Back to Journals » Therapeutics and Clinical Risk Management » Volume 14
Dendritic cell vaccines for high-grade gliomas
Authors Eagles ME ![]() , Nassiri F, Badhiwala JH, Suppiah S, Almenawer SA, Zadeh G, Aldape KD
, Nassiri F, Badhiwala JH, Suppiah S, Almenawer SA, Zadeh G, Aldape KD
Received 3 January 2018
Accepted for publication 11 April 2018
Published 26 July 2018 Volume 2018:14 Pages 1299—1313
DOI https://doi.org/10.2147/TCRM.S135865
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Professor Garry Walsh
Matthew E Eagles,1 Farshad Nassiri,2,3 Jetan H Badhiwala,2 Suganth Suppiah,2 Saleh A Almenawer,4 Gelareh Zadeh,3,5 Kenneth D Aldape3,6
1Section of Neurosurgery, Department of Clinical Neurosciences, University of Calgary, Calgary, AB, Canada; 2Division of Neurosurgery, Department of Surgery, University of Toronto, Toronto, ON, Canada; 3MacFeeters-Hamilton Neuro-Oncology Program, University Health Network, Toronto, ON, Canada; 4Division of Neurosurgery, Department of Surgery, McMaster University, Hamilton, ON, Canada; 5Division of Neurosurgery, University Health Network, Toronto, ON, Canada; 6Division of Pathology, University Health Network, Toronto, ON, Canada
Abstract: Glioblastoma (GBM) is the most common and fatal primary adult brain tumor. To date, various promising chemotherapeutic regimens have been trialed for use in GBM; however, temozolomide (TMZ) therapy remains the only US Food and Drug Administration-approved first-line chemotherapeutic option for newly diagnosed GBM. Despite maximal therapy with surgery and combined concurrent chemoradiation and adjuvant TMZ therapy, the median overall survival remains approximately 14 months. Given the failure of conventional chemotherapeutic strategies in GBM, there has been renewed interest in the role of immunotherapy in GBM. Dendritic cells are immune antigen-presenting cells that play a role in both the innate and adaptive immune system, thereby making them prime vehicles for immunotherapy via dendritic cell vaccinations (DCVs) in various cancers. There is great enthusiasm surrounding the use of DCVs for GBM with multiple ongoing trials. In this review, we comprehensively summarize the safety, efficacy, and quality of life results from 33 trials reporting on DCV for high-grade gliomas.
Keywords: glioma, vaccine, dendritic cells, glioblastoma
Introduction
Glioblastoma (GBM) is a lethal WHO grade IV diffuse glioma.1 It has an incidence of approximately 5 cases per 100,000 people,2,3 making it the most common malignant primary central nervous system tumor in adults. Current standard of care consists of maximal safe resection, adjuvant radiotherapy, and chemotherapy with temozolomide (TMZ), as outlined by Stupp et al.4–6 Despite this, the prognosis for these patients remains dismal with a median survival of 14.6 months and 5-year survival of approximately 5%.2,5,6
Current treatment for GBM and other high-grade gliomas (HGGs) fails for multiple reasons. The infiltrative nature of the tumor precludes surgery as a curative therapy.7 As such, GBM universally recurs, even at sites distant from the original resection. Furthermore, traditional chemotherapy has found little success in curing GBM; while TMZ is approved as a first-line therapy, the majority of tumors are MGMT unmethylated and show a diminished response to this drug.8–10 Radiotherapy has long been a mainstay in GBM treatment, but succeeds only in controlling the disease, not eradicating it.7 Moreover, prolonged radiotherapy is damaging to normal neural tissue, which may lead to clinically significant deterioration in cerebral function.11,12
The failure of current therapy to adequately treat GBM has prompted clinicians and scientists to look for novel means of treating this disease.13–15 As with most cancer therapies, there is a desire to find disease-specific treatments, which minimize harm to healthy cells. Over the past couple of decades, there has been a renewed interest in the role that the immune system plays in oncogenesis.16 As such, many novel cancer therapies aim to manipulate the immune response to target neoplastic cells.17–21 The aforementioned difficulty in treating GBM by conventional measures makes GBM a prime candidate for immunotherapy research,7 yet there are characteristics of this tumor that continue to pose a challenge.
GBM has an immune suppressive microenvironment as a consequence of several tumor-associated factors.7 There is an overexpression of immune inhibitory cytokines and checkpoint molecules,22–27 low levels of HLA,28 and elevated numbers of T-regulatory cells.29–31 To add to this, patients with GBM are often treated with steroids to help manage the tumor-associated vasogenic edema, and it has been reported that corticosteroid use is associated with decreased circulating T-cells and an abnormal local immune response.32 Hence, to be successful, an immunotherapy must induce a potent inflammatory response that is robust enough to overcome this baseline level of immune suppression within the tumor microenvironment.
Immunotherapies harness the power and specificity of the immune system (particularly the adaptive immune system) to target tumors. One area of immunotherapy that has been gaining traction in treating GBM and other HGGs is dendritic cell vaccination (DCV) therapy. Dendritic cells (DCs) are professional antigen-presenting cells that express MHC class 1 and 2 molecules. DCs also express many costimulatory molecules, and are equipped with receptors that promote their migration to lymph nodes (LNs) after antigen capture.33 Given these adaptive characteristics, DCs have been found to be the most efficient endogenous stimulus of new T- and B-cell responses.34,35 Mouse models have demonstrated the generation of antitumor immune responses that depend upon presentation of tumor antigens by DCs.36,37 Owing to the ability of DCs to control both immune tolerance and immunity, and their function in linking the innate and adaptive immune responses, DCs have become a promising target in efforts to generate immune response against various forms of cancer.18 DCVs appear well suited to meet the challenges posed by the tumor milieu in GBM, and consequently, there has been much enthusiasm surrounding the use of DCVs as a therapeutic adjuvant in GBM.7
In 2010, the US FDA approved the first DCV, sipuleucel-T (Provenge; Dendreon, Seattle, WA, USA), for the treatment of castration-resistant prostate cancer.38 As data from many other investigators indicate a potential for immunotherapy far beyond that of sipuleucel-T, immune-mediated approaches are being actively studied for several other tumor types, including breast, bladder, kidney, colorectal, head/neck, lung and melanoma, and for hematological malignancies.39
These collectively have paved the way for a number of clinical trials using DCVs in patients with GBM and other HGGs (Table 1).40–72 This review will begin by detailing work that has led to the only FDA-approved DCV across all cancer types. It will then move on to examining published clinical trials using DCVs to treat GBM and other HGGs. Emphasis will be placed on efficacy, safety/tolerability, and patient focused perspectives (eg, quality of life, satisfaction, adherence, etc.). Lastly, the review will draw conclusions regarding the future of DCVs in GBM and their role in treatment.
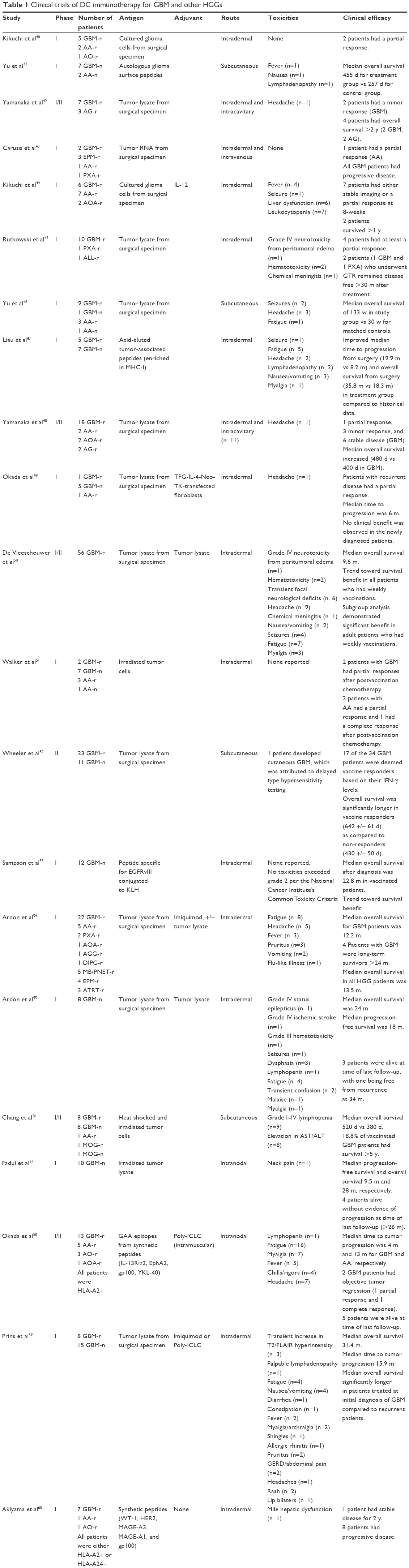 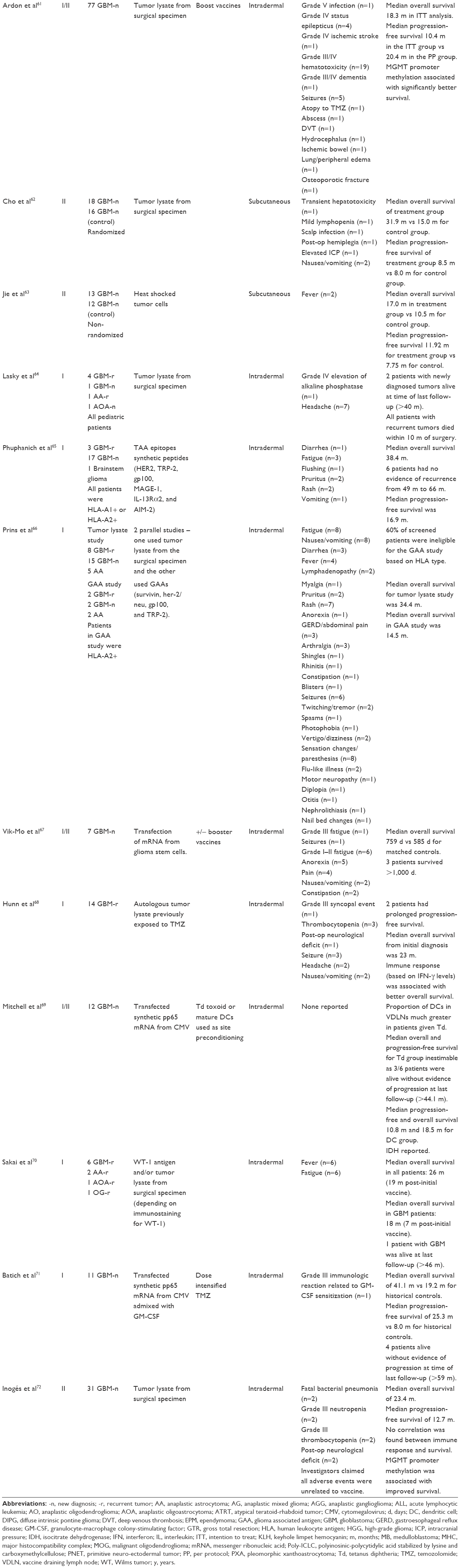 | Table 1 Clinical trials of DC immunotherapy for GBM and other HGGs |
DC generation in vitro
CD14+ monocytes are isolated from patient peripheral blood mononuclear cells. These monocytes are cultured with granulocyte macrophage colony-stimulating factor GM-CSF and interleukin (IL)-4 for 5–7 days to differentiate into immature DCs. The immature DCs are matured in a cytokine cocktail with GM-CSF, IL-4, tumor necrosis factor alpha, IL-1β, and IL-6 for 16–20 hours. The DCs are then loaded with tumor antigen, for example, peptides or tumor lysate; the DCs process these antigens and present epitopes on their MHC molecules at the cell surface. These are then injected back into the patient.34
Current FDA approved dendritic cell vaccines
To date, there is only one DCV that has been granted FDA approval.73 Sipuleucel-T (Dendreon) was accepted in 2010 for the treatment of metastatic, castration-resistant prostate cancer. The benefit of the vaccine was demonstrated in the Immunotherapy for Prostate Adenocarcinoma Treatment study.38 In this double-blind, placebo-controlled, multicenter trial, treatment with sipuleucel-T was associated with improved survival in patients with asymptomatic or minimally symptomatic, castration-resistant, metastatic prostate cancer. It should be noted that there was no objective evidence of delayed time to disease progression, but nonetheless the development of sipuleucel-T was a milestone in immunotherapy for cancer treatment. At the time, it was expected that a flood of DCVs would follow suit and that they would carve out their place in cancer treatment.74 While there have been multiple trials for DCVs in GBM, at this time none of them have been granted FDA approval, owing to the lack of a Phase III trial demonstrating efficacy.
Clinical efficacy
The first 2 studies using DCVs for treatment of patients with HGGs were published in 2001. In 1 paper, Kikuchi et al tested a DCV that used irradiated autologous glioma cells as the antigen on 8 patients with recurrent HGGs. Survival time was not reported in the study, but the authors did note that 2 patients exhibited a partial response to the treatment.40 The other paper published in the same year by Yu et al investigated a DCV using autologous tumor cell surface peptides as the antigen. Interestingly, they saw an increase in median overall survival for 9 patients with newly diagnosed HGGs (7 GBM) from 257 days to 455 days when compared to historical controls.41
In addition to the 2 abovementioned papers, there were 14 studies published from 2001 to 2010 detailing the use of DCVs for the treatment of GBM.42–55 Of these, 10 of the trials used either autologous tumor lysate or cultured tumor cells from the surgical specimen to create their DCV,42,44–46,48–50,52,54,55 1 used irradiated autologous tumor cells,51 1 used tumor RNA from the surgical specimen,43 1 used tumor associated peptides, and 1 used a peptide specific for EGFRvIII conjugated to keyhole limpet hemocyanin (KLH).53 The breakdown of published trials by antigen used in DCV creation is outlined in Figure 1. In this early era of DCV immunotherapy, 12 of the 16 published studies were Phase I trials. Published studies by phase of trial are shown in Figure 2. Nonetheless, there were early signs that DCV therapy might prove beneficial in the treatment of HGGs.
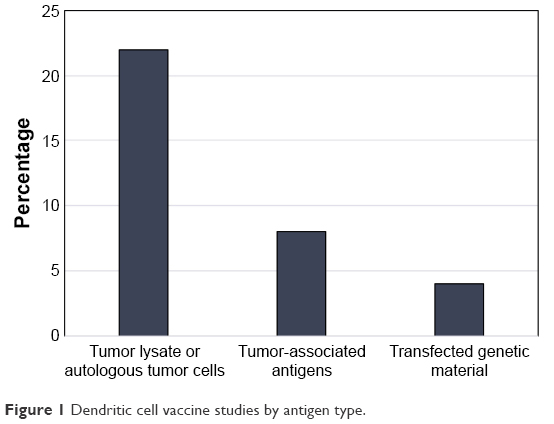 | Figure 1 Dendritic cell vaccine studies by antigen type. |
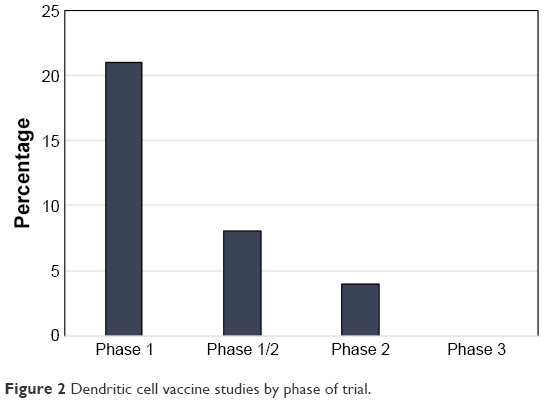 | Figure 2 Dendritic cell vaccine studies by phase of trial. |
In 2004, Kikuchi et al published a follow-up to their original paper, but in this study, they included an IL-12 adjuvant to the DCV. At the 8-week mark, 7 of 8 patients either had stable neuroimaging or a partial response to treatment. Yu et al also conducted another study of 14 patients with either new or recurrent HGGs who were treated with a DCV created from autologous tumor lysate. In this study, the authors saw a statistically significant increase in median survival for vaccinated patients (133 weeks) when compared to age, sex, and disease matched controls (30 weeks).46 Liau et al published the results of a Phase I trial, in which the investigators treated 12 patients with GBM (5 recurrent and 7 newly diagnosed) with a DCV that used acid-eluted, tumor-associated peptides as the antigen. Vaccine-treated patients had significantly increased progression-free (19.9 months vs 8.2 months) and overall survival (35.8 months vs 18.3 months) when compared to historical controls.47 Wheeler et al conducted a Phase II study in 2008 wherein they treated 34 patients with GBM (23 recurrent and 11 newly diagnosed) with a tumor lysate DCV. The authors also measured patient interferon gamma (IFN-γ) levels as an indication of patient response to the vaccine. Interestingly, there was a significant increase in median survival (642 days vs 430 days) in the 17 patients who were deemed vaccine responders based on the pre-treatment and post-treatment IFN-γ levels.52 Lastly, Ardon et al published a pair of studies in 2010. In the first of these, the authors treated 45 patients with a variety of central nervous system tumors, including 32 HGGs, with a DCV created using tumor lysate and adjuvant imiquimod +/- tumor lysate. HGG patients had a median survival of 13.5 months, and there were 4 of 22 patients with recurrent GBM who survived greater than 24 months. In their second paper, 8 patients with newly diagnosed GBM were treated with a DCV created from tumor lysate, with additional tumor lysate as an adjuvant therapy. Median progression-free survival was 18 months, and 3 patients remained alive at the time of last follow-up (>34 months).
Other studies published from 2001 to 2010 were less clear as to whether HGG patients treated with DCVs experienced any significant benefit. Yamanaka et al published 2 papers during this time. In their first study (2003), 2 of 7 patients with recurrent GBM survived greater than 2 years,42 while their Phase I/II study published in 2005 saw a small increase in median survival (480 days vs 400 days) when compared to age, sex, and disease matched controls. In the study published by Caruso et al in 2004,43 both patients who had recurrent GBM saw their disease progress, while Rutkowski et al reported that 1 of 10 patients with recurrent GBM remained disease free after 30 months. In 2007, Okada et al published a study in which they treated 7 patients (1 recurrent GBM, 1 recurrent anaplastic astrocytoma [AA], and 5 recurrent GBM) with a tumor lysate DCV along with TFG-IL4-Neo-TK-transfected fibroblasts as an adjuvant. Interestingly, there was no benefit to the patients with newly diagnosed GBM, but those with recurrent disease experienced a partial response. De Vleeschouwer et al published the largest trial during 2008. In this study, the investigators treated 56 patients with recurrent GBM using a tumor lysate DCV with adjuvant injections of tumor lysate on its own. There were various treatment schedules, and while there was no statistically significant increase in survival for the entire cohort, subgroup analysis revealed a survival benefit in adult patients who had weekly vaccinations. A study by Walker et al used irradiated tumor cells as the DCV antigen and subsequently treated 13 patients with HGGs (9 GBM and 4 AA). Only 2 patents with GBM and 2 patients with AA experienced partial responses, while 1 patient with AA had a complete response after treatment with the DCV. Finally, Sampson et al saw a trend toward survival benefit in their 2009 study where they used peptide specific for EGFRvIII conjugated to KLH as their DCV antigen.
From 2011 to 2017, there have been 17 trials published on DCVs for the treatment of HGGs (Figure 1). During this period, there were 9 Phase I, 5 Phase I/II, and 3 Phase II trials published (Figure 2). Lastly, the trials conducted during this period were slightly larger on average (21.5 patients vs 17.9 patients) than those that took place between 2001 and 2010. As knowledge surrounding DCV therapy advanced, published studies continued to elucidate the potential role for DCVs in the treatment of HGGs.
Tumor lysate was still the most common antigen used to create study vaccines during this time frame. Of these trials, the majority suggested that DCVs might benefit patients with HGGs. There were 3 such studies published in 2011. Chang et al treated 19 patients with HGGs using a DCV created from heat shocked and irradiated tumor cells. The participants had an impressive response, with a median survival of 520 days, and 18.8% of vaccinated patients surviving longer than 5 years. Fadul et al reported on a small trial of 10 newly diagnosed GBM patients who were treated with a DCV from irradiated tumor lysate. The participants had a median progression-free and overall survival of 9.5 and 28 months, respectively. Importantly, 4 of the 10 patients were alive at time of last follow-up (>26 months after their diagnosis).57 Prins et al also performed a small Phase I trial where they enrolled 8 recurrent and 15 newly diagnosed patients with GBM. Participants were treated with both the tumor lysate DCV, and adjuvant imiquimod or Poly-ICLC. Median time to tumor progression was an impressive 15.9 months, and median overall survival was 31.4 months. The authors found improved overall survival in patients treated at initial diagnosis, compared to those with recurrent disease.59 In 2012, Ardon et al enrolled 77 patients with newly diagnosed tumors in a Phase I/II study. Patients were initially treated with DCVs before radiochemotherapy, and boost injections were given throughout its course. Median overall survival was 18.3 months in the intention-to-treat group, which improved to 20.4 months using a per protocol analysis. Unsurprisingly, MGMT promoter methylation was associated with improved overall survival.61 Cho et al performed a randomized Phase II trial wherein 18 patients with a new diagnosis of GBM were assigned to receive DCV and 16 patients were assigned to conventional treatment alone. They reported a dramatic increase in overall survival among the treatment group when compared with the control group (31.9 months vs 15.0 months).62 In a different patient population, Lasky et al used DCVs to treat a small cohort of 7 pediatric patients with HGGs in a Phase I study. While 2 patients who had newly diagnosed tumors survived greater than 40 months, all patients with recurrent disease passed away within 10 months of repeat surgery.64 In 2015, Hunn et al attempted to circumvent TMZ resistance by treating 14 patients with recurrent GBM using a DCV created from autologous tumor lysate, which was exposed to TMZ before vaccine creation. Two of the patients had a prolonged progression-free survival of greater than 12 months. Patient IFN-γ levels were measured to assess immune response and correlated with patient survival.68 Lastly, Inogés et al published the results of a Phase II trial in 2017. The investigators treated 31 patients with newly diagnosed GBM using a DCV from autologous tumor lysate. Median overall survival was 23.4 months, but surprisingly, there was no association found between patient immune response and survival. Again, MGMT promoter sequence methylation was associated with increased survival.72
The use of tumor-associated antigens (TAAs) as target peptides for DCVs gained popularity during the period from 2011 to 2017. Okada et al published 1 such paper in 2011, where the authors treated 22 patients with recurrent HGGs, all of whom were HLA-A2+, with a DCV primed against 4 TAAs: IL-13Rα2, EphA2, gp100, and YKL-40. Patients also received adjuvant treatment with Poly-ICLC. Median time to tumor progression in vaccine-treated patients with GBM was 4 months, compared to 12 months for other malignant gliomas.58 The following year, Akiyama et al published the results of a Phase I trial where they treated 9 recurrent HGG patients, who were either HLA-A2+ or HLA-A24+, with a DCV created using a different set of TAAs: WT-1, HER2, MAGE-A3, MAGE-A1, and gp100. The results of this study left much to be desired, as 8 of 9 patients had disease progression despite the therapy.60 In 2013, Phuphanich et al published another Phase I trial, in which they treated 20 patients with GBM and 1 brainstem glioma patient with a DCV primed against various synthetic peptides (HER2, TRP-2, gp100, MAGE-1, IL-13Rα2, and AIM-2). Median progression-free and overall survival was an impressive 16.9 and 38.4 months, respectively.65 Lastly, Sakai et al performed another small Phase I trial in 10 patients with recurrent HGGs, which they published in 2015. In this study, the investigators performed immunostaining for WT-1 on the surgical specimens. Patients whose tumors stained positive were treated with a DCV primed against WT-1, whereas patients who stained negative were treated with a DCV borne from autologous tumor lysate. Median overall survival was 26 months in the entire cohort and 18 months in the 6 patients who had recurrent GBM.70
There were 3 studies from 2011 to 2017 where transfected mRNA was used as the antigen to create DCVs. Vik-Mo et al published the first of these in 2013. In their Phase I/II trial, the investigators treated 7 patients with GBM using a DCV primed against transfected tumor mRNA. The participants had a median overall survival of 759 days, which compared favorably to matched historical controls, who only had median survival of 585 days.67 In 2015, Mitchell et al, working under the supervision of Sampson et al, used transfected pp65 mRNA from cytomegalovirus as the DCV antigen to treat 12 patients with a new diagnosis of GBM in a Phase I trial. They also used tetanus-diphtheria (Td) toxoid or matured DCs as site preconditioning, prior to injecting the DCV. The authors reported that 3 of the 6 patients in the Td group were alive without evidence of disease at time of last follow-up (greater than 44.1 months). In the DC preconditioning group, median progression-free and overall survival was 10.8 and 18.5 months, respectively.69 The same author group, this time led by Batich et al, published another Phase I study using transfected pp65 mRNA as DCV antigen in 2017. This time the vaccine was admixed with GM-CSF, and patients received dose-intensified TMZ. For the 11 patients treated, median overall survival was 41.1 months, compared to 19.2 months for historical controls.71
There was one publication on DCV therapy in HGGs that combined the results of 2 parallel studies. In 2013, Prins et al performed a comparison of DCV therapy using tumor lysate vs TAAs in the creation of their DCVs. A total of 28 patients were treated with the tumor lysate DCV and 6 patients received the TAA DCV. All patients in the TAA group were HLA-A2+. Median overall survival in the tumor lysate group was 34.4 months, compared to 14.5 months in the TAA group. Of note, the authors reported that 60% of patients screened for the TAA group were ineligible based on HLA typing, which they cited as a limitation of this antigen choice.66
Recurrent vs de novo HGGs
Recurrent HGGs are met with an exceptionally poor prognosis. As such, many of the early trials for DCVs as a treatment for HGGs were conducted on patients with recurrent disease. Overall 12 of the 33 published trials were performed in patients whose disease recurred after initial treatment.40,42–45,48,50,54,58,60,68,70 Not surprisingly, success was limited in this patient population, with only 2 studies reporting survival benefit.48,50 However, many of the early studies on relapsed HGGs reported other findings, such as radiological response or immune response after vaccination, making any conclusions on survival benefit difficult.
There were 10 studies published that included both recurrent and de novo HGGs.46,47,49,51,52,56,59,64–66 Results in this group were more encouraging, as 5 of the trials reported a survival benefit.46,47,52,56,59 Importantly, 2 papers compared the benefit associated with DCV therapy between recurrent and de novo HGGs, with both seeing improved efficacy in newly diagnosed patients.59,64
Eleven studies looked exclusively at DCV therapy in de novo HGGs,41,53,55,57,61–63,67,69,71,72 and results from these trials were mostly positive as 7 trials reported a survival benefit associated with DCV use.41,53,61–63,67,71 Although it is possible that DCV therapy is more effective in newly diagnosed HGGs than recurrent disease, heterogeneity in the reporting of outcomes and conduct of trials limits meaningful comparison. Many of the early trials on recurrent patients were small Phase I studies designed to assess safety and feasibility of DCV therapy, whereas the newer trials that include de novo tumors are aimed at assessing survival. Furthermore, it is possible that survival benefit in the patients with new diagnoses of HGGs is due to a lead-time effect, as opposed to improved efficacy in tumors that have not been previously treated. Nonetheless, the most important takeaway from this qualitative analysis may be that DCVs have been reported to improve survival in both recurrent and de novo HGG patients, at least in a portion of the published trials. The underlying reasons why some patients respond, and others do not, regardless of the novelty of their tumor, remain to be elucidated.
Safety and tolerability
Five publications reported no toxicities related to the use of DCV therapy. In total, there were 399 adverse events reported, not including the expected minor delayed type hypersensitivity reaction at the injection site. The most common adverse event was fatigue, which occurred in 11% of the patients enrolled in the trials. There are several severe adverse events that warrant mention.
Three patients suffered from fatal adverse events during their study participation. One of these was reported as a grade V infection,61 and the other 2 were reported as fatal bacteria pneumonia.72 There was also a number of participants who suffered from significant neurotoxicity: 5 patients developed status epilepticus;55,61 2 patients had significant peritumoral edema, which caused grade IV neurotoxicity;45,50 and 2 patients suffered from ischemic strokes.55,61 Lastly, 1 patient developed an ischemic bowel,61 1 patient suffered from a deep venous thrombosis,61 and 1 patient had a grade IV elevation in their alkaline phosphatase.64
There was substantial variation among the publications in the reporting of adverse events. Authors rarely mentioned the grade of the adverse events (as specified by the National Cancer Institute – Common Terminology Criteria for the Reporting of Adverse Events) unless they were grade VI or greater. In addition, many of the reported adverse events may not have been related to DCV administration (eg, post-operative neurological deficit).62,68,72 Nonetheless, most adverse events reported were minor, and DCVs appear to have an acceptable safety profile, especially considering the potential clinical efficacy highlighted in the preceding section.
Quality of life
Of the 33 published studies on DCV treatment for HGGs, only 6 papers formally assessed quality of life as an outcome measure. Two papers published by Ardon et al in 2010 used the Fertigkeitenskala Münster-Heidelberg (FMH) questionnaire to assess quality of life during DCV treatment.54,55 This is a self-reported questionnaire that assesses one’s ability to carry out daily activities.75 One of these papers also used the QLQ-C30,55 a scale developed by the European Organization for Research and Treatment of Cancer that assesses functional disability, somatic symptoms, global health, and overall quality of life.76 In one of these studies, all patients reported FMH scores below the 35th percentile, meaning that they all had difficulty performing daily activities. However, the authors point out that their scores did not change significantly during DCV therapy, indicating that much of the disability may be secondary to their disease, and/or other therapies.54 The group’s other paper found FMH scores to remain quite high during their vaccine treatment, and most patients were able to continue carrying out their daily activities. This finding was supported by the QLQ-C30, as 6 out of 7 patients in the study rated their general health and overall quality of life as good.55
Two studies published in 2012 also assessed quality of life in patients undergoing DCV therapy. Cho et al, using the Karnofsky Performance Scale (KPS) as their outcome measure, found that median KPS was higher in patients undergoing DCV therapy (70) than those in the control group (50) at the end of the trial.62 Another study by Jie et al found patients undergoing DCV therapy to maintain their functional dependence better than those in control groups. At 6- and 9-month follow-up, a significantly higher proportion of patients in the DCV group were fully independent compared with patients who did not receive DCV.63
Lasky et al made a very brief mention of quality of life in their results of a study of 7 pediatric patients with HGGs. They simply pointed out that 2 patients who were among the 3 patients to receive DCV suffered mild hemiparesis from their tumor surgery but were cognitively well and alive at the time the paper was published. Lastly, Hunn et al reported on quality of life in their 2015 study. Using the QLQ-C30, the investigators found that patients’ general health and overall quality of life remained generally stable during their DCV therapy. They reasoned that DCV therapy was reasonably well tolerated and did not significantly increase the morbidity related to chemotherapy and surgery in the treatment of HGGs.
Perspectives and future directions
The positive results and relative safety seen with DCV use have spurred great interest and further investment in this novel therapy. Currently, the field of neuro-oncology is eagerly awaiting the results of its first Phase III trial using DCV treatment for GBM (clinical trial registration # NCT00045968). Two other Phase III trials are listed on ClinicalTrials.gov, one of which has suspended recruitment due to financial concerns (clinical trial registration # NCT02546102), while another is currently enrolling patients by invitation (clinical trial registration # NCT01759810). While the publication of large Phase III trials will more definitively answer the question of whether DCVs are a viable therapeutic option for patients with GBM, an examination of the previously discussed results provides reason for optimism. Seven of the 11 published studies using DCV as a treatment in newly diagnosed HGGs showed survival benefit, and although that was true in less than 20% of the trials evaluating DCVs in recurrent HGGs, these studies were largely aimed at proof of concept and evaluating safety for this novel therapy.
While DCV therapy has been approved for the treatment of castration-resistant metastatic prostate cancer,38 it remains the only one to secure FDA approval. On the other hand, immune checkpoint inhibitors have also found success in improving the prognosis of patients with various malignancies. In the past decade, large trials have shown immune checkpoint inhibitors such as anti-CTLA4 and anti-PD1 monoclonal antibodies (mAbs) to be effective as a treatment option in melanoma and non-small cell lung cancer.77–80 There are currently 2 Phase III trials investigating the use of Nivolumab (an anti-PD1 mAb) in patients with GBM (clinical trial registration # NCT02667587 and # NCT02617589). At present, it remains unclear what form of immunotherapy, alone or in combination, will reign supreme in improving prognosis and limiting treatment-related adverse events for patients with this terrible disease.
With regard to production, DCs normally take 5–7 days to mature. To improve accessibility of these vaccines to patients, so-called “fastDCs” have been developed that reduce the in vitro culture time to as short as 2 days, while retaining the potency of DCs to activate immunologic responses.81 FastDCs provide higher yields in culture and more effective priming of tumor-specific cytotoxic T cells.34,82
Further, less than 5% of injected DCs reach the LNs.83 It has been demonstrated that improving DC LN homing can improve the efficacy of tumor antigen-specific DCs.69 Mitchell et al69 used the Td toxoid as a potent recall antigen to pre-condition the vaccine site in GBM patients. The authors found that patients given Td had enhanced DC migration and significantly improved survival.
One potential avenue to overcome both the abovementioned limitations – lengthy DC production times and poor migration – is targeting DCs in vivo.84 In vivo DC vaccination strategies use mAbs to target DC-specific cell surface receptors. CD205 is a novel receptor that mediates antigen uptake and presentation to T cells. Injection of tumor (melanoma) antigens conjugated to anti-CD205-antibodies stimulated CD4+ and CD8+ T cells and slowed tumor growth in mice.85 The feasibility of an in vivo approach in humans has been demonstrated using a vaccine of human CD205 antibody fused to the tumor antigen NY-ESO-1.86 An in vivo approach involving mAb-mediated delivery of GBM antigens to DCs remains to be tested, and this certainly merits future investigation.
Strategies such as mentioned previously that optimize the production, targeting, and antigen loading of DCs and overcome the problem of limited migration, combined with immune checkpoint modulators that “release the brakes” on the immunosuppressed tumor microenvironment, will help improve the efficacy of DC vaccines for GBM. It is a probability that an optimized vaccine will reach Phase III trial and with promising results, bring this therapeutic approach into mainstream clinical use.
At the health care systems level, it is important to recognize that not every institution will be able to offer this therapy. Clinical care and referral pathways will need to be implemented to support a centralized model of care. Given the potential for serious adverse events related to cerebral edema and resultant elevated intracranial pressure, patients will need to be treated in close proximity to a neurosurgical center and monitored closely. Protocols will need to be instituted for handling complications.
Conclusion
The field of DC immunotherapy continues to evolve at an exponential rate. The FDA approval of sipuleucel-T for the treatment of metastatic prostate cancer marked an important milestone for the prospect of immunotherapy in oncology. As our understanding of physiologic immune surveillance in tumor control continues to grow, we can expect to see immune modulation make its way into standard therapeutic protocols in neuro-oncology as well. Indeed, with the promising results provided by clinical trials of DCV immunotherapy for GBM, it is perhaps more a probability than a possibility that in the near future, surgery, cytotoxic therapies (ie, radiochemotherapy), and immunotherapy will form a three-pronged therapeutic approach that will enhance clinical outcomes.
Disclosure
The authors report no conflicts of interest in this work.
References
Louis DN, Ohgaki H, Wiestler OD, et al. The 2007 WHO classification of tumours of the central nervous system. Acta Neuropathol. 2007;114(2):97–109. | ||
Ostrom QT, Gittleman H, Fulop J, et al. CBTRUS Statistical Report: primary brain and central nervous system tumors diagnosed in the United States in 2008–2012. Neuro Oncol. 2015;17(Suppl 4): iv1– iv62. | ||
Darlix A, Zouaoui S, Rigau V, et al. Epidemiology for primary brain tumors: a nationwide population-based study. J Neuro Oncol. 2017;131(3):525–546. | ||
Weller M, van den Bent M, Hopkins K, et al. EANO guideline for the diagnosis and treatment of anaplastic gliomas and glioblastoma. Lancet Oncol. 2014;15(9):E395–E403. | ||
Wen PY, Kesari S. Malignant gliomas in adults. N Engl J Med. 2008;359(5):492–507. | ||
Stupp R, Mason WP, van den Bent MJ, et al; European Organisation for Research and Treatment of Cancer Brain Tumor and Radiotherapy Groups; National Cancer Institute of Canada Clinical Trials Group. Radiotherapy plus concomitant and adjuvant temozolomide for glioblastoma. N Engl J Med. 2005;352(10):987–996. | ||
Badhiwala J, Decker WK, Berens ME, Bhardwaj RD. Clinical trials in cellular immunotherapy for brain/CNS tumors. Expert Rev Neurother. 2013;13(4):405–424. | ||
Thon N, Kreth S, Kreth FW. Personalized treatment strategies in glioblastoma: MGMT promoter methylation status. Onco Targets Ther. 2013;6:1363–1372. | ||
Hegi ME, Diserens AC, Gorlia T, et al. MGMT gene silencing and benefit from temozolomide in glioblastoma. N Engl J Med. 2005;352(10):997–1003. | ||
Esteller M, Garcia-Foncillas J, Andion E, et al. Inactivation of the DNA-repair gene MGMT and the clinical response of gliomas to alkylating agents. N Engl J Med. 2000;343(19):1350–1354. | ||
Belka C, Budach W, Kortmann RD, Bamberg M. Radiation induced CNS toxicity – molecular and cellular mechanisms. Br J Cancer. 2001;85(9):1233–1239. | ||
Monje ML, Palmer T. Radiation injury and neurogenesis. Curr Opin Neurol. 2003;16(2):129–134. | ||
Polivka J Jr, Polivka J, Holubec L, et al. Advances in experimental targeted therapy and immunotherapy for patients with glioblastoma multiforme. Anticancer Res. 2017;37(1):21–33. | ||
Wang G, Fu XL, Wang JJ, Guan R, Tang XJ. Novel strategies to discover effective drug targets in metabolic and immune therapy for glioblastoma. Curr Cancer Drug Targets. 2017;17(1):17–39. | ||
Clarke J, Butowski N, Chang S. Recent advances in therapy for glioblastoma. Arch Neurol. 2010;67(3):279–283. | ||
Kim R, Emi M, Tanabe K. Cancer immunoediting from immune surveillance to immune escape. Immunology. 2007;121(1):1–14. | ||
Mellman I, Coukos G, Dranoff G. Cancer immunotherapy comes of age. Nature. 2011;480(7378):480–489. | ||
Palucka K, Banchereau J. Cancer immunotherapy via dendritic cells. Nat Rev Cancer. 2012;12(4):265–277. | ||
Pardoll DM. The blockade of immune checkpoints in cancer immunotherapy. Nat Rev Cancer. 2012;12(4):252–264. | ||
Restifo NP, Dudley ME, Rosenberg SA. Adoptive immunotherapy for cancer: harnessing the T cell response. Nat Rev Immunol. 2012;12(4):269–281. | ||
Rosenberg SA, Yang JC, Restifo NP. Cancer immunotherapy: moving beyond current vaccines. Nat Med. 2004;10(9):909–915. | ||
Nitta T, Hishii M, Sato K, Okumura K. Selective expression of interleukin-10 gene within glioblastoma-multiforme. Brain Res. 1994;649(1–2):122–128. | ||
Sawamura Y, Diserens AC, de Tribolet N. In vitro prostaglandin-E2 production by glioblastoma cells and its effect on interleukin-2 activation of oncolytic lymphocytes. J Neuro Oncol. 1990;9(2):125–130. | ||
Couldwell WT, Yong VW, Dore Duffy P, Freedman MS, Antel JP. Production of soluble autocrine inhibitory factors by human glioma cell-lines. J Neurol Sci. 1992;110(1–2):178–185. | ||
Yang BC, Lin HK, Hor WS, et al. Mediation of enhanced transcription of the IL-10 gene in T cells, upon contact with human glioma cells, by Fas signaling through a protein kinase A-independent pathway. J Immunol. 2003;171(8):3947–3954. | ||
Rorive S, Belot N, Decaestecker C, et al. Galectin-1 is highly expressed in human gliomas with relevance for modulation of invasion of tumor astrocytes into the brain parenchyma. Glia. 2001;33(3):241–255. | ||
Parsa AT, Waldron JS, Panner A, et al. Loss of tumor suppressor PTEN function increases B7-H1 expression and immunoresistance in glioma. Nat Med. 2007;13(1):84–88. | ||
Facoetti A, Nano R, Zelini P, et al. Human leukocyte antigen and antigen processing machinery component defects in astrocytic tumors. Clin Cancer Res. 2005;11(23):8304–8311. | ||
Fecci PE, Mitchell DA, Whitesides JF, et al. Increased regulatory T-cell fraction amidst a diminished CD4 compartment explains cellular immune defects in patients with malignant glioma. Cancer Res. 2006;66(6):3294–3302. | ||
El Andaloussi A, Lesniak MS. An increase in CD4+CD25+FOXP3+ regulatory T cells in tumor-infiltrating lymphocytes of human glioblastoma multiforme. Neuro Oncol. 2006;8(3):234–243. | ||
Jacobs JF, Idema AJ, Bol KF, et al. Regulatory T cells and the PD-L1/PD-1 pathway mediate immune suppression in malignant human brain tumors. Neuro Oncol. 2009;11(4):394–402. | ||
Gustafson MP, Lin Y, New KC, et al. Systemic immune suppression in glioblastoma: the interplay between CD14(+)HLA-DRlo/neg monocytes, tumor factors, and dexamethasone. Neuro Oncol. 2010;12(7):631–644. | ||
Benencia F, Sprague L, McGinty J, Pate M, Muccioli M. Dendritic cells the tumor microenvironment and the challenges for an effective antitumor vaccination. J Biomed Biotechnol. 2012;2012:425476. | ||
Schaller TH, Sampson JH. Advances and challenges: dendritic cell vaccination strategies for glioblastoma. Expert Rev Vaccines. 2017;16(1):27–36. | ||
Ashley DM, Faiola B, Nair S, Hale LP, Bigner DD, Gilboa E. Bone marrow-generated dendritic cells pulsed with tumor extracts or tumor RNA induce antitumor immunity against central nervous system tumors. J Exp Med. 1997;186(7):1177–1182. | ||
Fuertes MB, Kacha AK, Kline J, et al. Host type I IFN signals are required for antitumor CD8+ T cell responses through CD8{alpha}+ dendritic cells. J Exp Med. 2011;208(10):2005–2016. | ||
Diamond MS, Kinder M, Matsushita H, et al. Type I interferon is selectively required by dendritic cells for immune rejection of tumors. J Exp Med. 2011;208(10):1989–2003. | ||
Kantoff PW, Higano CS, Shore ND, et al; IMPACT Study Investigators. Sipuleucel-T immunotherapy for castration-resistant prostate cancer. N Engl J Med. 2010;363(5):411–422. | ||
Dillman RO. Cancer immunotherapy. Cancer Biother Radiopharm. 2011;26(1):1–64. | ||
Kikuchi T, Akasaki Y, Irie M, Homma S, Abe T, Ohno T. Results of a phase I clinical trial of vaccination of glioma patients with fusions of dendritic and glioma cells. Cancer Immunol Immunother. 2001;50(7):337–344. | ||
Yu JS, Wheeler CJ, Zeltzer PM, et al. Vaccination of malignant glioma patients with peptide-pulsed dendritic cells elicits systemic cytotoxicity and intracranial T-cell infiltration. Cancer Res. 2001;61(3):842–847. | ||
Yamanaka R, Abe T, Yajima N, et al. Vaccination of recurrent glioma patients with tumour lysate-pulsed dendritic cells elicits immune responses: results of a clinical phase I/II trial. Br J Cancer. 2003;89(7):1172–1179. | ||
Caruso DA, Orme LM, Neale AM, et al. Results of a phase 1 study utilizing monocyte-derived dendritic cells pulsed with tumor RNA in children and young adults with brain cancer. Neuro Oncol. 2004;6(3):236–246. | ||
Kikuchi T, Akasaki Y, Abe T, et al. Vaccination of glioma patients with fusions of dendritic and glioma cells and recombinant human interleukin 12. J Immunother. 2004;27(6):452–459. | ||
Rutkowski S, De Vleeschouwer S, Kaempgen E, et al. Surgery and adjuvant dendritic cell-based tumour vaccination for patients with relapsed malignant glioma, a feasibility study. Br J Cancer. 2004;91(9):1656–1662. | ||
Yu JS, Liu GT, Ying H, Yong WH, Black KL, Wheeler CJ. Vaccination with tumor lysate-pulsed dendritic cells elicits antigen-specific, cytotoxic T-cells in patients with malignant glioma. Cancer Res. 2004;64(14):4973–4979. | ||
Liau LM, Prins RM, Kiertscher SM, et al. Dendritic cell vaccination in glioblastoma patients induces systemic and intracranial T-cell responses modulated by the local central nervous system tumor microenvironment. Clin Cancer Res. 2005;11(15):5515–5525. | ||
Yamanaka R, Homma J, Yajima N, et al. Clinical evaluation of dendritic cell vaccination for patients with recurrent glioma: results of a clinical Phase I/II trial. Clin Cancer Res. 2005;11(11):4160–4167. | ||
Okada H, Lieberman FS, Walter KA, et al. Autologous glioma cell vaccine admixed with interleukin-4 gene transfected fibroblasts in the treatment of patients with malignant gliomas. J Transl Med. 2007;5:67. | ||
De Vleeschouwer S, Fieuws S, Rutkowski S, et al. Postoperative adjuvant dendritic cell-based immunotherapy in patients with relapsed glioblastoma multiforme. Clin Cancer Res. 2008;14(10):3098–3104. | ||
Walker DG, Laherty R, Tomlinson FH, Chuah T, Schmidt C. Results of a Phase I dendritic cell vaccine trial for malignant astrocytoma: potential interaction with adjuvant chemotherapy. J Clin Neurosci. 2008;15(2):114–121. | ||
Wheeler CJ, Black KL, Liu GT, et al. Vaccination elicits correlated immune and clinical responses in glioblastoma multiforme patients. Cancer Res. 2008;68(14):5955–5964. | ||
Sampson JH, Archer GE, Mitchell DA, et al. An epidermal growth factor receptor variant III-targeted vaccine is safe and immunogenic in patients with glioblastoma multiforme. Mol Cancer Ther. 2009;8(10):2773–2779. | ||
Ardon H, De Vleeschouwer S, Van Calenbergh F, et al. Adjuvant dendritic cell-based tumour vaccination for children with malignant brain tumours. Pediatr Blood Cancer. 2010;54(4):519–525. | ||
Ardon H, Van Gool S, Lopes IS, et al. Integration of autologous dendritic cell-based immunotherapy in the primary treatment for patients with newly diagnosed glioblastoma multiforme: a pilot study. J Neuro Oncol. 2010;99(2):261–272. | ||
Chang CN, Huang YC, Yang DM, et al. A Phase I/II clinical trial investigating the adverse and therapeutic effects of a postoperative autologous dendritic cell tumor vaccine in patients with malignant glioma. J Clin Neurosci. 2011;18(8):1048–1054. | ||
Fadul CE, Fisher JL, Hampton TH, et al. Immune response in patients with newly diagnosed glioblastoma multiforme treated with intranodal autologous tumor lysate-dendritic cell vaccination after radiation chemotherapy. J Immunother. 2011;34(4):382–389. | ||
Okada H, Kalinski P, Ueda R, et al. Induction of CD8(+) T-cell responses against novel glioma-associated antigen peptides and clinical activity by vaccinations with alpha-type 1 polarized dendritic cells and polyinosinic-polycytidylic acid stabilized by lysine and carboxymethylcellulose in patients with recurrent malignant glioma. J Clin Oncol. 2011;29(3):330–336. | ||
Prins RM, Soto H, Konkankit V, et al. Gene expression profile correlates with T-cell infiltration and relative survival in glioblastoma patients vaccinated with dendritic cell immunotherapy. Clin Cancer Res. 2011;17(6):1603–1615. | ||
Akiyama Y, Oshita C, Kume A, et al. α-type-1 polarized dendritic cell-based vaccination in recurrent high-grade glioma: a phase I clinical trial. BMC Cancer. 2012;12:623. | ||
Ardon H, Van Gool SW, Verschuere T, et al. Integration of autologous dendritic cell-based immunotherapy in the standard of care treatment for patients with newly diagnosed glioblastoma: results of the HGG-2006 phase I/II trial. Cancer Immunol Immunother. 2012;61(11):2033–2044. | ||
Cho DY, Yang WK, Lee HC, et al. Adjuvant immunotherapy with whole-cell lysate dendritic cells vaccine for glioblastoma multiforme: a phase II clinical trial. World Neurosurg. 2012;77(5–6):736–744. | ||
Jie X, Hua L, Jiang W, Feng F, Feng G, Hua Z. Clinical application of a dendritic cell vaccine raised against heat-shocked glioblastoma. Cell Biochem Biophys. 2012;62(1):91–99. | ||
Lasky JL 3rd, Panosyan EH, Plant A, et al. Autologous tumor lysate-pulsed dendritic cell immunotherapy for pediatric patients with newly diagnosed or recurrent high-grade gliomas. Anticancer Res. 2013;33(5):2047–2056. | ||
Phuphanich S, Wheeler CJ, Rudnick JD, et al. Phase I trial of a multi-epitope-pulsed dendritic cell vaccine for patients with newly diagnosed glioblastoma. Cancer Immunol Immunother. 2013;62(1):125–135. | ||
Prins RM, Wang XY, Soto H, et al. Comparison of glioma-associated antigen peptide-loaded versus autologous tumor lysate-loaded dendritic cell vaccination in malignant glioma patients. J Immunother. 2013;36(2):152–157. | ||
Vik-Mo EO, Nyakas M, Mikkelsen BV, et al. Therapeutic vaccination against autologous cancer stem cells with mRNA-transfected dendritic cells in patients with glioblastoma. Cancer Immunol Immunother. 2013;62(9):1499–1509. | ||
Hunn MK, Bauer E, Wood CE, et al. Dendritic cell vaccination combined with temozolomide retreatment: results of a phase I trial in patients with recurrent glioblastoma multiforme. J Neuro Oncol. 2015;121(2):319–329. | ||
Mitchell DA, Batich KA, Gunn MD, et al. Tetanus toxoid and CCL3 improve dendritic cell vaccines in mice and glioblastoma patients. Nature. 2015;519(7543):366–369. | ||
Sakai K, Shimodaira S, Maejima S, et al. Dendritic cell-based immunotherapy targeting Wilms’ tumor 1 in patients with recurrent malignant glioma. J Neurosurg. 2015;123(4):989–997. | ||
Batich KA, Reap EA, Archer GE, et al. Long-term survival in glioblastoma with cytomegalovirus pp65-targeted vaccination. Clin Cancer Res. 2017;23(8):1898–1909. | ||
Inogés S, Tejada S, de Cerio AL, et al. A phase II trial of autologous dendritic cell vaccination and radiochemotherapy following fluorescence-guided surgery in newly diagnosed glioblastoma patients. J Transl Med. 2017;15(1):104. | ||
Garg AD, Vara Perez M, Schaaf M, et al. Trial watch: dendritic cell-based anticancer immunotherapy. Oncoimmunology. 2017;6(7):e1328341. | ||
Hovden AO, Appel S. The first dendritic cell-based therapeutic cancer vaccine is approved by the FDA. Scand J Immunol. 2010;72(6):554. | ||
Wolff JE, Däumling E, Dirksen A, Dabrock A, Hartmann M, Jürgens H. [Munster Heidelberg Abilities Scale – a measuring instrument for global comparison of illness sequelae]. Klin Padiatr. 1996;208(5):294–298. [Article in German]. | ||
Aaronson NK, Ahmedzai S, Bergman B, et al. The European Organization for Research and Treatment of Cancer QLQ-C30: a quality-of-life instrument for use in international clinical trials in oncology. J Natl Cancer Inst. 1993;85(5):365–376. | ||
Hodi FS, O’Day SJ, McDermott DF, et al. Improved survival with ipilimumab in patients with metastatic melanoma. N Engl J Med. 2010;363(8):711–723. | ||
Robert C, Long GV, Brady B, et al. Nivolumab in previously untreated melanoma without BRAF mutation. N Engl J Med. 2015;372(4):320–330. | ||
Hamid O, Robert C, Daud A, et al. Safety and tumor responses with lambrolizumab (anti-PD-1) in melanoma. N Engl J Med. 2013;369(2):134–144. | ||
Garon EB, Rizvi NA, Hui R, et al; KEYNOTE-001 Investigators. Pembrolizumab for the treatment of non-small-cell lung cancer. N Engl J Med. 2015;372(21):2018–2028. | ||
Ramadan G. Generation of functional monocyte-derived fast dendritic cells suitable for clinical application in the absence of interleukin-6. Cytotechnology. 2011;63(5):513–521. | ||
Kvistborg P, Boegh M, Pedersen AW, Claesson MH, Zocca MB. Fast generation of dendritic cells. Cell Immunol. 2009;260(1):56–62. | ||
De Vries IJ, Krooshoop DJ, Scharenborg NM, et al. Effective migration of antigen-pulsed dendritic cells to lymph nodes in melanoma patients is determined by their maturation state. Cancer Res. 2003;63(1):12–17. | ||
Radford KJ, Tullett KM, Lahoud MH. Dendritic cells and cancer immunotherapy. Curr Opin Immunol. 2014;27:26–32. | ||
Mahnke K, Qian Y, Fondel S, Brueck J, Becker C, Enk AH. Targeting of antigens to activated dendritic cells in vivo cures metastatic melanoma in mice. Cancer Res. 2005;65(15):7007–7012. | ||
Dhodapkar MV, Sznol M, Zhao B, et al. Induction of antigen-specific immunity with a vaccine targeting NY-ESO-1 to the dendritic cell receptor DEC-205. Sci Transl Med. 2014;6(232):232ra251. |
 © 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.