")
Back to Journals » International Journal of Nanomedicine » Volume 10 » Issue 1
Delivery of microRNA-146a with polyethylenimine nanoparticles inhibits renal fibrosis in vivo
Authors Morishita Y, Imai T, Yoshizawa H, Watanabe M, Ishibashi K, Muto S, Nagata D
Received 10 February 2015
Accepted for publication 31 March 2015
Published 11 May 2015 Volume 2015:10(1) Pages 3475—3488
DOI https://doi.org/10.2147/IJN.S82587
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 4
Editor who approved publication: Dr Thomas Webster
Yoshiyuki Morishita,1 Toshimi Imai,1 Hiromichi Yoshizawa,1 Minami Watanabe,1 Kenichi Ishibashi,2 Shigeaki Muto,1 Daisuke Nagata1
1Division of Nephrology, Department of Medicine, Jichi Medical University, Tochigi, 2Department of Medical Physiology, Meiji Pharmaceutical University, Tokyo, Japan
Abstract: Renal fibrosis is the final common pathway leading to end-stage renal disease. Although microRNA (miR) was recently shown to be involved in the development of renal fibrosis, few studies have focused on the effects on renal fibrosis of exogenous miR delivered in an in vivo therapeutic setting. The study reported here investigated the effects of miR-146a delivery using polyethylenimine nanoparticles (PEI-NPs) on renal fibrosis in vivo. PEI-NPs bearing miR-146 or control-miR (nitrogen/phosphate ratio: 6) were injected into the tail vein of a mouse model of renal fibrosis induced by unilateral ureteral obstruction. PEI-NPs bearing miR-146 significantly enhanced miR-146a expression in the obstructed kidney compared with the control group, while inhibiting the renal fibrosis area, expression of alpha-smooth muscle actin, and infiltration of F4/80-positive macrophages into the obstructed kidney. In addition, PEI-NPs bearing miR-146a inhibited the transforming growth factor beta 1–Smad and tumor necrosis factor receptor-associated factor 6–nuclear factor kappa B signaling pathways. Control-miR-PEI-NPs did not show any of these effects. These results suggest that the delivery of miR-146a attenuated renal fibrosis by inhibiting pro-fibrotic and inflammatory signaling pathways and that the delivery of appropriate miRs may be a therapeutic option for preventing renal fibrosis in vivo.
Keywords: miR, end-stage renal disease, pro-fibrotic signaling pathway, inflammatory signaling pathway
Introduction
Renal fibrosis is the final common pathway leading to end-stage renal disease, regardless of underling etiology.1,2 Renal fibrosis is characterized by the excessive accumulation of extracellular matrix components, such as collagen, in interstitial spaces.3 Inhibition of renal fibrosis is important in improving the prognosis of patients with chronic kidney diseases, but, to date, no method has been developed for the treatment of renal fibrosis. Thus, the development of therapeutic approaches and the identification of target molecules that can inhibit renal fibrosis are required.
Numerous studies have demonstrated that the transforming growth factor beta 1 (TGF-β1)–Smad signaling pathway plays a central role in the development of renal fibrosis.4–6 TGF-β1 receptor-activated Smad3 combines with a common Smad, Smad4, and this complex is translocated to the nucleus, where it binds to DNA elements to promote the transcription of various pro-fibrotic genes.4–6 Smad4 knockout in the kidney was found to enhance inflammation, as evidenced by leukocyte and macrophage infiltration.6 Unresolved inflammation characterized by macrophage infiltration has also been reported to promote progressive renal fibrosis.7–9 Infiltration by macrophages is invariably observed at sites of renal fibrosis, with the degree of infiltration correlated with the extent of fibrosis.10,11 Nuclear factor kappa B (NF-κB) is a major signaling pathway that activates and promotes macrophage infiltration.12,13 Blocking NF-κB signaling can suppress renal injury, whereas enhancing this pathway accelerates renal injury, including renal fibrosis, in vivo.12,13 Taken together, these findings suggest that blocking the TGF-β1–Smad signaling pathway and inflammatory signals, such as NF-κB, may be a potential strategy to inhibit renal fibrosis.
Gene therapy is a promising approach in the treatment of renal fibrosis because it can target specific molecules that are involved in or promote fibrosis. MicroRNAs (miRs) are small noncoding RNAs that inhibit the posttranscriptional processing of target messenger RNAs (mRNAs) by binding to certain specific sequences.14 Several miRs have been reported to suppress transcription of important mRNAs that regulate the development of renal fibrosis.15–24 However, few studies have focused on the effects of exogenous miR delivery on renal fibrosis in an in vivo therapeutic setting.15–18,24 miR-146a has been reported to inhibit inflammation by suppressing target mRNAs, such as those encoding tumor necrosis factor receptor-associated factor 6 (TRAF6), interleukin-1 receptor-associated kinase 1 (IRAK1), and Toll-like receptor 4 (TLR4), resulting in the inhibition of NF-κB in several organs.25–29 Recently, miR-146a was reported to inhibit the TGF-β1–Smad signaling pathway by suppressing Smad4.30 Increased miR-146a expression has been observed in kidney samples of diabetic nephropathy patients.31 Based on these findings, we hypothesized that miR-146a might strongly inhibit renal fibrosis by inhibiting the TGF-β1–Smad signaling pathway and inflammation in the kidney. To test this hypothesis, we used polyethylenimine (PEI) nanoparticles (NPs) (in vivo-jetPEI®), which have been reported effective in delivering oligonucleotides to the kidneys,32,33 to deliver synthetic miR-146a to the kidneys in vivo. Using a mouse model of renal fibrosis induced by unilateral ureteral obstruction (UUO), we tested the effects of miR-146 overexpression on renal fibrosis.
Materials and methods
Ethical considerations
Experimental protocols were approved by the Animal Ethics Committee of Jichi Medical University and were performed in accordance with the guide for use and care of laboratory animals of the Experimental Animal Commitee of the Jichi Medical University.
Mouse model of renal fibrosis
C57BL/6 male mice, aged 8 weeks and weighing 20–25 g, were purchased from CLEA Japan, Inc. (Tokyo, Japan). Mice were housed under antiviral and antibody-free micro-isolator conditions. To induce renal fibrosis, UUO was induced by left ureteral ligation, as described.34 Ureters were completely obstructed by double ligation with 4-0 silk sutures.
miR-146a
miR-146a and control-miR were purchased from Sigma-Aldrich Co (St Louis, MO, USA). The sequences of the miR-146a were 5′-UGAGAACUGAAUUCCAUGGGUUT-3′ (sense) and 5′-CCCAUGGAAUUCAGUUCUCAUU-3′ (antisense), and the sequences of the control-miR were 5′-UUCUCCGAACGUGUCACGUTT-3′ (sense) and 5′-ACGUGACACGUUCGGAGAATT-3′ (antisense).
Preparation of miR-146a-PEI-NPs
Linear PEI-based NPs (in vivo-jetPE®) were purchased from Polyplus-transfection SA (Illkirch-Graffenstaden, France) and dissolved in 5% glucose solution at a concentration of 50 μM. The solutions containing miR and PEI-NPs were mixed and incubated for 15 minutes at room temperature. A total of 200 μL of miR-PEI-NPs (miR: 5 nmol, the ratio of nitrogens (N) in the polymer to phosphate (P) in the nucleic acids =6) were intravenously injected via the tail vein into each mouse according to the manufacturer’s protocol. Control-miR-PEI-NPs were prepared using the same method.
Delivery of miR-146a-PEI-NPs to the kidneys
miR-146a-PEI-NPs (miR: 5 nmol, N/P ratio =6) dissolved in 200 μL of 5% glucose solution were injected via the tail vein into each mouse three times, on days -1, 1, and 3 relative to UUO treatment (UUO + miR-146a-PEI-NPs). The following groups served as controls: sham-operated mice (sham-operation), UUO mice with no injection (UUO + no injection), and UUO mice injected with control-miR-PEI-NPs (UUO + control-miR-PEI-NPs), using the same protocol. The mice were sacrificed 6 days after UUO, and their kidneys were removed and washed with phosphate-buffered saline (PBS; Thermo Fisher Scientific, Waltham, MA, USA) by reflux flow.
Quantitative real-time reverse-transcription polymerase chain reaction
Details of quantitative real-time reverse-transcription polymerase chain reaction (qRT-PCR) have been described.35 Briefly, kidney specimens were homogenized using a glass homogenizer and a filter column shredder (QIAshredder column; Qiagen NV, Venlo, The Netherlands). Total RNA from each kidney was isolated using an RNeasy Mini Kit (RNeasy®, Qiagen NV), with 250 ng of each isolated RNA reverse transcribed using the Life Technologies SuperScript® III First-Strand Synthesis System (Thermo Fisher Scientific). Real-time qRT-PCR was performed using Life Technologies SYBR® GreenER™ qPCR SuperMix (Thermo Fisher Scientific). Primers for mouse glyceraldehyde-3-phosphate dehydrogenase (GAPDH), alpha-smooth muscle actin (α-SMA), collagen 1A2, collagen 4, fibroblast-specific protein 1 (FSP-1), Smad4, TGF-β1, Snail1, Serpine1, F4/80 (also known as Emr1), IRAK1, TRAF6, NF-κB, TLR4, and monocyte chemotactic protein-1 (MCP-1) were purchased from Takara Bio Inc (Otsu, Shiga, Japan). The expression levels of each mRNA were normalized relative to that of GAPDH. To determine the expression levels of miR, homogenized kidney specimens were isolated using a miRNeasy Mini Kit (Qiagen NV), and 1 μg of each isolated RNA was reverse transcribed using the miScript® II RT Kit (Qiagen NV). Real-time qRT-PCR was performed using a miScript SYBR Green PCR Kit (Qiagen NV). Primers for U6 small nuclear RNA-2 (RNU6-2) and miR-146a were purchased from Qiagen NV. The expression levels of each mRNA were normalized relative to that of RNU6-2. Data are expressed relative to the sham-operation group.
Western blot analysis
Details of Western blot analysis to evaluate α-SMA have been described.35 Briefly, kidney samples were homogenized and equal amounts of protein samples (30 μg) were electrophoresed on Life Technologies NuPAGE® sodium dodecyl sulfate-polyacrylamide gels (Thermo Fisher Scientific), and transferred onto Immobilon™-P membranes (Merck Millipore, Billerica, MA, USA). After blocking, the membranes were incubated overnight at 4°C with appropriate dilutions of primary antibodies against α-SMA (Abcam plc, Cambridge, England), and GAPDH (Santa Cruz Biotechnology Inc, Dallas, TX, USA). After washing, the membrane was incubated for 1 hour at room temperature with a secondary antibody (horseradish peroxidase-conjugated donkey anti-rabbit immunoglobulin; GE Healthcare UK Ltd, Little Chalfont, UK). Antibody complexes were visualized using the Amersham ECL detection system (GE Healthcare UK Ltd) as directed by the manufacturer. Protein was quantified densitometrically using ImageJ 1.480 software. The expression levels of α-SMA were normalized relative to that of GAPDH. Data are expressed as relative to the sham-operation group.
Histological analysis
After perfusion with PBS, the kidneys were removed and fixed in 4% paraformaldehyde overnight at 4°C. They were then embedded in paraffin, sectioned, and subjected to azan staining and sirius red staining to evaluate fibrotic changes. The degree of fibrosis was quantitatively evaluated by examining ten fields per section positively stained for sirius red at 200-fold magnification. Fields for analysis were chosen randomly and the sirius red-stained areas quantified using computerized image analysis software (Image-Pro® 5.1; Media Cybernetics Inc, Rockville, MD, USA).
Fluorescent microscopy
To investigate the distribution of intravenously injected miR-PEI-NPs into the kidneys, liver, and spleen, Cy3-labeled double-strand oligos (Cy3-miR; Takara Bio Inc) were used to make a complex with PEI-NPs. These Cy3-miR-PEI-NPs were injected via the tail vein of mice with UUO-induced renal fibrosis. The kidneys, liver, and spleen were removed from each mouse 1 hour later. The tissue samples were embedded in Tissue-Tek® OCT (optimal cutting temperature) Compound and frozen in liquid nitrogen. Cryostat sections (5 μm thick) were mounted onto silane-coated glass slides (Matsunami Glass Ind Ltd, Osaka, Japan). Kidney tissues were incubated for 2 hours at room temperature with fluorescein-labeled Lotus tetragonolobus lectin (a proximal tubule marker) (Vector Laboratories Inc, Burlingame, CA, USA). Fluorescence was monitored by fluorescence microscopy (BH2-RFL-T3 and BX50; Olympus, Tokyo, Japan) and processed with Adobe Photoshop CS3 software (Adobe Systems Incorporated, San Jose, CA, USA).
Immunofluorescent microscopy
Details of immunofluorescent microscopy to evaluate cells positive for Smad4 and α-SMA have been described.35 Briefly, paraffin-embedded sections of kidneys from mice in each group were de-paraffinized, rehydrated, and autoclaved to retrieve antigens, followed by blocking with 10% normal goat serum. The sections were incubated overnight at 4°C with a polyclonal antibody to Smad4 (Abcam plc). After washing with PBS, the sections were incubated for 2 hours at room temperature with a monoclonal antibody to rabbit immunoglobulin (Ig) G conjugated to Alexa Fluor® 488 (Jackson ImmunoResearch Laboratories, Inc, West Grove, PA, USA).
To evaluate cells positive for α-SMA in each group, paraffin-embedded kidney sections were incubated overnight at 4°C with a monoclonal antibody to α-SMA conjugated to Cy3 (Sigma-Aldrich Co). The sections were mounted with VECTASHIELD® Mounting Medium with 4′,6-diamidino-2-phenylindole (Vector Laboratories) to stain the nuclei. Fluorescence was monitored by fluorescence microscopy (BH2-RFL-T3 and BX50) and processed with Adobe Photoshop software.
Immunohistochemistry
Immunohistochemical assays were performed using a Histofine® kit (Nichirei Biosciences Inc, Tokyo, Japan). Paraffin-embedded kidney sections were de-paraffinized, rehydrated, and autoclaved to retrieve antigens. Endogenous peroxidase activity was blocked by incubation in 3% H2O2 in methanol for 10 minutes at room temperature. Sections were incubated overnight at 4°C with a 1:100 dilution of anti-F4/80 monoclonal antibody (Abcam plc) in PBS. After rinsing in PBS, the sections were incubated with horseradish peroxidase-coupled goat antibody. The diaminobenzidine reaction was performed using a liquid diaminobenzidine substrate-chromogen system (Nichirei Biosciences Inc). After the tissues were rinsed in PBS, they were counterstained with hematoxylin and eosin. Immunolabeled sections were examined by light microscopy and processed with Adobe Photoshop software.
Statistical analysis
Data are expressed as mean ± standard error. Differences among groups were determined by analysis of variance; if statistically significant, Tukey’s test was performed as post-hoc analysis to compare means of two different groups. SPSS software (v 21; IBM Corporation, Armonk, NY, USA) was used for statistical analyses. A P-value <0.05 was considered statistically significant.
Results
Distribution of miR with PEI-NPs in the obstructed kidney
Tail vein injection of miR-PEI-NPs showed distribution of miR in the renal tubules of obstructed kidneys (Figure 1, red). Co-staining with a renal tubular cell marker, fluorescein-labeled L. tetragonolobus lectin (Figure 1, green), showed that miR was also distributed in the interstitial fibrous areas of obstructed kidneys, in which renal tubular cells did not have a regular form (Figure 1, arrow). miR-PEI-NPs were also present in the livers and spleens of these mice (Figure S1). Expression of signal transduction and activator of transcription 1 (STAT1) and 5′-oligoadenylate synthase (OAS) was significantly increased in the obstructed kidneys compared with in those of the sham-operated mice (Figure 2). However, the administration of miR-PEI-NPs did not further enhance the expression of these in the obstructed kidneys (Figure 2).
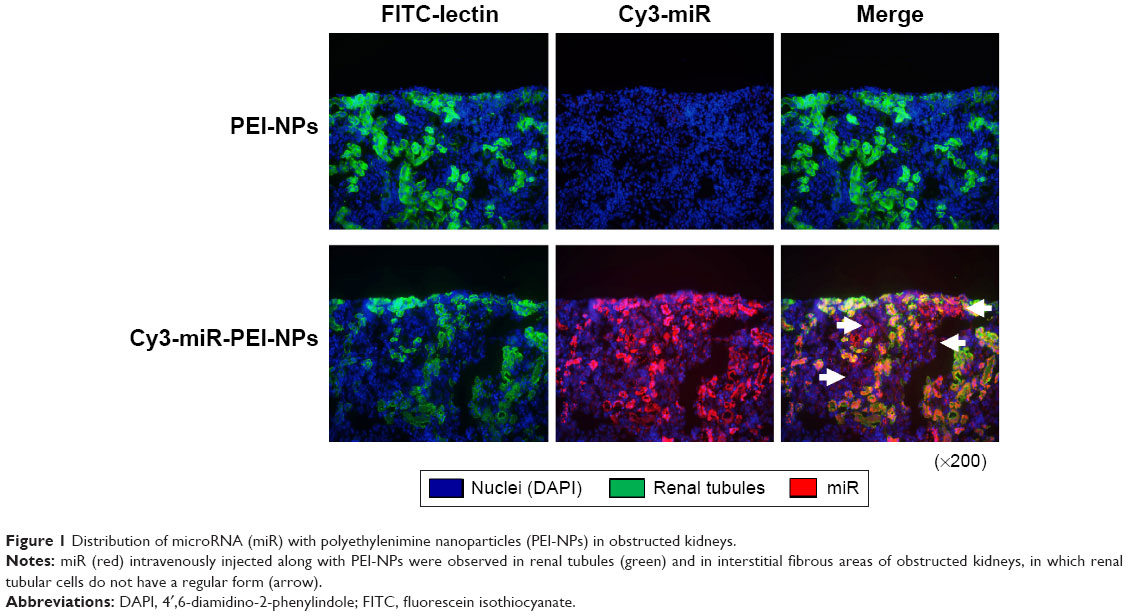 | Figure 1 Distribution of microRNA (miR) with polyethylenimine nanoparticles (PEI-NPs) in obstructed kidneys. |
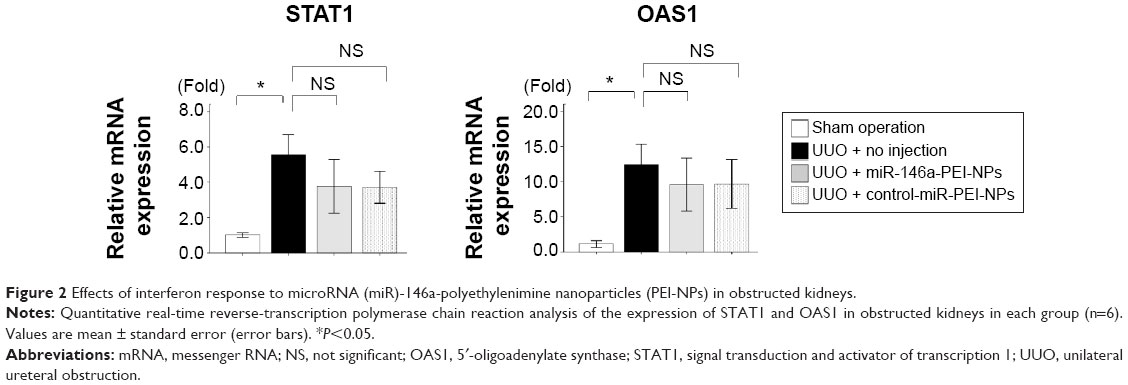 | Figure 2 Effects of interferon response to microRNA (miR)-146a-polyethylenimine nanoparticles (PEI-NPs) in obstructed kidneys. |
Overexpression of miR-146a in the obstructed kidneys by miR-146a-PEI-NPs
Administration of miR-146a-PEI-NPs, but not control-miR-PEI-NPs, significantly increased the in vivo expression of miR-146a in the obstructed kidneys (Figure 3).
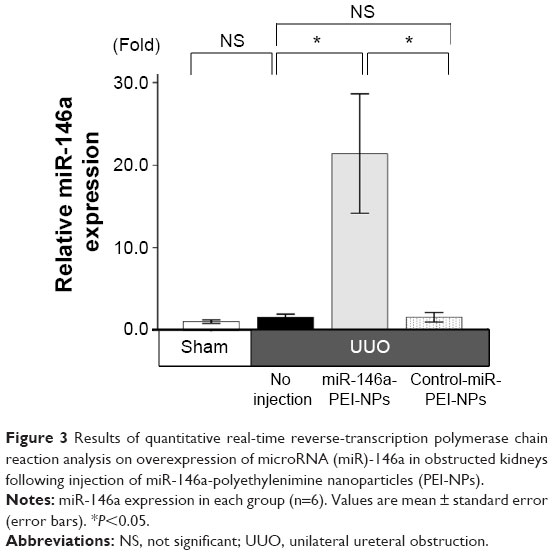 | Figure 3 Results of quantitative real-time reverse-transcription polymerase chain reaction analysis on overexpression of microRNA (miR)-146a in obstructed kidneys following injection of miR-146a-polyethylenimine nanoparticles (PEI-NPs). |
Effects of miR-146a-PEI-NPs on kidney weight
The ratios of the weight of the obstructed kidneys to that of the contralateral unobstructed kidneys in each group are shown in Figure 4. The ratio was significantly lower in the UUO than in the sham-operation group. This difference was inhibited by the administration of miR-146a-PEI-NPs, but not of control-miR-PEI-NPs.
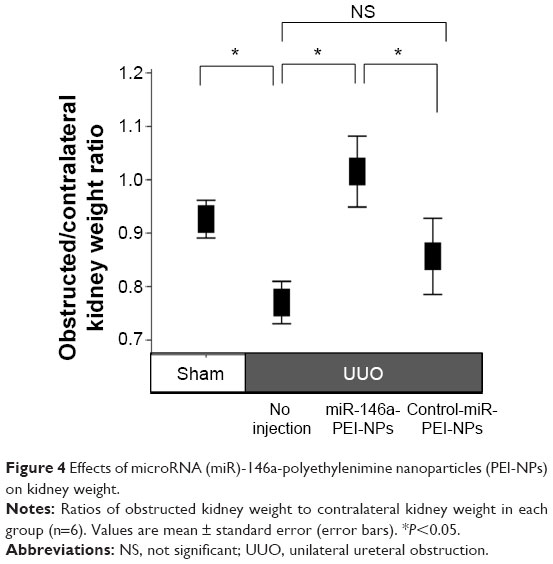 | Figure 4 Effects of microRNA (miR)-146a-polyethylenimine nanoparticles (PEI-NPs) on kidney weight. |
Inhibition of renal fibrosis by miR-146a-PEI-NPs
Positive blue-stained areas, showing collagen fibers with azan staining, were increased in the obstructed kidneys of the UUO + no injection group compared with in the sham-operation group (Figure 5A). These fibrotic areas were significantly inhibited by the administration of miR-146a-PEI-NPs, but not by control-miR-PEI-NPs. Fibrotic changes in each group were confirmed by sirius red staining and quantification, with the mean quantified fibrotic areas per field in the sham-operation, UUO + no injection, UUO + miR-146-PEI-NPs, and UUO + control-miR-PEI-NPs groups being 0.71%±0.20%, 7.83%±3.37%, 2.43%±0.89%, and 8.72%±4.86%, respectively (Figure 5B), again showing that the increase in fibrous changes in obstructed kidneys was significantly inhibited by miR-146a-PEI-NPs, but not by control-miR-PEI-NPs (Figure 5B).
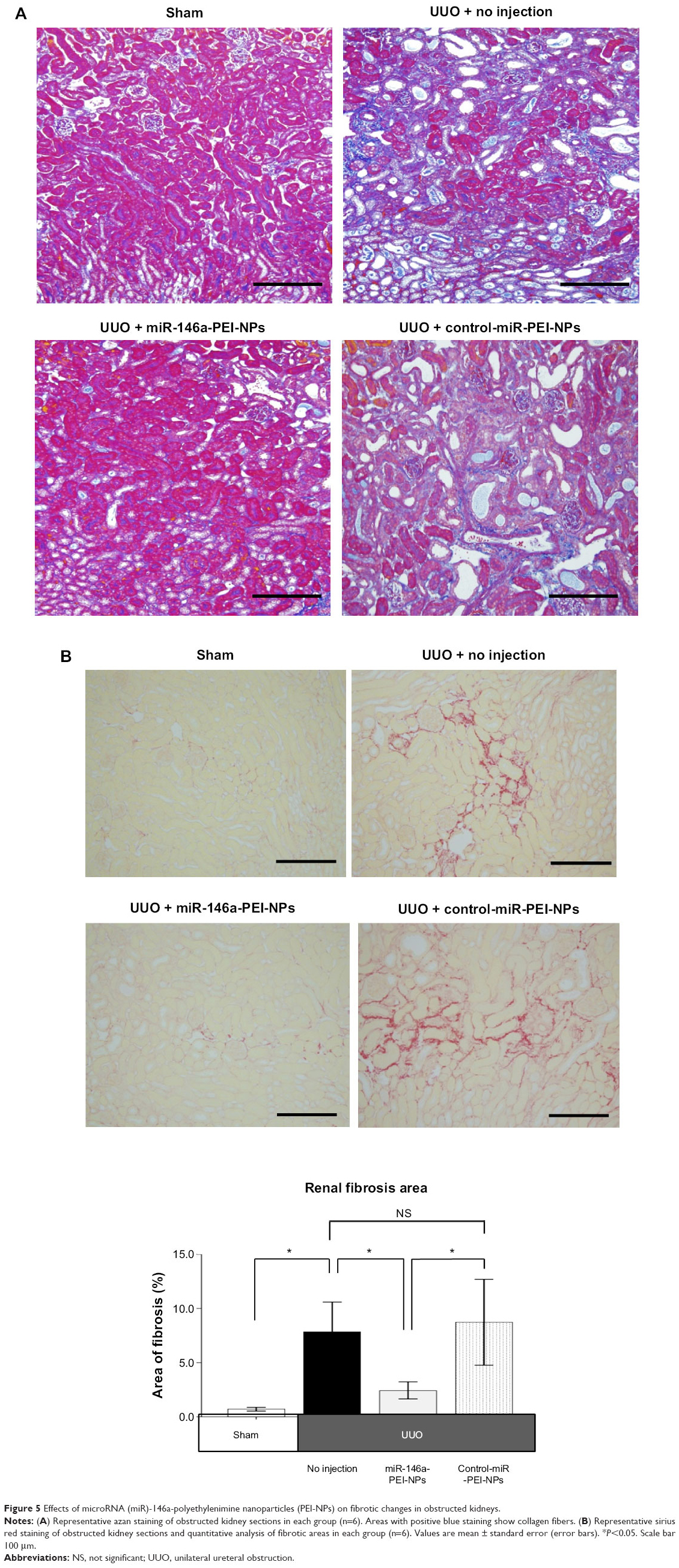 | Figure 5 Effects of microRNA (miR)-146a-polyethylenimine nanoparticles (PEI-NPs) on fibrotic changes in obstructed kidneys. |
Effects of miR-146a-PEI-NPs on fibrosis markers and pro-fibrotic signals in obstructed kidneys
qRT-PCR analysis showed that the expression of α-SMA, collagen 1A2, and FSP-1 mRNAs was higher in obstructed than in sham-operated kidneys (Figure 6). This increase was significantly inhibited by miR-146a-PEI-NPs, but not by control-miR-PEI-NPs. The level of expression of collagen IV mRNA showed similar trends, but these changes were not statistically significant.
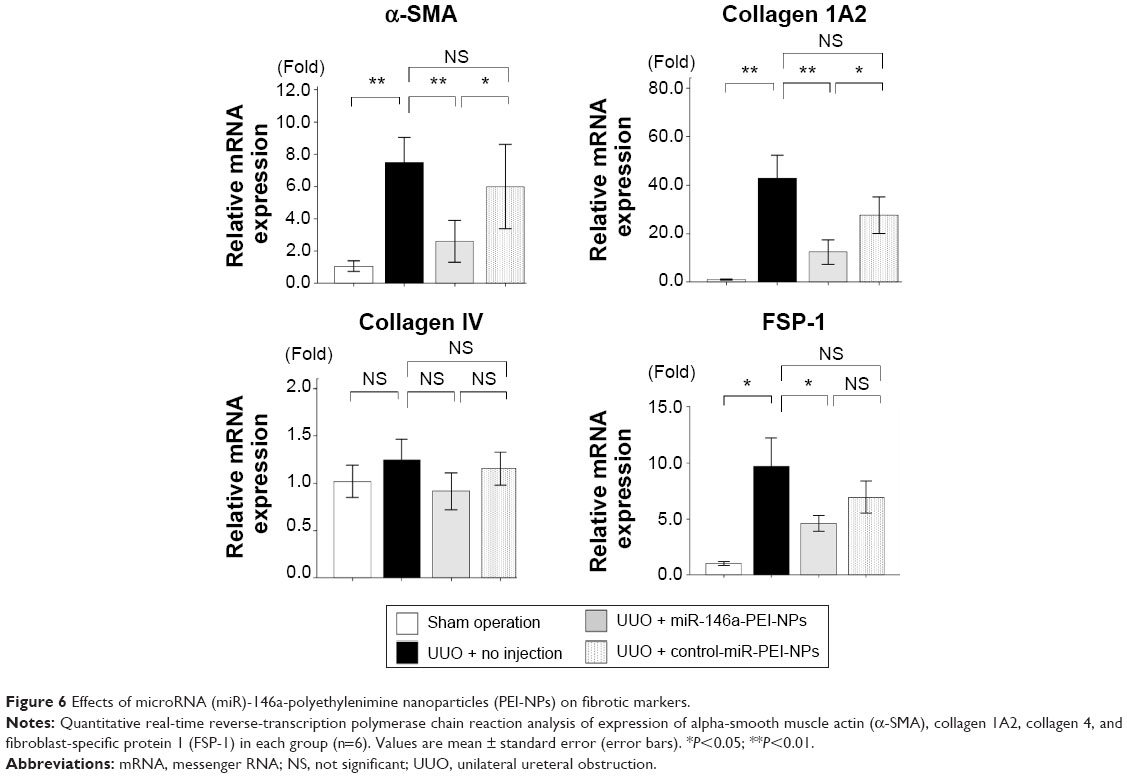 | Figure 6 Effects of microRNA (miR)-146a-polyethylenimine nanoparticles (PEI-NPs) on fibrotic markers. |
Western blot analysis showed that the expression of α-SMA was greater in obstructed than in sham-operated kidneys (Figure 7A). This increase was significantly inhibited by miR-146a-PEI-NPs, but not by control-miR-PEI-NPs (Figure 7A). Immunofluorescence analysis showed that the expression of α-SMA-positive myofibroblasts was higher in obstructed than in sham-operated kidneys, an increase significantly inhibited by miR-146a-PEI-NPs, but not by control-miR-PEI-NPs (Figure 7B).
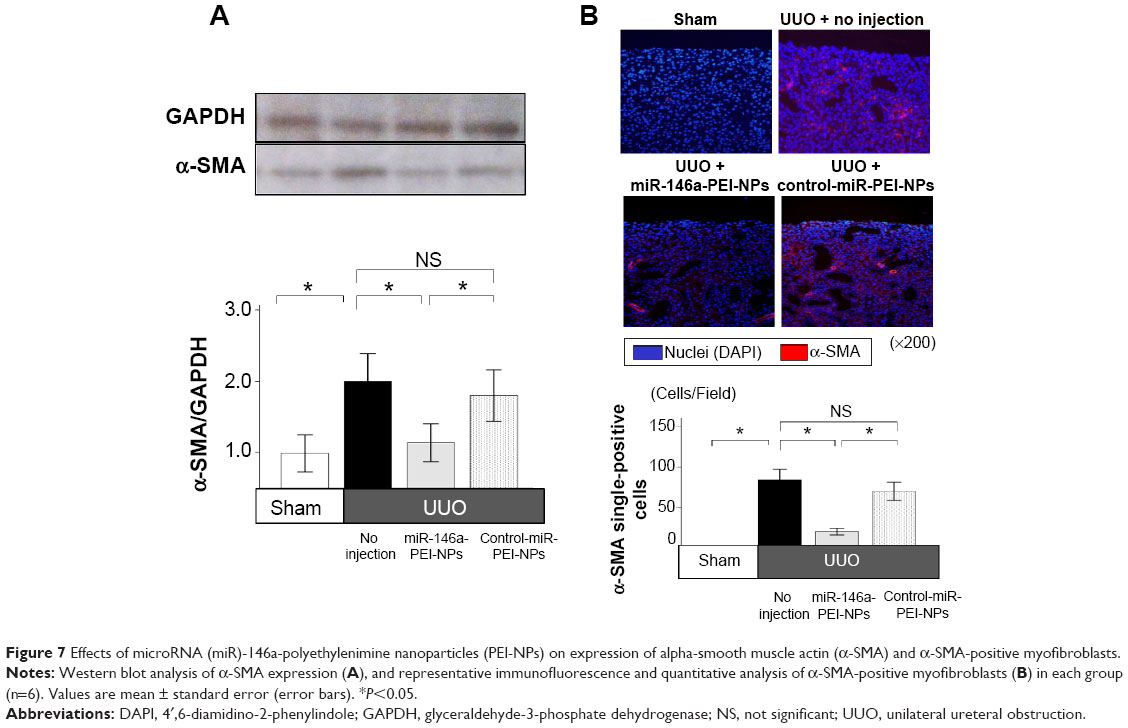 | Figure 7 Effects of microRNA (miR)-146a-polyethylenimine nanoparticles (PEI-NPs) on expression of alpha-smooth muscle actin (α-SMA) and α-SMA-positive myofibroblasts. |
Smad4 has been reported to be a target gene of miR-146a.30 qRT-PCR and immunofluorescence analysis showed that the expression of Smad4 mRNA and protein was higher in obstructed than in sham-operated kidneys (Figure 8A and B). These increases were also significantly inhibited by miR-146a-PEI-NPs, but not by control-miR-PEI-NPs. We also investigated changes in expression of TGF-β1, Snail1, and Serpine1, which are considered important signaling molecules in the development of renal fibrosis. The expression of TGF-β1 was higher in obstructed than in sham-operated kidneys (Figure 9). This increase was significantly inhibited by miR-146a-PEI-NPs, but not by control-miR-PEI-NPs (Figure 9). Expression levels of Snail1 and Serpine1 showed similar trends, but these changes were not statistically significant.
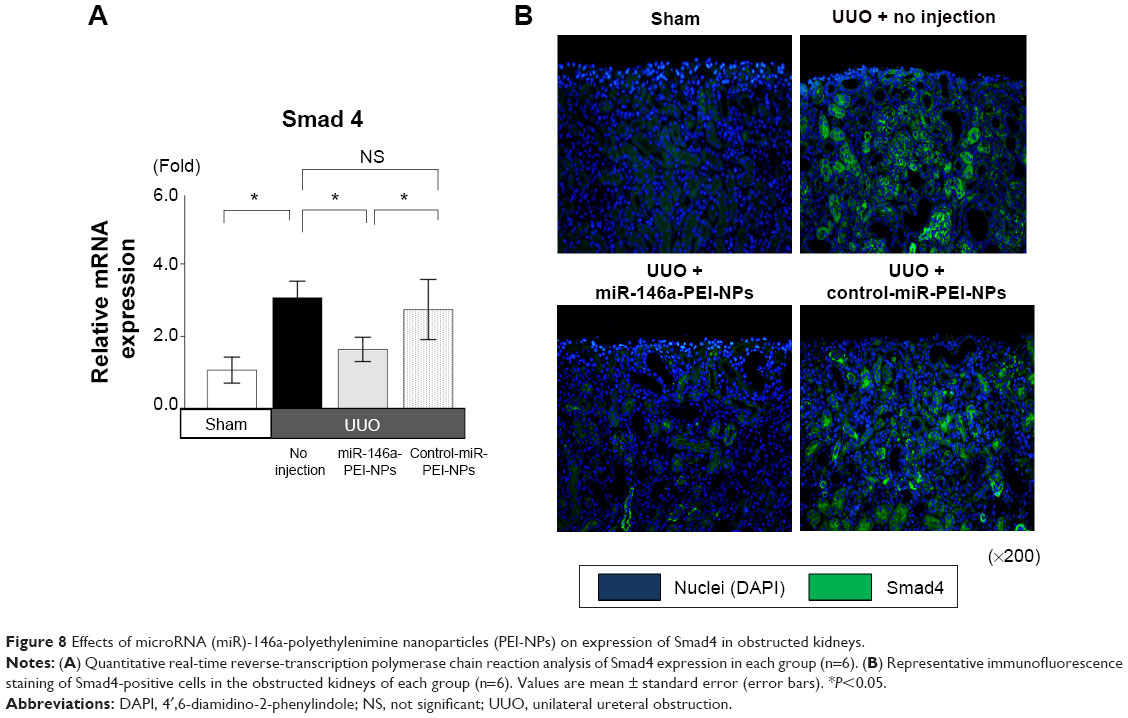 | Figure 8 Effects of microRNA (miR)-146a-polyethylenimine nanoparticles (PEI-NPs) on expression of Smad4 in obstructed kidneys. |
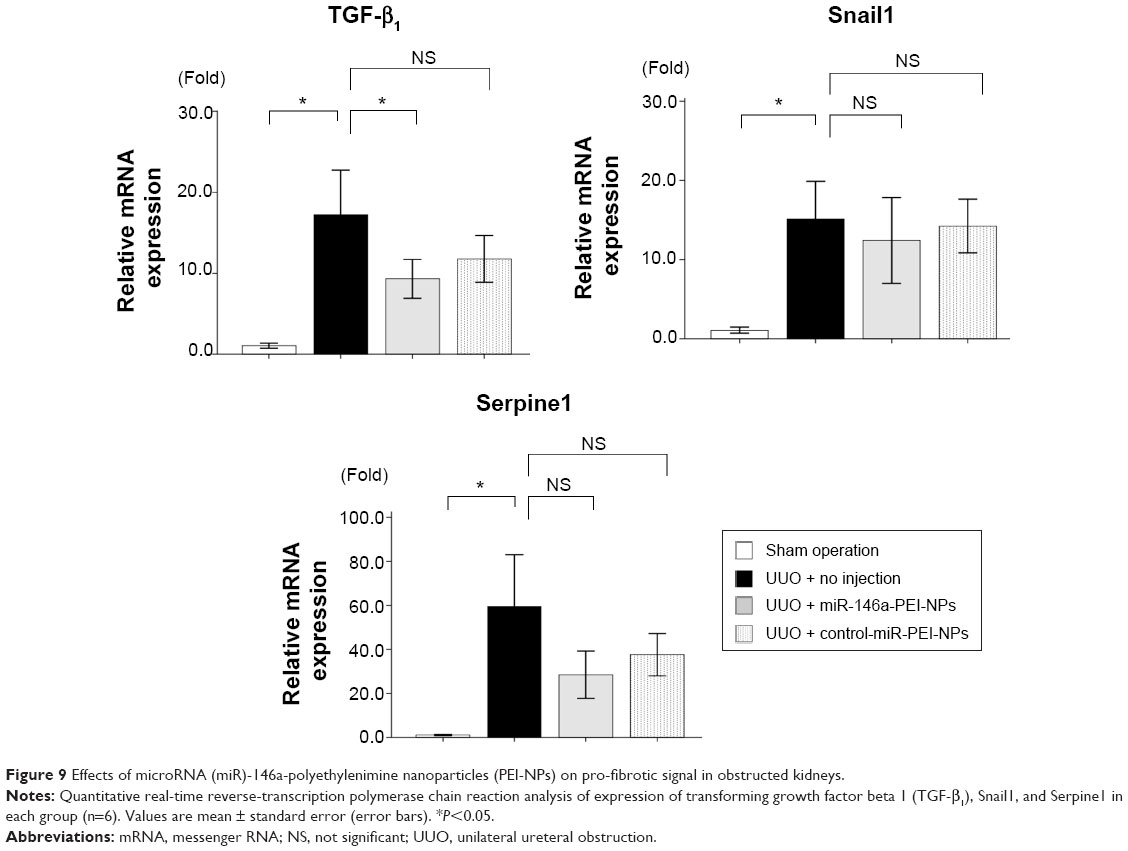 | Figure 9 Effects of microRNA (miR)-146a-polyethylenimine nanoparticles (PEI-NPs) on pro-fibrotic signal in obstructed kidneys. |
Effects of miR-146a-PEI-NPs on F4/80-positive macrophage infiltration and proinflammatory mediators in obstructed kidneys
qRT-PCR analysis and immunostaining showed that F4/80 expression and F4/80-positive macrophage infiltration were higher in obstructed than in sham-operated kidneys (Figure 10A and B). These increases were significantly inhibited by miR-146a-PEI-NPs, but not by control-miR-PEI-NPs. We also investigated changes in expression of TRAF6, IRAK1, and TLR4, which have been reported to be targets of miR-146a.25–29 Of these, TRAF6 expression was higher in obstructed than in sham-operated kidneys, an increase significantly inhibited by miR-146a-PEI-NPs, but not by control-miR-PEI-NPs (Figure 11). The expression levels of IRAK1 and TLR4 were also higher in obstructed than in sham-operated kidneys, but these increases were not significantly inhibited by either miR-146a-PEI-NPs or control-miR-PEI-NPs.
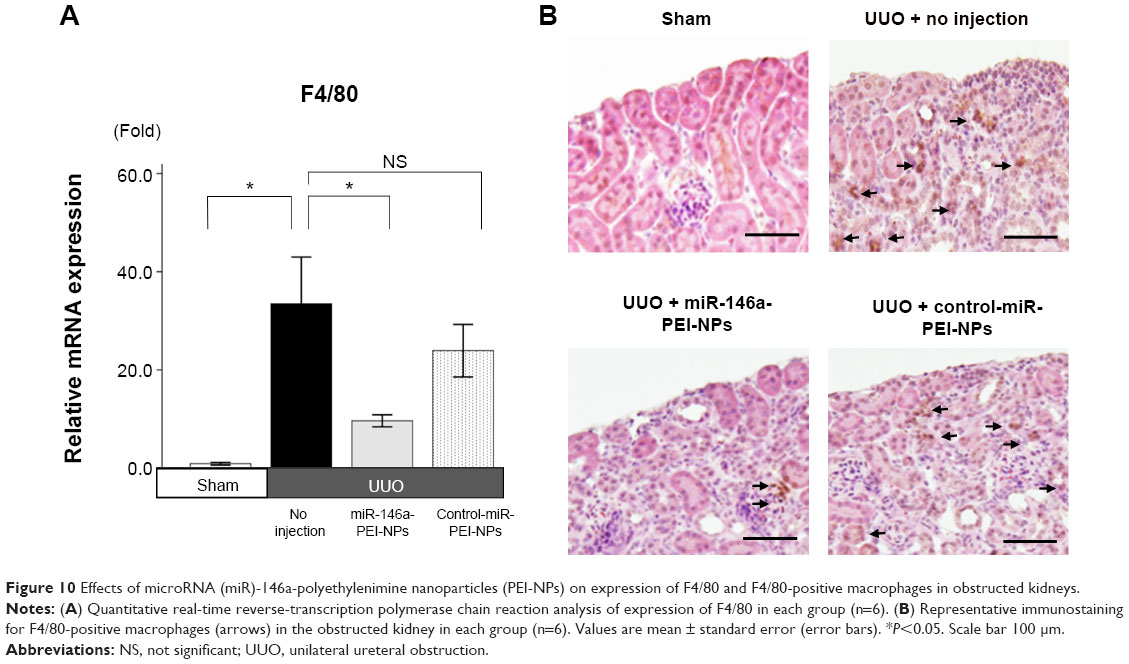 | Figure 10 Effects of microRNA (miR)-146a-polyethylenimine nanoparticles (PEI-NPs) on expression of F4/80 and F4/80-positive macrophages in obstructed kidneys. |
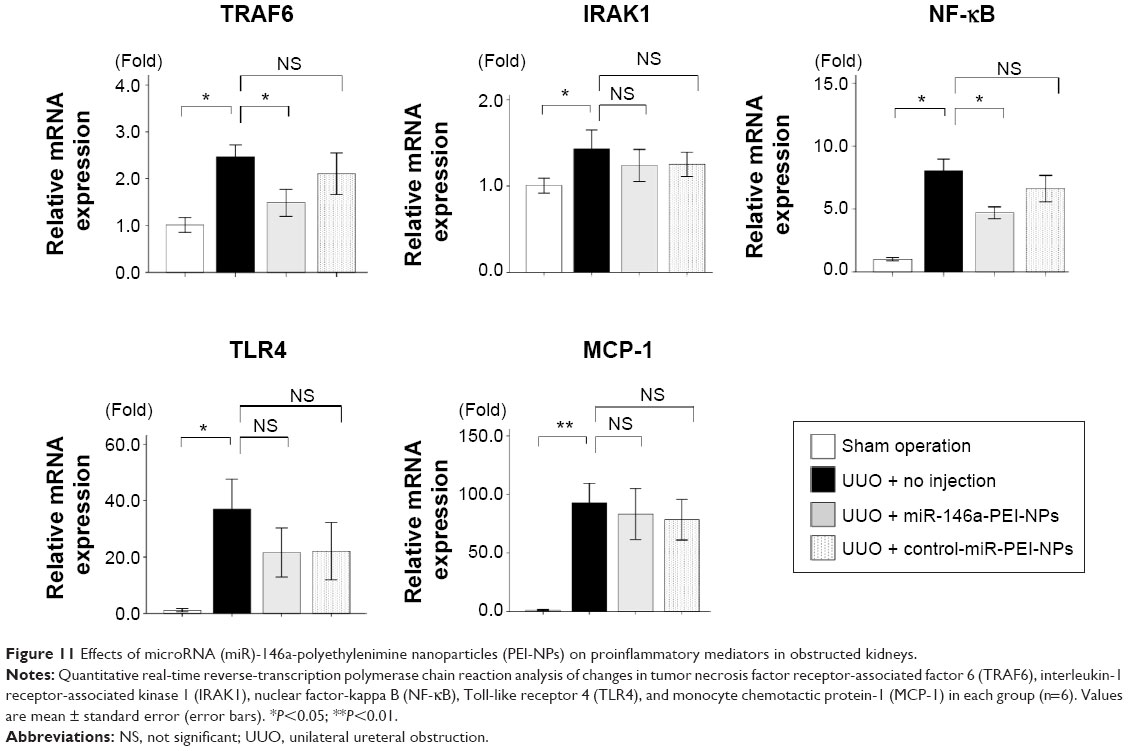 | Figure 11 Effects of microRNA (miR)-146a-polyethylenimine nanoparticles (PEI-NPs) on proinflammatory mediators in obstructed kidneys. |
We also investigated changes in expression of NF-κB, a major proinflammatory signaling molecule reported to be regulated by TRAF6.26 The expression of NF-κB was also higher in obstructed than in sham-operated kidneys, an increase significantly inhibited by miR-146a-PEI-NPs but not by control-miR-PEI-NPs (Figure 11). The expression of MCP-1, one of the main mediators of inflammation, was higher in obstructed than in sham-operated kidneys, but this increase was not significantly inhibited by either miR-146a-PEI-NPs or control-miR-PEI-NPs (Figure 11).
Discussion
The results presented here show the systemic administration of miR-146a-PEI-NPs inhibited renal fibrosis by suppressing pro-fibrotic (Smad4–TGF-β1) and inflammation (TRAF6–NF-κB) signaling pathways, resulting in the inhibition of F4/80-positive macrophage infiltration. These results suggest that miR-146a has a pivotal role in the development of renal fibrosis and may be a therapeutic target in patients with this condition. To our knowledge, this is the first report to show that delivery of exogenous miR-146a with PEI-NPs inhibits renal fibrosis in vivo.
miR is a small noncoding RNA that inhibits posttranscriptional processing of mRNA by binding to target sequences.14 Several miRs have been reported to suppress mRNAs that regulate the development of renal fibrosis.15–24 To date, however, few studies have assessed the effects of exogenous miR on renal fibrosis in an in vivo therapeutic setting.15–18,24 Tail vein injection of an miR-34c precursor with cationic lipid was found to inhibit renal fibrosis in a mouse model of UUO by suppressing the Notch/Jag1 signaling pathway.15 In addition, subcutaneous injection of locked nucleic acid-miR-192 inhibitor decreased renal fibrosis by increasing the expression of Zeb1/2 in streptozotocin-induced diabetic mice, as well as inhibiting arterial hypertension and increasing oxidative stress by suppressing the Akt signaling pathway.24 Overexpression of miR-29 and miR-21 by ultrasound-mediated gene transfer inhibited renal fibrosis in a mouse model of UUO by suppressing the TGF-β1–Smad signaling pathway.17,18 The results presented in this study show that miR-146a overexpression resulting from the tail vein injection of miR-146a-PEI-NPs inhibited renal fibrosis in UUO mice in vivo.
This study used PEI-NPs to deliver miR-146a. PEI-NPs are polymers that have been reported to effectively deliver oligonucleotides for modulating target gene expression in the kidney, as well as being considered preferable in preparing nonviral vectors because of their long-term safety and biocompatibility.32,33,36,37 This study found that PEI-NPs were effective in delivering miR-146a to the kidneys, significantly increasing miR-146a expression. In addition, they did not increase the interferon response in obstructed kidneys. These findings suggest that PEI-NPs can significantly deliver synthetic miR to the kidneys while having minimal immunotoxicity. We also found that miR-PEI-NPs were distributed in other organs, such as the liver and spleen, suggesting the need to assess the effects of miR-PEI-NPs on these other organs. In addition, pharmacokinetic analyses of PEI-NPs, such as its rates of excretion and degradation after injection, should be investigated, as should the PEI-NPs binding efficiency to miR and method of releasing miR in vivo.
This study shows that injection of miR-146a suppressed pro-fibrotic (Smad4–TGF-β1) and inflammation signaling (TRAF6–NF-κB) pathways, consistent with previous findings.25–27,30 These activities suggest that miR-146a-PEI-NPs may inhibit renal fibrosis in patients with kidney disease. miR-146a has also been reported to suppress expression of IRAK1 and TLR4, which are important genes in proinflammatory signaling.28,29 We found, however, that miR-146a-PEI-NPs did not significantly suppress the expression of these genes, nor did it suppress the expression of MCP-1, an important mediator of inflammation.38 These findings suggest that the expression of IRAK1, TLR4, and MCP-1 may be regulated by other miRs.
We also found that miR delivered by PEI-NPs was distributed in renal tubules and interstitial fibrotic areas, in which renal tubular cells did not have a regular form, in obstructed kidneys. Many types of cells have been reported associated with renal fibrosis, including epithelial cells, endothelial cells, bone marrow-derived cells, and pericytes, as the origins of myofibroblasts, as well as resident myofibroblasts.39–43 To date, however, the origins of myofibroblasts – the differentiation or proliferation of which was inhibited by miR-146a-PEI-NPs – remain undetermined, with experimental technology, such as fate tracing with genetically engineered mice, currently being used to determine this.39,43
Conclusion
The results of this study show that miR-146a-PEI-NPs inhibited renal fibrosis by suppressing pro-fibrotic (Smad4–TGF-β1) and inflammation (TRAF6–NF-κB) signaling pathways. miR-146a may play a pivotal role in the development of renal fibrosis, and may be a therapeutic target for treating or preventing renal fibrosis.
Acknowledgment
This work was supported by the Japanese Ministry of Education Culture, Sports, Science and Technology (MEXT)-Supported Program for Strategic Research Foundations at Private Universities (2013–2017) (grant number S1211029).
Author contributions
YM designed the study and coordinated the experiments, performed statistical analysis, and drafted the manuscript. HY performed all immunofluorescence analyses. MW performed all miR-PEI-NP injections and blood and tissue sampling. TI, KI, and SM participated in the study design. DN conceived the study and participated in its design and coordination. All authors contributed toward revising the paper and agree to be accountable for all aspects of the work.
Disclosure
The authors declare no conflicts of interest in this work.
References
Becker GJ, Hewitson TD. The role of tubulointerstitial injury in chronic renal failure. Curr Opin Nephrol Hypertens. 2000;9(2):133–138. | ||
Remuzzi G, Bertani T. Pathophysiology of progressive nephropathies. N Engl J Med. 1998;339(20):1448–1456. | ||
Campanholle G, Ligresti G, Gharib SA, Duffield JS. Cellular mechanisms of tissue fibrosis. 3. Novel mechanisms of kidney fibrosis. Am J Physiol Cell Physiol. 2013;304(7):C591–C603. | ||
Eddy AA. Molecular basis of renal fibrosis. Pediatr Nephrol. 2000; 15(3–4):290–301. | ||
Boor P, Floege J. Chronic kidney disease growth factors in renal fibrosis. Clin Exp Pharmacol Physiol. 2011;38(7):441–450. | ||
Meng XM, Huang XR, Xiao J, et al. Disruption of Smad4 impairs TGF-β/Smad3 and Smad7 transcriptional regulation during renal inflammation and fibrosis in vivo and in vitro. Kidney Int. 2012;81(3):266–279. | ||
Krensky AM, Ahn YT. Mechanisms of disease: regulation of RANTES (CCL5) in renal disease. Nat Clin Pract Nephrol. 2007;3(3): 164–170. | ||
Guijarro C, Egido J. Transcription factor-kappa B (NF-kappa B) and renal disease. Kidney Int. 2001;59(2):415–424. | ||
Lange-Sperandio B, Trautmann A, Eickelberg O, et al. Leukocytes induce epithelial to mesenchymal transition after unilateral ureteral obstruction in neonatal mice. Am J Pathol. 2007;171(3):861–871. | ||
Yang N, Wu LL, Nikolic-Paterson DJ, et al. Local macrophage and myofibroblast proliferation in progressive renal injury in the rat remnant kidney. Nephrol Dial Transplant. 1998;13(8):1967–1974. | ||
Eardley KS, Kubal C, Zehnder D, et al. The role of capillary density, macrophage infiltration and interstitial scarring in the pathogenesis of human chronic kidney disease. Kidney Int. 2008;74(4):495–504. | ||
Wilson HM, Chettibi S, Jobin C, Walbaum D, Rees AJ, Kluth DC. Inhibition of macrophage nuclear factor-kappaB leads to a dominant anti-inflammatory phenotype that attenuates glomerular inflammation in vivo. Am J Pathol. 2005;167(1):27–37. | ||
Anders HJ, Vielhauer V, Eis V, et al. Activation of toll-like receptor-9 induces progression of renal disease in MRL-Fas(lpr) mice. FASEB J. 2004;18(3):534–536. | ||
Benfey PN. Molecular biology: microRNA is here to stay. Nature. 2003;425(6955):244–245. | ||
Morizane R, Fujii S, Monkawa T, et al. miR-34c attenuates epithelial-mesenchymal transition and kidney fibrosis with ureteral obstruction. Sci Rep. 2014;4:4578. | ||
Zeisberg M, Kalluri R. The role of epithelial-to-mesenchymal transition in renal fibrosis. J Mol Med. 2004;82(3):175–181. | ||
Zhong X, Chung AC, Chen HY, Meng XM, Lan HY. Smad3-mediated upregulation of miR-21 promotes renal fibrosis. J Am Soc Nephrol. 2011;22(9):1668–1681. | ||
Qin W, Chung AC, Huang XR, et al. TGF-beta/Smad3 signaling promotes renal fibrosis by inhibiting miR-29. J Am Soc Nephrol. 2011;22(8):1462–1474. | ||
Chung AC, Huang XR, Meng X, Lan HY. miR-192 mediates TGF-beta/Smad3-driven renal fibrosis. J Am Soc Nephrol. 2010;21(8):1317–1325. | ||
Wang B, Komers R, Carew R, et al. Suppression of microRNA-29 expression by TGF-beta1 promotes collagen expression and renal fibrosis. J Am Soc Nephrol. 2012;23(2):252–265. | ||
Oba S, Kumano S, Suzuki E, et al. miR-200b precursor can ameliorate renal tubulointerstitial fibrosis. PLoS One. 2010;5(10):e13614. | ||
Macconi D, Tomasoni S, Romagnani P, et al. MicroRNA-324-3p promotes renal fibrosis and is a target of ACE inhibition. J Am Soc Nephrol. 2012;23(9):1496–1505. | ||
Li R, Chung AC, Dong Y, Yang W, Zhong X, Lan HY. The microRNA miR-433 promotes renal fibrosis by amplifying the TGF-beta/Smad3-Azin1 pathway. Kidney Int. 2013;84(6):1129–1144. | ||
Putta S, Lanting L, Sun G, Lawson G, Kato M, Natarajan R. Inhibiting microRNA-192 ameliorates renal fibrosis in diabetic nephropathy. J Am Soc Nephrol. 2012;23(3):458–469. | ||
Selvamani SP, Mishra R, Singh SK. Chikungunya virus exploits miR-146a to regulate NF-kappaB pathway in human synovial fibroblasts. PLoS One. 2014;9(8):e103624. | ||
Paik JH, Jang JY, Jeon YK, et al. MicroRNA-146a downregulates NFkappaB activity via targeting TRAF6 and functions as a tumor suppressor having strong prognostic implications in NK/T cell lymphoma. Clin Cancer Res. 2011;17(14):4761–4771. | ||
Comer BS, Camoretti-Mercado B, Kogut PC, Halayko AJ, Solway J, Gerthoffer WT. MicroRNA-146a and microRNA-146b expression and anti-inflammatory function in human airway smooth muscle. Am J Physiol Lung Cell Mol Physiol. 2014;307(9):L727–L734. | ||
Yang K, He YS, Wang XQ, et al. MiR-146a inhibits oxidized low-density lipoprotein-induced lipid accumulation and inflammatory response via targeting toll-like receptor 4. FEBS Lett. 2011;585(6):854–860. | ||
Ho BC, Yu IS, Lu LF, et al. Inhibition of miR-146a prevents enterovirus-induced death by restoring the production of type I interferon. Nat Commun. 2014;5:3344. | ||
Lv S, Sun B, Dai C, et al. The Downregulation of MicroRNA-146a Modulates TGF-β Signaling Pathways Activity in Glioblastoma. Mol Neurobiol. 2014. Epub October 19. | ||
Huang Y, Liu Y, Li L, et al. Involvement of inflammation-related miR-155 and miR-146a in diabetic nephropathy: implications for glomerular endothelial injury. BMC Nephrol. 2014;15:142. | ||
Ji SM, Shin YB, Park SY, Lee HJ, Oh B. Decreases in Casz1 mRNA by an siRNA Complex Do not Alter Blood Pressure in Mice. Genomics Inform. 2012;10(1):40–43. | ||
Hernandez-Vargas P, Lopez-Franco O, Sanjuan G, et al. Suppressors of cytokine signaling regulate angiotensin II-activated Janus kinase-signal transducers and activators of transcription pathway in renal cells. J Am Soc Nephrol. 2005;16(6):1673–1683. | ||
Chevalier RL, Forbes MS, Thornhill BA. Ureteral obstruction as a model of renal interstitial fibrosis and obstructive nephropathy. Kidney Int. 2009;75(11):1145–1152. | ||
Morishita Y, Yoshizawa H, Watanabe M, et al. siRNAs targeted to Smad4 prevent renal fibrosis in vivo. Sci Rep. 2014;4:6424. | ||
Lungwitz U, Breunig M, Blunk T, Gopferich A. Polyethylenimine-based non-viral gene delivery systems. Eur J Pharm Biopharm. 2005;60(2):247–266. | ||
Swami A, Kurupati RK, Pathak A, Singh Y, Kumar P, Gupta KC. A unique and highly efficient non-viral DNA/siRNA delivery system based on PEI-bisepoxide nanoparticles. Biochem Biophys Res Commun. 2007;362(4):835–841. | ||
Lloyd CM, Minto AW, Dorf ME, et al. RANTES and monocyte chemoattractant protein-1 (MCP-1) play an important role in the inflammatory phase of crescentic nephritis, but only MCP-1 is involved in crescent formation and interstitial fibrosis. J Exp Med. 1997;185(7):1371–1380. | ||
Humphreys BD, Lin SL, Kobayashi A, et al. Fate tracing reveals the pericyte and not epithelial origin of myofibroblasts in kidney fibrosis. Am J Pathol. 2010;176(1):85–97. | ||
Kalluri R, Neilson EG. Epithelial-mesenchymal transition and its implications for fibrosis. J Clin Invest. 2003;112(12):1776–1784. | ||
Li J, Qu X, Bertram JF. Endothelial-myofibroblast transition contributes to the early development of diabetic renal interstitial fibrosis in streptozotocin-induced diabetic mice. Am J Pathol. 2009;175(4): 1380–1388. | ||
Broekema M, Harmsen MC, van Luyn MJ, et al. Bone marrow-derived myofibroblasts contribute to the renal interstitial myofibroblast population and produce procollagen I after ischemia/reperfusion in rats. J Am Soc Nephrol. 2007;18(1):165–175. | ||
LeBleu VS, Taduri G, O’Connell J, et al. Origin and function of myofibroblasts in kidney fibrosis. Nat Med. 2013;19(8):1047–1053. |
Supplementary material
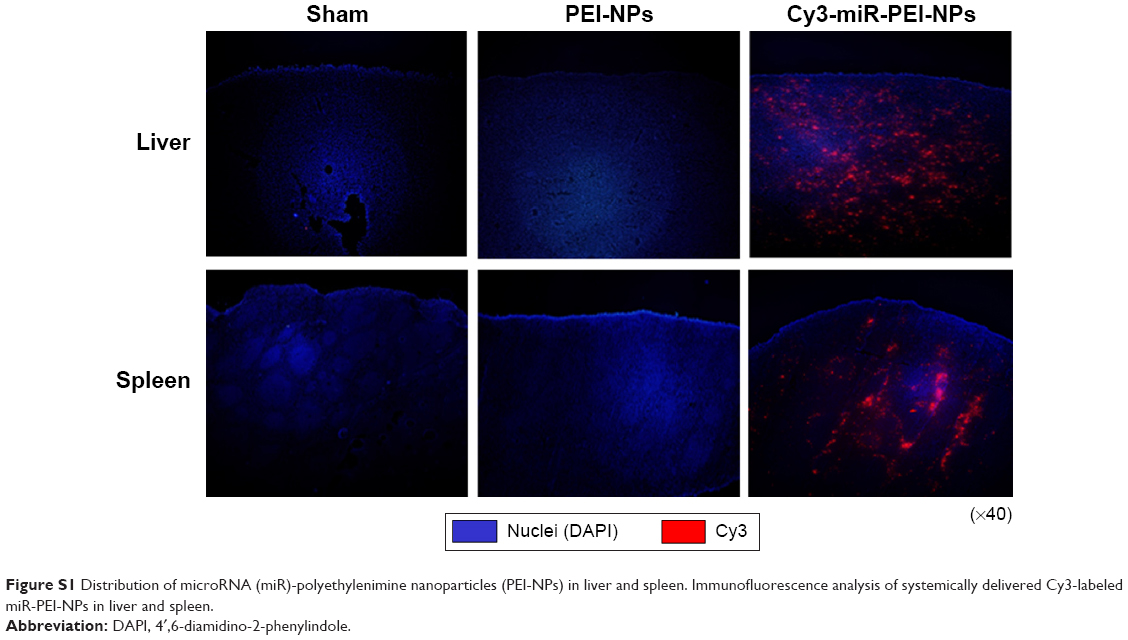 | Figure S1 Distribution of microRNA (miR)-polyethylenimine nanoparticles (PEI-NPs) in liver and spleen. Immunofluorescence analysis of systemically delivered Cy3-labeled miR-PEI-NPs in liver and spleen. |
 © 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms.php
and incorporate the Creative Commons Attribution
- Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms.php
and incorporate the Creative Commons Attribution
- Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.