Back to Journals » Drug Design, Development and Therapy » Volume 14
Design and Development of a Behaviorally Active Recombinant Neurotrophic Factor
Authors Pekas NJ ![]() , Petersen JL
, Petersen JL ![]() , Sathyanesan M, Newton SS
, Sathyanesan M, Newton SS ![]()
Received 29 July 2020
Accepted for publication 10 November 2020
Published 4 December 2020 Volume 2020:14 Pages 5393—5403
DOI https://doi.org/10.2147/DDDT.S274308
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 4
Editor who approved publication: Professor Anastasios Lymperopoulos
Nicholas J Pekas,1 Jason L Petersen,2,3 Monica Sathyanesan,1,2 Samuel S Newton1,2
1Department of Basic Biomedical Sciences, University of South Dakota Sanford School of Medicine, Vermilion, SD, USA; 2Sioux Falls VA Health Care System, Sioux Falls, SD, USA; 3Department of Internal Medicine, University of South Dakota Sanford School of Medicine, Vermillion, SD, USA
Correspondence: Samuel S Newton Attn: Lee Medical Building, 414 E. Clark Street, Vermillion, SD 57069, USA
Tel +1-605-658-6313
Email [email protected]
Introduction: Carbamoylated erythropoietin (CEPO) is a chemically engineered, nonhematopoietic derivative of erythropoietin (EPO) that retains its antidepressant and pro-cognitive effects, which are attributed to the increased expression of neurotrophic factors like brain derived neurotrophic factor (BDNF), in the central nervous system. However, the chemical modification process which produces CEPO from erythropoietin (EPO) requires pure EPO as raw material, is challenging to scale-up and can also cause batch-to-batch variability. To address these key limitations while retaining its behavioral effects, we designed, expressed and analyzed a triple, glutamine, substitution recombinant mimetic of CEPO, named QPO.
Methods and Materials: We employ a combination of computational structural biology, molecular, cellular and behavioral assays to design, produce, purify and test QPO.
Results: QPO was shown to be a nonhematopoietic polypeptide with significant antidepressant-like and pro-cognitive behavioral effects in rodent assays while significantly upregulating BDNF expression in-vitro and in-vivo. The in-silico binding affinity analysis of QPO bound to the EPOR/EPOR homodimer receptor shows significantly decreased binding to Active Site 2, but not Active Site 1, of EPOR.
Discussion: The results of the behavioral and gene expression analysis imply that QPO is a successful CEPO mimetic protein and potentially acts via a similar neurotrophic mechanism, making it a drug development target for psychiatric disorders. The decreased binding to Active Site 2 could imply that this active site is not involved in neuroactive signaling and could allow the development of a functional innate repair receptor (IRR) model. Substituting the three glutamine substitution residues with arginine (RPO) resulted in the loss of behavioral activity, indicating the importance of glutamine residues at those positions.
Keywords: carbamoylated erythropoietin, mimetic, antidepressant, innate repair receptor, erythropoietin receptor, nonhematopoietic
Introduction
Depression is the largest cause of disability in the world. Its detrimental effects on cognition and mood cause both a severe economic burden ($1 trillion/year according to the World Health Organization) and a danger to human life via suicidal ideation.1 Though many pharmacological options exist for this disorder, most work by the same or similar mechanisms (modulation of monoamine levels) which are ineffective in 30% of affected individuals.2 The global “years lived with disability” due to depression has been consistently increasing (rising 14.1% from 2011 to 2017), and it has become apparent that the field needs not just a new medication, but a new class of medications.3 Erythropoietin (EPO) and its derivatives have emerged as strong candidates for the development of this new “class” of neurotrophic therapeutic drugs.
EPO has robust pro-cognitive and antidepressant effects4–6 which are believed to be mediated by an increase in the expression of neurotrophic factors in the hippocampal region of the brain, particularly brain-derived neurotrophic factor (BDNF).7,8 In order to facilitate the use of EPO for CNS indications, it is necessary to eliminate its hematopoietic effects, which prevent it from being used long-term or in high doses in non-anemic individuals. In the past, both the hematopoietic and neuroactive effects were believed to be mediated by the same receptor complex: the erythropoietin receptor homodimer (EPOR/EPOR). EPO binds two EPOR monomers at distinct active sites, the high-affinity Active Site 1 (AS1, KD = ~1–5 nm) and the low-affinity Active Site 2 (AS2, KD = ~1 µM), to conform the extracellular domains of EPOR/EPOR into a specific angular configuration that activated its downstream signaling.9,10 However, recent studies have implied that the neuroactive effects of EPO may be mediated by a heterodimer receptor comprised of an EPOR monomer and CD131 (EPOR/CD131), forming what is called the Innate Repair Receptor (IRR).11
The existence of the IRR is supported by the binding affinity measurements of EPO’s carbamoylated form (CEPO),12 in which we found that seven surface lysine residues were carbamoylated.13 CEPO loses the ability to activate the classical EPOR/EPOR homodimer conformation and has no hematopoietic activity in-vivo or in-vitro.12 However, CEPO retains EPO’s neuroactive affects, implying that not only is EPOR/EPOR not mediating these effects but also that CEPO is a selective agonist of the IRR. While this would normally make it a strong candidate for clinical testing, its expensive nonenzymatic synthesis from EPO causes significant batch-to-batch variability, limiting the ability to scale up and produce large pure quantities of the molecule. CEPO is also challenging to computationally model, due to the lack of a solved crystal structure which addresses its n-linked glycosides. To address these limitations, we decided to express a CEPO mimetic protein in E. coli utilizing information obtained from our peptide mapping of CEPO.13 We substituted the crucial carbamoylated residues with a chemically similar amino acid (glutamine) and precluded glycosylation to facilitate faster systemic removal, better computational modeling and potentially improved transport across the blood–brain barrier.
The mimetic, called QPO, was designed to specifically mimic CEPO’s inability to activate the erythropoiesis, while retaining its ability to induce neurotrophic activity.13,14 Therefore, only the surface lysine residues involved in binding the homodimer were altered. Only three of the seven carbamoylated residues of CEPO are near the active sites of the EPO-EPOR/EPOR complex (K20, K45, K97), and so these were the only ones substituted with glutamine in QPO.
Using a neuronally differentiated Pheochromocytoma-12 (PC-12) cell line, stress-sensitive BALB/c mice, and molecular dynamics simulations based on previously reported high-resolution crystal structures of the active EPO-EPOR/EPOR complex, QPO was tested for its effects on antidepressant-like behavior, cognitive function, hematopoietic activity, BDNF regulation, and EPOR/EPOR binding modality and affinity.
Methods and Materials
Animals
Adult, male, BALB/C mice (ENVIGO) were housed in 36X15X12 cm mouse cages with free access to food and water except when undergoing behavioral testing. Animal use procedures and experimental protocols were approved by the University of South Dakota Sanford School of Medicine IACUC. They also strictly adhere to NIH guidelines.
Cell Maintenance and Treatment
Pheochromocytoma 12 (PC-12) cells (ATCC CRL-1721) were grown in RPMI 1640 (ATCC #30-2001) medium containing 5% fetal bovine serum (Gibco #A31604-01) and 10% inactivated horse serum (Gibco #26,050-070). Cells were plated on 60 mm Type IV Collagen plates (Corning #62,405-644) at a density of 3*105 cells/plate in 4 mL of RPMI 1640 medium and neuronally differentiated using 100ng/mL mouse NGF 7S (Alomone Labs #N-130) and 1% inactivated horse serum. The differentiation medium was replaced every 48 h for 10 days to allow cells to obtain maximum neurite outgrowth. On day 10, the cells were changed into medium containing no NGF for 24 h prior to treatment. Cells were treated with 100ng/mL of QPO in RPMI 1640 medium and allowed to incubate at 37°C for 3 h. Cells were then scraped, lysed and processed for RNA isolation and reverse transcriptase quantitative PCR analysis (RT-qPCR).
Vector Design and Expression
A DNA fragment was synthesized to encode QPO with an N-terminal 6x-Histidine tag and 5ʹ EcoRI and 3ʹ BamHI restriction sites (Integrated DNA Technologies). The 6x-His QPO DNA fragment was cloned into the pCR2.1 cloning plasmid (Invitrogen), sequenced for accuracy (Iowa State University, Office of Biotechnology), and subsequently cloned into pMal-c2 (New England Biolabs) using EcoRI and BamHI. The resulting plasmid was transformed into T7 Express Competent E. coli (New England Biolabs). Transformants were cultured in LB + ampicillin (50 µg/mL) and induced for expression with 0.4 mM Isopropyl β-D-1-thiogalactopyranoside (IPTG), resulting in a Maltose Binding Protein (MBP)/6x-His QPO fusion protein.
Purification and Analysis
MBP/6x-His QPO expression cell pellets from 300 mL cultures were resuspended and lysed in 10 mL Amylose Column Buffer (20mM Tris pH = 7.4, 200mM NaCl, 1mM EDTA in Type 1 H2O) using an Omni Bead Ruptor Homogenizer (OMNI International) (S = 6.00, T = 0:30, C = 03, D = 0:10) with 0.1 mm glass beads. Following centrifugation (16,000 x g, 15 min, 4°C), the soluble fraction was rocked at 4°C for 1 h with 5 mL amylose resin (New England Biolabs) for batch binding. The resin was washed 3x with 40 mL Amylose Column Buffer (centrifugation 500 x g, 5 min at 4°C) and transferred to a Poly-Prep Chromatography Column (BioRad) for elution of MBP/6x-His QPO with 10 mM maltose in Amylose Column Buffer. The eluate was concentrated using Vivaspin 10 kDa MWCO Polyethersulfone concentrator columns (Millipore Sigma) with centrifugation (4000 x g, 4°C, 5 x 10 min).
After dilution to 1 mg/mL, the MBP/6x-His QPO protein was digested with Factor Xa Protease (New England Biolabs) (18 h at 25°C) to cleave MBP from 6x-His QPO. The digested sample was loaded onto a Histrap FF IMAC column (General Electric) in binding buffer (20 mMol Imidazole in PBS, pH = 7.4). The manufacturer’s instructions were followed as written, with flow rates adjusted to 0.5mL/min to minimize backpressure. The protein was eluted with 0.5M Imidazole (pH = 7.4) in PBS, and each fraction was assayed for presence of the protein and level of purity by Coomassie and Silver Staining. All fractions containing the eluted protein were dialyzed for 15 h at 4°C into phosphate-buffered saline using a 7kDa MWCO Slide-A-Lyzer dialysis cassette (Thermo Scientific #66,370). Final protein concentration and yield were determined by performing a protein 280nm absorbance assay on a Nanodrop 2000 Spectrophotometer.
Western Blot Analysis
The isolated QPO was then characterized by Western blot using a BIORAD Trans-Blot SD Semi-Dry Transfer Cell to determine its reactivity to Anti-EPO (Abcam #AB126876, 1:1000 dilution) and Anti-6X Histidine (Abcam #AB9108, 1:1000 dilution) antibodies, with comparisons to ngEPO, deglycosylated CEPO (dgCEPO), EPO, and CEPO as positive and negative controls, respectively.
ngEPO and CEPO
ngEPO was produced from recombinant human EPO (Prospec Bio) and deglycosylated by PNGaseF digestion using manufacturer’s instructions (New England Biolabs P0704S). CEPO was produced as per our CEPO characterization study.13
RNA Isolation, cDNA Synthesis, and RT-qPCR
RNA isolation was carried out using an Invitrogen RNAqueous Phenol-Free Total RNA Isolation Kit (AM-1912) following the manufacturer’s instructions.
For cDNA synthesis, a microtube containing 500 ng RNA (volume of 1–12 µL), 1 µL Oligo DT20 Primer (Life Tech #18,418,020), and RNase-free water (Ambion #AM9937) for a final volume of 13 µL was heated for 10 min at 80°C. The solution was placed on ice for 5 min, after which 4 µL 5XRT buffer, 1 µL 10 mM dNTP mix (Life Tech #18,427,088), 1 µL SuperScript III Reverse Transcriptase (Life Tech #18,080,044), and 1 µL SUPERase Inhibitor (Life Tech #AM2696) were added to each sample. The solution was mixed in a microfuge before incubating for 2 h at 42°C. The synthesis was stopped through the addition of 3.5 µL of a 0.5M NaOH/50mM EDTA solution in nuclease-free water. The resulting mix was incubated at 65°C for 10 min to denature DNA/RNA hybrids before neutralizing with 5 µL of 1M Tris-HCL (Life Tech #15,567-027). At this point, 70.5 µL 10 mM Tris/1mM EDTA (Thermo Scientific #17,890) was added to each tube before adding 3 µL of 5mg/mL acrylamide, 4 µL 5 M NaCl, and 400 µL of 100% EtOH. The resulting mixture is incubated overnight at −20°C to precipitate the cDNA.
The precipitated cDNA was then pelleted in an Eppendorf 5804 R centrifuge (10,000 RPM, 15 min, 4°C), washed with 450 µL of cold 70% EtOH, pelleted again at the same settings, and dried at 65°C. The dry cDNA was then reconstituted in 100 µL nuclease-free water.
QPCR was performed using an Eppendorf Mastercycler Realplex 2. RT-qPCR was performed using an Eppendorf Mastercycler Realplex 2 for 40X cycles of 94.0°C for 2 s, 60.0°C for 30 s, and 72.0°C for 30 s. Primers were obtained from Integrated DNA technologies. BDNF rat primers - gctgcgcccatgaaagaagc, agacctctcgaacctgccct; mouse – agctgctggatgaggaccag, gaggaggctccaaaggcact. Quantification was performed using SYBR Green chemistry (Invitrogen #11,762-500).
Hippocampal Gene Expression
Male BALB/C mice were injected with QPO (40 µg/kg i.p. daily) for 4 days and sacrificed 5 h after the last injection. The hippocampus was dissected out and rapidly frozen on dry ice for RNA extraction, cDNA synthesis and QPCR analysis using BDNF gene-specific primers.
Hematocrit Measurement
After completion of the Object Recognition Memory Test, the mice were allowed 2 days of rest followed by 6 additional treatments of QPO (40 µg/kg i.p. daily) for a total of 10 doses. QPO-treated mice were then sacrificed 48 h after the final dose via rapid decapitation, during which truncal blood was collected for hematocrit measurement in Microvette CB 300 µL K2 EDTA tubes (Sarstedt #16.444). Hematocrit was measured using a Microhematocrit EZ Reader (LM Scientific # ZCP-EZRD-HEM7).
Object Recognition Memory/Novel Object Recognition (ORM/NOR) Test
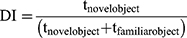
Male BALB/c mice were treated with QPO (40 µg/kilogram, i.p.) or an equivalent injection volume of PBS (~150 µL, i.p.) 1X/day for 4 days. Mice were then habituated to the 42X28X18cm testing cages under experimental conditions (no bedding, 40–50 lux) for 1 h. One day after habituation, mice were trained on two identical objects (either two 50 mL Falcon Tubes or two Lego brick towers of equivalent size) placed 6 cm from either end and 10–12 cm from either side for 30 min, at which point mice were returned to their home cages. After 24 h mice were placed in the testing cages containing their trained object and a novel object for 5 min. The test was recorded with a Basler acA 1300–60gm NIR camera and scored for the duration of time spent exploring each object in the 5-min testing period. Object exploration was determined by the length of time the mice spent with its nose pointed at and/or sniffing the object within 2 cm. Climbing behavior was not considered exploration. Data were expressed as a discrimination index determined by the amount of time spent exploring the novel and familiar objects (see Equation 1.1).
Forced Swim Test (FST)
The forced swim test (FST) was carried out in two clear cylindrical plastic tubs (20 cm diameter, 25 cm height, 16 cm water height, 26–28°C) placed in NIR chambers (180–200 lux/chamber). Male BALB/c mice were treated with QPO (40 µg/kg i.p.) or an equivalent injection volume of PBS (~150 µL, i.p.) 5 h before each 6-min testing session. Each testing session was recorded using a Basler acA 1300–60gm NIR camera and scored using automated tracking software (Ethovision XT 14, Noldus Information Technology, Leeburg, VA, USA) for behavioral mobility and immobility during the final 4 min of the test.
Open Field Test (OFT)
Mice were habituated to 40X40X50 cm black tubs in the testing room for 1 h (250–300 lux in the center of the tub, 150–250 lux at the edges) the day before testing. On the test day, mice were placed in the center of the tubs and allowed to move freely for 10 min. The trials were recorded with a Basler acA 1300–60gm NIR camera and were scored for total distance moved (in cm) using automated tracking software (Ethovision XT 14, Noldus Information Technology, Leeburg, VA, USA).
Novelty Induced Hypophagia (NIH) Test
The Novelty Induced Hypophagia Test was performed as previously described by Sampath, McWhirt, Sathyanesan, and Newton.15 Over the next 2 days, testing proceeded in their home cage (45–50 lux), then in a novel cage (550–600 lux, no bedding). Testing periods were 30 min long and recorded using a Basler acA Color Camera. The duration of time between introduction of the sweetened condensed milk to the first drink was recorded as “latency to drink”.
QPO Homology Model Design
The homology model of the triple-substitution mutant QPO (K-20,45,97-Q) used for molecular dynamics simulations was based on the solved crystal structure of non-glycosylated EPO (ngEPO) bound to EPOR/EPOR (Protein Data Bank: 1EER, Chain A, Resolution 1.9 Å) using Molecular Operating Environment 2018 (MOE).10 All protein models were protonated within MOE (pH = 7.4, T = 310K) and refined to an RMS gradient of 0.1 kCal/mol/Å before performing energy minimization to reduce torsional stress.
Molecular Dynamics Simulations
Molecular dynamics simulations were performed on the University of South Dakota’s “Lawrence” Supercomputing Cluster using Molecular Operating Environment v2018.1 (MOE) in tandem with Nanoscale Molecular Dynamics (NAMD v2.13) for Linux Multicore software.16,17 QPO and ngEPO were placed into the binding pocket of the active conformation of EPOR/EPOR in the same position and orientation by superposition onto Chain 1 of the 1EER pdb file. After placement, the complex was energy minimized to a 0.1 kCal/mol/Å threshold. All simulations were performed in explicit solvent (3D-RISM model, density: ~1 g/mL) with the following parameters: time step: 0.002 ps, heat: 3 ns to T=310K, equilibration time: 6 ns, production time: 26 ns, sample rate: 5.0 ps, Langevin damping: 5/dT, pressure: 101.325 bar. Simulations were run until equilibrium was achieved (between 24 and 34 ns), at which point they were compiled and analyzed using the MOE Molecular Dynamics Analysis suite.
Free Energy of Binding (∆GB) Estimation
Post-convergence, representative “snapshots” of each simulation were taken to perform the necessary calculations for the estimation of the ∆GB for each active site of the bound ngEPO/QPO-EPOR/EPOR complex (N = 4 measurements/trajectory, 4 trajectories/variant, at minimum 1 ns separation between each measurement). ∆GB and the associated dissociation constants (KD) were calculated as previously reported.18
Statistical Analysis
All data were presented as the mean ± standard error of the mean. The error and statistics were calculated for the raw data (∆ct for gene expression data, and raw hematocrit for Figure 1), but were transformed mathematically into a more easily interpreted form (fold-change in expression calculated from ∆∆ct and hematocrit % change compared to vehicle-treated controls). Any and all outliers were removed using Grubb’s tests, and statistical significance for behavioral data and computational tests were determined using the Student’s T-test. A p-value of <0.05 was considered statistically significant. Statistical tests were performed using Systat and graphs prepared with Microsoft Excel.
 |
Figure 1 Behavioral Assays and Hematocrit. (A) The treatment and testing schedule for the FST and OFT is shown. (B) The cumulative immobile duration over the scored portion of the FST is shown for both QPO-treated and vehicle-treated mice. QPO treatment resulted in a significantly decreased immobile duration compared to vehicle-treated controls (p < 0.05, N = 6). (C) The cumulative distance moved in the OFT is shown for QPO-treated and vehicle-treated mice. QPO showed no significant difference in total distance moved during the OFT compared to vehicle-treated mice (p > 0.05, N = 6). (D) The treatment and testing schedule for the ORMT is shown. (E) The preference of QPO-treated vs vehicle-treated mice for a novel object instead of a trained object is shown here as a discrimination index. QPO showed a significantly increased preference for the novel object compared to vehicle-treated controls (p < 0.05, N = 6 for treated, 8 for vehicle control). (F) The comparison of total hematocrit (as a percentage) between QPO-treated mice (10 doses, 40µg/kg i.p., over 14 days) and vehicle-treated mice is shown. QPO treatment caused neither a significant increase nor decrease in hematocrit. (G) Treatment and testing schedule for mice undergoing Novelty-Induced Hypophagia Testing. (H) NIHT results for QPO-treated mice are presented in comparison to vehicle-treated controls in terms of the duration of time between introduction of sweetened condensed milk and the first drink in seconds. Statistical significance is denoted by *p < 0.05. Abbreviations: Idv.Cages, day that mice were separated into individual housing; HC, home cage; NC, novel cage. |
Results
For orientation purposes, Figure 2 displays the ribbon structure of the bound state of non-glycosylated erythropoietin (ngEPO, in green) to EPOR/EPOR (gray molecular surface representation) is shown from two views, front and top (Figure 2A and B), with AS1 and AS2 colored salmon and blue, respectively. The three surface lysine residues at position 20, 45 and 97, that were substituted to glutamine in QPO, are indicated by red spheres and their distance from EPOR residues are shown (Figure 2C and D).
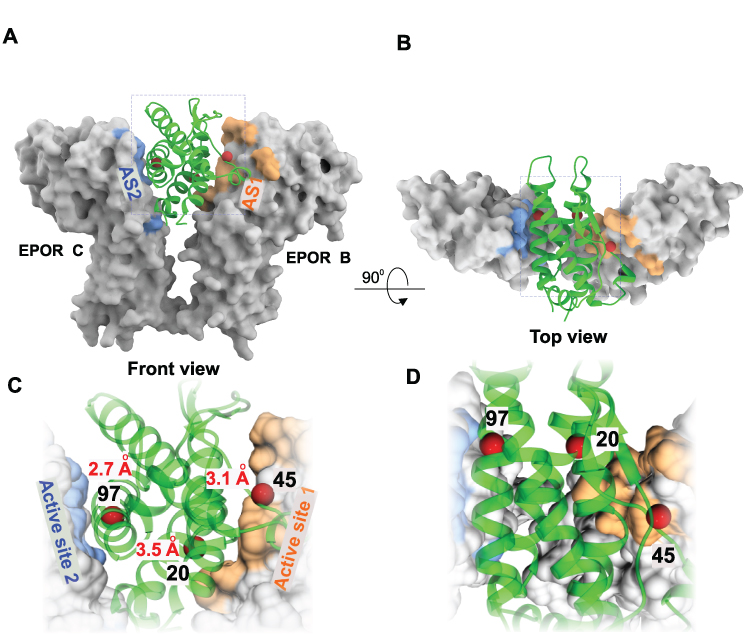 |
Figure 2 Design of QPO from EPO-EPOR complex. (A) EPO (green ribbon) is shown bound to EPOR (molecular surface representation) – PDB ID-1EER. The high-affinity active site 1(AS1) is colored salmon and the low-affinity active site 2 (AS2) is colored blue. (B) The front view in A is rotated 90̊ toward the viewer. (C) Magnified view of boxed region in A is shown. The 3 amino acid residues that were chosen for substitution mutagenesis are indicated by red spheres and the corresponding residue number in the sequence is shown. The distance between the residue atoms and the nearest receptor atom in the active sites are indicated in red. (D) Magnified view of the boxed region in the top view is shown. |
Purification and Characterization
The purification procedure for QPO was verified by silver staining. The progress in protein isolated from each purification step (see Materials and Methods) is shown left to right (Figure 3A), ending with the final QPO-6XHis product in 1X PBS (last lane). The isolated QPO was then characterized by Western blot to determine its reactivity to Anti-EPO (Figure 3B) and Anti-6X Histidine (Figure 3C) antibodies, with comparisons to ngEPO, deglycosylated CEPO (dgCEPO), EPO, and CEPO.
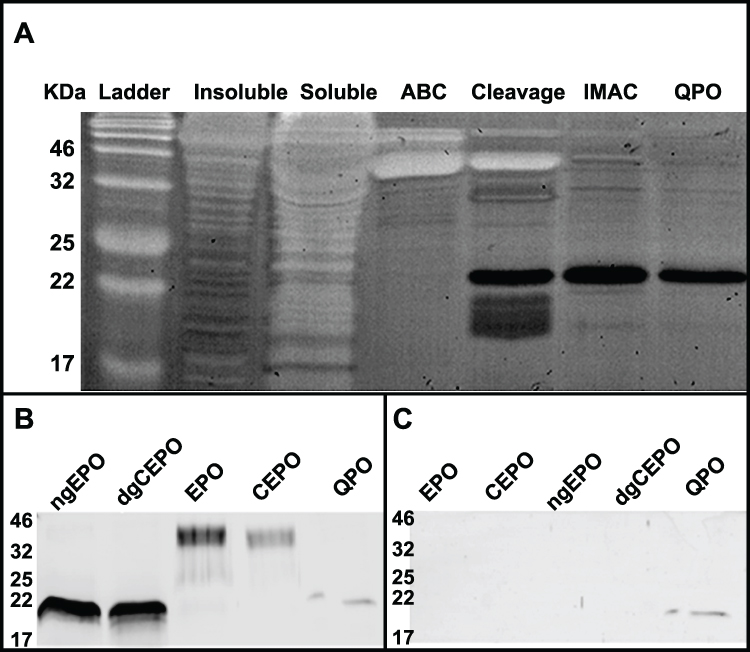 |
Figure 3 Purification Silver Stain and Western Blot Analyses. (A) A silver stain depicting each step of the purification procedure is shown. From left to right with the well # in parentheses: (1) broadband protein ladder (2) the insoluble fraction of the whole cell lysate (3) the soluble fraction of the whole cell lysate (4) the eluate of the amylose binding column, containing a mixture of MBP-QPO and MBP (5) cleavage of MBP-QPO using factor Xa, resulting in the disappearance of the MBP-QPO band, and appearance of the QPO band at approximately 22 kDa (6) eluate from immobilized metal affinity chromatography column, containing the histidine-tagged QPO polypeptide with imidazole (7) dialysis of QPO polypeptide into 1X phosphate-buffered saline (B) A Western blot in which QPO’s reactivity with an anti-EPO antibody was compared with ngEPO, dgCEPO, EPO, and CEPO is shown. (C) A Western blot in which QPO’s reactivity with an anti-6X histidine antibody was compared with ngEPO, dgCEPO, EPO, and CEPO is shown. |
Behavioral Assays and Hematopoiesis
First, to screen for potential antidepressant-like activity, BALB/c mice treated with QPO (40 µg/kg, i.p.) were administered the Forced swim test. The treatment and testing schedule, and the resulting changes to immobility duration, are shown (Figure 1A and B). QPO-treated mice showed a significantly decreased immobility duration compared to vehicle-treated controls (p < 0.05, N = 6). To examine generalized locomotion and emotional behavior, the same cohort of mice were administered the Open filed test (Figure 1C). There was no significant difference in total distance moved over the testing period, implying that the FST results reflect specific antidepressant-like effects (p > 0.05, N = 6). To test the effects of QPO on cognitive function, the Object recognition memory test/Novel object recognition (ORMT/NOR) was performed. The training and testing schedule, as well as the results, are presented (Figure 1D and E). QPO-treated mice showed significantly increased preference for the novel object compared to vehicle-treated controls (p < 0.05, N = 6 for QPO, 8 for vehicle). After 10 doses of the drug over the course of 2 weeks, hematocrit was measured from truncal blood to determine QPO’s in-vivo hematopoietic activity. There was no significant difference in hematocrit (Figure 1F) between QPO treated and vehicle-treated mice (p > 0.05, N = 6). To further confirm the antidepressant-like effects of QPO, we employed the novelty-induced hypophagia test. Unlike FST, which is influenced by acute antidepressant drug administration, the NIH test requires chronic drug administration. The treatment and testing schedule, and the results are shown in Figure 1G and H, respectively. In the home cage, there was no significant difference between the latency-to-drink of QPO-treated and vehicle-treated mice (p > 0.05, N = 6). However, in the novel cage, QPO-treated mice showed a significantly decreased latency-to-drink compared to vehicle-treated controls, indicating antidepressant-like activity (p < 0.05, N = 6).
In contrast to the significant improvements in behavioral performance seen in QPO-treated mice, the arginine substitution recombinant (RPO) produced no change and behavioral response was comparable to vehicle administered controls in FST, NIH and OFT (Figure 4A–C). We therefore did not test RPO further.
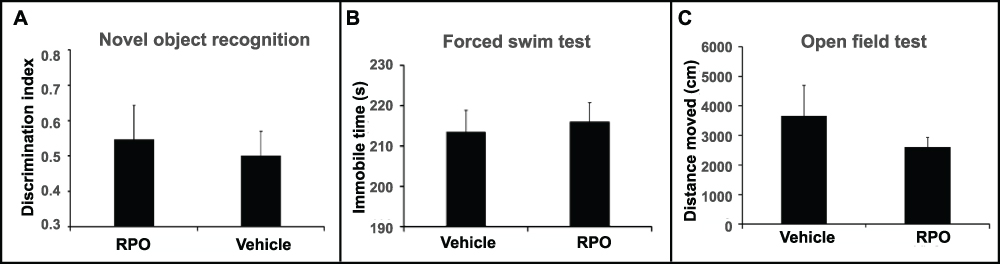 |
Figure 4 Behavioral analysis of arginine substitution recombinant. (A) Discrimination index of RPO and vehicle in the novel object recognition test. (B) Immobility time in the forced swim test. (C) Distance moved in the open field test in vehicle and RPO treated mice. RPO was dosed at 40µg/kg/day i.p for 4 days (N = 6). |
Binding Affinity Assay
The in-silico binding affinity (ΔGB) of QPO and ngEPO to the EPOR/EPOR homodimer was calculated for each active site. The AS1 values for both are shown in Figure 5A, while the AS2 values are shown in Figure 5B. QPO showed significantly decreased ΔGB at AS2 compared to ngEPO, but not at AS1 (p < 0.05, N = 16). These ΔGB were used to calculate the AS1 and AS2 dissociation constants for QPO and ngEPO and compared them to known experimental constants for glycosylated EPO (Figure 5C). QPO and ngEPO both show AS1 binding affinities that are similar to that of EPO, while ngEPO shows increased binding affinity to AS2 compared to EPO.
 |
Figure 5 Binding Affinity Assessment and Dissociation Constant Calculation. This figure presents the average calculated free energies of binding (ΔGB) of ngEPO and QPO to (A) Active Site 1 of EPOR/EPOR and (B) Active Site 2 of EPOR/EPOR. Statistical significance is denoted by *p < 0.05 by Student’s T-test. (C) This table provides calculated dissociation constants (nM) for ngEPO and QPO. Experimentally determined EPO dissociation constants taken from Goldwasser/Wilson and Jolliffe.9 (D) The fold change in expression levels of BDNF mRNA in neuronally differentiated PC-12 cells are shown in comparison to vehicle-treated controls. BDNF shows a significant upregulation in expression after treatment with QPO (p < 0.01, N = 5 vehicle/6 treated). (E) The fold change in the expression of BDNF in the hippocampus of BALB/c mice. BDNF shows a statistically significant 50% increase in expression compared to vehicle-treated controls (p < 0.05, N = 5). |
BDNF Gene Expression
QPO treatment of neuronally differentiated PC-12 cells (100 ng/mL, 3 h incubation) caused an approximate 50% increase in BDNF expression (Figure 5D) when compared to vehicle-treated controls (p < 0.01, N = 6). A comparable increase in BDNF occurs in-vivo in the mouse hippocampus (Figure 5E) after QPO treatment (40µg/kg/day i.p.) (p < 0.05, N = 6). All gene expression results are presented as fold change in expression compared to vehicle-treated controls.
Discussion
To determine if QPO is a successful mimetic of CEPO, it is important to assess its effects on cognition, mood, and neurotrophic gene regulation. First, in terms of cognitive effects the object recognition memory has been previously shown to be highly specific to the perirhinal and hippocampal regions of the brain, where improved performance (increased preference for the novel object) has been associated with increased cognitive function. QPO treatment at doses equivalent to those used for CEPO administration results in a significantly increased preference for the novel object. This effect of QPO in the novel object recognition assay is comparable to the results of our previous CEPO experiments in rats.13
Second, the results of the FST and NIHT (reduced immobility duration and decreased latency-to-drink in the novel cage, respectively) imply that QPO has functional antidepressant-like activity. Like CEPO,13,14 treatment with QPO caused significant upregulation in BDNF expression in-vitro and in-vivo, which has previously been demonstrated as one of the more promising neurotrophic factors associated with the symptomatic improvement of depressive behavior in animal models.7 Thirdly, QPO is completely nonhematopoietic in-vivo while retaining neuroactivity, which when coupled with the behavioral data, gene expression data, and the substituted residues’ with similar physiochemical properties to homocitrulline, it can be stated that QPO is a successful mimetic of CEPO and appears to mediate its effects through the same neurotrophic mechanism. Non-erythropoietic neuroprotective peptides designed using EPO crystal structure information have been shown to produce cognitive effects when administered at high doses (5 mg/kg) despite half-life of only 2 min.19,20 However, unlike EPO, antidepressant effects were not observed in clinical testing.21 Additional work is necessary to understand the differences in receptor binding and behavioral effects produced by short peptides and full-length proteins.
Tsuda et al showed that as the glycosylation level of EPO decreases, the affinity of EPO for the EPOR/EPOR homodimer receptor increases.22 Surprisingly, molecular dynamics simulations of ngEPO revealed that complete removal of EPO’s glycan groups results not in an overall receptor affinity increase, but rather in a selective increase in AS2 affinity compared to the glycosylated form’s experimental values. This may mean that the glycosylation sites of EPO serve to sterically hinder binding to AS2 in a selective manner. When comparing the binding affinity of QPO vs ngEPO to the EPOR dimer, the data suggest that the substitution of lysine residues at positions 20, 45 and 97 to glutamine causes a significant decrease in binding only at the lower affinity AS2, with AS1 affinity being largely unaffected. This likely suggests that the K20 and K45 residues of EPO do not play a major role in receptor binding at AS1, but that K97 has a significant impact on binding affinity to AS2. Substitution mutagenesis studies followed by biochemical analysis of in vitro biological activity have reported that alanine substitution at K20 and K45 is without impact, but at K97 (AS2) there is substantial loss of activity, indicating the importance of this position in AS2.10 As QPO mimics CEPOs behavioral effects it is useful to consider the role of carbamoylated K97 in CEPO’s interactions with AS2, keeping in mind that CEPO is glycosylated unlike QPO which has no sugars. It is conceivable that there is a further decrease in AS2 affinity, which could provide a clue to the structure of the theoretical IRR.
Although there is robust data associating IRR activity with EPO’s tissue protective effects,23 there is currently insufficient evidence establishing that the IRR mediates EPO’s behavioral effects in the CNS.24,25 Our understanding that QPO, and by extension CEPO, binds AS1 as strongly as EPO indicates that at least one half of the IRR complex involves EPO bound to AS1 of an EPOR monomer. In addition, it also implies that the residues of EPO associated with AS2 might not play a significant role. Whether the residues facing AS2 are involved in binding to CD131/betacommon receptor is yet to be shown. However, it is known that a dimer configuration is necessary for activation of signaling.26 Future behavioral studies with brain region-specific deletion of the betacommon receptor can provide insight into the importance of this receptor for the CNS effects of non-erythropoietic but neurotrophic variants of EPO. The ability to safely employ well-understood neurotrophic mechanisms to produce antidepressant and cognitive enhancing effects in the clinic has the potential to add a much-needed novel approach to the treatment of neuropsychiatric disorders.
Conclusion
A triple glutamine substitution, carbohydrate-free, recombinant molecule, QPO, designed using EPO and CEPO as the template was produced in a bacterial expression system. QPO upregulated the expression of BDNF, a neurotrophic factor known for its antidepressant properties. In behavioral studies, treatment with QPO showed beneficial cognitive and antidepressant-like effects in the ORMT, FST, and NIHT. The computational binding assays for ngEPO and QPO provide evidence that AS2 binding is unnecessary for the activation of neurotrophic signaling by EPO through the IRR. QPO showed actions similar to CEPO, implying that the production of a polypeptide mimetic of CEPO was successful, and therefore has translational potential as a neurotrophic drug molecule.
Acknowledgments
This work was supported by US Public Health Service grants MH106640 (SSN), the University of South Dakota Center for Brain and Behavioral Research, South Dakota Board of Regents and the use of instrumentation and facilities at the Sioux Falls VA Healthcare system. The funding agencies had no role in the design of the study and collection, analysis, and interpretation of data and in writing the manuscript.
Disclosure
Nicholas Pekas (NP), Dr. Jason L Peterson (JLP), Monica Sathyanesan (MS), and Samuel S Newton (SSN) all report no current financial relationships with commercial interests. JLP, MS and SSN are co-inventors on a provisional patent application for the invention of the triple substitution recombinant. The authors report no other potential conflicts of interest for this work.
References
1. Organization WH. Mental Health in the Workplace. May, 2019.
2. Al-Harbi KS. Treatment-resistant depression: therapeutic trends, challenges, and future directions. Patient Prefer Adherence. 2012;6:369–388. doi:10.2147/PPA.S29716
3. James SL, Abate D, Abate KH, et al. Global, regional, and national incidence, prevalence, and years lived with disability for 354 diseases and injuries for 195 countries and territories, 1990–2017: a systematic analysis for the Global Burden of Disease Study 2017. Lancet. 2018;392(10159):1789–1858.
4. Miskowiak KW, Vinberg M, Harmer CJ, Ehrenreich H, Kessing LV. Erythropoietin: a candidate treatment for mood symptoms and memory dysfunction in depression. Psychopharmacology. 2012;219(3):687–698. doi:10.1007/s00213-011-2511-1
5. Miskowiak KW, Vinberg M, Christensen EM, et al. Recombinant human erythropoietin for treating treatment-resistant depression: a double-blind, randomized, placebo-controlled Phase 2 trial. Neuropsychopharmacology. 2014;39(6):1399–1408. doi:10.1038/npp.2013.335
6. Miskowiak KW, Vinberg M, Macoveanu J, et al. Effects of erythropoietin on hippocampal volume and memory in mood disorders. Biol Psychiatry. 2015;78(4):270–277. doi:10.1016/j.biopsych.2014.12.013
7. Shirayama Y, Chen AC-H, Nakagawa S, Russell DS, Duman RS. Brain-derived neurotrophic factor produces antidepressant effects in behavioral models of depression. J Neurosci. 2002;22(8):3251–3261. doi:10.1523/JNEUROSCI.22-08-03251.2002
8. Girgenti MJ, Hunsberger J, Duman CH, Sathyanesan M, Terwilliger R, Newton SS. Erythropoietin induction by electroconvulsive seizure, gene regulation, and antidepressant-like behavioral effects. Biol Psychiatry. 2009;66(3):267–274. doi:10.1016/j.biopsych.2008.12.005
9. Goldwasser E. Erythropoietin and its mode of action. Blood Cells. 1984;10(2–3):147–162.
10. Syed RS, Reid SW, Li C, et al. Efficiency of signalling through cytokine receptors depends critically on receptor orientation. Nature. 1998;395(6701):511–516. doi:10.1038/26773
11. Punnonen J, Miller JL, Collier TJ, Spencer JR. Agonists of the tissue-protective erythropoietin receptor in the treatment of parkinson’s disease. Curr Top Med Chem. 2015;15(10):955–969. doi:10.2174/156802661510150328224527
12. Leist M, Ghezzi P, Grasso G, et al. Derivatives of erythropoietin that are tissue protective but not erythropoietic. Science. 2004;305(5681):239–242.
13. Sathyanesan M, Watt MJ, Haiar JM, et al. Carbamoylated erythropoietin modulates cognitive outcomes of social defeat and differentially regulates gene expression in the dorsal and ventral hippocampus. Transl Psychiatry. 2018;8(1):113. doi:10.1038/s41398-018-0168-9
14. Tiwari NK, Sathyanesan M, Schweinle W, Newton SS. Carbamoylated erythropoietin induces a neurotrophic gene profile in neuronal cells. Prog Neuropsychopharmacol Biol Psychiatry. 2019;88:132–141. doi:10.1016/j.pnpbp.2018.07.011
15. Sampath D, McWhirt J, Sathyanesan M, Newton SS. Carbamoylated erythropoietin produces antidepressant-like effects in male and female mice. Prog Neuropsychopharmacol Biol Psychiatry. 2020;96:109754. doi:10.1016/j.pnpbp.2019.109754
16. Phillips JC, Wang W, Gumbart J, et al. Scalable molecular dynamics with NAMD. J Comput Chem. 2005;26:1781–1802. doi:10.1002/jcc.20289
17. Molecular Operating Environment (MOE) 2013;08. Chemical Computing Group ULC SSWS. Montreal, QC, Canada; 2018:H3A 2R7.
18. Pekas NJ, Newton SS. Computational analysis of ligand-receptor interactions in wild-type and mutant erythropoietin complexes. Adv Appl Bioinform Chem. 2018;11:1–8. doi:10.2147/AABC.S177206
19. Dmytriyeva O, Belmeguenai A, Bezin L, et al. Short erythropoietin-derived peptide enhances memory, improves long-term potentiation, and counteracts amyloid beta-induced pathology. Neurobiol Aging. 2019;81:88–101. doi:10.1016/j.neurobiolaging.2019.05.003
20. Brines M, Patel NS, Villa P, et al. Nonerythropoietic, tissue-protective peptides derived from the tertiary structure of erythropoietin. Proc Natl Acad Sci U S A. 2008;105(31):10925–10930. doi:10.1073/pnas.0805594105
21. Cerit H, Veer IM, Dahan A, et al. Testing the antidepressant properties of the peptide ARA290 in a human neuropsychological model of drug action. Eur Neuropsychopharmacol. 2015;25(12):2289–2299. doi:10.1016/j.euroneuro.2015.09.005
22. Tsuda E, Kawanishi G, Ueda M, Masuda S, Sasaki R. The role of carbohydrate in recombinant human erythropoietin. Eur J Biochem. 1990;188(2):405–411. doi:10.1111/j.1432-1033.1990.tb15417.x
23. Dahan A, Brines M, Niesters M, Cerami A, van Velzen M. Targeting the innate repair receptor to treat neuropathy. Pain Rep. 2016;1(1):e566. doi:10.1097/PR9.0000000000000566
24. Cheung Tung Shing KS, Broughton SE, Nero TL, et al. EPO does not promote interaction between the erythropoietin and beta-common receptors. Sci Rep. 2018;8(1):12457. doi:10.1038/s41598-018-29865-x
25. Nadam J, Navarro F, Sanchez P, et al. Neuroprotective effects of erythropoietin in the rat hippocampus after pilocarpine-induced status epilepticus. Neurobiol Dis. 2007;25(2):412–426. doi:10.1016/j.nbd.2006.10.009
26. Zhang YL, Radhakrishnan ML, Lu X, Gross AW, Tidor B, Lodish HF. Symmetric signaling by an asymmetric 1 erythropoietin: 2 erythropoietin receptor complex. Mol Cell. 2009;33(2):266–274. doi:10.1016/j.molcel.2008.11.026
 © 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.