of RIV obtained with CD was 0.0041, 0.0033, and 0.0028 for 0.5% Soluplus, 0.5% HPMC, and CD-alone solutions. The solubilization effect of CD further increased when the polymer was present. In poorly soluble drugs with a solubility of 1 mg/mL or less, a large error appears due to the inaccuracy of S0; therefore, the CE value representing the ratio of complexed CD to free CD is more reliable.37 The CE of the solution containing CD alone, 0.5% Soluplus, and 0.5% HPMC 2208 was 0.0028, 0.0038, and 0.0033, respectively. The CE value was also clearly higher in the polymer-added solution than in the CD solution, implying that the RIV solubilizing effect of CD further increased synergistically on adding the polymer. However, in the previous screening test (Figures 1 and 2), the solubilization effect and the dissolution rate were not directly proportional; therefore, Soluplus and HPMC 2208 were used as variables when studying the formulation of the R-C-P complex using DoE.
 |
Figure 3 Phase solubility profile with RIV, CD, and different polymers. Total CD concentration range: (A) 0–160 mM. (B) 0–8 mM (mean ± standard deviation, n = 3). |
Full Factorial Design and Response Optimization of the R-C-P Complex
The preparation and evaluation of the R-C-P complex were performed according to the DoE run order. The values of the responses for each DoE experiment are listed in Table 3. When factor regression analysis was performed using the Minitab software, the effect was beyond the standard point of the significance level of the Pareto chart shown in Figure 4. As a value affecting the solubility of RIV (Y1, Figure 4A), the variables satisfying P-value ≤ 0.05 were B (Amount of polymer), A (Amount of CD), and C (Type of polymer). Other interaction effects were not significant. Because the coefficients of A, B, and C were positive, the solubility of RIV increased as the amounts of CD and polymer increased, especially in the case of HPMC 2208. Unlike previous phase solubility test results, higher solubility was obtained for the R-C-P complex with HPMC 2208. This is because, unlike the phase solubility test, the amount of polymer and CD were limited in the DoE study. In addition, in the DoE study, an R-C-P complex was manufactured, and RIV, CD, and polymer were physically combined. However, there was a difference in the phase solubility study because the RIV, CD, and polymer were separately dissolved and mixed. Figure 4B and C show the effect of each variable on the dissolution rate at 45 min in pH 4.5 and 1.2 medium. The pH of 4.5 represents the pH after meals, whereas the pH of 1.2 represents the pH before meals. Significant results were obtained for the variables C (type of polymer), B (amount of polymer), and BC (interaction effect between B and C) in both media. These results imply that the R-C-P complex using HPMC 2208 improved the solubility of RIV, and the dissolution rate was also high, regardless of whether a meal was taken. In addition, it was confirmed that the type and amount of polymer had a significant synergistic effect in BC. Neither A (amount of CD) nor any other interaction effects were significant. The same result as the dissolution test in the screening step was obtained. To be precise, the higher the polymer, the higher the dissolution rate, especially when using HPMC 2208.
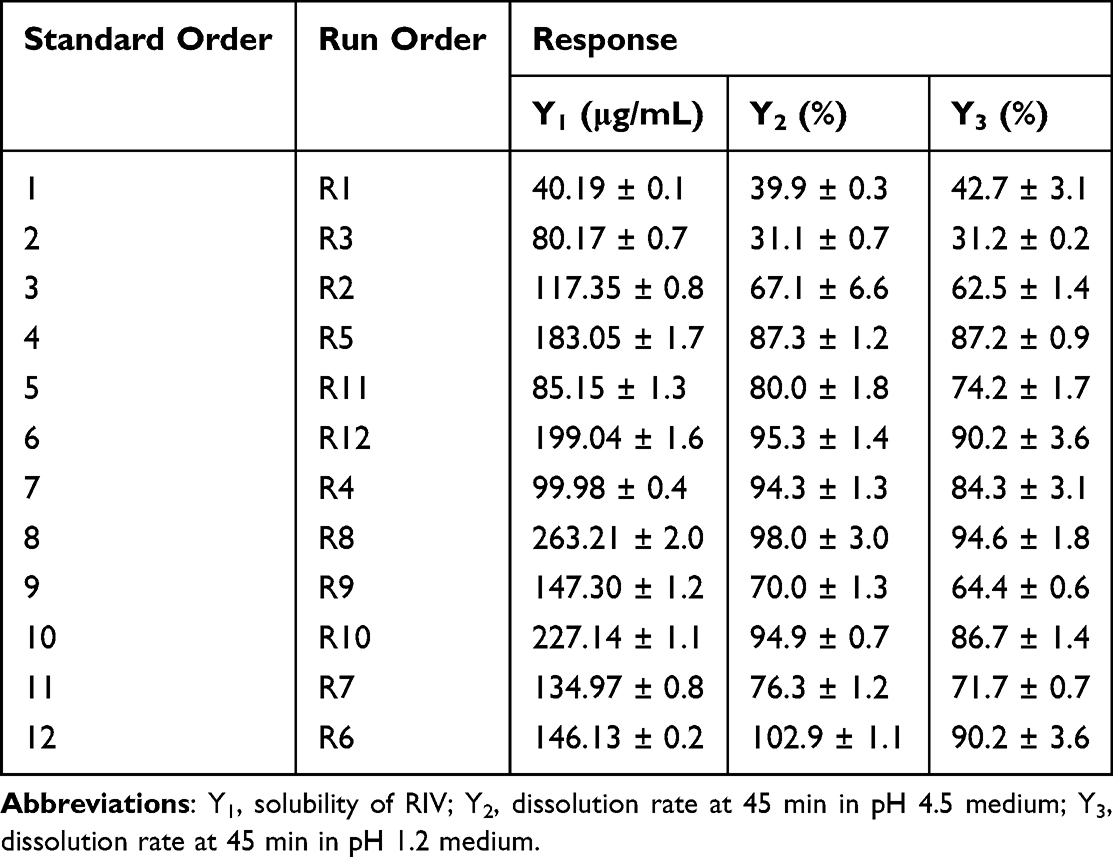 |
Table 3 Response of the R-C-P Complex in Each Experiment Designed Using 23 Full Factorial Design |
 |
Figure 4 Pareto charts with the standardized effect of each variable. (A) RIV solubility of the R-C-P complex (Y1). (B) Dissolution rate of the R-C-P complex at 45 min in pH 4.5 medium (Y2). (C) Dissolution rate of the R-C-P complex at 45 min in pH 1.2 medium (Y3). |
Evaluation of the Optimal R-C-P Complex
After removing insignificant variables from the Pareto chart, a more accurate regression equation was obtained by reducing the model using the pooling process. The higher the responses (Y1, Y2, and Y3), the smaller the amount of dose and the more efficient the prescription. Therefore, we set the standard and attempted to obtain the optimal R-C-P complex formulation using the response optimization tool of Minitab. The solubility of RIV in the R-C-P complex (Y1) was set to 80 μg/mL or more based on the BCS class, and the dissolution rate at 45 min in pH 4.5 and 1.2 media was set to 85% or more. Among the compositions satisfying these conditions, the formulation with the smallest total amount of a single dose was obtained using Minitab. As a result, similar to the standard orders 10 and 12 formulations, CD 129.4 mg and HPMC 2208 25 mg were selected as the optimal R-C-P complex formulations. Therefore, we prepared an optimal R-C-P complex formulation according to the above composition and performed the physicochemical evaluation and dissolution tests.
Figure 5A shows the DSC thermogram. In raw RIV, a melting point of approximately 230 °C was found, and in raw HPMC 2208 and raw CD, only a broadened water peak at approximately 100 °C was found. In the optimal R-C-P complex, the peak of raw RIV was not found, and a water peak around 100 °C was mainly found. However, a small endothermic event was observed at approximately 190 °C. This small peak is the glass transition temperature of RIV in the amorphous state.42 Similarly, amorphous features of RIV were observed (Figure 5B). Raw RIV exhibited high and sharp peaks, whereas only hollow peaks were observed for HPMC 2208 and CD. In particular, peaks of RIV in the physical mixture of the same composition as optimal R-C-P complex were found around 2 theta values of 14.5° to 26.5°, but no sharp peaks were found in optimal R-C-P complex. However, broad hills at 12.9° and 21.6°, which did not exist in raw RIV, were observed. Some RIV may partially retain crystals, similar to the 2 theta value of the polymorph form F in reference.43 However, because the intensity is very low and broad, there is a high possibility that very few particles are retained as crystals. Figure 5C shows the FTIR spectra. Most of the optimal R-C-P complexes showed a pattern very similar to that of HPMC 2208 or CD, and it seemed to have a C=O stretch peak of raw RIV at 1726 cm−1. Through physicochemical property evaluation, it was confirmed that most of the amorphous RIV were present in the optimal R-C-P complex, and it was confirmed that solubilization could occur due to this.
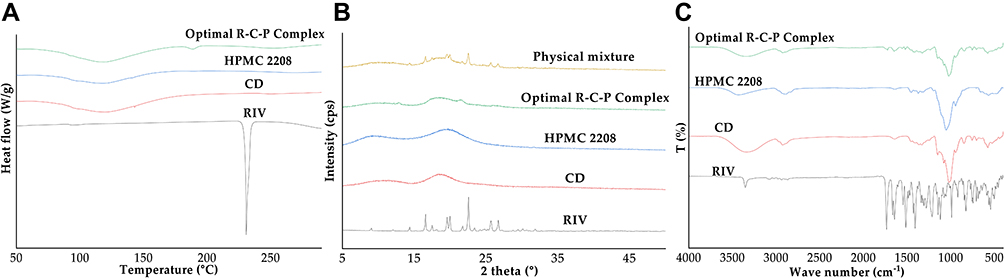 |
Figure 5 Physicochemical properties of the optimal R-C-P complex. (A) DSC thermograms of the raw materials and optimal R-C-P Complex. (B) XRD patterns of the raw materials, physical mixture, and optimal R-C-P complex. (C) FT-IR spectra of the raw materials and optimal R-C-P complex. |
Finally, the dissolution pattern of the optimal R-C-P complex was determined (Figure 6). The dissolution test was carried out in two media, pH 4.5, representing the pH after meals, and pH 1.2, representing fasting, as described above. In addition, comparative dissolution was performed using 20 mg of Xarelto, a commercial product, to confirm the RIV solubilization effect of the optimal R-C-P complex. At both pH 4.5 and 1.2, Xarelto showed a dissolution rate of approximately 40%, but the optimal R-C-P complex completed drug release in approximately 45 min. The commercial product was finally released at 40%, whereas the optimal R-C-P complex was dissolved up to 100%. These results showed that the solubilization effect of the optimal R-C-P complex was sufficient. In addition, it can be expected that even if 20 mg of RIV is administered, it is not necessary to administer the optimal R-C-P complex formulation with meals, unlike commercial products. These results will not only improve patient convenience but also ensure consistency in the therapeutic effect of RIV.
 |
Figure 6 The release profile of the optimal R-C-P complex and commercial product in (A) pH 4.5 medium and (B) pH 1.2 medium (mean ± standard deviation, n = 3). |
Conclusion
In this study, a triple combination of RIV-CD-polymer was applied to secure a formulation capable of administering high-dose RIV regardless of whether the patient was eating. After selecting the polymer by screening for the solubilization effect of the water-soluble polymer, the effect of each factor was evaluated using the phase solubility test and DoE, and the optimal R-C-P complex formulation was derived. Unlike commercial products, the optimal R-C-P complex formulation had sufficient solubility to reach 100% release quickly and was not affected by pH. These results suggest that limiting the dose of 20 mg RIV after meals is unnecessary; hence, so the patient’s dosing convenience can be improved.
Funding
This study was supported by a National Research Foundation of Korea grant provided by the Korean government (NRF-2021R1A2C4002746 and 2017R1A5A2015541). This research was supported by “Regional Innovation Strategy (RIS)” through the National Research Foundation of Korea(NRF) funded by the Ministry of Education(MOE)(2021RIS-001).
Disclosure
The authors declare no conflicts of interest in this work.
References
1. Elshafei MN, Mohamed MFH, El-Bardissy A, et al. Comparative effectiveness and safety of direct oral anticoagulants compared to warfarin in morbidly obese patients with acute venous thromboembolism: systematic review and a meta-analysis. J Thromb Thrombolysis. 2021;51:388–396. doi:10.1007/s11239-020-02179-4
2. Ageno W, Gallus AS, Wittkowsky A, Crowther M, Hylek EM, Palareti G. Oral anticoagulant therapy: antithrombotic therapy and prevention of thrombosis, 9th ed: American College of Chest Physicians evidence-based clinical practice guidelines. Chest. 2012;141(2):e44S–e88S. doi:10.1378/chest.11-2292
3. Park JK, Kim J, Baek YS, et al. 2018 KHRS practical guide on the use of non-vitamin K antagonist oral anticoagulants in Korean patients with atrial fibrillation: how to initiate and organize the follow-up. Korean J Med. 2019;94:17–39. doi:10.3904/kjm.2019.94.1.17
4. Thomas I, EncisoSilva J, Schlueter M, Greenberg B. Anticoagulation therapy and NOACs in heart failure. Handb Exp Pharmacol. 2017;243:515–535. doi:10.1007/164_2016_126
5. Samama MM. The mechanism of action of Rivaroxaban—An oral, direct Factor Xa inhibitor--compared with other anticoagulants. Thromb Res. 2011;127:497–504. doi:10.1016/j.thromres.2010.09.008
6. Stampfuss J, Kubitza D, Becka M, Mueck W. The effect of food on the absorption and pharmacokinetics of Rivaroxaban. Int J Clin Pharmacol Ther. 2013;51:549–561. doi:10.5414/CP201812
7. Reçber T, Haznedaroğlu İC, Çelebier M. Review on characteristics and analytical methods of rivaroxaban. Crit Rev Anal Chem. 2022;52:865–877. doi:10.1080/10408347.2020.1839735
8. Raymond J, Imbert L, Cousin T, et al. Pharmacogenetics of direct oral anticoagulants: a systematic review. J Pers Med. 2021;11:37. doi:10.3390/jpm11010037
9. Mueck W, Stampfuss J, Kubitza D, Becka M. Clinical pharmacokinetic and pharmacodynamic profile of Rivaroxaban. Clin Pharmacokinet. 2014;53:1–16. doi:10.1007/s40262-013-0100-7
10. Samama MM, Contant G, Spiro TE, et al. Laboratory assessment of Rivaroxaban: a review. Thromb J. 2013;11:11. doi:10.1186/1477-9560-11-11
11. Xue X, Cao M, Ren L, Qian Y, Chen G. Preparation and optimization of Rivaroxaban by Self-Nanoemulsifying Drug Delivery System (SNEDDS) for enhanced oral bioavailability and no food effect. AAPS PharmSciTech. 2018;19:1847–1859. doi:10.1208/s12249-018-0991-6
12. Metre S, Mukesh S, Samal SK, Chand M, Sangamwar AT. Enhanced biopharmaceutical performance of Rivaroxaban through polymeric amorphous solid dispersion. Mol Pharm. 2018;15:652–668. doi:10.1021/acs.molpharmaceut.7b01027
13. Anwer MK, Mohammad M, Iqbal M, et al. Sustained release and enhanced oral bioavailability of Rivaroxaban by PLGA nanoparticles with no food effect. J Thromb Thrombolysis. 2020;49:404–412. doi:10.1007/s11239-019-02022-5
14. Siepmann J, Siepmann F. Mathematical modeling of drug dissolution. Int J Pharm. 2013;453:12–24. doi:10.1016/j.ijpharm.2013.04.044
15. Noyes AA, Whitney WR. The rate of solution of solid substances in their own solutions. J Am Chem Soc. 1897;19:930–934. doi:10.1021/ja02086a003
16. Sekiguchi K, Obi N. Studies on absorption of eutectic mixture. I. A comparison of the behavior of eutectic mixture of sulfathiazole and that of ordinary sulfathiazole in man. Chem Pharm Bull. 1961;9:866–872. doi:10.1248/cpb.9.866
17. Sammour OA, Hammad MA, Megrab NA, Zidan AS. Formulation and optimization of mouth dissolve tablets containing rofecoxib solid dispersion. AAPS PharmSciTech. 2006;7:E55. doi:10.1208/pt070255
18. Cid AG, Simonazzi A, Palma SD, Bermúdez JM. Solid dispersion technology as a strategy to improve the bioavailability of poorly soluble drugs. Ther Deliv. 2019;10:363–382. doi:10.4155/tde-2019-0007
19. Pund S, Borade G, Rasve G. Improvement of anti-inflammatory and anti-angiogenic activity of berberine by novel rapid dissolving nanoemulsifying technique. Phytomedicine. 2014;21:307–314. doi:10.1016/j.phymed.2013.09.013
20. Pund S, Shete Y, Jagadale S. Multivariate analysis of physicochemical characteristics of lipid based nanoemulsifying cilostazol—Quality by design. Colloids Surf B Biointerfaces. 2014;115:29–36. doi:10.1016/j.colsurfb.2013.11.019
21. Taha E, Ghorab D, Zaghloul AA. Bioavailability assessment of vitamin A self-nanoemulsified drug delivery systems in rats: a comparative study. Med Princ Pract. 2007;16:355–359. doi:10.1159/000104808
22. Jansook P, Maw PD, Soe HM, et al. Development of amphotericin B nanosuspensions for fungal keratitis therapy: effect of self-assembled γ-cyclodextrin. J Pharm Investig. 2020;50:513–525. doi:10.1007/s40005-020-00474-z
23. Sander C, Holm P. Porous magnesium Aluminometasilicate tablets as carrier of a cyclosporine self-emulsifying formulation. AAPS PharmSciTech. 2009;10:1388–1395. doi:10.1208/s12249-009-9340-0
24. Jacob S, Nair AB. Cyclodextrin complexes: perspective from drug delivery and formulation. Drug Dev Res. 2018;79:201–217. doi:10.1002/ddr.21452
25. Loftsson T, Brewster ME. Pharmaceutical applications of cyclodextrins: basic science and product development. J Pharm Pharmacol. 2010;62:1607–1621. doi:10.1111/j.2042-7158.2010.01030.x
26. Loftsson T, Jarho P, Másson M, Järvinen T. Cyclodextrins in drug delivery. Expert Opin Drug Deliv. 2005;2:335–351. doi:10.1517/17425247.2.1.335
27. Budhwar V. Cyclodextrin complexes: an approach to improve the physicochemical properties of drugs and applications of cyclodextrin complexes. Asian J Pharm. 2018;12. doi:10.22377/ajp.v12i02.2367
28. Sherje AP, Jadhav M. β-cyclodextrin-based inclusion complexes and nanocomposites of Rivaroxaban for solubility enhancement. J Mater Sci Mater Med. 2018;29:186. doi:10.1007/s10856-018-6194-6
29. Miranda JC, Martins TEA, Francisco V, Ferraz HG. Cyclodextrins and ternary complexes: technology to improve solubility of poorly soluble drugs. Braz J Pharmacol. 2011;47:665–681. doi:10.1590/S1984-82502011000400003
30. Loftsson T, Frikdriksdóttir H, Sigurkdardóttir AM, Ueda H. The effect of water-soluble polymers on drug-cyclodextrin complexation. Int J Pharm. 1994;110:169–177. doi:10.1016/0378-5173(94)90155-4
31. Lahiani-Skiba M, Barbot C, Bounoure F, Joudieh S, Skiba M. Solubility and dissolution rate of progesterone-cyclodextrin-polymer systems. Drug Dev Ind Pharm. 2006;32:1043–1058. doi:10.1080/03639040600897093
32. Medarević D, Kachrimanis K, Djurić Z, Ibrić S. Influence of hydrophilic polymers on the complexation of carbamazepine with hydroxypropyl-β-cyclodextrin. Eur J Pharm Sci. 2015;78:273–285. doi:10.1016/j.ejps.2015.08.001
33. Taupitz T, Dressman JB, Buchanan CM, Klein S. Cyclodextrin-water soluble polymer ternary complexes enhance the solubility and dissolution behaviour of poorly soluble drugs. Case example: itraconazole. Eur J Pharm Biopharm. 2013;83:378–387. doi:10.1016/j.ejpb.2012.11.003
34. Erdoğar N, Akkın S, Nielsen TT, et al. Development of oral aprepitant-loaded chitosan–polyethylene glycol-coated cyclodextrin nanocapsules: formulation, characterization, and pharmacokinetic evaluation. J Pharm Investig. 2021;51:297–310. doi:10.1007/s40005-020-00511-x
35. Tao C, Huo T, Zhang Q, Song H. Effect of Soluplus on the supersaturation and absorption of tacrolimus formulated as inclusion complex with dimethyl-β-cyclodextrin. Pharm Dev Technol. 2019;24:1076–1082. doi:10.1080/10837450.2019.1630651
36. Higuchi T, Connors KA. Phase solubility techniques. Adv Anal Chem Instrum. 1965;4:117–122.
37. Loftsson T, Hreinsdóttir D, Másson M. Evaluation of cyclodextrin solubilization of drugs. Int J Pharm. 2005;302:18–28. doi:10.1016/j.ijpharm.2005.05.042
38. Kim DH, Lee SE, Pyo YC, Tran P, Park JS. Solubility enhancement and application of cyclodextrins in local drug delivery. J Pharm Investig. 2020;50:17–27. doi:10.1007/s40005-019-00434-2
39. Loftsson T, Brewster ME. Cyclodextrins as functional excipients: methods to enhance complexation efficiency. J Pharm Sci. 2012;101:3019–3032. doi:10.1002/jps.23077
40. Kou W, Cai C, Xu S, et al. In vitro and in vivo evaluation of novel immediate release carbamazepine tablets: complexation with hydroxypropyl-β-cyclodextrin in the presence of HPMC. Int J Pharm. 2011;409:75–80. doi:10.1016/j.ijpharm.2011.02.042
41. Saokham P, Muankaew C, Jansook P, Loftsson T. Solubility of cyclodextrins and drug/cyclodextrin complexes. Molecules. 2018;23:1161. doi:10.3390/molecules23051161
42. Lee JH, Jeong HS, Jeong JW, et al. The development and optimization of hot-melt extruded amorphous solid dispersions containing Rivaroxaban in combination with polymers. Pharmaceutics. 2021;13:344. doi:10.3390/pharmaceutics13030344
43. Zhai L, Zhang Z, Guo L, Zhu Z, Hu C, Zhang G. Synthesis, characterization, and properties of Rivaroxaban new crystalline forms. Cryst Res Technol. 2021;56:2000243. doi:10.1002/crat.202000243
 © 2022 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2022 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.