")
Back to Journals » International Journal of Nanomedicine » Volume 10 » Issue 1
Design and activity of a cyclic mini-β-defensin analog: a novel antimicrobial tool
Authors Scudiero O , Nigro E, Cantisani M, Colavita I, Leone M, Mercurio F, Galdiero M, Pessi A, Daniele A, Salvatore F, Galdiero S
Received 30 May 2015
Accepted for publication 23 July 2015
Published 15 October 2015 Volume 2015:10(1) Pages 6523—6539
DOI https://doi.org/10.2147/IJN.S89610
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 4
Editor who approved publication: Dr Thomas Webster
Olga Scudiero,1,2 Ersilia Nigro,1 Marco Cantisani,3 Irene Colavita,1 Marilisa Leone,4 Flavia Anna Mercurio,4 Massimiliano Galdiero,5 Antonello Pessi,1 Aurora Daniele,1,6 Francesco Salvatore,1,2,7 Stefania Galdiero3,4
1CEINGE-Biotecnologie Avanzate Scarl, Naples, Italy; 2Dipartimento di Medicina Molecolare e Biotecnologie Mediche, 3Dipartimento di Farmacia, Università di Napoli Federico II, Naples, Italy; 4Istituto di Biostrutture e Bioimmagini, CNR, Naples, Italy; 5Dipartimento di Medicina Sperimentale, Seconda Università di Napoli, Naples, Italy; 6Dipartimento di Scienze e Tecnologie Ambientali Biologiche Farmaceutiche, Seconda Università di Napoli, Caserta, Italy; 7IRCCS Fondazione SDN, Naples, Italy
Abstract: We have designed a cyclic 17-amino acid β-defensin analog featuring a single disulfide bond. This analog, designated “AMC” (ie, antimicrobial cyclic peptide), combines the internal hydrophobic domain of hBD1 and the C-terminal charged region of hBD3. The novel peptide was synthesized and characterized by nuclear magnetic resonance spectroscopy. The antimicrobial activities against gram-positive and gram-negative bacteria as well as against herpes simplex virus type 1 were analyzed. The cytotoxicity and serum stability were assessed. Nuclear magnetic resonance of AMC in aqueous solution suggests that the structure of the hBD1 region, although not identical, is preserved. Like the parent defensins, AMC is not cytotoxic for CaCo-2 cells. Interestingly, AMC retains the antibacterial activity of the parent hBD1 and hBD3 against Pseudomonas aeruginosa, Enterococcus faecalis, and Escherichia coli, and exerts dose-dependent activity against herpes simplex virus type 1. Moreover, while the antibacterial and antiviral activities of the oxidized and reduced forms of the parent defensins are similar, those of AMC are significantly different, and oxidized AMC is also considerably more stable in human serum. Taken together, our data also suggest that this novel peptide may be added to the arsenal of tools available to combat antibiotic-resistant infectious diseases, particularly because of its potential for encapsulation in a nanomedicine vector.
Keywords: antimicrobial activity, cyclic mini-peptide, human beta-defensin
Introduction
Antimicrobial peptides (AMPs) play a key role in many biological and physiological functions of living organisms and are an attractive starting point for the development of novel drugs.1,2 These peptides of the innate immune defense system are attracting widespread attention given the growing resistance to antibiotics.3 Among the most promising AMPs is the human β-defensins (hBDs) family that displays potent antimicrobial activity against gram-positive and gram-negative bacteria, fungi, viruses, and parasites.4 hBDs exert their effect by contacting and permeabilizing the cell membrane structure of microorganisms, and inhibiting DNA synthesis and deranging metabolism turnover.5 Consequently, defensins are an excellent springboard for the development of new molecules able to fight infectious diseases, even though they are inhibited in the presence of high concentrations of salt.5,6
In this scenario, we previously designed a number of β-defensin analogs that have increased potency and/or reduced sensitivity to high ionic strength, and investigated their antibacterial, antiviral, and chemotactic activities, as well as their salt resistance.7,8 The results showed that the charged C-terminal domain (RRKK) of hBD3 and the internal domain of hBD1 (PIFTKIQGT) are crucial for defensin activity and that deletion of six residues at the N-terminus of hBD3 did not reduce the activity.7,8 Consequently, the internal region of hBD1 and the C-terminus of hBD3 are attractive domains in terms of engineering functional truncated defensin analogs. Notwithstanding these promising features, natural defensins have several properties that detract from their suitability as drug candidates, such as the fact that they are rapidly cleaved in human fluids by proteases.9 Another factor limiting the potential therapeutic use of defensins is the need for a single molecule with the correct pattern of disulfide bonds which is extremely complicated and expensive, while linear analogs with no disulfide bridges often occur in an ensemble of conformations that reduce their specificity for biological targets.10,11
In an attempt to overcome the above drawbacks, structural restraints can be introduced to reduce the number of conformations, thus resulting in a higher affinity for the target and improved protease stability, bioavailability, and specificity. In particular, cyclization is frequently used by the pharmaceutical industry to reduce the flexibility of peptides and lock them into well-defined secondary structures.1,3 Moreover, cyclic peptides typically have a much higher stability and enhanced biopharmaceutical properties compared to their linear counterparts. Cyclization through disulfide bonds occurs naturally in AMPs such as hBDs that, despite the drawbacks mentioned above, display highly potent and broad antimicrobial activity.
We were inspired by the properties of θ-defensins, which are the only known ribosomally synthesized cyclic peptides in mammals and are hence protease-resistant and stable, and represent ideal scaffolds for peptide drug design. They present a cyclic cystine ladder motif, comprising a cyclic peptide backbone. The ease of synthesis, the stability, and the great diversity of cyclic peptides that can be produced make them suitable for a wide range of applications as drug scaffolds for bioactive peptides.
We have designed and synthesized an antimicrobial cyclic peptide (AMC) carrying the crucial active regions of hBD1 and hBD3. This new analog is not cytotoxic, it exerts effective antibacterial and antiviral activity, and is stable in blood. Given these features, this molecule may be considered a novel weapon in the fight against infectious diseases that has the potential of being encapsulated as a nanomedicine.
Materials and methods
General procedures
Fmoc-protected amino acids, coupling reagents, and Rink amide p-methylbenzhydrylamine resin were purchased from Calbiochem-Novabiochem (San Diego, CA, USA). All other chemicals were purchased from Sigma-Aldrich Co. (St Louis, MO, USA). Analytical and semi-prep reverse-phase high-performance liquid chromatography (HPLC) was performed on a Shimadzu LC8 pump setup using Phenomenex (Phenomenex, Aschaffenburg, Germany) C18 column (22×250 mm, 5 μm; or 4.6×250 mm, 5 μm). Serum stability was assessed by Liquid chromatography–Mass spectrometry (LC/MS) (Gilson, Middleton, WI, USA) interfaced with a Flexar SQ 300 mass spectrometer with electrospray ionization (PerkinElmer Inc., Waltham, MA, USA) on a Jupiter C18 analytical column (5 μm particle size, 300 Å pore size, 4.6×250 mm) (Phenomenex). Electron spray ionization LC/MS (electrospray ionization [ESI] LC/MS) was performed using a Thermo Electron MSQ Surveyor. Compounds were characterized by nuclear magnetic resonance (NMR) on a VarianUNITY INOVA 600 spectrometer, equipped with a cold-probe and an INOVA 400 spectrometer provided with Z-axis pulsed-field gradients and a triple resonance probe.
Peptide synthesis
The peptide was synthesized using the standard solid-phase 9-fluorenylmethoxycarbonyl (Fmoc) method as previously reported.12 The Wang resin (Sigma-Aldrich Co.) (substitution 1.0 mmol/g) was used as solid-phase support, and syntheses were performed on a scale of 100 μmol. The peptide was fully deprotected and cleaved from the resin with trifluoroacetic acid (TFA) with 5% thioanisole, 3% ethanedithiol, and 2% anisole as scavengers. The crude peptide was precipitated with ice-cold ethyl ether, filtered, dissolved in water, lyophilized, and reduced with dithiothreitol. It was purified to homogeneity by preparative reverse-phase high-pressure liquid chromatography (HPLC). The sample was injected on a Phenomenex C18 column (22 mm ×25 cm, 5 μm) eluted by: 1) H2O/0.1% TFA and 2) CH3CN/0.1% TFA solvent mixture. A linear gradient from 5% to 50% of CH3CN/0.1% TFA solvent mixture over 17 minutes at a flow rate of 20 mL/min was used. The collected fractions were lyophilized to dryness and analyzed by analytical reverse-phase HPLC using a Phenomenex C18 analytical column (4.6×250 mm, 5 mm). The identity of the purified peptide was confirmed by electron spray ionization LC/MS using a Thermo Electron MSQ Surveyor. The purified peptide was oxidized at a maximum concentration of 1 mg/mL in order to favor intramolecular interactions and avoid the formation of dimeric species; then, the oxidized peptide was further purified. Briefly, peptide oxidation was performed by dissolving 1 mg/mL of the crude peptide in a 0.1 M solution of ammonium bicarbonate at pH 8.2, leaving the mixture in contact with air for 24 hours. Reactions were monitored by LC/MS analysis. The oxidized AMC analog was purified by preparative reverse-phase HPLC (Shimadzu SPD-10A VP UV-Vis detector) using a gradient of acetonitrile (0.1% TFA) in water from 5% to 70% for 20 minutes and a Jupiter column (10 μm, Proteo 90 Å, 250×21 mm). The purity of the cyclic defensin analog AMC is over 99% as checked by LC/MS.
NMR
NMR spectra were acquired at 298 K on a VarianUNITY INOVA 600 spectrometer, equipped with a cold-probe and an INOVA 400 spectrometer provided with Z-axis pulsed-field gradients and a triple resonance probe. NMR samples consisted of the AMC peptide (1 mg) dissolved in either 600 μL of H2O/deuterium oxide (D2O) (99.8% d; Armar Scientific, Döttingen, Switzerland) (90/10 v/v) or a mixture of H2O/2-2-2 trifluoroethanol-d3 (TFE) (98% d; Armar Chemicals, Döttingen, Switzerland) (20/80 v/v). Trimethylsilyl-3-propionic acid sodium salt-d4 (99% d; Armar Scientific) was used as internal standard for chemical shift referencing. Water suppression was achieved with the double pulsed field gradient selective echo sequence.13 1D [1H] proton experiments were recorded with a relaxation delay of d1=1.5 seconds and 64–128 scans. The process of proton resonance assignments (Tables S1 and S2) was achieved with a standard method14 based on combined analysis of 2D [1H, 1H] total correlation spectroscopy (TOCSY)15 (70 ms mixing time) and Nuclear Overhauser enhancement spectroscopy (NOESY)16 (200 and 300 ms mixing times) experiments. The 2D [1H, 1H] double-quantum-filter correlation spectroscopy17 spectrum also served to confirm proton side-chain assignments. The NMR data were processed with the VNMRJ software (Agilent Technologies, Santa Clara, CA, USA). Two-dimensional NMR spectra were analyzed with NEASY,18 as implemented in CARA (http://www.nmr.ch/). Temperature coefficients (Δδ/ΔT) (Tables S3 and S4) were evaluated by 2D [1H, 1H] TOCSY experiments that were recorded for peptide samples at the following temperatures: 298, 301, 304, 307, and 310 K. The software CYANA (version 2.1)19 was used for structure calculations (Tables S5 and S6); distance constraints were generated from 2D NOESY 300 spectra.
Cell culture and cytotoxicity studies: 3-[4.5-dimethylthiazol-2-yl]-2.5-diphenyltetrazolium bromide test
Parental CaCo-2 human cells stored in liquid nitrogen were thawed by gentle agitation of their vials for 2 minutes in a water bath at 37°C. After thawing, the content of each vial was transferred to a 75 cm2 surface area tissue culture flask and diluted with 90% Dulbecco’s Modified Eagle’s Minimal Essential Medium (Sigma-Aldrich Co.) supplemented with 10% fetal bovine serum (Lonza, Basel, Switzerland) and 1% L-glutamine (Sigma-Aldrich Co.). Then the cells were incubated for 24 hours at 37°C in 5% CO2 to allow them to grow and form a monolayer in the flask. Cells grown to 80%–95% confluency were washed with phosphate-buffered saline (PBS), trypsinized with 3 mL of trypsin-EDTA solution (1X) (Sigma-Aldrich Co.), diluted, counted, and seeded (4×103 cells/200 μL per well) into 96-well microtiter tissue culture plates for 24 hours in triplicate. The reduction in the proliferation of the cells was evaluated by the 3-[4.5-dimethylthiazol-2-yl]-2.5-diphenyltetrazolium bromide (MTT) (Sigma-Aldrich Co.) assay which enables the measurement of metabolic changes. On the next day, cells were incubated at 37°C with or without wild-type hBD1 and hBD3 and the AMC analog according to different schemes: a) 2.5, 12.5, and 25 μM for 4, 8, 12, 16, 20, 24, 48, 72, and 168 hours; and b) 125 μM for 8, 24, and 72 hours. Adherent cells were stained with MTT dye solution, ie, 20 μL of 1:10 diluted MTT stock solution (5 mg/mL) and incubated for 4 hours. After incubation, we verified the presence of the violet crystals that normally indicate metabolization of MTT. Then the medium was removed (180 μL) and 180 μL of dimethyl sulfoxide (Sigma-Aldrich Co.) was added to dissolve the MTT crystals. The eluted specific stain was measured with a spectrophotometer (550 nm). The proliferation index of the untreated cells was compared to that of the negative control (cell plus medium without peptides). The experiments were performed in duplicate. Ethical approval was not sought for the use of this type of human cell lines.
Antibacterial activity
A colony-forming unit assay (CFU) of the antibacterial activity of hBDs against Pseudomonas aeruginosa ATCC (American Type Culture Collection, Manassas, VA, USA) 27853, Enterococcus faecalis ATCC 29212, and Escherichia coli ATCC 25922 was performed. The strains were grown in aerobic conditions in tryptic soy broth (Difco Laboratories, Detroit, MI, USA) at 37°C and incubated in the presence of hBDs for 2 hours at 37°C in PBS. We used two concentrations of peptides, ie, 2.5 μM and 12.5 μM. For salt-dependence assay, 0, 50, 100, and 200 mM NaCl was included in the incubation PBS buffer for 2 hours at 37°C under non-proliferating conditions (see also Scudiero et al7 and Scudiero et al8). A CFU assay was used to measure the antibacterial activity of hBD1, hBD3, and the AMC analog against E. coli, P. aeruginosa, and E. faecalis also in the presence of various concentrations of Ca2+ and Mg2+ ions. The strains were incubated for 2 hours at 37°C with two peptide concentrations (2.5 and 12.5 μM) in the presence of Ca2+ and Mg2+ ions (0, 0.1, and 1 mM) in 150 mM TRIS buffer. Each assay was performed in triplicate. Bactericidal activity (mean and standard deviation of three assays) is expressed as the ratio between colonies counted and the number of colonies on a control plate.
The minimal inhibitory concentration of the new molecule AMC was determined with a modified version of the microbroth dilution assay of the National Committee for Clinical Laboratory Standards using a final inoculum of 105 CFU/mL. The following peptide concentrations were used: 100.0, 50.0, 25.0, 12.5, 6.25, 3.12, and 1.56 μM.
The growth performance of P. aeruginosa ATCC 27853, E. faecalis ATCC 29212, and E. coli ATCC 25922 was also tested. Bacterial absorbance of the overnight culture was determined and adjusted to 0.2 at OD600. Bacteria were then incubated in the presence or absence of the AMC analog at 37°C (5% CO2). Duplicate absorbance readings (OD600) of bacterial cultures were taken every 30 minutes for 21 hours. Absorbance readings were plotted against time to generate bacterial growth curves.
Antiviral activity
Vero cells were grown in Dulbecco’s Modified Eagle’s Medium supplemented with 10% fetal calf serum. Herpes simplex virus (HSV)-1 carrying a lacZ gene driven by the cytomegalovirus IE-1 promoter to express beta-galactosidase was propagated as described elsewhere.20 All experiments were conducted in parallel with no-peptide controls. The effect of peptides on inhibition of HSV infectivity on cell monolayers20 was assessed as follows:
- For “co-exposure” experiments, the cells were incubated with increasing concentrations of the peptides (1, 5, 10.0, 20.0, 50.0, 100.0, and 150.0 μM) and with the viral inoculum for 45 minutes at 37°C. Non-penetrated viruses were inactivated by citrate buffer at pH 3.0. Monolayers were fixed, stained with 5-bromo-4-chloro-3-indolyl-beta-D-galactopyranoside and plaque numbers were scored. Experiments were performed in triplicate and the percentage of inhibition was calculated with respect to no-peptide control experiments.
- For “virus pre-exposure” experiments, approximately 2×104 plaque-forming unit (PFU) of HSV-1 were incubated with 20 μM of peptides for 45 minutes at 37°C then titrated on Vero cell monolayers.
- For “cell pre-exposure” experiments, Vero cells were incubated with 20 μM of peptides for 30 minutes at 4°C and infected with serial dilutions of HSV-1 for 45 minutes at 37°C.
Serum stability
The serum stability of AMC peptides, in reduced and oxidized form, was assessed at 0, 0.5, 1, 2, 3, 6, 8, 24, and 72 hours as reported elsewhere.8 Supernatants were analyzed by LC/MS using a combined system constituted by a reverse-phase HPLC (Gilson, Middleton, USA) interfaced with a Flexar SQ 300 mass spectrometer with electrospray ionization (PerkinElmer Inc.). Components were separated using a reverse-phase Jupiter C18 analytical column (5 μm particle size, 300 Å pore size, 4.6×250 mm) (Phenomenex) with a linear gradient of 5%–60% acetonitrile in acidified water (0.1% TFA) over 30 minutes. Peptide cleavage in serum was assessed by electron spray ionization mass spectrometry analysis. Mass spectrometry was performed in the mass range from 200 to 3,000 m/z.
Results and discussion
Design and synthesis of the new cyclic peptide
A novel molecule, designated AMC, containing the internal hydrophobic domain of hBD1 and the C-terminal charged region of hBD3 was obtained from the cyclization of a 17 amino acid sequence (Table 1). The internal domain was cyclized using two cysteine residues which were easily oxidized to obtain the molecule containing the charged hBD3 domain at its C-terminus. In this molecule, the cycle and the charged region are separated by two glycine residues (GG spacer) in order to allow sufficient flexibility of the two domains. The peptide was synthesized using the standard solid-phase-Fmoc method as previously reported,10 and the purified reduced peptide yield was higher (>60%) than the yields usually obtained for defensin peptides. The purified peptide was oxidized to obtain the final molecule and further purified.
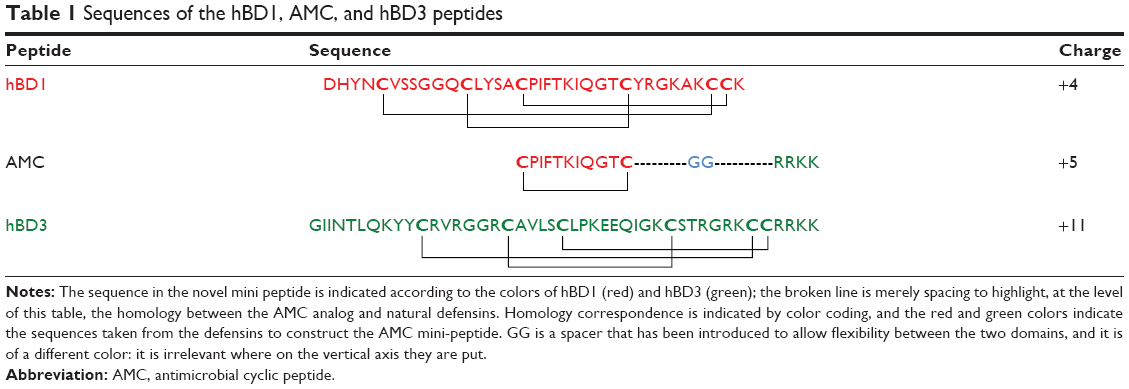 | Table 1 Sequences of the hBD1, AMC, and hBD3 peptides |
Characterization of the AMC structure
We first investigated the conformational properties of the AMC peptide in H2O/D2O (90/10 v/v) at pH =4.2. The Nuclear Overhauser effect (NOE) diagram (Figure S1A) is not consistent with the presence of ordered secondary structure elements.14 The high absolute values of temperature coefficients (|Δδ/ΔT| higher than 4 ppb/K) (Table S3) obtained for the majority of residues indicate that most amide protons are solvent-exposed and are unlikely to be involved in the formation of hydrogen bonds.21
In the NOESY16 spectrum of the AMC peptide in H2O, we clearly identified the NOE contact between the Hα proton of Cys1 and the Hδδ’ protons of Pro2, which is indicative of a proline in a trans configuration.14 In the TOCSY15 and NOESY16 spectra, besides the unambiguously assigned spin systems (Table S1), there were sets of lower-intensity signals. This feature could result from proline cis–trans isomerization; however, because of spectral overlaps and closeness of the chemical shifts of the Hα protons to the water signal, we could not clearly distinguish NOE contacts characteristic of a proline in a cis configuration. For structural calculations, only the higher-intensity spin systems were considered and thus the proline was set in a trans configuration. The NMR solution structure was calculated with the torsion angle dynamics algorithm of the CYANA software19 (Table S5). The NMR conformers appeared rather disordered, also in the cyclic portion Cys1–Cys11, as indicated by the high root mean square deviation (RMSD) values, and were subjected to a clusterization procedure (Figure 1A–D; Table S7). Clusters were generated with the Chimera program22 by superimposing the final 20 NMR structures on the backbone atoms (residues 1–11). This analysis revealed three clusters that represent conformationally related subfamilies (Figure 1A–D); the most abundant cluster includes ten structures, and the other two clusters are populated each by five structures (Table S7).
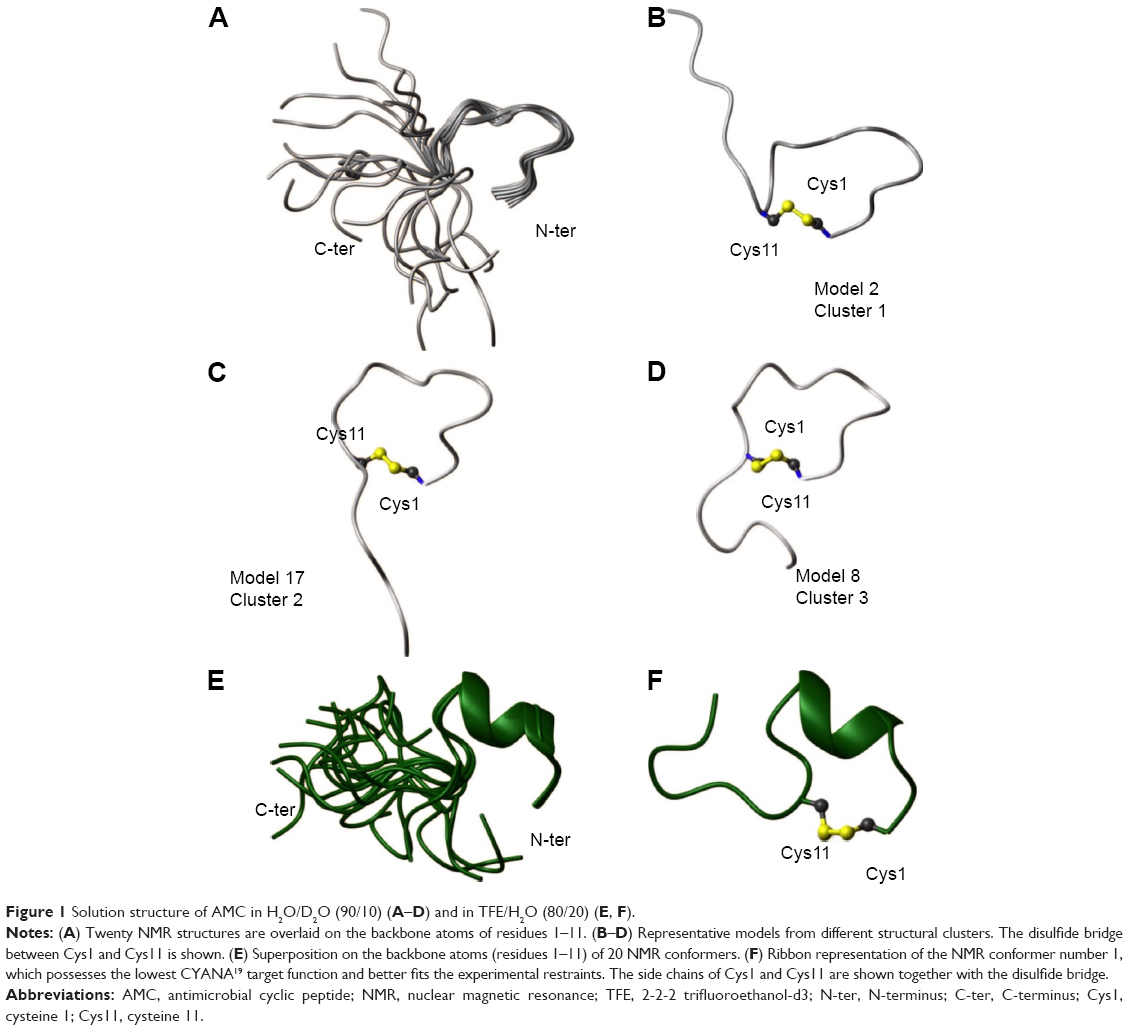 | Figure 1 Solution structure of AMC in H2O/D2O (90/10) (A–D) and in TFE/H2O (80/20) (E, F). |
We also conducted NMR studies in a membrane mimetic environment (ie, in TFE/H2O 80/20 v/v) (Table S2).23 The NOESY16 spectrum recorded in the presence of TFE contains more high-intensity contacts than in an H2O environment, thereby indicating a clear increase in ordered conformations. The NOE pattern (ie, a stretch of strong HNi−HNi+1; Hαi−HNi+2; Hαi−HNi+3; Hαi−Hβi+3 connectivities) indicates the presence of a helix in the peptide segment encompassing residues Ile3 and Gln8 (Figure S1B).
A 3D model of AMC in the presence of TFE was constructed (Figure 1E and F; Table S6). The peptide cyclic region encompassing the Cys1–Cys11 segment is well defined as indicated by the low RMSD values (Table S6), and the region-spanning residues Phe4–Ile7 adopt a 3.10 helix conformation (Figure 1E and F). Indeed, low absolute values of temperature coefficients, below 4 ppb/K, which is indicative of scarce solvent exposure,21 were measured for residues in the helical region, namely Phe4, Thr5, and Lys6, as well as for Gly9 and Cys11 (Table S4). In the NOESY spectrum, we unambiguously assigned the NOE contacts Hα Cys1–Hδδ’ Pro2 that are diagnostic of a proline in a trans configuration.14 However, as discussed above for the peptide sample in H2O, we cannot exclude some cis–trans pro-isomerization also in the presence of TFE.
In summary, when analyzed by NMR in H2O, the structure of the AMC peptide was found to be rather disordered in both the cyclic and C-terminal regions (Figure 1). A strict comparison between the conformation assumed by AMC and those of the corresponding regions of hBD1 and hBD3 is not feasible due to the high flexibility of the peptide in aqueous solution. However, as can be clearly seen in Figure 2, where a representative AMC structure is taken into account, the insertion of the disulfide bridge between the two cysteines likely constrains the AMC peptide backbone which thereby assumes a structure that resembles the corresponding region of hBD1. NMR analysis in a membrane mimetic environment like TFE revealed an increase of order in the AMC cyclic region, which gained a small helical turn encompassing residues Phe4–Ile7 (Figure 1). It cannot be excluded that a structural rearrangement may occur in the peptide once it interacts with membranes, and in this respect, the structure observed in TFE may represent a bioactive AMC conformation.
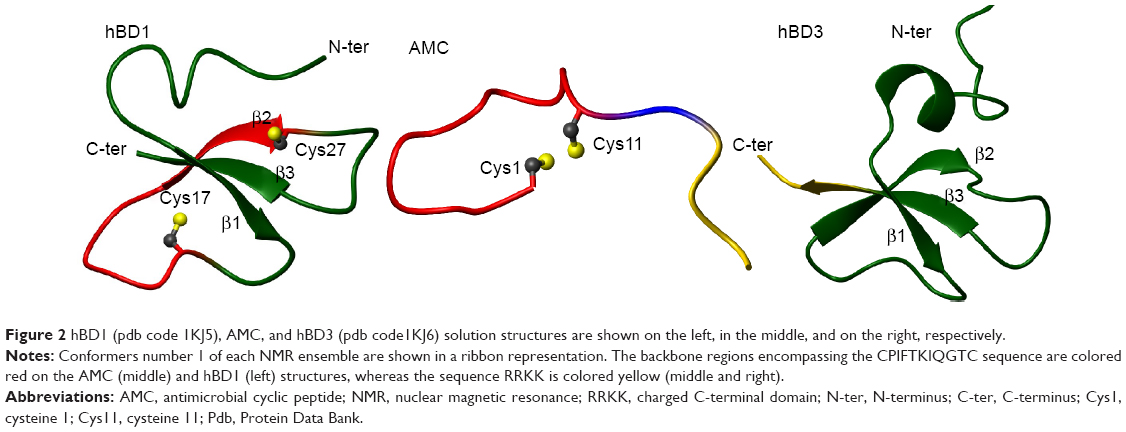 | Figure 2 hBD1 (pdb code 1KJ5), AMC, and hBD3 (pdb code1KJ6) solution structures are shown on the left, in the middle, and on the right, respectively. |
Cytotoxicity of wild-type hBD1 and hBD3 and analog AMC in CaCo-2 cells
The MTT test was performed on CaCo-2 cells using wild-type hBD1 and hBD3, and their analog AMC at concentrations of 2.5, 12.5, and 25.0 μM. Exposure times were 4, 8, 12, 16, 20, 24, 48, and 72 hours. Since similar results were obtained with the three concentrations, we report only the results obtained with the highest concentration. Figure 3A shows the percentage of viability of CaCo-2 cells exposed to the peptides at a concentration of 25.0 μM. These results confirm our previous results obtained with hBD1 and hBD3.7 At all peptide concentrations used, cell viability was only slightly reduced after 24 hours (5%–20%), whereas it was reduced by approximately 25% after 48 and 72 hours (Figure 3A). Moreover, we performed toxicity assays at a peptide concentration of 125 μM for 8, 24, and 72 hours of incubation (Figure 3B). In these conditions, we found a more pronounced reduction in cell viability; for all tested peptides, cell viability was reduced by 40% after 8 hours and by approximately 50% after 24 and 72 hours. In conclusion, this set of experiments shows that the wild-type hBDs and AMC do not induce relevant cytotoxic effects in vitro at concentrations of 2.5, 12.5, and 25.0 μM.
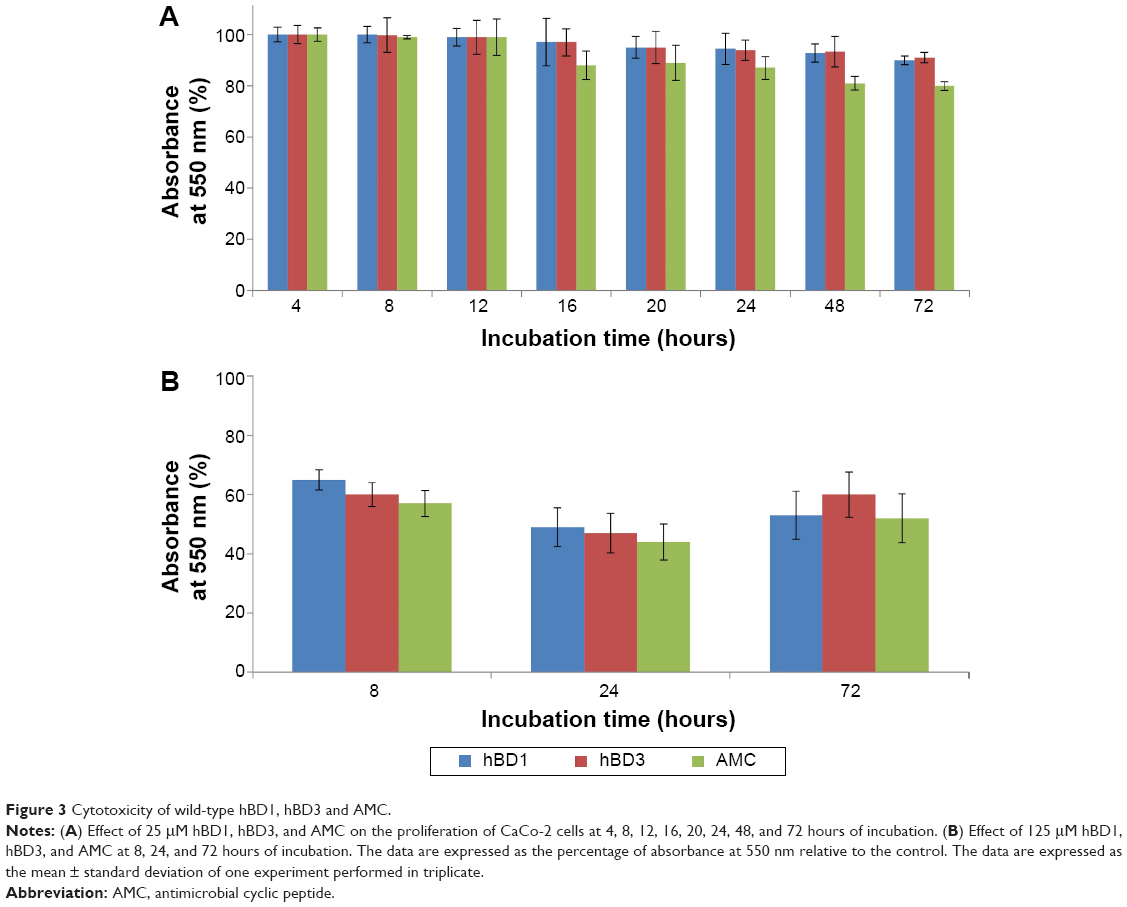 | Figure 3 Cytotoxicity of wild-type hBD1, hBD3 and AMC. |
Antibacterial activity of the AMC analog compared to the parent hBDs
We analyzed the antibacterial activity of the AMC analog and of hBD1 and hBD3 as reference peptides.7,8 We tested two concentrations of each peptide (2.5 and 12.5 μM) and four concentrations of NaCl (0, 50, 100, and 200 mM) against P. aeruginosa, E. coli, and E. faecalis, which are the most frequent causes of many human infections. Firstly, we tested the antibacterial properties of AMC in the reduced and oxidized forms against E.coli. The reduced form of AMC was less active than the oxidized form (data not shown). This behavior may be due to the structural features of AMC and its flexibility. Therefore, we decided to test only the oxidized form for the other bacterial strains (P. aeruginosa and E. faecalis). The results confirmed the antibacterial properties of hBD1 and the salt resistance of hBD3.7,8 Tested against P. aeruginosa at 2.5 μM, AMC exerted similar antibacterial activity to hBD3 in the presence of 200 mM NaCl; at a concentration of 12.5 μM, AMC was more active at 200 mM NaCl (Figure 4A). Interestingly, the antibacterial activity of AMC against E. coli and E. faecalis was similar to that of hBD3 in the presence of up to 100 mM salt at both 2.5 and 12.5 μM of peptide. AMC exerted the best antibacterial activity at 12.5 μM; at this concentration it was more effective than hBD3 in the presence of the highest salt concentration (200 mM) (Figure 4B and C).
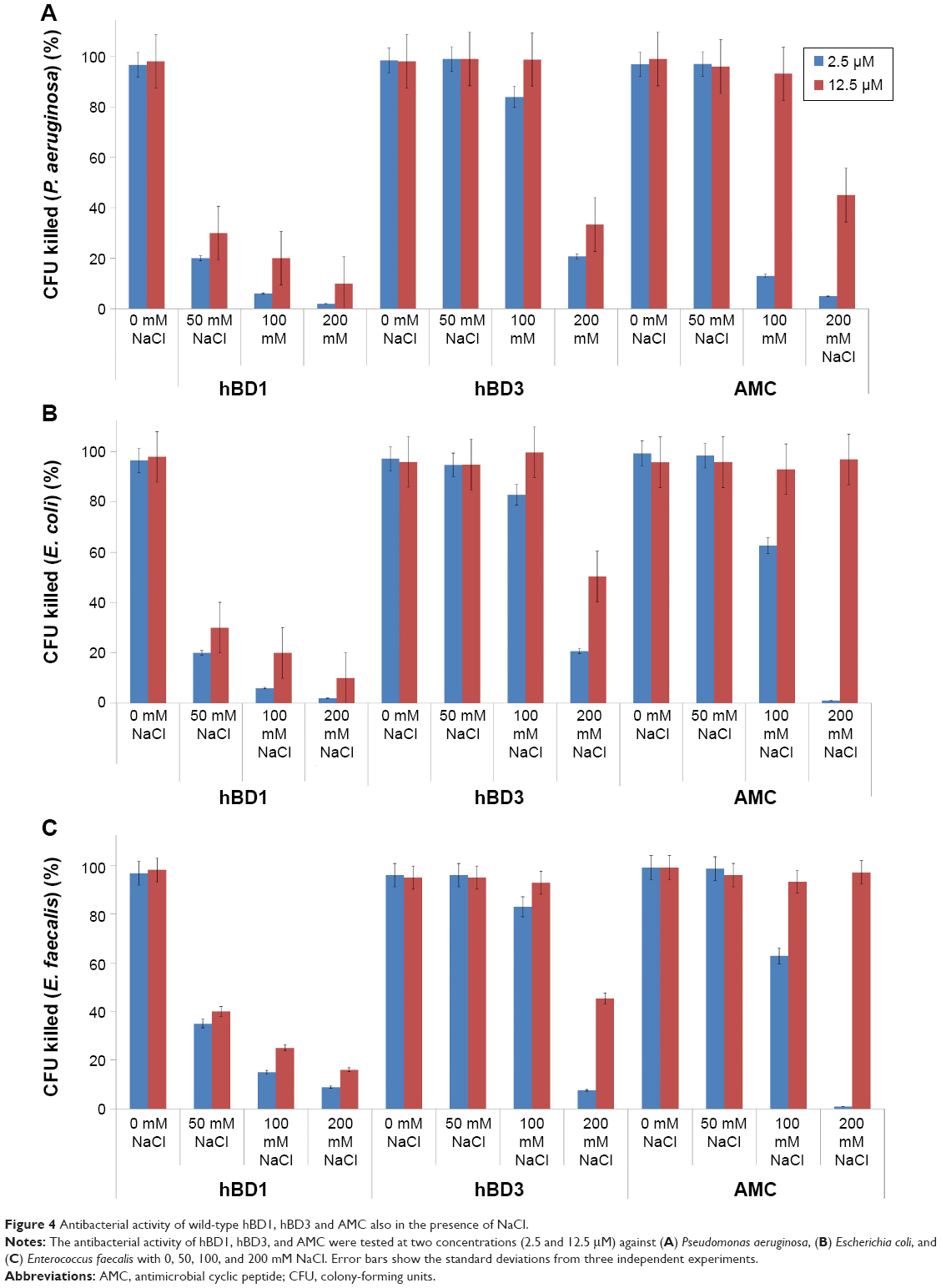 | Figure 4 Antibacterial activity of wild-type hBD1, hBD3 and AMC also in the presence of NaCl. |
Since divalent cations (magnesium and calcium) decrease the antimicrobial activity of peptides, we performed a CFU assay in the presence of Ca2+ and Mg2+ (0.1 and 1 mM, respectively) against P. aeruginosa, E. coli, and E. faecalis. The results are shown in Figure 5. Against P. aeruginosa and E. coli, hBD1, hBD3, and AMC lose their activity to a similar extent in the presence of Ca2+ and Mg2+ (Figure 5A and B). Interestingly, the antibacterial activity of hBD1, hBD3, and AMC against E. faecalis in the presence of Ca2+ and Mg2+ was retained compared with the samples without these ions indicating that the influence of cations strictly depends on the bacteria strains (Figure 5C). Moreover, we evaluated AMC minimal inhibitory concentration, which ranged between 12.5 and 25.0 μM in the tested microorganisms.
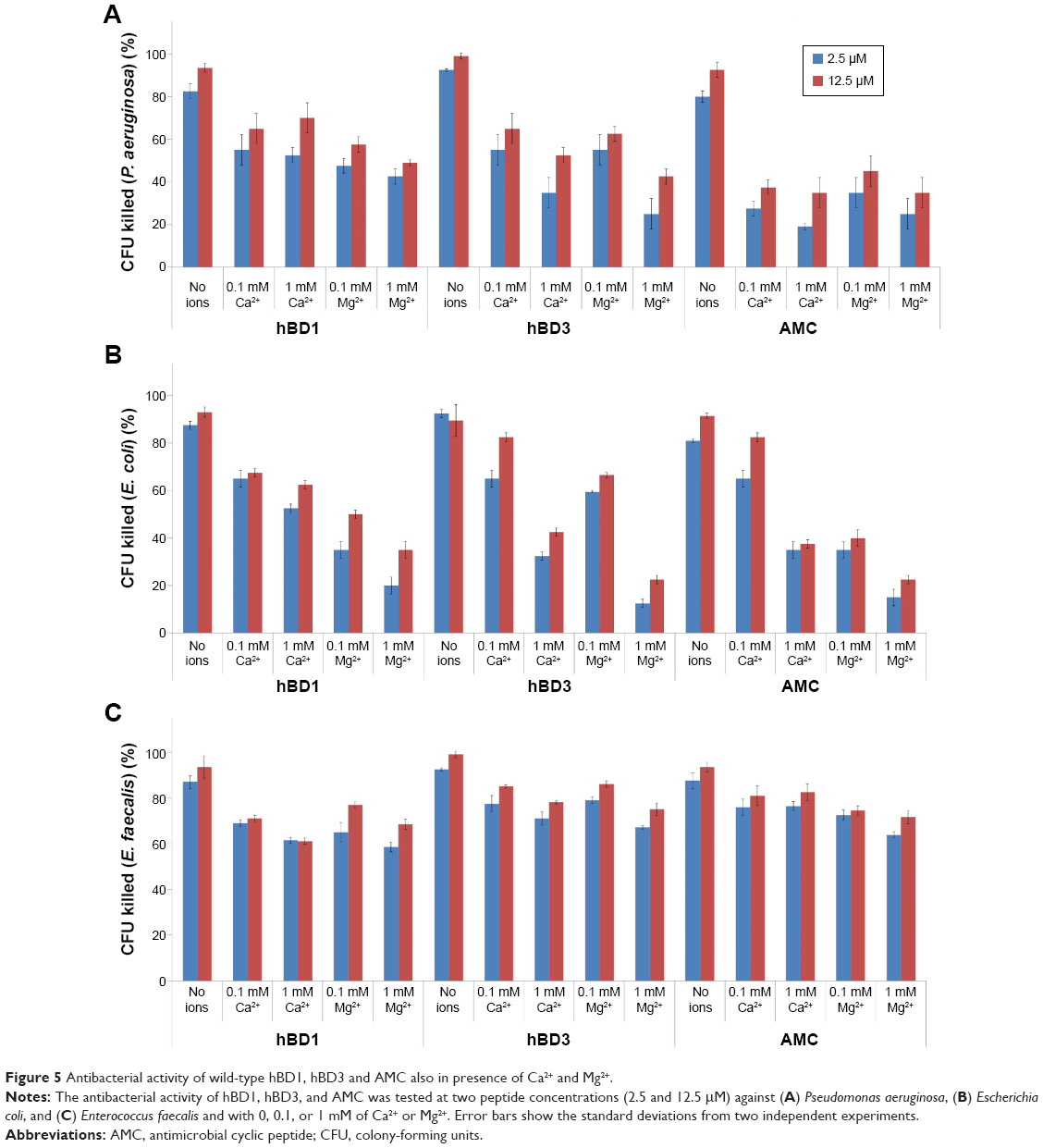 | Figure 5 Antibacterial activity of wild-type hBD1, hBD3 and AMC also in presence of Ca2+ and Mg2+. |
Finally, we tested the effect exerted by AMC on the growth performance of the indicator strains, and found that it prolonged the lag phase and reduced the maximum population level after 48 hours of incubation (Figure 6). These antibacterial tests provide an indication about the effects of the new synthetized AMC; in fact, we observed a delay in the lag phase of the three bacteria tested in the presence of 2.5 μM, with a longer lag phase of 7.5 hours versus 4 hours.
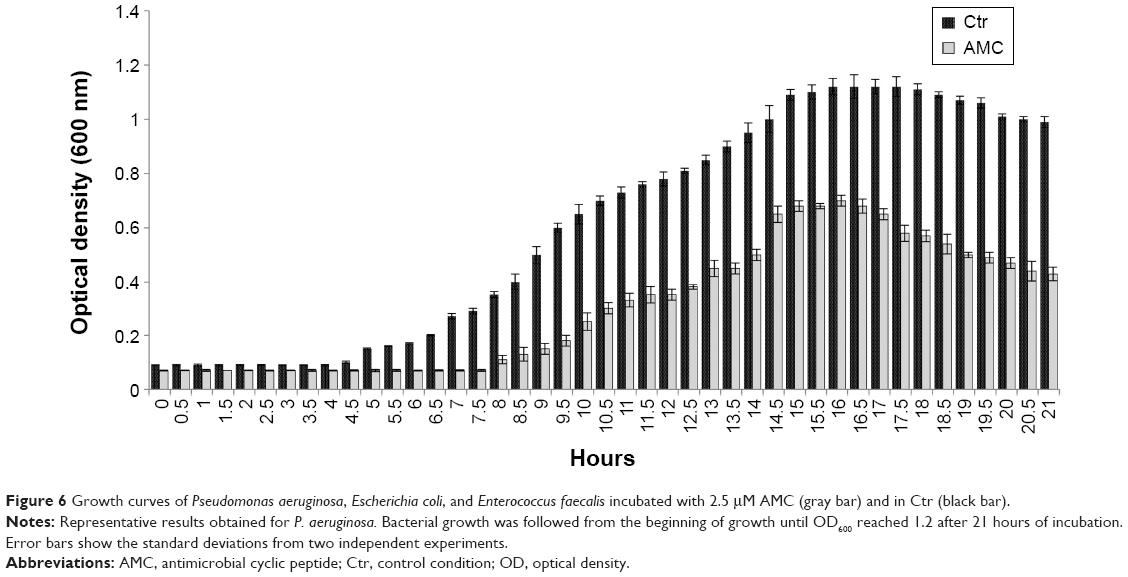 | Figure 6 Growth curves of Pseudomonas aeruginosa, Escherichia coli, and Enterococcus faecalis incubated with 2.5 μM AMC (gray bar) and in Ctr (black bar). |
AMC exerts good antiviral activity against HSV type 1
We evaluated the antiviral activity of AMC, both oxidized and reduced, and compared it to the activity of hBD3. All peptides inhibited HSV infectivity in a dose-dependent manner (Figure 7). We evaluated the antiviral activities in co-exposure experiments (Figure 7A) (with cell, virus, and peptide at zero time), in virus pre-exposure experiments (Figure 7B) (virus plus peptide before cell infection), in cell pre-exposure experiments (Figure 7C) (cell plus peptide before virus infection), and in post-treatment (cell plus virus before peptide addition). This allowed us to evaluate if and in which phase the peptides inhibited virus infectivity. The reduced form of AMC was always less active than the oxidized form. Notably, we previously reported that oxidized and reduced hBD3 had the same antiviral activity.7 This discrepancy may reflect the small size of AMC and its flexibility. The disulfide bridge may be necessary to make the molecule more rigid and assume the correct folding for interaction. Both in the co-exposure and virus pre-treatment experiments, the oxidized AMC reduced virus infectivity by 60% at 150 μM, while infectivity was reduced by 30% in the cell-pre-treatment experiment, and no effect was found in the post-treatment experiment. These results suggest that AMC can interfere with viral attachment and entry; however, we found no proof of inhibition when cells were already infected. The oxidized AMC has a half maximal inhibitory concentration of 100 μM and hBD3 has a half maximal inhibitory concentration of 10 μM. This difference is probably due to the small size of AMC (17 residues versus 45 residues of hBD3), and AMC may thus represent a good starting point for next-generation analogs.
 | Figure 7 Antiviral activity of AMC against Herpes simplex virus type 1. |
Human serum stability
To evaluate the stability of oxidized AMC to the proteolytic effect of serum, peptide aliquots were incubated with 20% (v/v) human serum for different times and then analyzed by liquid chromatograph/mass spectrometry for peptide integrity (Table 2; Figure 8A–F). The reduced AMC peptide was quickly degraded after 0.5 hours of incubation (data not shown). On the contrary, the integral oxidized peptide was detected, mixed with its degradation product, after 0.5 hours of incubation. Peptide degradation started at the C-terminus, –K after 0.5 hours of incubation and –KKR after 2 hours of incubation, as detected by mass spectrometric analysis (Table 2; Figure 8B and C). The integral peptide disappeared after 2 hours of incubation (Table 2; Figure 8C) and peptide degradation continued at the C-terminus –GRRKK up to 24 hours (Table 2; Figure 8F), which suggests that degradation is mediated by a carboxypeptidase. At 72 hours, peptide degradation did not seem to proceed further, suggesting that serum peptide sensitivity is reduced by the presence of the disulfide bridge (data not shown).
 | Table 2 Time-course of the incubation of oxidized AMC in human serum |
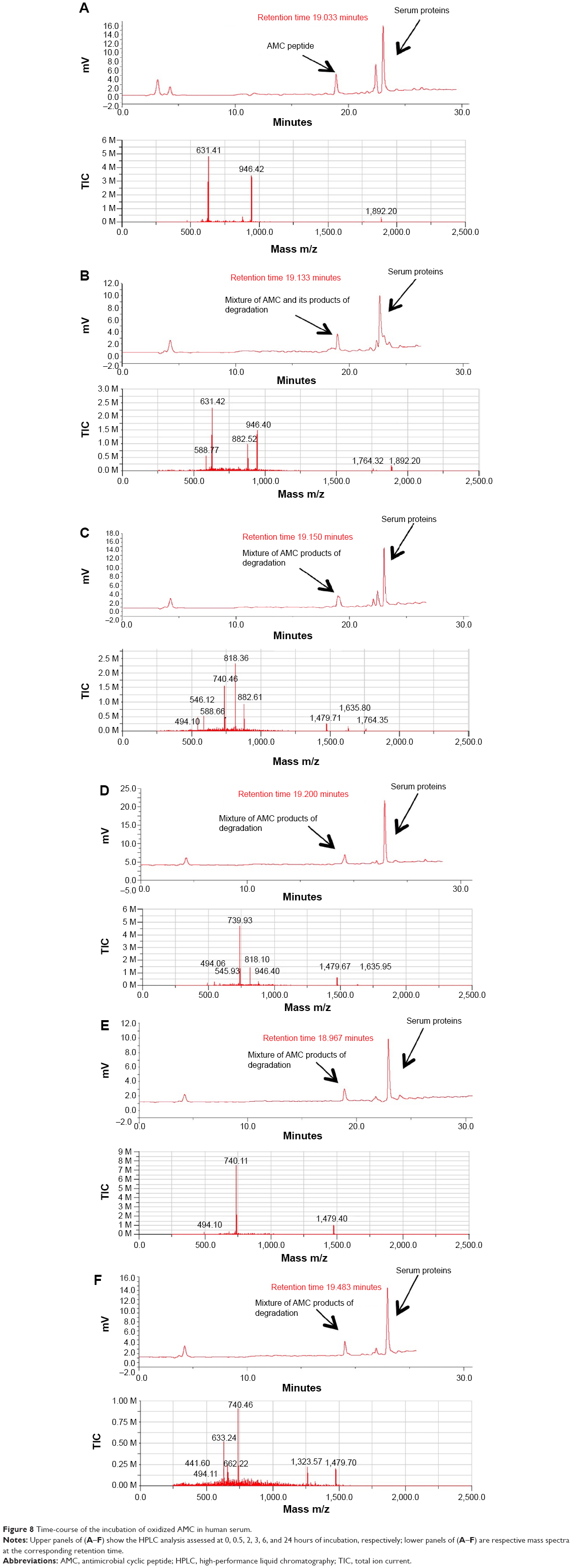 | Figure 8 Time-course of the incubation of oxidized AMC in human serum. |
Discussion
The development of peptidomimetics which mimic the active conformation of a peptide by incorporating non-peptidic features has recently received much attention, resulting in many cases in improved pharmacological properties. Introduction of structural restraints is frequently used by the pharmaceutical industry to reduce the number of conformations and may result in higher affinity for the target and improved protease stability, bioavailability, and specificity. In fact, cyclization though disulfide bonds is naturally found in AMPs, such as the hBDs, which display highly potent and broad antimicrobial activity. The highly conserved triple-disulfide scaffold characteristic of hBDs makes their accessibility through chemical synthesis difficult and poor-yielding. In addition to the high cost, this complexity limits the number of analogs that can be produced to identify better therapeutic candidates.
In an effort to preserve the antimicrobial properties of hBDs while simplifying the structure of the molecule, we took advantage of our previous study of chimerae between the hBDs 1–3. Having established that the key domains involved in the antibacterial and antiviral activities were the RRKK of hBD3 and PIFTKIQGT, we combined them in a single-disulfide peptide, AMC. Analog AMC is an effective, highly stable, low-toxic, cyclic mini-β-defensin AMP. It recapitulates the antibacterial and antiviral properties of hBDs in a considerably smaller sequence (only 17 amino acids). Moreover, its ease of synthesis will enable the ready preparation of analogs to further improve biological potency and in vivo duration of action. Therefore, this molecule should be considered a new weapon with which to combat infectious diseases. Additional values that may be considered are its potential use in case of antibiotic resistance and its delivery by way of nanovector devices.
Acknowledgments
We thank Jean Ann Gilder (Scientific Communication srl, Naples, Italy) for revising and editing the text; and Vittorio Lucignano, CEINGE-Biotecnologie Avanzate for technical assistance.
Disclosure
This work was supported by Grant (RF-2010-2318372) from the Italian Ministry of Health (to FS); Protocollo d’intesa Regione Campania-CEINGE (D.C. n.11 dated February 28, 2014), Italy (to FS); Grants POR Campania FSE 2007/2013: CAMPUS-Bioframe, C.RE.ME, and DIAINTECH from the Regione Campania, Italy (to FS). The authors report no other conflicts of interest in this work.
References
Aoki W, Kuroda K, Ueda M. Next generation of antimicrobial peptides as molecular targeted medicines. J Biosci Bioeng. 2012;114:365–370. | ||
Aoki W, Ueda M. Characterization of antimicrobial peptides toward the development of novel antibiotics. Pharmaceuticals (Basel). 2013;6(8):1055–1081. | ||
Giuliani A, Pirri G, Nicoletto S. Antimicrobial peptides: an overview of a promising class of therapeutics. Open Life Sciences. 2007;2:1–33. | ||
Pazgier M, Hoover DM, Yang D, Lu W, Lubkowski J. Human beta-defensins. Cell Mol Life Sci. 2006;63:1294–1313. | ||
Klüver E, Adermann K, Schulz A. Synthesis and structure-activity relationship of beta-defensins, multi-functional peptides of the immune system. J Pept Sci. 2006;12:243–257. | ||
Taylor K, Clarke DJ, McCullough B, et al. Analysis and separation of residues important for the chemoattractant and antimicrobial activities of beta-defensin 3. J Biol Chem. 2008;283:6631–6639. | ||
Scudiero O, Galdiero S, Cantisani M, et al. Novel synthetic, salt-resistant analogs of human beta-defensins 1 and 3 endowed with enhanced antimicrobial activity. Antimicrob Agents Chemother. 2010;54:2312–2322. | ||
Scudiero O, Galdiero S, Nigro E, et al. Chimeric beta-defensin analogs, including the novel 3NI analog, display salt-resistant antimicrobial activity and lack toxicity in human epithelial cell lines. Antimicrob Agents Chemother. 2013;57:1701–1708. | ||
Nguyen LT, Chau JK, Perry NA, de Boer L, Zaat SA, Vogel HJ. Serum stabilities of short tryptophan- and arginine-rich antimicrobial peptide analogs. PLoS One. 2010;5(9):e12684. | ||
Schibli DJ, Hunter HN, Aseyev V, et al. The solution structures of the human beta-defensins lead to a better understanding of the potent bactericidal activity of HBD3 against Staphylococcus aureus. J Biol Chem. 2002;277:8279–8289. | ||
Bai Y, Liu S, Jiang P, et al. Structure-dependent charge density as a determinant of antimicrobial activity of peptide analogues of defensin. Biochemistry. 2009;48:7229–7239. | ||
Galdiero S, Capasso D, Vitiello M, D’Isanto M, Pedone C, Galdiero M. Role of surface-exposed loops of Haemophilus influenzae protein P2 in the mitogen-activated protein kinase cascade. Infect Immun. 2003;71:2798–2809. | ||
Hwang TL, Shaka AJ. Water suppression that works. Excitation sculpting using arbitrary wave-forms and pulsed-field gradients. J Magn Reson A. 1995;112:275–279. | ||
Wüthrich K. NMR of Proteins and Nucleic Acids. New York: John Wiley & Sons; 1986. | ||
Griesinger C, Otting G, Wuethrich K, Ernst RR. Clean TOCSY for proton spin system identification in macromolecules. J Am Chem Soc. 1988;110:7870–7872. | ||
Kumar A, Ernst RR, Wüthrich K. A two-dimensional nuclear Overhauser enhancement (2D NOE) experiment for the elucidation of complete proton-proton cross-relaxation networks in biological macromolecules. Biochem Biophys Res Commun. 1980;95:1–6. | ||
Piantini U, Sorensen OW, Ernst RR. Multiple quantum filters for elucidating NMR coupling networks. J Am Chem Soc. 1982;104:6800–6801. | ||
Bartels C, Xia TH, Billeter M, Güntert P, Wüthrich K. The program XEASY for computer-supported NMR spectral analysis of biological macromolecules. J Biomol NMR. 1995;6(1):1–10. | ||
Herrmann T, Güntert P, Wüthrich K. Protein NMR structure determination with automated NOE assignment using the new software CANDID and the torsion angle dynamics algorithm DYANA. J Mol Biol. 2002;319:209–227. | ||
Tarallo R, Carberry TP, Falanga A, et al. Dendrimers functionalized with membrane-interacting peptides for viral inhibition. Int J Nanomedicine. 2013;8:521–534. | ||
Baxter NJ, Williamson MP. Temperature dependence of 1H chemical shifts in proteins. J Biomol NMR. 1997;9:359–369. | ||
Pettersen EF, Goddard TD, Huang CC, et al. UCSF Chimera – a visualization system for exploratory research and analysis. J Comput Chem. 2004;25:1605–1612. | ||
Buck M. Trifluoroethanol and colleagues: cosolvents come of age. Recent studies with peptides and proteins. Q Rev Biophys. 1998;31:297–355. |
Supplementary materials
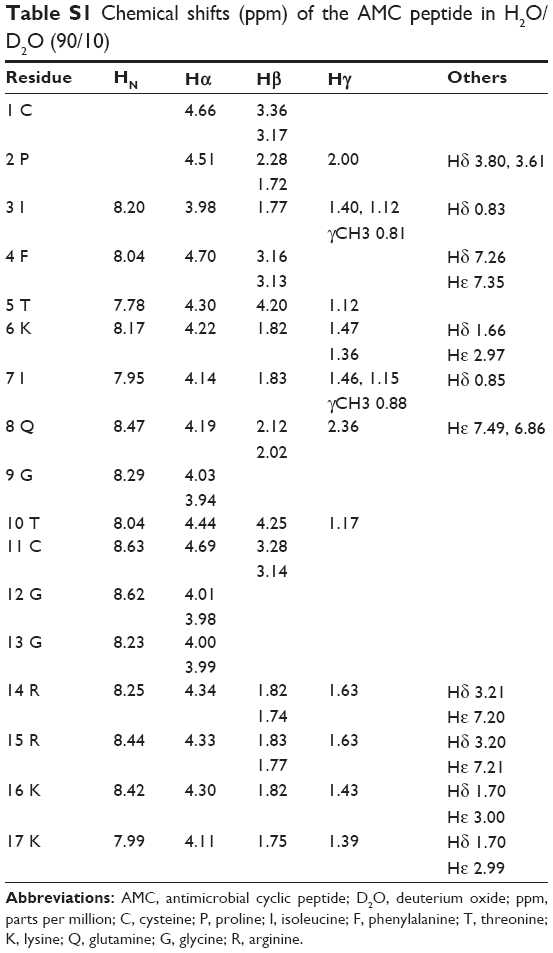 | Table S1 Chemical shifts (ppm) of the AMC peptide in H2O/D2O (90/10) |
 | Table S2 Chemical shifts (ppm) of the AMC peptide in TFE/H2O (80/20) |
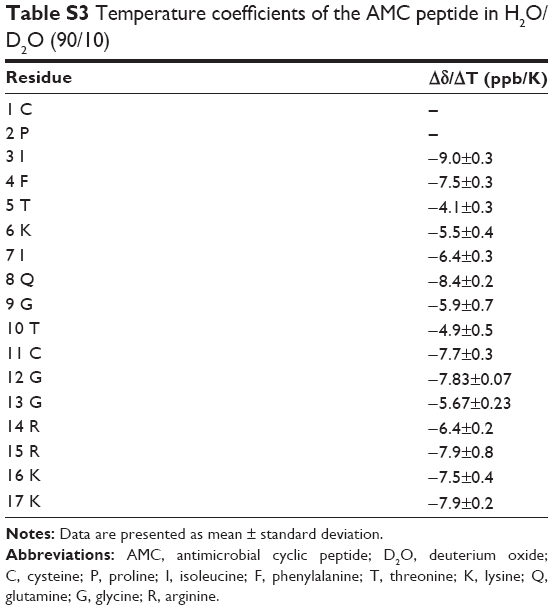 | Table S3 Temperature coefficients of the AMC peptide in H2O/D2O (90/10) |
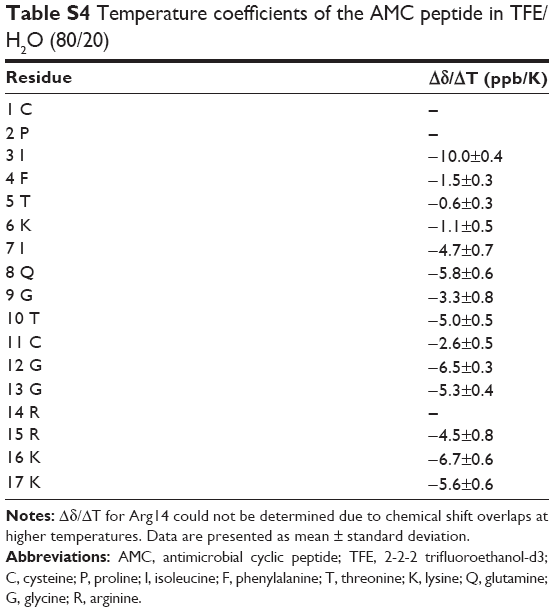 | Table S4 Temperature coefficients of the AMC peptide in TFE/H2O (80/20) |
 | Table S5 Structure statistics for the AMC NMR conformers in H2O/D2O (90/10) |
 | Table S6 Structure statistics for AMC NMR ensemble of structures in TFE/H2O (80/20) |
 | Table S7 Cluster analysis of the AMC NMR ensemble calculated in H2O/D2O |
 | Figure S1 Summary of main short and medium range NOEs. |
References
Herrmann T, Güntert P, Wüthrich K. Protein NMR structure determination with automated NOE assignment using the new software CANDID and the torsion angle dynamics algorithm DYANA. J Mol Biol. 2002;319:209–227. | ||
Doreleijers JF, Sousa da Silva AW, Krieger E, et al. CING: an integrated residue-based structure validation program suite. J Biomol NMR. 2012;54:267–283. | ||
Laskowski RA, Rullmannn JA, MacArthur MW, Kaptein R, Thornton JM. AQUA and PROCHECK-NMR: programs for checking the quality of protein structures solved by NMR. J Biomol NMR. 1996;8:477–486. | ||
Koradi R, Billeter M, Wüthrich K. MOLMOL: a program for display and analysis of macromolecular structures. J Mol Graph. 1996;14:51–55, 29–32. | ||
Pettersen EF, Goddard TD, Huang CC, et al. UCSF Chimera – a visualization system for exploratory research and analysis. J Comput Chem. 2004;25:1605–1612. | ||
Kelley LA, Gardner SP, Sutcliffe MJ. An automated approach for clustering an ensemble of NMR-derived protein structures into conformationally related subfamilies. Protein Eng. 1996;9:1063–1065. |
 © 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms.php
and incorporate the Creative Commons Attribution
- Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms.php
and incorporate the Creative Commons Attribution
- Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.