Back to Journals » OncoTargets and Therapy » Volume 12
Dehydrocorydaline inhibits cell proliferation, migration and invasion via suppressing MEK1/2-ERK1/2 cascade in melanoma
Authors Hu H, Dong Z ![]() , Wang X, Bai L
, Wang X, Bai L ![]() , Lei Q, Yang J, Li L, Li Q, Liu L, Zhang Y, Ji Y, Guo L, Liu Y, Cui H
, Lei Q, Yang J, Li L, Li Q, Liu L, Zhang Y, Ji Y, Guo L, Liu Y, Cui H
Received 16 November 2018
Accepted for publication 1 May 2019
Published 2 July 2019 Volume 2019:12 Pages 5163—5175
DOI https://doi.org/10.2147/OTT.S183558
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Faris Farassati
Huanrong Hu 1,2 Zhen Dong, 2– 4 Xianxing Wang, 2– 4 Longchang Bai, 2– 4 Qian Lei, 2– 4 Jie Yang, 2– 4 Lin Li, 2– 4 Qian Li 1, 2, Lichao Liu 1,2 Yanli Zhang, 1 Yacong Ji, 1 Leiyang Guo, 1 Yaling Liu, 1 Hongjuan Cui 2– 4
1Department of Dermatology, the Third Hospital of Hebei Medical University, Shijiazhuang 050000, People’s Republic of China; 2State Key Laboratory of Silkworm Genome Biology, Southwest University, Chongqing 400715, People’s Republic of China; 3Engineering Research Center for Cancer Biomedical and Translational Medicine, Southwest University, Chongqing 400715, People’s Republic of China; 4Chongqing Engineering and Technology Research Center for Silk Biomaterials and Regenerative Medicine, Southwest University, Chongqing 400715, People’s Republic of China
Purpose: Alkaloids are naturally occurring chemical compounds that are widely distributed in plants, and have pharmaceutical values and low toxicity. In recent years, some of them have been demonstrated to be promising therapeutic drug candidates for cancer treatment. Herein, we tried to explore the antitumor effect of dehydrocorydaline (DHC), a natural alkaloid isolated from Corydalis, on malignant melanoma.
Methods: We treated two malignant metastatic melanoma cell lines, A375 and MV3, and a normal melanocyte cell line, PIG1, with various concentrations of DHC for set amounts of time, and detected cell proliferation, migration, and invasion by using MTT, BrdU, transwell, Western blot and soft agar assay in vitro and tumorigenicity in the xenografts in vivo.
Results: Our results showed that DHC dramatically blocked cell proliferation and led to cell cycle arrest at G0/G1 phase and downregulated the expressions of cell cycle regulators CDK6 and Cyclin D1 in melanoma cells. However, DHC had little inhibitory effect on normal melanocyte cell line PIG-1. Meanwhile, DHC suppressed cell invasion and migration through modulating the epithelial–mesenchymal transition (EMT) markers including E-cadherin, vimentin, as well as β-catenin. In addition, DHC also significantly attenuated tumor growth in vivo. The expressions of cell cycle-related and metastasis-related proteins were further confirmed by immunohistochemical staining in the xenografts. Importantly, MEK1/2-ERK1/2 cascade was inactivated after DHC treatment and ERK activator t-butylhydroquinone (tBHQ) treatment rescued DHC-induced cell proliferation inhibition.
Conclusions: Our results indicated that DHC inhibited cell proliferation and migration/invasion via inactivating MAPK signaling, and showed that DHC might be a potential novel drug to treat malignant melanoma.
Keywords: dehydrocorydaline, melanoma, cell cycle, migration and invasion, MAPK
Corrigendum for this paper has been published
Introduction
Malignant melanoma (MM) is the most aggressive form of skin cancer. Over the past few decades, the number of MM cases has increased worldwide.1,2 In the absence of new interventions, from 2011 to 2030, the annual expenses of the treatment for newly diagnosed MM cases are estimated to rise by 252.4%.3 Importantly, metastasis has been shown to be the main reason for the increasing morbidity and mortality of MM. Metastasis occurs even in patients with thin small primary MM.4 Until recently, the prognosis of patients with MM remains very poor, despite conventional therapy of surgical resection, radiotherapy, and chemotherapy.5 Therefore, it is urgent to find some efficient drugs with minimal toxicity for the treatment of MM.
Recent reports showed that alkaloids such as berberine,6–10 tetrahydropalmatine,11 and vinblastine12 from the plants had various anticancer activities. The functions and mechanisms of these alkaloids showed multiformity in different kinds of cancers. For example, growth inhibition of human hepatoma cells induced by berberine was mediated via mitochondria damage-induced apoptosis.7 But in lung cancer cells, berberine attenuated the invasiveness through decreasing the expressions of urokinase-plasminogen activator (uPA) and MMP2.10 In breast cancer cells, tetrahydropalmatine can reverse the multidrug resistance of MCF-7 cells by attenuating the activity of P-glycoprotein.11 Another alkaloid, vinblastine, could inhibit the cell proliferation of leukemia cells through reversing multi-drug resistance (MDR).12 This evidence dramatically indicated that alkaloids are promising candidates for tumor therapy.
As an alkaloid, dehydrocorydaline (C21H21NO4, DHC) was isolated from Corydalis yanhusuo, Corydalis tuber or Corydalis bulbosa,and was originally identified to block the release of noradrenaline in the taenia caecum and pulmonary artery from the adrenergic nerve terminals.13 In addition, DHC inhibited antibody-mediated and cell-mediated allergic reactions14 and suppressed the expression of pro-inflammatory cytokines, including IL-1β and IL-6.15 Moreover, DHC was known to have biological effects in the treatment of coronary artery disease,16 anti-acetylcholinesterase17 and anthelmintic features.18 A recent study showed that DHC promoted myogenic differentiation via p38 MAPK activation.19 Interestingly, DHC also had some bioactivity that could inhibit tumor progression. For example, DHC inhibited cell proliferation through inducing apoptosis in breast cancer cells.20 Also, DHC exerted anti-metastatic potential by suppressing MMPs and Bcl-2 in non-small cell lung carcinoma (NSCLC) cells.21 However, the effect of DHC in melanoma cells remained unknown.
In this paper, we explored the function of DHC in MM progression and metastasis. Our studies showed that DHC inhibited cell proliferation, cell cycle progression, and migration/invasion by inactivating the MAPK (MEK1/2-ERK1/2) cascade in MM. This evidence indicated that DHC could act as a potential candidate drug in the treatment of metastatic MM.
Materials and methods
Cell culture
Human metastatic melanoma cell line A375 and normal melanocyte PIG1 were obtained from the American Type Culture Collection (ATCC, Manassas, VA, USA). Another human metastatic melanoma cell line, MV3, was described previously,22 and was obtained from the Army Medical University (previously termed as the Third Military Medical University). Briefly, A375 and PIG1 cells were maintained in DMEM (Thermo Fisher Scientific, Waltham, MA, USA). MV3 cells were cultured in Roswell Park Memorial Institute-1640 (RPMI-1640; Gibco, Thermo Fisher Scientific). Both were supplemented with 10% fetal bovine serum (FBS; Gibco) and 1% penicillin-streptomycin (P/S; Gibco). Cells were cultured at 37°C with 5% CO2 in a humidified incubator (Sanyo, Osaka, Japan). The use of these cells was approved by the Academic Board of Southwest University.
Drug treatment
DHC, with purity higher than 99%, was obtained from the Chinese National Institutes for Food and Drug Control (NIFDC, Beijing, China) and was dissolved in dimethyl sulfoxide (DMSO; Sigma-Aldrich, Merck, Shanghai, China). A375 and MV3 were treated with DHC at indicated concentrations or times, with DMSO as control. t-Butylhydroquinone (tBHQ; HY-100,489) was purchased from MedChemExpress (Shanghai, China) and was dissolved in DMSO. The cell morphology was taken by the Olympus microscopy (Olympus, Japan). Cell viability was performed by trypan blue assay, described previously.23
MTT assays
Cell proliferation was performed by using the thiazolyl blue tetrazolium bromide (MTT) assay, which was described previously.23 1,000 cells were used to determine the growth curve of A375 and MV3 cells and 5,000 cells were used to determine the cell proliferation rate of PIG1 cells. Each experiment was independently performed for three times, and a two-tailed unpaired Student’s t-test was performed to analyze the significance.
BrdU staining
For BrdU staining, 1×104 cells were cultured in the 24-well plates for 8 h and treated with either DMSO or DHC for another 24 h, and then incubated with 10 μg/mL 5-bromo-2-deoxyuridine (BrdU; Sigma-Aldrich Co.) for 0.5 h; then, the BrdU assay was employed as previously described.24 Each experiment was independently performed three times, and a two-tailed unpaired Student’s t-test was performed to analyze the significance.
Cell cycle assay
For the cell cycle assay, 3×105 cells were cultured in 60-mm dishes for 24 h and then treated with 40 μM DHC or isometric DMSO. After 48 h treatment, cells were washed with cold PBS and then fixed in 70% ethyl alcohol at 4°C for more than 24 h. Subsequently, the cell cycle was analyzed by using a BD Accuri C6 cytometer (San Jose, CA, USA). Detailed information was described previously.25 The cell cycle and sub-G1 phase were further analyzed by using the FlowJo Software version 7.6.1 (FlowJo LLC, Ashland, OR, USA). Each sample in this experiment was performed in triplicate, and a two-tailed unpaired Student’s t-test was performed to analyze the significance.
Wound-healing assays
For wound-healing assays, 1×106 cells were cultured in 2 mL DMEM supplemented with 1% FBS, in 6-well plates. After the cells reached full confluence, we used a yellow pipette tip to scratch a linear wound in the monolayer of the cells. Subsequently, floating and damaged cells were removed by cold PBS washing three times. Then, serum-free DMEM with 40 μM DHC or DMSO, respectively, were added to the plates. After the indicated time, cells that migrated over the denuded area were observed under an inverted microscope (Nikon Corporation, Tokyo, Japan), and pictures were taken by a charge coupled device (CCD). The corporation of wound closure was calculated according to the migration over the denuded area. Each sample in this experiment was performed in triplicate, and a two-tailed unpaired Student’s t-test was performed to analyze the significance.
Cell migration and invasion
The migration assay was conducted with the treatment of 40 μM DHC or DMSO as previously described.24 Each experiment was performed in triplicate and the migration/invasion rate was normalized by the proliferation rate. A two-tailed unpaired Student’s t-test was performed to analyze the significance.
Western blot assay
The Western blot assay was conducted as previously described.26 All the primary antibodies used in this study were purchased from the Cell Signaling Technology (CST, Danvers, MA, USA) and are listed here: GAPDH (51132), CDK6 (3136), cyclin D1 (2978), E-cadherin (3195), vimentin (5741), β-catenin (8480), MEK1/2 (4694), p-MEK1/2 (Ser221, 2338), ERK1/2 (4695) or p-ERK1/2 (Thr202/Tyr204, 4370). HRP (horseradish peroxidase)-conjugated secondary antibodies including goat anti-mouse IgG (H+L) (A0216, 1:2,000) and goat anti-rabbit IgG (H+L) (A0208 1:2,000) were purchased from Beyotime (Taicang, Jiangsu, China). Proteins were visualized by the BeyoECL Plus (Beyotime) and Western blotting detection instruments (Clinx Science, Shanghai, China). The gray levels of the protein bands were calculated by using Image J 1.8.0 software (developed at the US National Institutes of Health and available on the Internet at http://rsb.info.nih.gov/nih-image/), and the relative protein levels were normalized by grey levels of GAPDH. Each blot was analyzed for 3 times, and a two-tailed unpaired Student’s t-test was performed to analyze the significance.
Soft agar assay
The ability of colony formation was conducted on A375 and MV3 cells. 1×103 A375 or MV3 cells in the soft agar contained 40 μM DHC or DMSO were used in this study. The detailed method was described previously.27 Each sample in this experiment was performed in triplicate, and a two-tailed unpaired Student’s t-test was performed to analyze the significance.
Tumor xenografts
Six 4-week old female nude mice (BALB/c-nu) were purchased from Beijing HFK Bioscience Lo., Ltd (Beijing, China) and housed in a pathogen-free room to acclimate for about 7 days. Then, 1×106 A375 and MV3 cells suspended in 100 μL medium were subcutaneously injected into both sides of the mice’s back, respectively. After two weeks, the tumors were formed, and the mice were randomly divided into two groups. Each group was orally administrated with 100 mg/kg DHC or isometric DMSO every 24 h for 12 days. Tumor length and width were measured by a digital caliper every day to calculate the tumor volume with the formula [volume= (π/6)×length×width2]. Finally, the mice were sacrificed and formed tumors were removed and weighed. All animal experiments were pre-approved by the Experimental Animal Care and Use Committees of the Institute of Sericulture and Systems Biology and the Institutional Animal Care and Use Committees of the Southwest University, with the principles of the Declaration of Helsinki, Experimental Animal Management Regulations (State Scientific and Technological Commission of China [1988] No. 2, 2017 version) and Chongqing Experimental Animal Management Regulations (Chongqing Municipal People’s Government [2006] No.195).
Immunohistochemistry staining
The immunohistochemistry (IHC) staining assay was conducted as previously reported.28 Each sample in this experiment was performed in triplicate.
Results
Dehydrocorydaline inhibits cell proliferation in melanoma cells
To assess the effect of DHC in cell proliferation inhibition, we tested the cell viability using the MTT assay with six different doses of DHC treatment for 48 h in melanoma cell lines A375 and MV3 as well as a normal cell line PIG1. According to the results, we calculated the IC50 of DHC in inhibition of cell proliferation in A375, MV3, and PIG1 cells, and the results showed that the IC50 of DHC in A375, MV3 and PIG1 were 39.73, 42.34, and 262.6 μM, respectively (Figure 1). This result indicated that melanoma cells are more sensitive to DHC treatment than normal melanocytes. Then, different concentrations of DHC were used in two different human melanoma cell lines A375 and MV3 for 48 h. Under the microscope, cells treated with different concentrations (20, 40 and 80 μM) of DHC resulted in cell proliferation inhibition in a dose-dependent manner, determined by using cell counting under a microscope (Figure 2A–C). However, the MTT assay showed that DHC did not induce significant cell proliferation inhibition in PIG1 cells after 20 and 40 μM DHC treatment for 48 h (Figure 2D). We further investigated the cell growth curve by MTT assay for 7 days after different concentrations of DHC treatment. The results showed DHC at 40 and 80 μM dramatically suppressed cell proliferation in these two cell lines (Figure 2E and F). BrdU staining assay further showed that 40 μM DHC treatment for 48 h showed a significant decline in the ratio of BrdU-positive cells compared to the control groups (Figure 2G). These results suggested that DHC dramatically blocked cell proliferation and growth in melanoma cells.
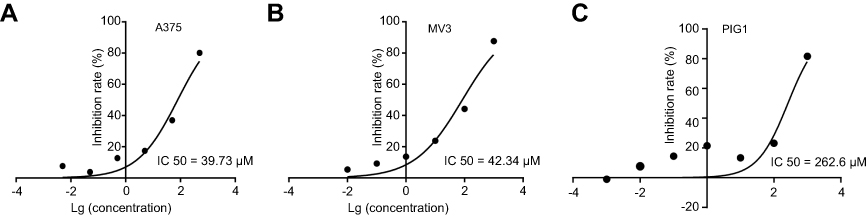 |
Figure 1 IC50 of dehydrocorydaline treatment for 48 h in the inhibition of cell proliferation in (A) A375 melanoma cells; (B) MV3 melanoma cells; and (C) PIG1 normal melanocytes determined by MTT assay. |
 |
Figure 2 Dehydrocorydaline inhibits cell growth and proliferation in human melanoma cells. (A) Cell morphology of A375 and MV3 melanoma cells after treating with DMSO or the indicated concentrations of DHC for 48 h. Scale bar, 100 μm. (B and C) The effect of DHC on the proliferation rates of A375 and MV3 cells determined by cell counting in the microscope. (D) The effect of different concentrations of DHC treatment for 48 h on the proliferation rate of PIG1 cells determined by MTT assay. (E and F) The effect of DHC on the viability of A375 and MV3 cells. (G) Images and quantifications of A375 and MV3 cells positive for BrdU staining after treating with DMSO or 40 μM DHC for 24 h. Scale bar, 100 μm. All data are shown as the mean ± SD. A two-tailed unpaired Student’s t-test was carried out. *p<0.05, **p<0.01, ***p<0.001, ****p<0.0001.Abbreviations: DHC, dehydrocorydaline; DMSO, dimethyl sulfoxide; MTT, 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl tetrazolium bromide. |
Dehydrocorydaline induces cell cycle arrest at G0/G1 phase in melanoma cells
Cell proliferation was suggested to be tightly related to cell cycle progression. To detect the alterations of DHC on the cell cycle, A375 and MV3 cells were stained with a fluorescent intercalating agent, propidium iodide (PI), for DNA staining, and then cells were analyzed by BD Accuri C6 flow cytometry. The results showed that 20, 40, and 80 μM DHC-treated cells led to a significant G0/G1 phase arrest in a dose-dependent manner, compared with the DMSO-treated cells (Figure 3A and B). Furthermore, we measured the expression of CDK6 and cyclin D1, which could promote cell cycle progression. The results showed that protein levels of cyclin D1 and CDK6 were decreased in DHC-treated cells in both dose- and time-dependent manners (Figure 3C and D). In addition, we also checked the effect of DHC on the cell cycle of PIG1 cells, and the result showed that 40 μM DHC treatment for 48 h did not induce significant cell cycle arrest in PIG1 cells (Figure 3E). These results demonstrated that the DHC-induced cell cycle arrest might be the reason for DHC-inhibited cell proliferation in human melanoma cells.
 |
Figure 3 Dehydrocorydaline induces cell cycle arrest at G1 phase in human melanoma cells. (A and B) The cell cycle of A375 and MV3 cells was analyzed by flow cytometry after treating with DMSO or the indicated concentrations of DHC for 48 h. (C and D) Western blot assay was performed to assess the cell cycle-related protein levels in A375 and MV3 cells, respectively. Protein levels were calculated based on the grayscale value of protein bands and normalized with the grayscale value of GAPDH bands. Cells were treated with the indicated concentrations (0, 20, 40, 80 μM) of DHC for 48 h or with 40 μM DHC treatment for indicated times (0, 24, 48, 72 h) of DHC; GAPDH was used as a control. (E) The cell cycle of PIG1 cells was analyzed by flow cytometry after treating with DMSO or 40 μM DHC for 48 h. All data are shown as the mean ± SD. A two-tailed unpaired Student’s t-test was carried out. **p<0.01, ***p<0.001.Abbreviations: n.s.=not significant; DHC, dehydrocorydaline; DMSO, dimethyl sulfoxide; GAPDH, glyceraldehyde 3-phosphate dehydrogenase. |
Dehydrocorydaline inhibits cell migration and invasion in melanoma cells
As metastasis is an important reason for the malignancy of melanoma, we next explored the effect of DHC on the regulation of cell migration and invasion in human melanoma cells. Firstly, cell migration abilities were tested by wound-healing assay, and the results revealed that cells treated with 40 μM DHC treatment significantly blocked the rate of wound closure, compared with control groups (Figure 4A and B). Consistently, the transwell migration assay further showed that cells treated with 40 μM DHC exerted remarkable inhibition of the cellular trans-migration ability compared with the DMSO-treated cells (Figure 4C). Similarly, in the transwell invasion assay, we further confirmed that DHC treatment significantly downregulated the number of cells that passed through the Matrigel-coated membrane (Figure 4D). Consistent with the above, the expression of E-cadherin, an epithelial marker, was upregulated, and the expression of the mesenchymal markers, including vimentin and β-catenin, was downregulated, in both dose- and time-dependent manners (Figure 4E and F). Therefore, these results demonstrated that DHC inhibited cell migration/invasion possibly via downregulating epithelial–mesenchymal transition (EMT) in melanoma cells.
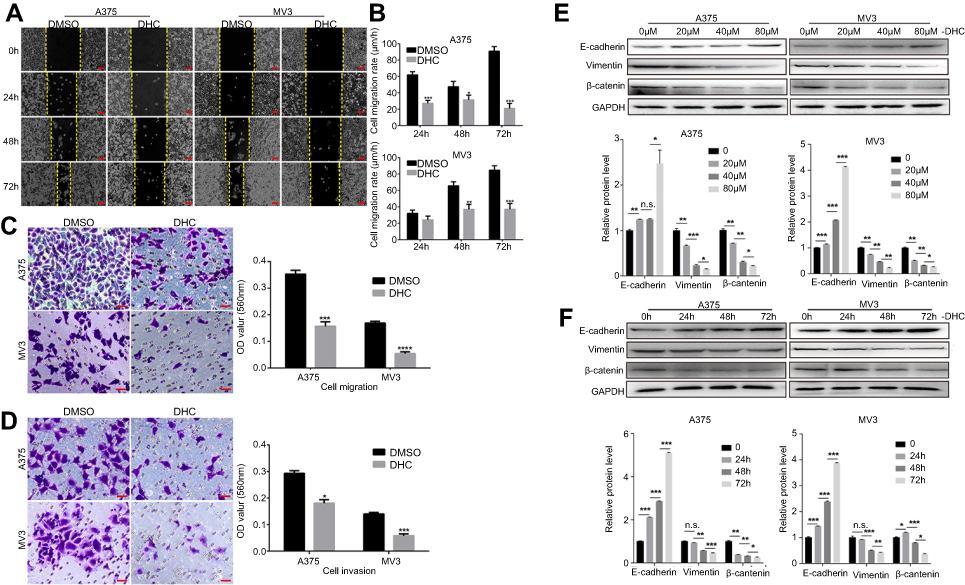 |
Figure 4 Dehydrocorydaline inhibits cell migration and invasion in melanoma cells. (A) Cell migration rate detected by wound-healing assay of A375 and MV3 cells after treating with DMSO or 40 μM DHC for the indicated time. Scale bar, 100 μm. (B) The effect of 40 μM DHC on the wound closure in A375 and MV3 cells. (C) The effect of transwell migration assays in A375 and MV3 cells after treating with DMSO or 40 μM DHC for 24 h. Scale bar, 100 μm. Migration rates were normalized by proliferation. (D) The effect of transwell invasion assays in A375 and MV3 cells after treating with DMSO or 40 μM DHC for 72 h. Scale bar, 100 μm. Invasion rates were normalized by proliferation. (E and F) Western blot analysis of the metastasis-related protein levels in A375 and MV3 cells, respectively. Protein levels were calculated based on the grayscale value of protein bands and normalized with the grayscale value of GAPDH bands. Cells were treated with the indicated concentrations (0, 20, 40, 80 μM) of DHC for 48 h or with 40 μM DHC treatment for indicated times (0, 24, 48, 72 h) of DHC; GAPDH was used as a control. All data are shown as the mean ± SD. A two-tailed unpaired Student’s t-test was carried out. *p<0.05, **p<0.01, ***p<0.001, ****p<0.0001. Abbreviations: DHC, dehydrocorydaline; DMSO, dimethyl sulfoxide; GAPDH, glyceraldehyde 3-phosphate dehydrogenase. |
Dehydrocorydaline suppresses tumor growth in xenograft model of melanoma cells
To further assess the impacts of DHC on colony formation of melanoma cells, we employed the soft agar assay. After 2 or 3 weeks (MV3 for 2 weeks and A375 for 3 weeks), the colonies in DHC-treated cells were smaller and less numerous than in the control groups (Figure 5A and B). Then, A375 and MV3 melanoma cells were transplanted subcutaneously into both flanks of the female BALB/c nude mice. After about a week, the tumor plumped. The mice were randomly divided into two groups and orally administrated with 100 mg/kg DHC or isometric DMSO every day 12 times. The results of tumor volume showed that DHC treatment dramatically suppressed tumor growth in vivo (Figure 5E and F). Then, the mice were sacrificed on the final day, and the tumors in the mice were excised and weighed. Consistent with the results above, DHC dramatically blocked the weight of tumors that formed in the nude mice (Figure 5G and H). Importantly, DHC treatment did not affect the mean weight, the appearance or the behavior of the mice, which suggested that DHC had little toxicity to the mice (Figure 5C and D). H&E staining showed that cell proliferation was inhibited in DHC-treated xenograft tumor samples (Figure 6). Besides, the expression of Ki-67, a cell proliferation marker, was downregulated in the A375 and MV3 xenograft tumor samples (Figure 6). Furthermore, immunohistochemical staining revealed that Cyclin D1 and vimentin expressions were significantly reduced in the melanoma tumor samples (Figure 6), which were consistent with previous results. These results further suggested that DHC treatment led to tumor growth retardation in xenograft models of melanoma cells.
 |
Figure 5 Dehydrocorydaline suppresses tumor growth in xenograft model of melanoma cells. (A and B) The colony formation was examined by soft agar assay (1,000 cells/well) in A375 or MV3 cells after treating with DMSO or 40 μM DHC for 14 days. Scale bar, 1 mm. (C and D) A375 and MV3 cells were injected into the flank of BALB/c nude mice. When tumors were palpable, mice were orally administrated with 100 mg/kg DHC or DMSO every day 12 times after the tumor plumped, and mice body weight was measured. (E and F) The tumor volume of xenograft tumors formed by the A375 and MV3 cells which were treated with DHC (100 mg/kg) or DMSO was measured. (G and H) The weight of the tumor was measured after DHC or DMSO treatment. All data are shown as the mean ± SD. A two-tailed unpaired Student’s t-test was carried out. *p<0.05, ***p<0.001, ****p<0.0001.Abbreviations: DHC, dehydrocorydaline; DMSO, dimethyl sulfoxide. |
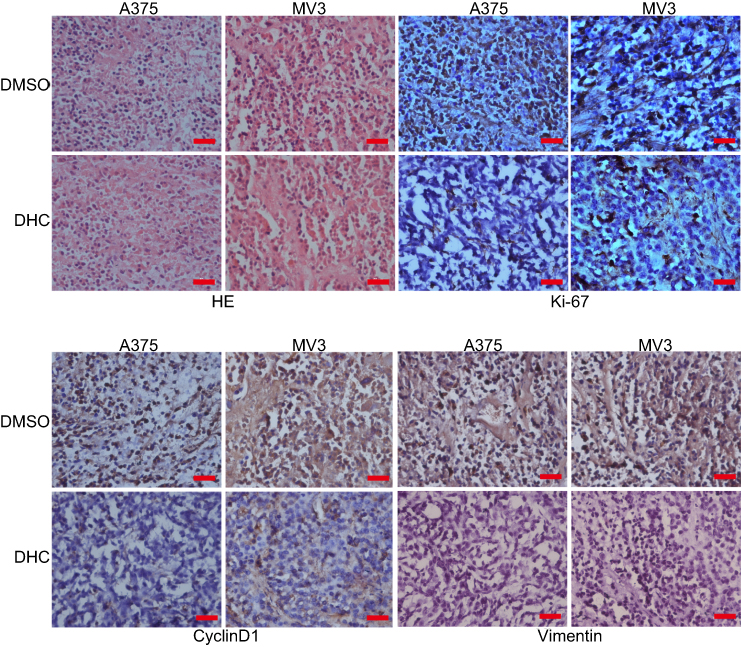 |
Figure 6 Dehydrocorydaline decreases the expression of cell cycle and metastatic markers in xenograft model of melanoma cells. The images of the H&E staining and immunohistochemistry analysis of d Ki-67, cyclin D1, and vimentin expression are presented. Abbreviations: DHC, dehydrocorydaline; DMSO, dimethyl sulfoxide. |
Dehydrocorydaline downregulates MEK1/2-ERK1/2 cascade in melanoma cells
The mitogen-activated protein kinase (MAPK) signaling network is one of the most important major signalings that contribute to cell proliferation and survival of cancer cells.29 The MAPK pathway is comprised of MAP3 kinase (MAP3K, BRAF), MAP-extracellular signal-regulated kinase 1/2 (MEK1/2) as well as extracellular signal-regulated kinase 1/2 (ERK1/2).30 Since the BRAF-MEK-ERK pathway plays a central role in determining cell fate, they have become primary targets for the treatment of tumors.31 For example, dabrafenib, a BRAF inhibitor, and trametinib, a MEK inhibitor, are significantly more active in patients with BRAF mutant melanoma.32 In our study, we found that DHC treatment induced downregulation of phosphorylation of MEK1/2 (Ser221, 2338) and ERK1/2 (Thr202/Tyr204, 4370), both in a dose- and time-dependent manner, compared with the control groups (Figure 7A and B). Besides, ERK activator t-butylhydroquinone (tBHQ, 50 μM) treatment recovered DHC induced cell proliferation inhibition (Figure 7C). This evidence indicated that DHC downregulated MAPK cascade and might be a candidate drug to treat melanoma.
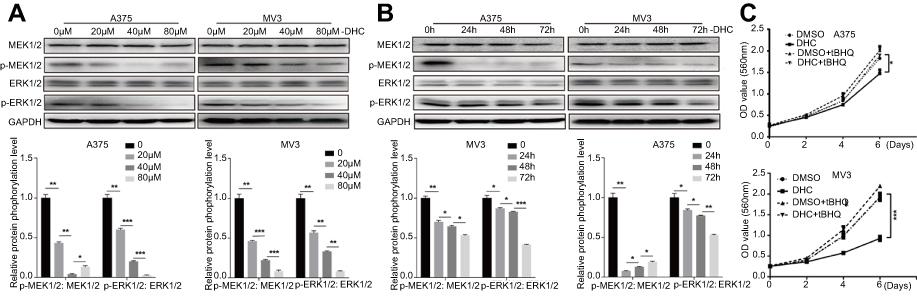 |
Figure 7 Dehydrocorydaline downregulates MEK1/2-ERK1/2 cascade in melanoma cells. (A and B) Western blot analysis of the expression of MAPK cascade in A375 and MV3 cells, respectively. Protein phosphorylation levels were calculated based on the grayscale value of phosphorylated protein bands and normalized with the grayscale value of total protein bands. Cells were treated with the indicated concentrations (0, 20, 40, 80 μM) of DHC for 48 h or with 40 μM DHC treatment for indicated times (0, 24, 48, 72 h) of DHC; GAPDH was used as a control. (C) The effect of 40 μM DHC and 50 μM ERK1/2 activator t-butylhydroquinone (tBHQ) on the viability of A375 and MV3 cells, respectively. All data are shown as the mean ± SD. A two-tailed unpaired Student’s t-test was carried out. *p<0.05, **p<0.01, ***p<0.001.Abbreviations: DHC, dehydrocorydaline; DMSO, dimethyl sulfoxide; GAPDH, glyceraldehyde 3-phosphate dehydrogenase; MAPK, mitogen-activated protein kinase; tBHQ, t-butylhydroquinone. |
Discussion
Metastatic melanoma still remains as a highly aggressive disease, despite the breakthrough introduction of targeted therapies such as BRAF inhibitors33 and immune checkpoint blockade therapies such as PD-1 and CTLA-4 inhibitors.34,35 There is a need to find novel potential drugs and therapies for further improving the treatment outcomes of melanoma malignancies. Alkaloids are novel treatment strategies that are emerging as an effective and promising treatment option against several types of cancers. The alkaloid DHC inhibited cells proliferation in breast cancer cells20 and suppressed the metastatic potential of NSCLC cells.21 However, the effect of DHC in melanoma remains to be further explored.
In this study, we tried to evaluate the activities of DHC in melanoma cells. The results showed that DHC inhibited melanoma cell growth both in vitro and in vivo (Figures 1, 2 and 5). Besides, DHC treatment induced conspicuous cell cycle arrest at G0/G1 phase in melanoma cells (Figures 3 and 6). However, we did not see any cellular apoptosis after DHC treatment in melanoma cells (Figure S1). In breast cancer MCF-7 cells, DHC dose-dependently promoted Bax expression and decreased Bcl-2 expression.20 Another study showed that DHC exerted little cytotoxicity and inhibited the protein expression of Bcl-2 in NSCLC cells.21 However, in our previous study, Bcl-2 expression was extremely high in MV3 melanoma cells while very low in A375 cells.26 In MV3 cells, overexpressed Bcl-2 suppressed apoptosis induced by DHC treatment; while in A375 cells, Bcl-2 was not a fateful factor. That might be the reason why DHC did not induce apoptosis in MV3 and A375 melanoma cells.
EMT is a cellular process in which cells lose cell polarity and release cell–cell adhesion, while obtaining metastatic properties to show mesenchymal features, which are more likely to migrate from primary focus to a metastatic focus.36,37 Cells in EMT procession upregulate many factors, such as N-cadherin, vimentin, and β-catenin, and downregulate E-cadherin, so these can be used as markers of EMT.38 Importantly, we found that DHC remarkably inhibited the migration or invasion of melanoma cells and promoted the expression of E-cadherin, while attenuating the expression of vimentin and β-catenin (Figures 4 and 6). A previous study also showed that DHC could inhibit the metastatic potential of NSCLC cells by downregulating MMP7 and MMP9.21 These indicated that DHC might inhibit cell migration/invasion via influencing the EMT.
Nearly 50% of metastatic melanoma patients have constitutively activated BRAFV600 mutations, which mostly excite the ERK1/2 signaling pathway to drive cancer cell proliferation and metastasis.39 Vemurafenib, dabrafenib, and trametinib are effective drugs for BRAFV600-mutant metastatic melanoma.40 However, resistance to these drugs has occurred in some melanoma patients hitherto.41 In our study, we found that DHC reduced phosphorylation of MEK and ERK in a dose- and time-dependent manners both in BRAFV600E mutant A375 cells and in BRAF wild MV3 cells (Figure 7A and B), and activated ERK1/2 by tBHQ retrieved DHC-induced cell proliferation inhibition (Figure 7C). Activated ERK also directly or indirectly regulated cell cycle-related proteins, such as CDKs and cyclins controlling G1 to S-phase transition42 and pro-metastatic factors, such as E-cadherin,43 and β-catenin.44 Phosphorylation of ERK was also facilitated by vimentin,45 which was also downregulated by DHC in our study (Figures 4E and 4F). This evidence suggested that DHC might be a new therapeutic drug to block MEK-ERK signaling and to treat melanoma, especially BRAFV600E mutant metastatic melanoma. However, more efforts should be made to evaluate its effects and mechanisms.
In summary, we found that DHC inhibited the progression of metastatic melanoma cells and led to cell cycle arrest via suppressing MEK-ERK cascade (Figure 8). Our results indicated that DHC might be a promising and effective therapeutic agent for the treatment of patients with metastatic malignant melanoma.
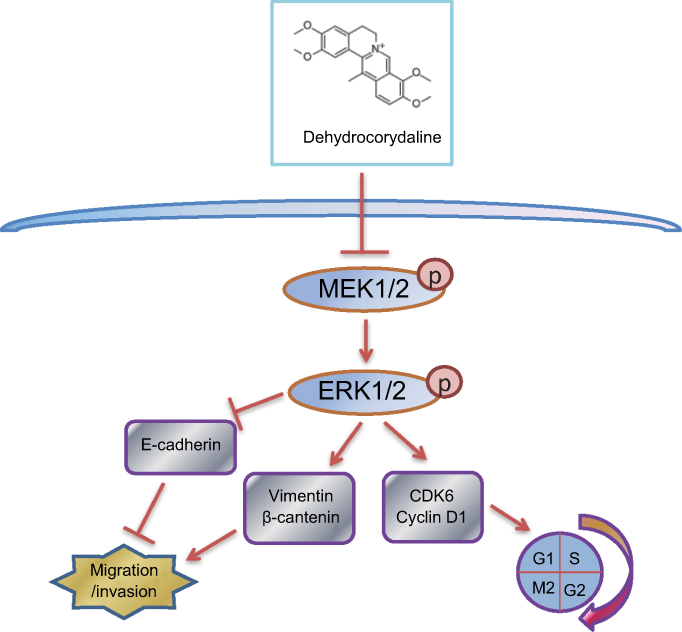 |
Figure 8 Overview of the molecular mechanism of dehydrocorydaline in the inhibition of cell proliferation, migration, and invasion of melanoma. |
Abbreviation list
MM, malignant melanoma; DHC, dehydrocorydaline; tBHQ, t-butylhydroquinone; EMT, epithelial–mesenchymal transition; MMP2, matrix metalloproteinase-2; NSCLC, non-small cell lung carcinoma; IL-1β, interleukin-1β; IL-6, interleukin-6; MEK, mitogen-activated protein kinase kinase; ERK, extracellular regulated protein kinase; MAPK, mitogen-activated protein kinase; GAPDH, glyceraldehyde 3-phosphate dehydrogenase; CDK6, cell division protein kinase 6; MTT, 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl tetrazolium bromide; CTLA-4, cytotoxic T-lymphocyte-associated protein 4; PD-1, programmed cell death protein 1; P/S, penicillin–streptomycin; DMSO, dimethyl sulfoxide; BrdU, 5-bromo-2ʹ-deoxyuridine.
Acknowledgments
Huanrong Hu and Zhen Dong are co-first authors. This study was supported by the Project Funded by Chongqing Special Postdoctoral Science Foundation (No. XmT2018080 to Z. Dong), the Fundamental Research Funds for the Central Universities (XDJK2019C013 to Z. Dong), the National Key Research and Development Program of China (No. 2016YFC1302204 and 2017YFC1308600 to H. Cui) and the National Natural Science Foundation of China (No. 81672502 to H. Cui). We thank Dr Muhammad Nadeem Abbas and Dr Saima Kausar from the State Key Laboratory of Silkworm Genome Biology, Southwest University for looking through the manuscript and giving some suggestions. We also thank Miss Li Tan from the State Key Laboratory of Silkworm Genome Biology, Southwest University for the assistance in performing some experiments during revision.
Disclosure
The authors report no conflicts of interest in this work.
References
1. Gray-Schopfer V, Wellbrock C, Marais R. Melanoma biology and new targeted therapy. Nature. 2007;445(7130):851–857. doi:10.1038/nature05661
2. DeSantis CE, Lin CC, Mariotto AB, et al. Cancer treatment and survivorship statistics, 2014. CA Cancer J Clin. 2014;64(4):252–271. doi:10.3322/caac.21235
3. Guy GP
4. Bedrosian I, Faries MB, Guerry D, et al. Incidence of sentinel node metastasis in patients with thin primary melanoma (< or =1 mm) with vertical growth phase. Ann Surg Oncol. 2000;7(4):262–267.
5. Shtivelman E, Davies MQ, Hwu P, et al. Pathways and therapeutic targets in melanoma. Oncotarget. 2014;5(7):1701–1752. doi:10.18632/oncotarget.1892
6. Tsang CM, Lau EP, Di K, et al. Berberine inhibits rho GTPases and cell migration at low doses but induces G2 arrest and apoptosis at high doses in human cancer cells. Int J Mol Med. 2009;24(1):131–138.
7. Hwang JM, Kuo HC, Tseng TH, Liu JY, Chu CY. Berberine induces apoptosis through a mitochondria/caspases pathway in human hepatoma cells. Arch Toxicol. 2006;80(2):62–73. doi:10.1007/s00204-005-0014-8
8. Lin CC, Ng LT, Hsu FF, Shieh DE, Chiang LC. Cytotoxic effects of coptis chinensis and epimedium sagittatum extracts and their major constituents (berberine, coptisine and icariin) on hepatoma and leukaemia cell growth. Clin Exp Pharmacol Physiol. 2004;31(1–2):65–69. doi:10.1111/j.1440-1681.2004.03951.x
9. Letasiova S, Jantova S, Cipak L, Muckova M. Berberine-antiproliferative activity in vitro and induction of apoptosis/necrosis of the U937 and B16 cells. Cancer Lett. 2006;239(2):254–262. doi:10.1016/j.canlet.2005.08.024
10. Peng PL, Hsieh YS, Wang CJ, Hsu JL, Chou FP. Inhibitory effect of berberine on the invasion of human lung cancer cells via decreased productions of urokinase-plasminogen activator and matrix metalloproteinase-2. Toxicol Appl Pharmacol. 2006;214(1):8–15. doi:10.1016/j.taap.2005.11.010
11. Chen J, Shen L, Zhou R, et al. [Reversal effect and mechanism of lobeline on the multidrug-resistance of human breast cancer cells MCF-7/ADM]. Zhong Nan Da Xue Xue Bao Yi Xue Ban. 2009;34(8):738–743.
12. He L, Liu GQ. Effects of various principles from Chinese herbal medicine on rhodamine123 accumulation in brain capillary endothelial cells. Acta Pharmacol Sin. 2002;23(7):591–596.
13. Kurahashi K, Fujiwara M. Adrenergic neuron blocking action of dehydrocorydaline isolated from Corydalis bulbosa. Can J Physiol Pharmacol. 1976;54(3):287–293.
14. Matsuda H, Tokuoka K, Wu J, Shiomoto H, Kubo M. Inhibitory effects of dehydrocorydaline isolated from corydalis tuber against type I-IV allergic models. Biol Pharm Bull. 1997;20(4):431–434. doi:10.1248/bpb.20.431
15. Ishiguro K, Ando T, Maeda O, Watanabe O, Goto H. Dehydrocorydaline inhibits elevated mitochondrial membrane potential in lipopolysaccharide-stimulated macrophages. Int Immunopharmacol. 2011;11(9):1362–1367. doi:10.1016/j.intimp.2011.04.022
16. Jiang XR, Wu QX, Shi HL, et al. [Pharmacological actions of dehydrocorydaline on cardiovascular system (author’s transl)]. Yao Xue Xue Bao. 1982;17(1):61–65.
17. Xiao HT, Peng J, Liang Y, et al. Acetylcholinesterase inhibitors from corydalis yanhusuo. Nat Prod Res. 2011;25(15):1418–1422. doi:10.1080/14786410802496911
18. Satou T, Akao N, Matsuhashi R, Koike K, Fujita K, Nikaido T. Inhibitory effect of isoquinoline alkaloids on movement of second-stage larvae of toxocara canis. Biol Pharm Bull. 2002;25(12):1651–1654. doi:10.1248/bpb.25.1651
19. Yoo M, Lee SJ, Kim YK, et al. Dehydrocorydaline promotes myogenic differentiation via p38 MAPK activation. Mol Med Rep. 2016;14(4):3029–3036. doi:10.3892/mmr.2016.5653
20. Xu Z, Chen X, Fu S, et al. Dehydrocorydaline inhibits breast cancer cells proliferation by inducing apoptosis in MCF-7 cells. Am J Chin Med. 2012;40(1):177–185. doi:10.1142/S0192415X12500140
21. Lee J, Sohn EJ, Yoon SW, et al. Anti-metastatic effect of dehydrocorydaline on H1299 non-small cell lung carcinoma cells via inhibition of matrix metalloproteinases and B cell lymphoma 2. Phytother Res. 2017;31(3):441–448. doi:10.1002/ptr.5766
22. van Muijen GN, Jansen KF, Cornelissen IM, Smeets DF, Beck JL, Ruiter DJ. Establishment and characterization of a human melanoma cell line (MV3) which is highly metastatic in nude mice. Int J Cancer. 1991;48(1):85–91.
23. Dong Z, Lei Q, Yang R, et al. Inhibition of neurotensin receptor 1 induces intrinsic apoptosis via let-7a-3p/Bcl-w axis in glioblastoma. Br J Cancer. 2017;116(12):1572–1584. doi:10.1038/bjc.2017.126
24. Hu H, Dong Z, Tan P, et al. Antibiotic drug tigecycline inhibits melanoma progression and metastasis in a p21CIP1/Waf1-dependent manner. Oncotarget. 2016;7(3):3171–3185. doi:10.18632/oncotarget.6419
25. Yang R, Yi L, Dong Z, et al. Tigecycline inhibits glioma growth by regulating miRNA-199b-5p-HES1-AKT pathway. Mol Cancer Ther. 2016;15(3):421–429. doi:10.1158/1535-7163.MCT-15-0709
26. Liu L, Dong Z, Lei Q, et al. Inactivation/deficiency of DHODH induces cell cycle arrest and programed cell death in melanoma. Oncotarget. 2017;8(68):112354–112370. doi:10.18632/oncotarget.19379
27. Zhao Y, He J, Li J, et al. Demethylzeylasteral inhibits cell proliferation and induces apoptosis through suppressing MCL1 in melanoma cells. Cell Death Dis. 2017;8(10):e3133. doi:10.1038/cddis.2017.529
28. Hou J, Deng Q, Zhou J, et al. CSN6 controls the proliferation and metastasis of glioblastoma by CHIP-mediated degradation of EGFR. Oncogene. 2017;36(8):1134–1144. doi:10.1038/onc.2016.280
29. Johnson GL, Stuhlmiller TJ, Angus SP, Zawistowski JS, Graves LM. Molecular pathways: adaptive kinome reprogramming in response to targeted inhibition of the BRAF-MEK-ERK pathway in cancer. Clin Cancer Res. 2014;20(10):2516–2522. doi:10.1158/1078-0432.CCR-13-1081
30. Dhanasekaran DN, Johnson GL. MAPKs: function, regulation, role in cancer and therapeutic targeting. Oncogene. 2007;26(22):3097–3099. doi:10.1038/sj.onc.1210395
31. De Luca A, Maiello MR, D’Alessio A, Pergameno M, Normanno N. The RAS/RAF/MEK/ERK and the PI3K/AKT signalling pathways: role in cancer pathogenesis and implications for therapeutic approaches. Expert Opin Ther Targets. 2012;16(Suppl 2):S17–S27. doi:10.1517/14728222.2011.639361
32. Long GV, Stroyakovskiy D, Gogas H, et al. Dabrafenib and trametinib versus dabrafenib and placebo for Val600 BRAF-mutant melanoma: a multicentre, double-blind, phase 3 randomised controlled trial. Lancet (London, England). 2015;386(9992):444–451. doi:10.1016/S0140-6736(15)60898-4
33. Fedorenko IV, Abel EV, Koomen JM, et al. Fibronectin induction abrogates the BRAF inhibitor response of BRAF V600E/PTEN-null melanoma cells. Oncogene. 2016;35(10):1225–1235. doi:10.1038/onc.2015.188
34. Chauvin JM, Pagliano O, Fourcade J, et al. TIGIT and PD-1 impair tumor antigen-specific CD8(+) T cells in melanoma patients. J Clin Invest. 2015;125(5):2046–2058. doi:10.1172/JCI80445
35. Eggermont AM, Chiarion-Sileni V, Grob JJ, et al. Prolonged survival in stage III melanoma with ipilimumab adjuvant therapy. N Engl J Med. 2016;375(19):1845–1855. doi:10.1056/NEJMoa1611299
36. Thiery JP, Acloque H, Huang RY, Nieto MA. Epithelial-mesenchymal transitions in development and disease. Cell. 2009;139(5):871–890. doi:10.1016/j.cell.2009.11.007
37. Acloque H, Adams MS, Fishwick K, Bronner-Fraser M, Nieto MA. Epithelial-mesenchymal transitions: the importance of changing cell state in development and disease. J Clin Invest. 2009;119(6):1438–1449. doi:10.1172/JCI38019
38. Zeisberg M, Neilson EG. Biomarkers for epithelial-mesenchymal transitions. J Clin Invest. 2009;119(6):1429–1437. doi:10.1172/JCI36183
39. Lito P, Pratilas CA, Joseph EW, et al. Relief of profound feedback inhibition of mitogenic signaling by RAF inhibitors attenuates their activity in BRAFV600E melanomas. Cancer Cell. 2012;22(5):668–682. doi:10.1016/j.ccr.2012.10.009
40. Hauschild A, Grob JJ, Demidov LV, et al. Dabrafenib in BRAF-mutated metastatic melanoma: a multicentre, open-label, phase 3 randomised controlled trial. Lancet (London, England). 2012;380(9839):358–365. doi:10.1016/S0140-6736(12)60868-X
41. McArthur GA, Chapman PB, Robert C, et al. Safety and efficacy of vemurafenib in BRAF(V600E) and BRAF(V600K) mutation-positive melanoma (BRIM-3): extended follow-up of a phase 3, randomised, open-label study. Lancet Oncol. 2014;15(3):323–332. doi:10.1016/S1470-2045(14)70012-9
42. Chambard JC, Lefloch R, Pouyssegur J, Lenormand P. ERK implication in cell cycle regulation. Biochim Biophys Acta. 2007;1773(8):1299–1310. doi:10.1016/j.bbamcr.2006.11.010
43. Tashiro E, Henmi S, Odake H, Ino S, Imoto M. Involvement of the MEK/ERK pathway in EGF-induced E-cadherin down-regulation. Biochem Biophys Res Commun. 2016;477(4):801–806. doi:10.1016/j.bbrc.2016.06.138
44. Gortazar AR, Martin-Millan M, Bravo B, Plotkin LI, Bellido T. Crosstalk between caveolin-1/extracellular signal-regulated kinase (ERK) and beta-catenin survival pathways in osteocyte mechanotransduction. J Biol Chem. 2013;288(12):8168–8175. doi:10.1074/jbc.M112.437921
45. Perlson E, Michaelevski I, Kowalsman N, et al. Vimentin binding to phosphorylated Erk sterically hinders enzymatic dephosphorylation of the kinase. J Mol Biol. 2006;364(5):938–944. doi:10.1016/j.jmb.2006.09.056
Supplementary material
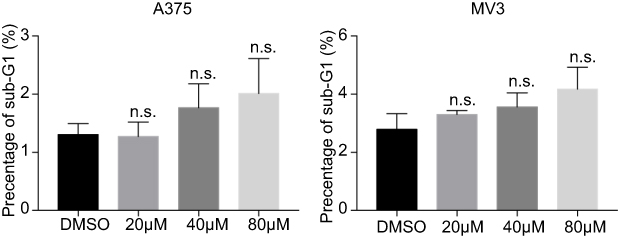 |
Figure S1 DHC treatment induces no significant cellular apoptosis (sub-G1 phase) in A375 and MV3 melanoma cells.Abbreviations: n.s.=not significant; DHC, dehydrocorydaline; DMSO, dimethyl sulfoxide. |
 © 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.