")
Back to Journals » Hepatic Medicine: Evidence and Research » Volume 8
Defibrotide in the treatment of hepatic veno-occlusive disease
Authors Fulgenzi A, Ferrero ME
Received 5 July 2016
Accepted for publication 8 September 2016
Published 31 October 2016 Volume 2016:8 Pages 105—113
DOI https://doi.org/10.2147/HMER.S79243
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Dr Gerry Lake-Bakaar
Alessandro Fulgenzi, Maria Elena Ferrero
Department of Biomedical Sciences for Health, University of Milan, Milan, Italy
Abstract: Hepatic veno-occlusive disease (VOD), also known as sinusoidal obstruction syndrome (SOS), represents the most frequent complication in patients in early phase following hematopoietic stem-cell transplantation (HSCT). In its severe form, VOD/SOS can be associated with multiorgan failure and with a mortality rate >80% by day +100. Defibrotide (DF) (a mixture of 90% single-stranded phosphodiester oligonucleotides and 10% double-stranded phosphodiester oligonucleotides derived from controlled depolarization of porcine intestinal mucosal DNA) has been proposed for the treatment of SOS due to its ability to restore thrombo-fibrinolytic balance and protect endothelial cells. The present review highlights why the mechanisms of action of DF allow its successful use in the prevention and treatment of SOS following HSCT.
Keywords: hepatic veno-occlusive disease, hematopoietic stem-cell transplantation, defibrotide
Introduction to hepatic veno-occlusive disease
Definition and causes
Veno-occlusive disease (VOD) is obliterative inflammation of the terminal hepatic venules. Now known as hepatic sinusoidal obstruction syndrome (SOS), since damage to the sinusoidal endothelium is known to be a primary event,1 it has various causes. Senecio poisoning in humans, causing conspicuous sinusoidal congestion and hemorrhagic necrosis in the centrilobular area, has been described since 1920.2 Liver terminal hepatic vein lesions in Jamaican drinkers of bush tea were seen in 1954.3 More recently, herbal remedies using plants containing pyrrolizidine alkaloids have been seen to be hepatotoxic and involved in SOS.4 The liver is the most common site of metastases in patients with colorectal cancer: SOS may occur following treatment with alkylating agents prior to surgical resection (eg. partial hepatectomy),5,6 or following antibody therapy with bevacizumab or cetuximab.7 It might also affect patients who undergo radiation therapy.8,9 Indeed, radiation-induced SOS might occur following treatment for liver metastases. A low incidence of SOS has been noted in patients affected by acute myeloid leukemia treated with the cytotoxic chemotherapeutic agent gemtuzumab ozogamicin.10,11 The rare occurrence of SOS due to liver graft dysfunction following liver transplantation,12 and SOS associated with immunodeficiency syndrome related to a mutation of the immunoregulatory gene SP110 have also been described.13 Finally, SOS typically develops following allogeneic hematopoietic stem-cell transplantation (HSCT).14 Some drugs that cause bone marrow suppression, which are responsible for SOS development, have recently been described: 6-mercaptopurine, 6-thioguanine, actinomycin D, azathioprine, busulfan, cytosine arabinoside, cyclophosphamide, dacarbazine, melphalan, oxaliplatin, urethane, and the above-cited gemtuzumab ozogamicin.15
Histologic and clinical features
Histology changes are related to the duration of SOS and can exhibit acute, subacute, and chronic features: 1) Acute features are characterized by extensive sinusoidal dilatation and congestion, and by centrilobular hepatocellular necrosis. 2) In subacute features (days to weeks), collagen deposition occurs in and around the affected terminal hepatic venules. 3) Chronic lesions (weeks to months) present dense perivenular fibrosis radiating out into the parenchyma; hepatocytes undergo severe destruction, and evolution into cirrhosis can occur.16
Clinically, in the acute variant, patients display massive abdominal swelling and pain, associated with hemorrhagic centrilobular necrosis. In the subacute type, recurrent ascites, splenomegaly, and hepatomegaly are associated with extensive fibrosis in centrilobular areas. In a clinical context, the chronic variant is difficult to distinguish from cirrhosis of other origins but shows a venocentric type of cirrhosis at histologic examination.15 The onset of SOS is characterized by painful hepatomegaly, jaundice, and weight gain. Late onset of SOS is typically associated with multiorgan failure (MOF) and high mortality rates (>80%).14,17 Clinical grading of SOS (mild, moderate, or severe) is based on the evaluation of bilirubin, liver enzymes (aspartate transferase and alanine transferase), weight above baseline in percent, and serum creatinine levels.16,18 Revision of diagnosis and severity criteria for SOS in adult patients, with a new classification from the European Society for Blood and Marrow Transplantation, has recently been published.18
The clinical criteria for SOS have recently been reported. They are Seattle criteria, and modified Seattle criteria and Baltimore criteria.19 In Seattle criteria, at least two of the three following need to be present during the first month following HSCT: jaundice, hepatomegaly and right upper quadrant pain, ascites, and/or unexplained weight gain. In modified Seattle criteria, during the first 20 days after HSCT, two or all of the following should be present: bilirubin >2 mg/dL, hepatomegaly or pain in right upper quadrant, and/or weight gain (>2% basal). In Baltimore criteria, the presence of bilirubin >2 mg/dL should be accompanied by two or all of the following during the first 21 days following HSCT: painful hepatomegaly, ascites, and/or weight gain (>5% basal).
Laboratory data
Some authors have reported that baseline levels of naturally occurring anticoagulants (eg. antithrombin III [AT III] and plasminogen) were lower in patients who developed SOS compared to those who did not develop SOS.20,21 In addition, plasminogen activator inhibitor-1 (PAI-1) levels were significantly higher in SOS patients than in those with other forms of posttransplantation liver injury.22 The N-terminal fragment of type III procollagen, which is a marker of fibrinogenesis, was elevated in patients affected by SOS,23 as well as the tissue inhibitor of metalloproteinases (TIMP-1).24
Pathophysiology of SOS
Initially, damage to liver sinusoidal endothelial cells (ECs) takes place. Endothelial injury is mainly induced during the HSCT procedure. Direct effects on ECs are provoked by irradiation: clinically relevant doses of radiotherapy lead to apoptosis with the involvement of endothelial transmembrane tumor necrosis factor alpha (TNFα).25 In cultured human umbilical vein ECs, irradiation increases inducible nitric oxide synthase (NOS) and nitrotyrosine levels; the latter is an indirect chemical indicator of peroxynitrite-induced cellular injury.26 In addition, irradiation can promote the expression of endothelial adhesion molecules and the enhancement of vascular permeability.27 Other direct effects on ECs are induced by chemotherapeutic drugs used in patients undergoing HSCT (cyclophosphamide, methotrexate, busulfan), and by prophylaxis with immunosuppressive treatment to prevent graft-versus-host disease (GVHD) in allogeneic HSCT (cyclosporine A, tacrolimus, sirolimus), and use of granulocyte colony-stimulating factor to accelerate recovery from post-chemotherapy neutropenia. In particular, tacrolimus, a macrolide lactone with potent immunosuppressive activity that is effective in the prophylaxis of organ rejection following kidney, heart, and liver transplantation, has been associated with SOS in lung and pancreatic transplantation, and has also been observed in liver transplant recipients.28 The above-mentioned treatment methods all affect ECs directly by increasing expression of adhesion molecules (intracellular cell adhesion molecule 1 [ICAM-1], vascular cell adhesion molecule 1 [VCAM-1]) and procoagulants (von Willebrand factor [vWF], PAI-1).29 Subsequent activation of the fibrinolytic pathway leads to sinusoidal fibrosis, followed by perivascular hepatocyte necrosis, and the venular blockage that characterizes SOS. Irradiation, chemotherapy, and immunosuppressive treatment also induce indirect effects on ECs. In fact, such therapies damage many organs and tissues, which release TNFα and interleukin (IL)-1 to promote dead-cell clearance and tissue repair. Breaks in mucosal barrier favor an increase in circulating lipopolysaccharides. These factors interfere with the antithrombotic properties of ECs, increasing PAI-1 and vWF and decreasing thrombomodulin expression, as well as increasing permeability and apoptosis.30
At a molecular level, under normal conditions drugs are metabolized by the cytochrome P450 enzymatic system, leading to the production of toxic metabolites, which are converted to stable nontoxic metabolites by the glutathione enzymatic system (GSH) and then eliminated. Centrilobular regions of the liver are rich in P-450 but poor in GSH, making them more sensitive to toxic agents. In patients with previous liver diseases or treated with alkylating agents, liver GSH levels are impaired, and toxic metabolites accumulate, thus explaining the predominant damage of centrilobular regions. Accumulated toxins impair vascular tone by inducing a decrease in prostaglandin E2 (PGE2) concentration, and an increase in matrix metalloproteinase 9 expression in ECs, leading to their detachment. Depletion of endothelial GSH seems to be more important for toxicity than hepatocyte GSH depletion.29
In conclusion, injury provoked by previous damaged liver conditions or by the direct effects of HSCT, for example, irradiation, chemotherapy, and immunosuppressive treatment, leads to endothelial activation followed by inflammation, increased permeability, vasoconstriction, coagulation, thrombosis, and EC death. The indirect effects of HSCT lead to organ damage, for example, the liver, lung, skin, kidney, brain, and heart, and to the delivery of bacterial products. Both endothelial activation and the indirect effects of HSCT can induce endothelial-associated syndromes, such as capillary leak syndrome, engraftment syndrome, GVHD, pulmonary VOD, transplant-associated microangiopathy, as well as late cardiovascular events.29
Incidence of SOS, risk factors, and mortality
More than 20 years ago, SOS was described as a common life-threatening complication of toxicity related to preparative regimen for bone-marrow transplantation (BMT), with its rate varying from 1 to 2% in centers performing pediatric BMT for thalassemia to over 50% in some centers carrying out BMT for hematologic malignancies.31 Pharmacokinetic measurement of busulfan as a conditioning agent has shown correlation between high plasma busulfan concentrations and liver toxicity. The development of SOS has frequently been associated with renal and cardiopulmonary failure.32 The significance of risk factors in determining the incidence of SOS and the application of diagnostic criteria after BMT have been highlighted.33 Late-onset SOS after HSCT (3 or more weeks after high-dose chemotherapy and HSCT) has been identified.34 The occurrence of SOS following HSCT was reported in 2010 with the analysis of 135 studies performed between 1979 and 2007: mortality rate was elevated (84.3%) only when severe SOS developed, and evolution into MOF was the most frequent cause of death.14 In 2011, analysis of 845 allogeneic HSCT cases showed that technical and therapeutic progress had notably reduced the incidence of SOS and improved outcome.35 Common “pretransplantation” patient-related SOS risk factors of HSCT include young recipient age (<1 year), liver damage, liver infection (eg. active viral hepatitis) or prior liver transplantation, abdominal or liver radiation and exposure to hepatotoxic drugs, and prior use of parenteral nutrition.19 Genetic factors are also involved: children with osteopetrosis have a high risk of SOS. Transplantation-related factors seem to be important for SOS development. In fact, risk factors are elevated in the presence of allogeneic HSCT, matched unrelated donors, and non-T-cell-depleted transplantation, as well as with the use of certain drugs for GVHD prophylaxis during HSCT (cyclosporine A, sirolimus, methotrexate).19 Additional factors that might be implicated in the development of SOS include elevation of serum alanine aminotransferase, thrombocytopenia with rapid platelet consumption, attenuated or reversed hepatic venous flow detected by ultrasound, and esophageal varices and their rate of change.
Management issues and treatment options
SOS prevention
Heparin, low-molecular weight heparin, prostaglandin E1 (PGE1), and ursodeoxycholic acid have been used, alone or in combination, after HSCT.36 There seem to be no differences between the efficacy of heparin plus ursodiol and heparin alone in the possible prevention of SOS.37 Prophylactic low-dose heparin or lipo-PGE1 might be able to prevent severe SOS and decrease mortality in children undergoing HSCT.38 The combined prophylactic regimen of intravenous heparin, oral glutamine, and ursodiol determined a low incidence of SOS in pediatric patients with various diagnoses at a single transplant institution.39 The side effects of heparin treatment were hemorrhagic complications; treatment with PGE1 provoked toxicity (fluid retention, pain, hypotension). Ursodeoxycholic acid protected against cholestasis,36 and parenteral glutamine protected hepatic function by maintaining GSH levels.40
Treatment of established SOS
Orthotopic liver transplantation has been carried out in a small number of marrow-transplant patients who developed severe SOS. Several patients have received surgical or transvenous portosystemic shunts or transjugular intrahepatic portosystemic shunts to treat SOS after HSCT.36 Early thrombolytic therapy with recombinant human tissue plasminogen activator (tPA) was seen to improve the outcome in patients who developed signs of hepatotoxicity following HSCT.36,41
High-dose methylprednisolone has also been proposed for the treatment of SOS,42 although it needs to be used with caution due to the risk of infection.
Finally, the use of defibrotide (DF) was proposed.
Defibrotide: review of pharmacokinetics, mode of action, and pharmacology
DF is a mixture of 90% single-stranded phosphodiester oligonucleotides (molecular mass 16.5±2.5 kDa) and 10% double-stranded phosphodiester oligonucleotides derived from controlled depolymerization of mammalian (typically porcine) intestinal mucosa DNA.43,44 Inside the mixture of oligonucleotides have been found certain aptamers, as well as cathepsin G, which can inhibit thrombin through the recurrence of alternating TG repeats.45–47
Pharmacokinetics of 125I-DF following intravenous or oral administration in rats or in man showed that half-life of the distribution phase (T1/2α) was in the range of minutes, while half-life of the elimination phase (T1/2β) was in the range of hours, and bioavailability in the range of 50–70%.44,48–50
Many studies, both in vitro and in vivo, have highlighted the activities of DF, underlining its anti-inflammatory, anti-ischemic, antithrombotic, and thrombolytic properties. Experimental studies have shown the pharmacologic role of DF in avoiding loss of vascular integrity: it reduces vascular permeability (by inhibiting TNF release and deactivating platelets) and inflammation (by reducing nuclear factor kappa B [NFκB] and by impairing cysteinyl-leukotriene release). Downregulating the expression of adhesion molecules, DF reduces P-selectin on activated endothelium, and interferes with ICAM/lymphocyte function-associated antigen 1 (LFA-1) adhesion system. Antithrombotic and thrombolytic effects of DF are mediated by an increase of tPA, tissue factor pathway inhibitor, and thrombomodulin, and by a decrease of PAI, platelet activating factor, and thrombin.
Modulating the production of some inflammatory mediators, including cytokines, DF increases PGE2 and prostacyclin 2 (PGI2), and reduces IL-6, vascular endothelial growth factor, thromboxane (TX) A2, leukotriene B4, and TNF. Reducing MHC class 1 molecule expression, DF also antagonizes the vasoconstrictor activity of endothelin-1, and enhances the production of nitric oxide (NO) and NOS, thus favoring the maintenance of vascular tone.44
Clinical studies on the pharmacological properties of DF have demonstrated its antiischemic and antithrombotic activities. It has been used in the antithrombotic prophylaxis of atherosclerosis50 and to improve the walking distance in patients with intermittent claudication.51 More recently, DF has been considered as a new potential treatment approach in antiphospholipid syndrome, which is a hypercoagulable state characterized by arterial and venous thromboses and pregnancy morbidity in the presence of antiphospholipid antibodies. The role of DF can be compared with emerging therapies using new oral anticoagulants and immunoregulatory agents, such as direct thrombin or factor Xa inhibitors, hydroxychloroquine, statins, B cell inhibitors, complement inhibitors, NFκB and 38 mitogen-activated kinase (38 MAPK) inhibitors, abciximab, and mammalian target of rapamycin inhibitors.52
Studies on the efficacy of DF in the treatment and prevention of SOS
More recently, DF has been proposed for the treatment of patients with SOS after HSCT. It was administered intravenously in 88 patients affected by severe SOS complicated by MOF in doses ranging from 5 to 60 mg/kg per day for a median of 15 days. Complete resolution of SOS was seen in 36% of patients, with 35% survival at day +100.
A decrease in mean creatinine and PAI-1 levels during DF therapy predicted better survival.53
In pediatric HSCT patients who developed SOS, high-dose DF (60 mg/day) and AT III replacement therapy were used in combination. Patients with SOS receiving combined therapy achieved complete remission, and 93% (13/14) survived up to day + 100, whereas preemptive AT III administration alone did not alter the incidence of SOS.54 In another multicenter trial, patients with severe SOS and MOF following HSCT showed significantly lower mortality when they received intravenous DF 6.25 mg/kg every 6 hours compared to controls.55
A single-center experience based on 273 allogeneic HSCT review over a 45-month period has been reported.56 The patients received DF therapy (25 mg/kg/day) within 24 hours of diagnosis of SOS, and aggressive supportive management for fluid balance. The median duration of DF therapy was 14 days: early treatment with DF was able to completely resolve SOS in 11 out of 12 evaluable patients.56
More recently, a phase-3 trial on DF treatment of severe SOS and advanced MOF has been reported. Patients (n=102) given DF 25 mg/kg/day were compared with 32 historical controls identified from 6867 medical charts of HSCT patients by blinded independent reviewers. The baseline characteristics between groups were well balanced. Survival at day +100 post-HSCT was 38.2% in the DF group and 25% in the controls. Day +100 complete response rates were 25.5% for DF and 12.5% for controls.57 The efficacy of DF in the treatment of severe SOS was also evaluated using observational data from the Center for International Blood and Marrow Transplant Research. Day +100 survival was 39% in patients receiving DF and 30.9% in those not receiving DF. Resolution rate of SOS at day +100 was 51% in the DF group and 29% in the non-DF group. The results of the study suggest that DF is effective in the treatment of severe SOS.58
Studies on the efficacy of DF in the prophylaxis of SOS have also been performed.
An open-label phase-3 randomized controlled trial was reported in 2012, in which eligible patients were enrolled at 28 European university hospitals or academic medical centers. The patients aged under 18 years had undergone myeloablative conditioning before allogeneic or autologous HSCT. The DF prophylaxis was well tolerated and reduced the incidence of SOS.59
Thirty-four patients (69.4%) were classified as a high-risk group for developing SOS. DF was well tolerated, without any grade 3 or 4 toxicity. The median value of maximum total bilirubin within 100 days after HSCT was within the normal range. SOS was diagnosed in only one patient, who underwent autologous HSCT due to relapsed medulloblastoma. There was no day 100 treatment-related mortality in our study.60
A summary of clinical studies is reported in the Table 1.
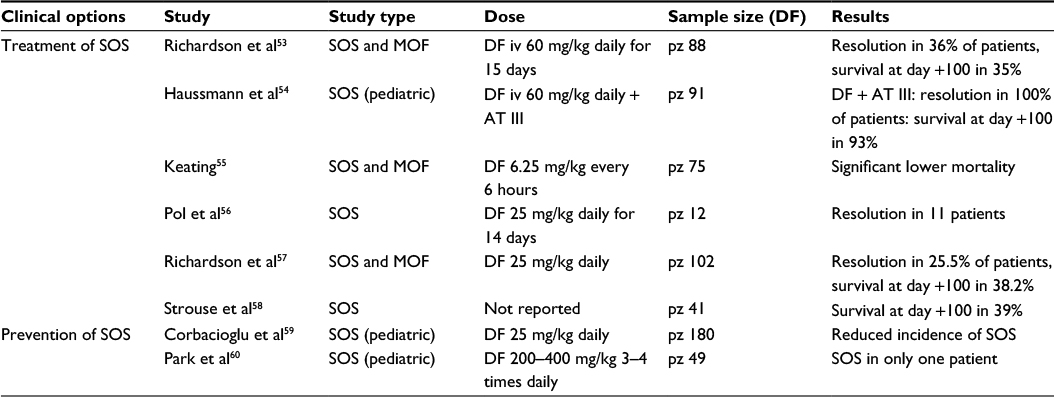 | Table 1 Summary of studies regarding treatment and prevention of SOS by DF Abbreviations: SOS, sinusoidal obstruction syndrome; DF, defibrotide; MOF, multiorgan failure; pz, patients; iv, intravenous; AT III, antithrombin III. |
Clinical recommendations
A joint working group, established by the Haemato-oncology subgroup of the British Committee for Standards in Haematology and the British Society for Blood and Marrow Transplantation, recently reviewed the available literature and made recommendations for the diagnosis and management of post-HSCT SOS. Such guidelines included recommendations for both prophylaxis and treatment in children and adults undergoing HSCT.61 Diagnosis of SOS was based primarily on established clinical criteria (modified Seattle or Baltimore criteria). Regarding prophylaxis, DF is recommended at a dose of 6.25 mg/kg intravenously four times daily for the prevention of SOS in children and adults with the following risk factors that need to undergo HSCT: preexisting hepatic disease, second myeloablative transplant, allogeneic transplant for leukemia beyond second relapse, conditioning with busulfan-containing regimens, prior treatment with gemtuzumab ozogamicin, diagnosis of primary hemophagocytic lymphohistiocytosis, adrenoleukodystrophy, or osteopetrosis. Pentoxiphylline, heparin (unfractionated and low molecular weight), and AT III are not suggested for the prophylaxis of SOS, unlike ursodeoxycholic acid. Regarding treatment, DF is recommended in SOS therapy in both adults and children.61
Safety and tolerability
DF was generally well tolerated with manageable toxicity. Related diverse events included hemorrhage or hypotension. In one reported study, the incidence of common hemorrhagic adverse events (including pulmonary, alveolar, and gastrointestinal bleeding) was similar in the DF and control groups.57 DF is considered a safe and effective treatment when dosed at 25 mg/kg/day. Escalation to high-dose DF has been evaluated.62 Increased toxicity has not been observed until doses were increased beyond 100 mg/kg/day. Patients receiving doses between 10 and 100 mg/kg/day have experienced an average of three bleeding episodes per 100 days of treatment, whereas those receiving doses >100 mg/kg/day have experienced 13.2 bleeding episodes per 100 days. Due to toxicity, dose reduction was more often necessary at 110 mg/kg/day than at lower doses.62 A case of severe SOS has been reported in a child undergoing chemotherapy (vincristine, actinomycin D, and cyclophosphamide) for rhabdomyosarcoma: the child was successfully treated with DF without sequelae to the liver.63 A review of the current clinical findings concerning DF, primarily regarding its safety in the treatment and prophylaxis of SOS, together with relevant safety data regarding its use in other diseases, shows that DF is generally well tolerated. The safety profile of DF is largely favorable, with toxicity being comparable to control populations in the setting of HSCT complicated by SOS.64
Pathophysiology of successful treatment with DF after HSCT
As shown in Figure 1, alkylating chemotherapy and radiation cause damage to sinusoidal ECs and hepatocytes. EC activation, induced by locally released cytokines as TNFα, favors the expression of adhesion molecules such as ICAM-1 and the increase in endothelial permeability. Endothelial barrier loss permits the passage of monocytes expressing the adhesion molecule LFA-1 and red blood cells that penetrate the space of Disse, causing obstruction of the sinusoidal blood flow. Release of vWF, TXA2, endothelin 1, and PAI-1 by damaged endothelium activates the coagulation pathway, mediated by platelet activation, and the subsequent fibrinolytic pathway. Progressive removal of the endothelial lining, loss of Kupffer cells, increased recruitment of monocytes (able to remove necrotic hepatocytes), and finally venular blockage occur. DF treatment seems to protect ECs from activation, as shown by the release of mediators, indicating good endothelial physiological function (NO, PGE2, PGI2, tPA) and the inhibition of mediator production that is typical of endothelial dysfunction (TNFα, PAI, thrombin).27–30,44
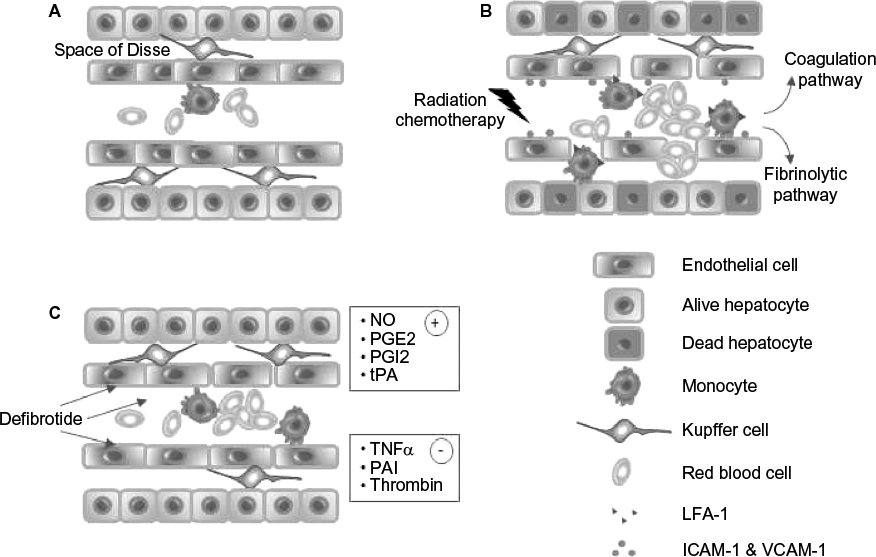 | Figure 1 Effects of defibrotide on hepatic sinusoidal endothelium. (A) Normal sinusoid. (B) Endothelial cells are damaged by radiation and chemotherapeutic treatment carried out before HSCT. Endothelial cell activation promotes, through the expression of endothelial molecules (VCAM-1, ICAM-1), the binding and diapedesis of monocytes expressing LFA-1. The increased permeability of endothelium permits the flow of red blood cells to the space of Disse, with loss of Kupffer cells, and increased recruitment of monocytes. Activation of coagulation pathway following endothelial release of von Willebrand factor favors vessel obstruction, and hepatocyte necrosis occurs. (C) Treatment with defibrotide abrogates endothelial activation and preserves sinusoidal blood flow function by favoring (+) and inhibiting (–) the release of some mediators. Abbreviations: HSCT, hematopoietic stem-cell transplantation; VCAM-1, vascular cell adhesion molecule 1; ICAM-1, intracellular cell adhesion molecule 1; LFA-1, lymphocyte function-associated antigen 1; NO, nitric oxide; PGE2, prostaglandin E2; PGI2, prostacyclin 2; tPA, tissue plasminogen activator; TNFα, tumor necrosis factor alpha; PAI, plasminogen activator inhibitor-1. |
The role of DF as an endothelial protective agent was recently described.65 It was shown how DF interacts with ECs by attaching to the external cell membrane, and then being internalized by ECs. The study provides direct evidence that DF’s interaction with the cell membrane guarantees at least two of the actions attributed to the drug, that is, anti-inflammatory and antioxidant effects. Inhibition of the inflammatory reaction is mediated by the decrease of VCAM-1 expression and the activation of p38MAPK, even when macropinocytotic entrance of DF was inhibited. In addition, DF has been shown to attenuate the generation of ROS and to restore endothelial NOS levels in the face of oxidative stress.65
Other authors support the hypothesis that prophylaxis with DF for SOS can lead to a delay in the engraftment of polymorphonuclear neutrophils in pediatric patients undergoing HSCT.66
Regarding post-HSCT intracellular mechanisms, preexisting liver diseases or the actions of alkylating agents impair GSH activity:28 toxic compounds accumulate in the liver, and the physiological functions of EC are impaired.
DF might abrogate GSH activity impairment through a reduction of ROS generation.
Focused patient perspectives
Important changes in the management of patients requiring HSCT, including reduced-intensity conditioning, more effective agents, and better supportive care, have resulted in reduced toxicity and improved outcome. Rapid and accurate diagnosis, together with risk assessment and the early initiation of effective treatment, helps to prevent progression to SOS and MOF, and improve survival. Nevertheless, SOS remains a frequent cause of post-HSCT mortality. Based on data from several studies carried out over the last 15 years, in the European Union, DF was approved for the treatment of severe post-HSCT VOD/SOS in October 2013. In the US, DF therapies for SOS have been approved with criteria based on the phase-3 study results of Richardson et al57 presented to the US Food and Drug Administration in 2015 as part of a New Drug Application.67 At present, there are not sufficient data to indicate patient perspectives, such as quality of life, satisfaction, preference, and adherence to the use of DF before or after HSCT. In the phase-3 study, severe SOS showed a 23% improvement in day +100 survival following HSCT in patients treated with DF compared to historical controls.57. In this context, we stress the necessity to identify preventive intervention rather than prophylactic intervention in HSCT patients. A study describing a panel of biomarkers useful to stratify patients at risk of SOS as early as the day of HSCT has recently been published.68 One marker, L-Ficolin, was particularly interesting in terms of SOS pathophysiology because it was seen to be involved in homeostatic clearance of mitochondria in the liver.69
Conclusion: a place in therapy
Although recent changes in the management of patients requiring HSCT have resulted in improved survival, SOS remains an important cause of post-HSCT mortality. It has been suggested that the onset of SOS might be predicted using biomarkers of endothelial injury.70 Indeed, the pathophysiological mechanisms involved in SOS are related to the HSCT-specific and patient-specific risk factors described in the present review. Such risks can produce endothelial injury in both sinusoids and small hepatic venules.71 The primary role of EC activation in the vascular bed has been described as an early complication of HSCT, which, other than SOS, also includes thrombotic microangiopathy, capillary leak syndrome, and acute GVHD.67 DF is known to exert various functions that are especially related to hemostasis. The protective effect of DF on the endothelium was recently highlighted, underlining how the anti-inflammatory and antioxidant properties of the drug seem to be caused by its interaction with the EC membrane.65 The application of DF as an endothelial protectant to help avoid the high risk of post-HSCT SOS is therefore highly feasible. Finally, the efficacy and safety of DF for the treatment of SOS has recently been published as the final results of the International Compassionate Use Program.72
Acknowledgments
This review is in honor of Dr Gianfranco Ferro, who founded the Crinos SpA and promoted the development of the drug DF. The authors would like to thank Michael John of the Vita-Salute University in Milan, Italy, for the English language editing of this paper.
Disclosure
The authors report no conflicts of interest in this work.
References
DeLeve LD, McCuskey RS, Wang X, et al. Characterization of a reproducible rat model of hepatic veno-occlusive disease. Hepatology. 1999;29(6):1779–1791. | ||
Stuart KL, Bras G. Veno-occlusive disease of the liver. Q J Med. 1957;26(103):291–315. | ||
Bras G, Jelliffe DB, Stuart KL. Veno-occlusive disease of liver with nonportal type of cirrhosis, occurring in Jamaica. AMA Arch Pathol. 1954;57(4):285–300. | ||
Abdualmjid RJ, Sergi C. Hepatotoxic botanicals - an evidence-based systematic review. J Pharm Pharm Sci. 2013;16(3):376–404. | ||
Rubbia-Brandt L. Sinusoidal obstruction syndrome. Clin Liver Dis. 2010;14(4):651–668. | ||
Nakano H, Oussoultzoglou E, Rosso E, et al. Sinusoidal injury increases morbidity after major hepatectomy in patients with colorectal liver metastases receiving preoperative chemotherapy. Ann Surg. 2008;247(1):118–124. | ||
Tamandl D, Klinger M, Eipeldauer S, et al. Sinusoidal obstruction syndrome impairs long-term outcome of colorectal liver metastases treated with resection after neoadjuvant chemotherapy. Ann Surg Oncol. 2011;18(2):421–430. | ||
Reed GB Jr, Cox AJ Jr. The human liver after radiation injury. A form of veno-occlusive disease. Am J Pathol. 1966;48(4):597–611. | ||
Lawrence TS, Robertson JM, Anscher MS, Jirtle RL, Ensminger WD, Fajardo LF. Hepatic toxicity resulting from cancer treatment. Int J Radiat Oncol Biol Phys. 1995;31(5):1237–1248. | ||
Tallman MS, McDonald GB, Deleve LD, et al. Incidence of sinusoidal obstruction syndrome following mylotarg (gemtuzumab ozogamicin): a prospective observational study of 482 patients in routine clinical practice. Int J Hematol. 2013;97(4):456–464. | ||
Rajvanshi P, Shulman HM, Sievers EL, McDonald GB. Hepatic sinusoidal obstruction after gemtuzumab ozogamicin (Mylotarg) therapy. Blood. 2002;99(7):2310–2314. | ||
Izaki T, Inomata Y, Asonuma K, et al. Early graft failure due to a veno-occlusive disease after a pediatric living donor liver transplantation. Pediatr Transplant. 2004;8(3):301–304. | ||
Cliffe ST, Bloch DB, Suryani S, et al. Clinical, molecular, and cellular immunologic findings in patients with SP110-associated veno-occlusive disease with immunodeficiency syndrome. J Allergy Clin Immunol. 2012;130(3):735–742.e6. | ||
Coppell JA, Richardson PG, Soiffer R, et al. Hepatic veno-occlusive disease following stem cell transplantation: incidence, clinical course, and outcome. Biol Blood Marrow Transplant. 2010;16(2):157–168. | ||
Valla D-C, Cazals-Hatem D. Sinusoidal obstruction syndrome. Clin Res Hepatol Gastroenterol. 2016;40(4):378–385. | ||
Fan CQ, Crawford JM. Sinusoidal obstruction syndrome (hepatic veno-occlusive disease). J Clin Exp Hepatol. 2014;4(4):332–346. | ||
Richardson PG, Ho VT, Cutler C, Glotzbecker B, Antin JH, Soiffer R. Hepatic veno-occlusive disease after hematopoietic stem cell transplantation: novel insights to pathogenesis, current status of treatment, and future directions. Biol Blood Marrow Transplant. 2013;19(1 Suppl):S88–S90. | ||
Chao N. How I treat sinusoidal obstruction syndrome. Blood. 2014;123(26):4023–4026. | ||
Dalle JH, Giralt SA. Hepatic veno-occlusive disease after hematopoietic stem cell transplantation: risk factors and stratification, prophylaxis, and treatment. Biol Blood Marrow Transplant. 2016;22(3):400–409. | ||
Park YD, Yasui M, Yoshimoto T, et al. Changes in hemostatic parameters in hepatic veno-occlusive disease following bone marrow transplantation. Bone Marrow Transplant. 1997;19(9):915–920. | ||
Harper PL, Jarvis J, Jennings I, Luddington R, Marcus RE. Changes in the natural anticoagulants following bone marrow transplantation. Bone Marrow Transplant. 1990;5(1):39–42. | ||
Salat C, Holler E, Kolb HJ, et al. Plasminogen activator inhibitor-1 confirms the diagnosis of hepatic veno-occlusive disease in patients with hyperbilirubinemia after bone marrow transplantation. Blood. 1997;89(6):2184–2188. | ||
Rio B, Bauduer F, Arrago JP, Zittoun R. N-terminal peptide of type III procollagen: a marker for the development of hepatic veno-occlusive disease after BMT and a basis for determining the timing of prophylactic heparin. Bone Marrow Transplant. 1993;11(6):471–472. | ||
Schuppan D, Farrand A, Oesterling C, Gehrmann M, McDonald GB. Circulating markers of hepatic fibrosis predict evolution of venocclusive disease after marrow transplantation. Hepatology. 1997;26:452. | ||
Eissner G, Kohlhuber F, Grell M, et al. Critical involvement of transmembrane tumor necrosis factor-alpha in endothelial programmed cell death mediated by ionizing radiation and bacterial endotoxin. Blood. 1995;86(11):4184–4193. | ||
Hong CW, Kim YM, Pyo H, et al. Involvement of inducible nitric oxide synthase in radiation-induced vascular endothelial damage. J Radiat Res. 2013;54(6):1036–1042. | ||
Panés J, Anderson DC, Miyasaka M, Neil Granger D. Role of leukocyte-endothelial cell adhesion in radiation-induced microvascular dysfunction in rats. Gastroenterology. 1995;108(6):1761–1769. | ||
Shen T, Feng XW, Geng L, Zheng SS. Reversible sinusoidal obstruction syndrome associated with tacrolimus following liver transplantation. World J Gastroenterol. 2015;21(20):6422–6426. | ||
Vion AC, Rautou PE, Durand F, Boulanger CM, Valla DC. Interplay of inflammation and endothelial dysfunction in bone marrow transplantation: focus on hepatic veno-occlusive disease. Semin Thromb Hemost. 2015;41(6):629–643. | ||
Mallat Z, Tedgui A. Apoptosis in the vasculature: mechanisms and functional importance. Br J Pharmacol. 2000;130(5):947–962. | ||
Shulman HM, Hinterberger W. Hepatic veno-occlusive disease--liver toxicity syndrome after bone marrow transplantation. Bone Marrow Transplant. 1992;10(3):197–214. | ||
McDonald GB, Hinds MS, Fisher LD, et al. Veno-occlusive disease of the liver and multiorgan failure after bone marrow transplantation: a cohort study of 355 patients. Ann Intern Med. 1993;118(4):255–267. | ||
Carreras E, Granena A, Rozman C. Hepatic veno-occlusive disease after bone marrow transplant. Blood Rev. 1993;7(1):43–51. | ||
Toh HC, McAfee SL, Sackstein R, Cox BF, Colby C, Spitzer TR. Late onset veno-occlusive disease following high-dose chemotherapy and stem cell transplantation. Bone Marrow Transplant. 1999;24(8):891–895. | ||
Carreras E, Díaz-Beyá M, Rosiñol L, Martínez C, Fernández-Avilés F, Rovira M. The incidence of veno-occlusive disease following allogeneic hematopoietic stem cell transplantation has diminished and the outcome improved over the last decade. Biol Blood Marrow Transplant. 2011;17(11):1713–1720. | ||
Richardson P, Bearman SI. Prevention and treatment of hepatic venocclusive disease after high-dose cytoreductive therapy. Leuk Lymphoma. 1998;31(3–4):267–277. | ||
Park SH, Lee MH, Lee H, et al. A randomized trial of heparin plus ursodiol vs. heparin alone to prevent hepatic veno-occlusive disease after hematopoietic stem cell transplantation. Bone Marrow Transplant. 2002;29(2):137–143. | ||
Joon SS, Jong JS, Hyung NM, Ghim T, Ho JI. Prophylactic low-dose heparin or prostaglandin E1 may prevent severe veno-occlusive disease of the liver after allogeneic hematopoietic stem cell transplantation in Korean children. J Korean Med Sci. 2006;21(5):897–903. | ||
Lakshminarayanan S, Sahdev I, Goyal M, Vlachos A, Atlas M, Lipton JM. Low incidence of hepatic veno-occlusive disease in pediatric patients undergoing hematopoietic stem cell transplantation attributed to a combination of intravenous heparin, oral glutamine, and ursodiol at a single transplant institution. Pediatr Transplant. 2010;14(5):618–621. | ||
Brown SA, Goringe A, Fegan C, et al. Parenteral glutamine protects hepatic function during bone marrow transplantation. Bone Marrow Transplant. 1998;22(3):281–284. | ||
Schriber J, Milk B, Shaw D, et al. Tissue plasminogen activator (tPA) as therapy for hepatotoxicity following bone marrow transplantation. Bone Marrow Transplant. 1999;24(12):1311–1314. | ||
Myers KC, Lawrence J, Marsh RA, Davies SM, Jodele S. High-dose methylprednisolone for veno-occlusive disease of the liver in pediatric hematopoietic stem cell transplantation recipients. Biol Blood Marrow Transplant. 2013;19(3):500–503. | ||
Palmer KJ, Goa KL. Defibrotide. A review of its pharmacodynamic and pharmacokinetic properties, and therapeutic use in vascular disorders. Drugs. 1993;45(2):259–294. | ||
Pescador R, Capuzzi L, Mantovani M, Fulgenzi A, Ferrero ME. Defibrotide: properties and clinical use of an old/new drug. Vascul Pharmacol. 2013;59(1–2):1–10. | ||
Bracht F, Schror K. Isolation and identification of aptamers from defibrotide that act as thrombin antagonists in vitro. Biochem Biophys Res Commun. 1994;200(2):933–937. | ||
Evangelista V, Piccardoni P, Maugeri N, De Gaetano G, Cerletti C. Inhibition by heparin of platelet activation induced by neutrophil-derived cathepsin G. Eur J Pharmacol. 1992;216(3):401–405. | ||
Gatto B, Vianini E, Lucatello L, et al. Effective DNA inhibitors of cathepsin G by in vitro selection. Int J Mol Sci. 2008;9(6):1008–1023. | ||
Fisher J, Johnston AM, Holland TK, et al. Pharmacokinetics, absorption, distribution and disposition of [125I]-defibrotide following intravenous or oral administration in the rat. Thromb Res. 1993;70(1):77–90. | ||
Fisher J, Holland TK, Pescador R, Porta R, Ferro L. Study on pharmacokinetics of radioactive labelled defibrotide after oral or intravenous administration in rats. Thromb Res. 1996;81(1):55–63. | ||
Patrassi GM, Sartori MT, Viero ML, Scapinello MP, Boeri G, Girolami A. Fibrinolytic effects of defibrotide in atherosclerotic patients. Semin Thromb Hemost. 1991;17(1 Suppl):101–105. | ||
Violi F, Marubini E, Coccheri S, Nenci GG. Improvement of walking distance by defibrotide in patients with intermittent claudication. Results of a randomized, placebo-controlled study (the DICLIS study). Thromb Haemost. 2000;83(5):672–677. | ||
Andrade D, Tektonidou M. Emerging therapies in antiphospholipid syndrome. Curr Rheumatol Rep. 2016;18(4):22. | ||
Richardson PG, Murakami C, Jin Z, et al. Multi-institutional use of defibrotide in 88 patients after stem cell transplantation with severe veno-occlusive disease and multisystem organ failure: response without significant toxicity in a high-risk population and factors predictive of outcome. Blood. 2002;100(13):4337–4343. | ||
Haussmann U, Fischer J, Eber S, Scherer F, Seger R, Gungor T. Hepatic veno-occlusive disease in pediatric stem cell transplantation: impact of pre-emptive antithrombin III replacement and combined antithrombin III/defibrotide therapy. Haematologica. 2006;91(6):795–800. | ||
Keating GM. Defibrotide: a review of its use in severe hepatic veno-occlusive disease following haematopoietic stem cell transplantation. Clin Drug Investig. 2014;34(12):895–904. | ||
Pol RR, Russell N, Das-Gupta E, Watson L, Rachael L, Byrne J. Incidence and management of hepatic severe veno-occlusive disease in 273 patients in a single centre with defibrotide. Bone Marrow Transplant. 2016;51(9):1262–1264. | ||
Richardson PG, Riches ML, Kernan NA, et al. Phase 3 trial of defibrotide for the treatment of severe veno-occlusive disease and multi-organ failure. Blood. 2016;127(13):1656–1665. | ||
Strouse C, Richardson P, Prentice G, et al. Defibrotide for treatment of severe veno-occlusive disease in pediatrics and adults: an exploratory analysis using data from the center for international blood and marrow transplant research. Biol Blood Marrow Transplant. 2016;22(7):1306–1312. | ||
Corbacioglu S, Cesaro S, Faraci M, et al. Defibrotide for prophylaxis of hepatic veno-occlusive disease in paediatric haemopoietic stem-cell transplantation: an open-label, phase 3, randomised controlled trial. Lancet. 2012;379(9823):1301–1309. | ||
Park M, Park HJ, Eom HS, et al. Safety and effects of prophylactic defibrotide for sinusoidal obstruction syndrome in hematopoietic stem cell transplantation. Ann Transplant. 2013;18:36–42. | ||
Dignan FL, Wynn RF, Hadzic N, et al. BCSH/BSBMT guideline: diagnosis and management of veno-occlusive disease (sinusoidal obstruction syndrome) following haematopoietic stem cell transplantation. Br J Haematol. 2013;163(4):444–457. | ||
Triplett BM, Kuttab HI, Kang G, Leung W. Escalation to high-dose defibrotide in patients with hepatic veno-occlusive disease. Biol Blood Marrow Transplant. 2015;21(12):2148–2153. | ||
Choi A, Kang YK, Lim S, Kim DH, Lim JS, Lee JA. Severe hepatic sinusoidal obstruction syndrome in a child receiving vincristine, actinomycin-d, and cyclophosphamide for rhabdomyosarcoma: successful treatment with defibrotide. Cancer Res Treat. Epub 2016 Mar 30. | ||
Richardson PG, Corbacioglu S, Ho VT, et al. Drug safety evaluation of defibrotide. Expert Opin Drug Saf. 2013;12(1):123–136. | ||
Palomo M, Mir E, Rovira M, Escolar G, Carreras E, Diaz-Ricart M. What is going on between defibrotide and endothelial cells? Snapshots reveal the hot spots of their romance. Blood. 2016;127(13):1719–1727. | ||
Maximova N, Pizzol A, Giurici N, Granzotto M. Does defibrotide induce a delay to polymorphonuclear neutrophil engraftment after hematopoietic stem cell transplantation? Observation in a pediatric population. Adv Ther. 2015;32(4):381–386. | ||
Arai S. Long-awaited news for hepatic veno-occlusive disease. Blood. 2016;127(13):1630–1631. | ||
Akil A, Zhang Q, Mumaw CL, et al. Biomarkers for diagnosis and prognosis of sinusoidal obstruction syndrome after hematopoietic cell transplantation. Biol Blood Marrow Transplant. 2015;21(10):1739–1745. | ||
Brinkmann CR, Jensen L, Dagnaes-Hansen F, et al. Mitochondria and the lectin pathway of complement. J Biol Chem. 2013;288(12):8016–8027. | ||
Cutler C, Kim HT, Ayanian S, et al. Prediction of veno-occlusive disease using biomarkers of endothelial injury. Biol Blood Marrow Transplant. 2010;16(8):1180–1185. | ||
Senzolo M, Germani G, Cholongitas E, Burra P, Burroughs AK. Veno occlusive disease: update on clinical management. World J Gastroenterol. 2007;13(29):3918–3924. | ||
Corbacioglu S, Carreras E, Mohty M, et al. Defibrotide for the treatment of hepatic veno-occlusive disease: final results from the international compassionate-use program. Biol Blood Marrow Transplant. 2016;22(10):1874–1882. |
 © 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.