")
Back to Journals » International Journal of Nanomedicine » Volume 13
Deepened cellular/subcellular interface penetration and enhanced antitumor efficacy of cyclic peptidic ligand-decorated accelerating active targeted nanomedicines
Authors Shi NQ, Li Y, Zhang Y , Li ZQ , Qi XR
Received 28 April 2018
Accepted for publication 3 July 2018
Published 19 September 2018 Volume 2018:13 Pages 5537—5559
DOI https://doi.org/10.2147/IJN.S172556
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Dr Linlin Sun
Nian-Qiu Shi,1 Yan Li,2 Yong Zhang,3 Zheng-Qiang Li,4 Xian-Rong Qi5
1School of Pharmacy, Jilin Medical University, Jilin, Jilin, 132013, China; 2Immunology Department, Laboratory Medical College, Jilin Medical University, Jilin, Jilin, 132013, China; 3College of Life Science, Jilin University, Changchun, Jilin, 130012, China; 4Key Laboratory for Molecular Enzymology and Engineering, Ministry of Education, College of Life Science, Jilin University, Changchun, Jilin, 130012, China; 5Department of Pharmaceutics, School of Pharmaceutical Science, Peking University, Beijing, 100191, China
Introduction: Acceleration and improvement of penetration across cell-membrane interfaces of active targeted nanotherapeutics into tumor cells would improve tumor-therapy efficacy by overcoming the issue of poor drug penetration. Cell-penetrating peptides, especially synthetic polyarginine, have shown promise in facilitating cargo delivery. However, it is unknown whether polyarginine can work to overcome the membrane interface in an inserted pattern for cyclic peptide ligand-mediated active targeting drug delivery. Here, we conducted a study to test the hypothesis that tandem-insert nona-arginine (tiR9) can act as an accelerating component for intracellular internalization, enhance cellular penetration, and promote antitumor efficacy of active targeted cyclic asparagine–glycine–arginine (cNGR)-decorated nanoliposomes.
Methods: Polyarginine was coupled with the polyethylene glycol (PEG) chain and the cNGR moiety, yielding a cNGR–tiR9–PEG2,000–distearoylphosphatidylethanolamine conjugate.
Results: The accelerating active targeted liposome (Lip) nanocarrier (cNGR-tiR9-Lip–doxorubicin [Dox]) constructed in this study held suitable physiochemical features, such as appropriate particle size of ~150 nm and sustained-release profiles. Subsequently, tiR9 was shown to enhance cellular drug delivery of Dox-loaded active targeted systems (cNGR-Lip-Dox) significantly. Layer-by-layer confocal microscopy indicated that the tandem-insert polyarginine accelerated active targeted system entry into deeper intracellular regions based on observations at marginal and center locations. tiR9 enhanced the penetration depth of cNGR-Lip–coumarin 6 through subcellular membrane barriers and caused its specific accumulation in mitochondria, endoplasmic reticulum, and Golgi apparatus. It was also obvious that cNGR-tiR9-Lip-Dox induced enhanced apoptosis and activated caspase 3/7. Moreover, compared with cNGR-Lip-Dox, cNGR-tiR9-Lip-Dox induced a significantly higher antiproliferative effect and markedly suppressed tumor growth in HT1080-bearing nude mice.
Conclusion: This active tumor-targeting nanocarrier incorporating a tandem-insert polyarginine (tiR9) as an accelerating motif shows promise as an effective drug-delivery system to accelerate translocation of drugs across tumor-cell/subcellular membrane barriers to achieve improved specific tumor therapy.
Keywords: accelerating active targeting nanotherapeutics, tandem-insert nona-arginine, tiR9, cyclic asparagine–glycine–arginine, cNGR, penetration and tumor therapy, multifunctional nanomedicines
Introduction
Cancer is still one of the world’s most devastating diseases, and over 10 million new cases appear every year.1,2 Nanocarriers have great potential to revolutionize cancer diagnosis and therapy. Some therapeutic nanocarriers, including liposomes (Lip), polymeric micelles, and nanoparticles, have been approved for clinical application.3,4 Targeted drug delivery by nanocarriers is currently of great interest to cancer researchers, because nanotargeting can improve distribution within tumor tissue and reduce side effects involving nontumor sites. Active targeting approaches depend on biomarkers that are specifically oversecreted by tumor cells, while passive targeting, known as the enhanced permeability and retention (EPR) effect, depends on tumor-cell characteristics that increase uptake relative to nontumor cells.5,6 Active targeting systems may further deliver substances to diseased tissue via interaction between coupled ligands and corresponding functionalized receptor biomarkers to induce cellular uptake via endocytosis. In the past few years, various targeting ligands have been employed, including proteins (eg, antibodies, antibody fragments, growth factors, and transferrin), peptides (eg, RGD, asparagine–glycine–arginine [NGR], octreotide, tLyp1, and iRGD peptide), polysaccharides (eg, hyaluronic acid), and small biomolecules (eg, folic acid, biotin, galactose, and bisphosphonates).7–9
Peptides composed of a few amino acids (from three to ten) as active targeting ligands have emerged. These peptides are cell-specific and have been already used in many therapeutic assays.10 Recently, a new target within tumor vasculature or endothelial cells has been identified, along with its corresponding targeting peptide ligand. The target includes a form of aminopeptidase N (APN), which specifically binds peptides containing the NGR motif.11 APN is a membrane-bound, zinc-dependent metalloproteinase that plays a critical role in tumor invasion and angiogenesis.12 NGR-mediated targeting delivery has been confirmed and extensively used for transporting various types of agents into APN-rich tumors in many studies.13–18 Several NGR-conjugated agents are now in preclinical and clinical trials.13 For example, Luo et al14 developed NGR-decorated sterically stabilized liposomes (NGR-SSL–paclitaxel) and demonstrated that these enhanced treatment efficacy. In another study, Dunne et al13 evaluated the in vivo performance of several NGR-conjugated liposomes using computed tomography and found that targeting tumor angiogenic vasculature in APN-secreted tumors was a viable approach for cancer imaging and therapy. Meanwhile, other confirmed reports have shown that cyclic NGR (cNGR) presents more effective and specific targeting capacity to enhance delivery of antitumor agents.11,15–18
In spite of their promise, ligand-mediated active targeted systems still face many challenges. On one hand, the absolute level of biomarkers (receptors) in the target site is relatively low, despite their overexpression. These low levels have a strong impact on the efficiency of active targeting.19 On the other hand, irregular blood flow,20 compression of blood and lymphatic vessels,21 disorganized vascular network,22 increased interstitial fluid pressure,23 and the composition of the extracellular matrix can slow or disrupt the ligand-receptor-binding process in tumor microenvironments. Therefore, poor drug penetration in tumor cells or solid-tumor masses is an intrinsic restriction of chemotherapy that often leads to cancer-treatment failure.24,25 Accelerating and improving the penetration of peptide-ligand-mediated active targeted nanocarriers in tumor cells is a formidable obstacle for enhancing their intracellular entry and achieving enhanced tumor-therapy efficacy.
Cell-penetrating peptides (CPPs), also termed protein-transduction domains or “Trojan horse” peptides, are a diverse set of short peptide sequences that typically consist of 30 or fewer amino acids.26 The first highlighted advantage of CPPs is their ability to transport very diverse cargoes (in terms of size and biological nature) into cells.27,28 Notably, numerous preclinical and clinical evaluations of CPP–cargo conjugates have shown their potential for therapeutic use with drugs.26 Meanwhile, conjugation of CPP to antitumor agents has been demonstrated to be an attractive approach to overcome various biological barriers and achieve improved therapeutic efficacy for a series of tumors.26,29,30 A second important characteristic of CPPs is the apparent cellular ubiquity of this transduction function.31 Furthermore, the nonselectivity of CPPs underscores their theoretical potential for penetrating any cells or tumor tissue without involving any specific markers (eg, receptors or antigens) commonly used in traditional passive or active targeting strategies. The use of CPPs is commonly established by unilateral conjugation with payloads or directive attachment on the surface of nanocarriers (CPP–).27,29 However, it is unknown whether a tandem-insert pattern of CPPs (–CPP–) can work to overcome membrane barriers in peptide-ligand-mediated active targeted delivery of nanomedicines.
Nona-arginine (R9), a well-known synthetic arginine-rich CPP, increases the transportation of cargo across biological barriers or interfaces to achieve intracellular entry.26,32 Using the barrier-penetrating function of R9, the oversecretion of APN by some tumor cells, and the availability of the NGR peptide for effective tumor-cell targeting, a rational design was used to fabricate cyclic NGR-modified nanoliposomes loaded with doxorubicin (Dox)-containing tandem-insert R9 (tiR9) CPPs, yielding a cyclic peptide ligand-mediated accelerating active targeting nanotherapeutics (ie, cNGR-tiR9-Lip-Dox). tiR9 was coupled with a polyethylene glycol (PEG) chain and cNGR moiety, yielding a polyarginine-insert cNGR-tiR9-PEG2,000–distearoylphosphatidylethanolamine (DSPE) conjugate. The main purpose of this work was to demonstrate the notion that tiR9 (tandem-insert polyarginine) in cNGR-tiR9-Lip-Dox (Figure 1) could work to overcome membrane barriers and upgrade antitumor efficacy of cNGR-modified nanosystems (cNGR-Lip-Dox) for active targeted drug delivery. This system incorporates tiR9, which acts an accelerating motif to penetrate cells better to accelerate intracellular delivery, and is expected to exert increased antitumor effects compared with active targeted liposomes lacking the tandem-insert polyarginine (cNGR-Lip-Dox). Figure 1A illustrates the deepened intracellular penetration and enhanced antitumor efficacy of the cNGR-tiR9-Lip-Dox system, and Figure 1B shows its surface-structure design. To test tiR9 as an accelerating component, functional conjugates (cNGR-PEG2,000-DSPE and cNGR-tiR9-PEG2,000-DSPE) were synthesized and characterized by matrix-assisted laser desorption/ionization (MALDI) time-of-flight mass spectrometry (TOF-MS) for the construction of targeted nanoliposomes (cNGR-Lip-Dox and cNGR-tiR9-PEG2,000-DSPE). Penetration effects were studied in various cellular locations using layer-by-layer scanning confocal microscopy. Furthermore, the endocytic pathway used by cNGR-tiR9-Lip-Dox was also explored by employing chemical inhibitors. In addition, in vitro and in vivo antitumor characteristics of cNGR-tiR9-Lip-Dox were investigated.
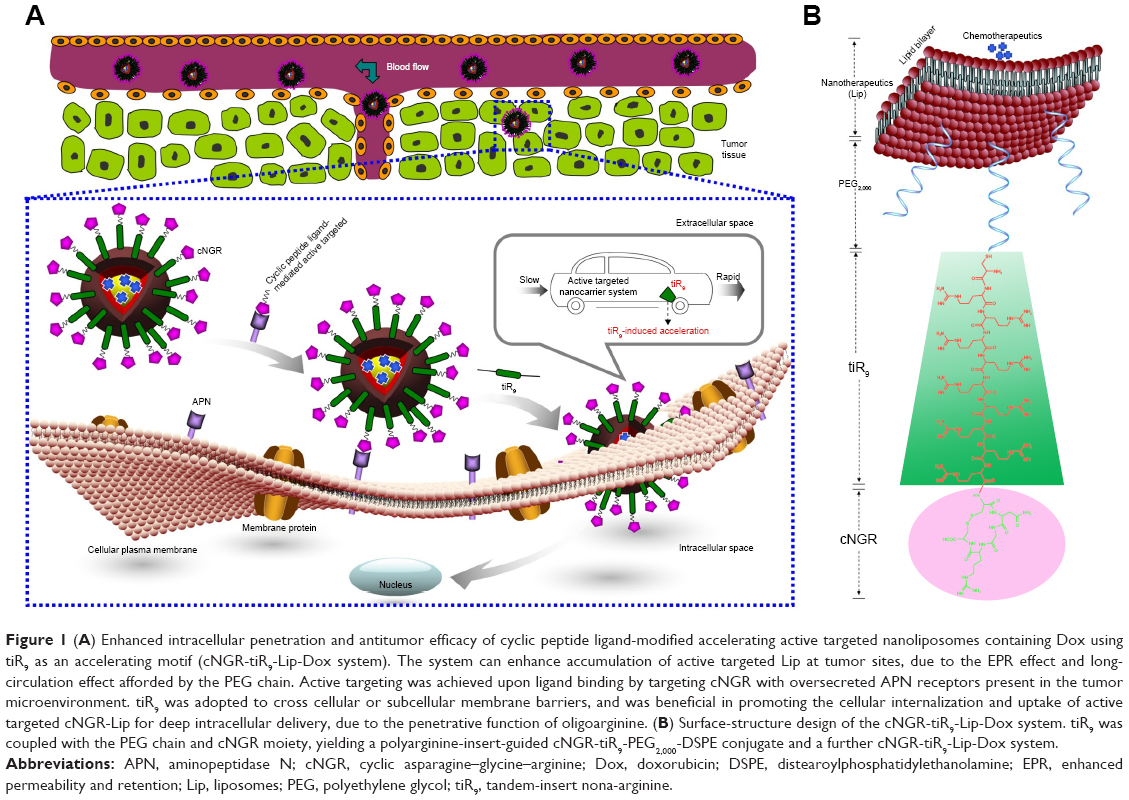 | Figure 1 (A) Enhanced intracellular penetration and antitumor efficacy of cyclic peptide ligand-modified accelerating active targeted nanoliposomes containing Dox using tiR9 as an accelerating motif (cNGR-tiR9-Lip-Dox system). The system can enhance accumulation of active targeted Lip at tumor sites, due to the EPR effect and long-circulation effect afforded by the PEG chain. Active targeting was achieved upon ligand binding by targeting cNGR with oversecreted APN receptors present in the tumor microenvironment. tiR9 was adopted to cross cellular or subcellular membrane barriers, and was beneficial in promoting the cellular internalization and uptake of active targeted cNGR-Lip for deep intracellular delivery, due to the penetrative function of oligoarginine. (B) Surface-structure design of the cNGR-tiR9-Lip-Dox system. tiR9 was coupled with the PEG chain and cNGR moiety, yielding a polyarginine-insert-guided cNGR-tiR9-PEG2,000-DSPE conjugate and a further cNGR-tiR9-Lip-Dox system. |
Methods and materials
Materials
N-[(3-Maleimide-1-oxopropyl) aminopropyl polyethyleneglycol-carbamyl] distearoylphosphatidyl-ethanolamine (Mal-PEG2000-DSPE) was purchased from NOF (Tokyo, Japan). Soybean phosphatidylcholine (SPC) was obtained from Lipoid (Ludwigshafen am Rhein, Germany). Dox was supplied by Hisun Pharmaceutical (Taizhou, China). Trypsin was obtained from VWR (Radnor, PA, USA). MTT and cholesterol (Chol) were purchased from Sigma-Aldrich Co. (St Louis, MO, USA). Penicillin, streptomycin, and fluorescent-probe Hoechst 33258 were provided by MacGene (Beijing, China). All other chemicals were of analytic or high-performance liquid-chromatography grade. The cNGR (Cys-Cys-Asn-Gly-Arg-Cys) and cNGR-tiR9 (Cys-Arg-Arg-Arg-Arg-Arg-Arg-Arg-Arg-Arg-Cys-Asn-Gly-Arg-Cys) peptides were custom-synthesized via standard fluorenylmethyloxycarbonyl chloride solid-phase peptide synthesis by KareBay Biochem (Monmouth Junction, NJ, USA). The purity of peptides was 98.93% (cNGR) and 98.11% (cNGR-tiR9). The correct product was demonstrated by electrospray-ionization MS: cNGR, m/z, 654.8 (theoretical), 654 (practical, [M+H]+: 655.0); cNGR-tiR9, m/z, 1,526.85 (theoretical), 1,525.9 (practical, [M+H]+: 1,526.9).
Cells and animals
HT1080 (human fibrosarcoma) cells were provided by the China Center for Type Culture Collection at Wuhan University (Wuhan, China). Cells were cultured in MEM with Earle’s salts, L-glutamine, nonessential amino acid (MacGene), 10% FBS, 100 U/mL penicillin, and 100 μg/mL streptomycin. Female BALB/c nude mice weighing 22–24 g were supplied by Vital River Laboratory Animal Technology (Beijing, China) and maintained in specific-pathogen-free conditions for 1 week before the investigation. During this period, the animals were free to consume standard food and water. All care and handling of animals received the approval of the ethics committee of Jilin Medical University and was performed in accordance with the requirements of the Institutional Authority for Laboratory Animal Care of Jilin Medical University.
Synthesis and characterization of cNGR-PEG2,000-DSPE
For generation of active targeted cNGR-Lip-Dox, cNGR peptide was conjugated with Mal-PEG-DSPE by the Michael addition principle to achieve a targeting conjugate of cNGR-PEG2,000-DSPE. cNGR (2.0 mg) and Mal-PEG-DSPE (10 mg) with a molar ratio of 1:1 were dissolved in 2 mL HEPES buffer solution (pH 7.2, 20 mM) that had been deoxidized for 30 minutes in advance. The reactive solution was agitated mildly for 24 hours via the protection of N2 gas at room temperature. After 24 hours’ incubation, the reaction solution was transferred to a dialysis bag with a molecular weight (MW) cutoff of 2,000 Da and subjected to a dialysis procedure against deionized water for 48 hours to eliminate unreacted ligand. The dialysate was freeze-dried and preserved at 20°C. The linkage of cNGR with PEG2,000-DSPE was demonstrated by thin-layer chromatography and MALDI-TOF-MS (Bruker, Billerica, MA, USA).
Synthesis and characterization of cNGR-tiR9-PEG2,000-DSPE
To generate a cNGR-tiR9-Lip-Dox accelerated targeting system, cNGR-tiR9 peptide was conjugated with Mal-PEG2,000-DSPE by Michael addition to achieve the tumor-guiding conjugate cNGR-tiR9-PEG2,000-DSPE. cNGR-tiR9 (6.2 mg) and Mal-PEG2,000-DSPE (10 mg) with a molar ratio of 1:1 were dissolved in 2 mL previously deoxidized HEPES buffer solution. The products were monitored with thin-layer chromatography and MALDI-TOF-MS, followed by dialysis and lyophilization.
Preparation of various Dox-loaded nanoliposomes
Liposomes contained SPC and Chol or other functional conjugates. Lipids of SPC–Chol (20:10 w:w), SPC–Chol–cNGR-DSPE2,000-PEG (20:10:1 w:w:w) and SPC–Chol–cNGR-tiR9-DSPE2,000-PEG (20:10:1 w:w:w) were the components of Lip-Dox, cNGR-Lip-Dox, and cNGR-tiR9-Lip-Dox, respectively. Thin-lipid-film hydration followed by sonication was used to prepare liposome formulations. Lipids were dissolved in chloroform and dried until a thin lipid film had formed on a rotary evaporator (RE52; Yarong, Shanghai, China) via reduced pressure. Ammonium sulfate (300 mM) was used to hydrate the dried lipid film under sonication with a bath-type sonicator. To generate an ammonium sulfate gradient, a Sephadex G50 column pre-equilibrated with a 20 mM HEPES buffer solution in the presence of 150 mM NaCl (HBS, pH 7.4) was used to elute Lip. Remote loading of Dox into liposomes was performed through the ammonium sulfate-gradient approach.33
Characterization of various liposomes
Particle size and ζ-potential
A Zetasizer Nano ZS (Malvern Instruments, Malvern, UK) was used to detect the particle size and ζ-potential of various formulations, including Lip-Dox, cNGR-Lip-Dox, and cNGR-tiR9-Lip-Dox, on the basis of dynamic light scattering at 25°C. Each sample was determined for three times. Transmission electron microscopy (JEM-1230; JEOL, Tokyo, Japan) using an acceleration voltage of 80 kV was employed to observe the morphological shapes of various liposomes. A carbon-coated copper grid was used to load sample solutions, and a 1% uranyl acetate solution was adopted to stain samples negatively. Finally, samples were air-dried at room temperature.
Encapsulation efficiency
The Sephadex G50 column was applied to pass through liposomes to eliminate free Dox. The liposomes were disrupted with 10% Triton X-100 (v:v). Dox in formulations was analyzed with a spectrofluorometry (RF-5301PC; Shimadzu, Kyoto, Japan). The same amount of liposomes was not passed through the column and was determined as the total Dox concentration. Encapsulation efficiency was calculated:
|

In vitro release
We measured the in vitro drug-release profile of the formulation by dialysis. A dialysis bag with MW cutoff of 3,500 Da was adopted to load 1 mL formulation and then sealed at both ends. Dialysis was performed against PBS (pH 7.4) with continuous agitation at 37°C. Aliquots were removed and substituted with fresh medium at each measured time point. Drug concentration in formulations was determined according to the fluorescence intensity of drug excitation at 485 nm. Cumulative drug release was calculated over 24 hours.
Cellular uptake by flow cytometry
Six-well plates were seeded by APN-rich tumor cell lines (HT1080 cells) at a density of 5×105 cells/well. Plates were maintained at 37°C and cultured for 24 hours. PBS (pH 7.4) was used to wash cells twice for removal of residual culture medium. Cells were incubated with different formulations mixed in serum-free medium at a Dox concentration of 10 μg/mL. Cells without any drug treatment were employed as a negative control group. After 12 hours’ treatment, cold PBS was used to wash cells three times and resuspend them. The fluorescence value of Dox was detected by FACSCalibur (Becton Dickinson, Mountain View, CA, USA).
Confocal laser-scanning microscopy
For various formulations, cell uptake and intracellular dispersion were observed in HT1080 cells. After adherent culture of the cells for 24 hours on petri dishes, free Dox, Lip-Dox, cNGR-Lip-Dox, or cNGR-tiR9-Lip-Dox was diluted in culture medium and incubated with cells for 24 hours at 37°C under a Dox concentration of 2.5 μg/mL. Then, cells were subjected to washing three times and fixing for 10 minutes by 4% paraformaldehyde in PBS at room temperature. After 15 minutes’ staining of cell nuclei with Hoechst 33258, HT1080 cells were visualized by confocal laser-scanning microscopy (Leica, Heidelberg, Germany).
Subcellular localization
We carried out triple-labeling measurements by confocal microscopy in HT1080 cells to observe which subcompartments were associated with the cytoplasmic dispersion of functional liposomes. Localization of coumarin 6 (Cou6) or various Cou6 nanoliposomes in subcellular organelles was observed by marking cells with fluorescent probes specific to each subcellular organelle. HT1080 cells were seeded onto a petri dish and cultured for 24 hours at 37°C in the presence of 5% CO2, followed by addition of 1.0 μg/mL free Cou6, Lip-Cou6, cNGR-Lip-Cou6, or cNGR-tiR9-Lip-Cou6. Cells were subjected to further incubation for an additional 2 hours, then the drug-containing medium was removed and cells washed with PBS. Next, cells were stained with organelle-selective dyes (Molecular Probes, Eugene, OR, USA). Lysosomes, mitochondria, endoplasmic reticulum (ER), and Golgi apparatus were observed by staining the cells with 50 nM LysoTracker Red DND-99, 200 nM MitoTracker Red CMXRos, 1 mM ER-Tracker Red (boron–dipyrromethene), and 5 mM Golgi-Tracker Red (boron–dipyrromethene C5-ceramide) for 30 minutes. PBS was used to wash cells with organelle markers, and staining of cell nuclei was carried out using 1 μM Hoechst 33258 solution with 3 minutes’ incubation. Then, cells were washed twice with PBS and imaged using confocal laser-scanning microscopy. All procedures were carried out as gently as possible to avoid any detachment of adherent cells from the culture dishes.
Apoptosis detection
An annexin V–fluorescein isothiocyanate (FITC)/propidium iodide (PI) kit was adopted to measure apoptotic and necrotic cells caused by various samples. Annexin V–FITC was capable of binding with phosphatidyl serine that traversed from the inner to outer plasma-membrane leaflet during the apoptotic stage. Necrotic cells were stained using PI. HT1080 cells were incubated with free Dox, Lip-Dox, cNGR-Lip-Dox, or cNGR-tiR9-Lip-Dox, each at concentrations of 0.5 μg/mL, 5 μg/mL, and 15 μg/mL, for 24 hours. Next, the supernatant medium was collected. Attached cells were detached by adding trypsin solution and suspended in fresh medium. After mixing both cell suspensions, centrifugation of cells at 4,000 rpm was done for 5 minutes before resuspension in 150 μL fresh medium. The cells were mixed with annexin V–FITC and PI (50 μL) solutions and kept in the dark at room temperature for 20 minutes. Cells were detected by flow cytometry (Muse; Merck, Darmstadt, Germany).
Caspase 3/7 activation
Caspase 3/7 activation was evaluated using the Muse caspase 3/7 kit specifically designed for use with the Muse cell analyzer. HT1080 cells were incubated for 24 hours with free Dox, Lip-Dox, cNGR-Lip-Dox, or cNGR-tiR9-Lip-Dox, each at concentrations of 5 μg/mL and 15 μg/mL. Next, the medium was collected, and treated cells were detached using trypsin solution and suspended in fresh medium. Cells were centrifuged at 4,000 rpm for 5 minutes and resuspended in 50 μL medium. Next, 5 μL Muse caspase 3/7 working solution was added to the 50 μL cell suspension and the suspension incubated at 37°C for 30 minutes. 7-Aminoactinomycin D (150 μL) working solution was added and mixed thoroughly. Cells were analyzed using the Muse device.
Endocytic routes of cell entry of cNGR-tiR9-Lip-Dox
Endocytic routes of cNGR-tiR9-Lip-Dox cell entry were investigated using various treatments. To suppress energy-dependent endocytosis completely, cells were subjected to incubation at 4°C for 30 minutes in advance, exposed to complete MEM, and maintained for another 12 hours at 4°C with cNGR-tiR9-Lip-Dox. Flow cytometry was used to detect fluorescence levels of cells. To inhibit specific endocytic routes, cells were pretreated with chlorpromazine (20 μM), chloroquine (100 μM), amiloride (50 μM), β-cyclodextrin (100 μM), or heparin (10 μM). All chemical dilutions were prepared in serum-free medium and incubated for 30 minutes, followed immediately by incubation with cNGR-tiR9-Lip-Dox at 37°C for an additional 12 hours. Next, cells were subjected to flow-cytometry analysis. All tests were carried out in triplicate.
Cytotoxicity assay
In vitro cytotoxicity of samples was determined by MTT.25 Plates (96 wells) were seeded with HT1080 cells at a density of 5,000 cells/well and subjected to adherent culture for 24 hours. Altered concentrations of free Dox, Lip-Dox, cNGR-Lip-Dox, or cNGR-tiR9-Lip-Dox were added into cells at 37°C. Cells were treated for 24 hours and survival measured by MTT. MTT/PBS solution (5 mg/mL) was added to each well. An additional 4 hours’ incubation of the plates at 37°C was performed and then medium removed. Furthermore, each well had 200 mL dimethyl sulfoxide added with vigorous stirring to dissolve the crystals generated. The absorbance of each well was determined by a microplate reader (680; Bio-Rad, Hercules, CA, USA) at 570 nm. All measurements were carried out using six replicate tests. Results are presented as cell survival. Cell-growth inhibition was calculated: survival rate = A570sample/A570control × 100%.
Animal models
Female BALB/c nude mice were employed for in vivo investigation. An animal model of human fibrosarcoma was established by inoculation of 0.2 mL HT1080 cell suspensions (5×106 cells) to the right armpits of animals. In accordance with previous confirmed studies,13,14,17,34 HT1080 tumor was chosen as an in vivo tumor model due to its overexpression of the APN receptor.
In vivo antitumor efficacy
Antitumor efficacy was compared using animals bearing HT1080 tumors. When tumor size reached approximately 50 mm3, animals (n=8) were given saline, Lip-Dox, cNGR-Lip-Dox, or cNGR-tiR9-Lip-Dox via injection into the tail vein. Administration dosage was 1.0 mg Dox/kg every 2 days under observation for 15 days. During this period, tumor volume (V) was measured and calculated: V = [m×n2]/2, where m represents tumor-mass length and n tumor-mass width. Body weights were recorded during the period.
Histology analysis
Major organs were fixed with 4% formalin, embedded in paraffin, and then sectioned. Sections of 7 μm thickness were mounted on glass slides, stained with H&E, and examined by light microscopy.
Data analysis
Data are presented as mean ± SEM. The difference between any two groups was determined by Students’ t-test. P<0.05 was considered statistically significant.
Results and discussion
Design strategy and synthesis of functional conjugates
CPPs can be divided into three types according to their origin: protein-derived CPPs, model CPPs, and designed CPPs.35 For efficient intracellular translocation, the guanidinium group of some arginine-rich CPPs is important and essential. CPPs with guanidinium groups are thought to translocate across cell membranes under the driving force of membrane potential via ion-pair formation stemming of bidentate hydrogen-bond effects among the groups and negative residues on the cell surface.36 Meanwhile, oligoarginine is a synthetic CPP that has been shown to enhance delivery of cargo across biological barriers for improving intracellular access, while itself also entering subcellular compartments or intracellular organelles.37,38 Oligoarginine has been reported to exhibit better translocation ability (~20-fold) than the popular peptide TAT49–57 (RKKRRQRRR) originating from HIV1.39 More specifically, oligoarginine CPPs have been used in several studies, besides of their excellent transmembrane-translocation ability.26,39,40 However, it is unknown whether polyarginine can work in a tandem-insert pattern in active targeted drug delivery. In this work, tiR9 was adopted for its excellent transduction ability and incorporated into active targeted cNGR-modified liposome systems. By conjugating the tiR9 (acting as an accelerating motif) to the C-terminal of cNGR, the constructed tandem peptide cNGR-tiR9 was generated. cNGR-tiR9 was then evaluated for its ability to accelerate membrane translocation after being specifically recognized by APN receptors in tumor cells.
The C-terminal cysteine in cNGR and cNGR-tiR9 peptides is a synthetically designed linker that offers a chemically reactive thiol group for later linking to the maleimide group of Mal-PEG2,000-DSPE. Mal-PEG2,000-DSPE was next covalently coupled to the cysteine sulfur of cNGR-tiR9 (cNGR as a control) via maleimide by Michael addition (nucleophilic addition) to generate functional conjugates (cNGR-PEG2,000-DSPE and cNGR-tiR9-PEG2,000-DSPE). Figure 2 shows the synthesis of cNGR-PEG2,000-DSPE and cNGR-tiR9-PEG2,000-DSPE. At pH 7.2 in HEPES solution, the maleimide group of Mal-PEG2,000-DSPE efficiently reacted with the sulfhydryl group of cysteine-containing peptides. N2 gas and ultrasonic sound under low-temperature conditions were applied during the reaction to promote formation of functional products by preventing Mal-PEG2,000-DSPE oxidation. Next, MALDI-TOF-MS was employed to measure MW to verify the correctness of synthetic products. The theoretical MWs of cNGR (Figure S1) and cNGR-tiR9 (Figure S2) were 653 Da and 2,059 Da, respectively. In Figure S3, Mal-PEG2,000-DSPE exhibited peaks at 2,900–3,100 Da that approximate the calculated MW of 2,984 Da. After linkage, the observed central MWs of cNGR-PEG2,000-DSPE (Figure 3) and cNGR-tiR9-PEG2,000-DSPE (Figure 4) were 3,600.44 Da and 4,997.88 Da, respectively. These observed values were close to the calculated MWs of 3,596 Da (cNGR-PEG2,000-DSPE) and 5,002 Da (cNGR-tiR9-PEG2,000-DSPE). Therefore, by means of the well-designed synthetic route and confirmation using MALDI-TOF-MS, functional cNGR-PEG2,000-DSPE and cNGR-tiR9-PEG2,000-DSPE polymers had been synthesized successfully.
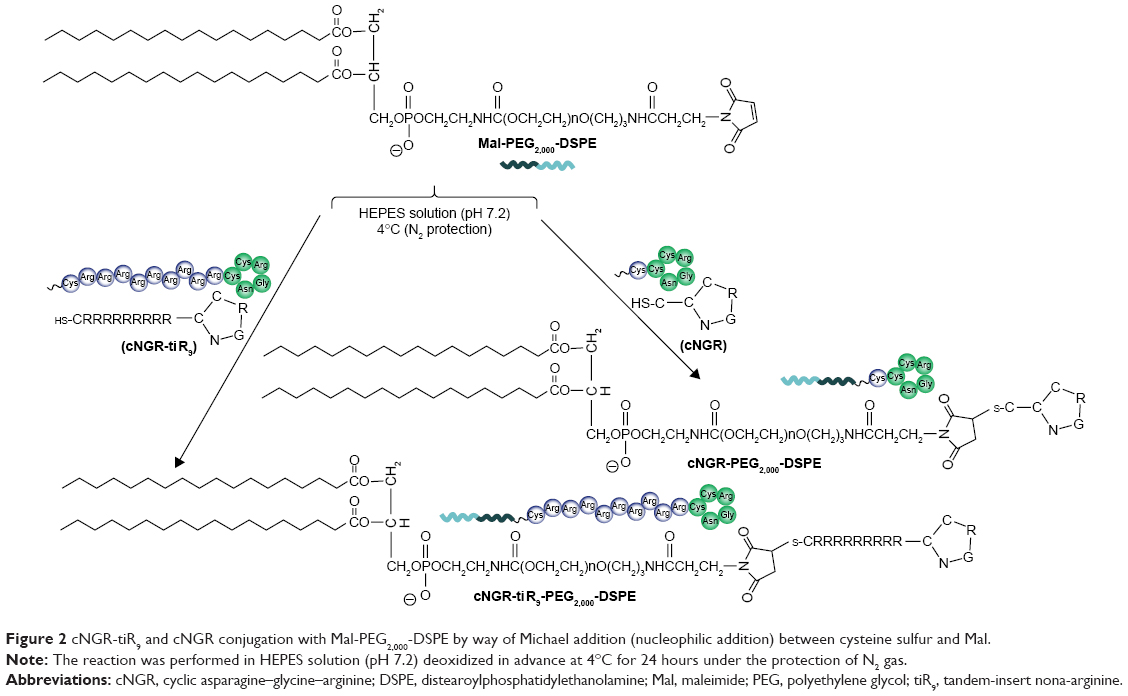 | Figure 2 cNGR-tiR9 and cNGR conjugation with Mal-PEG2,000-DSPE by way of Michael addition (nucleophilic addition) between cysteine sulfur and Mal. |
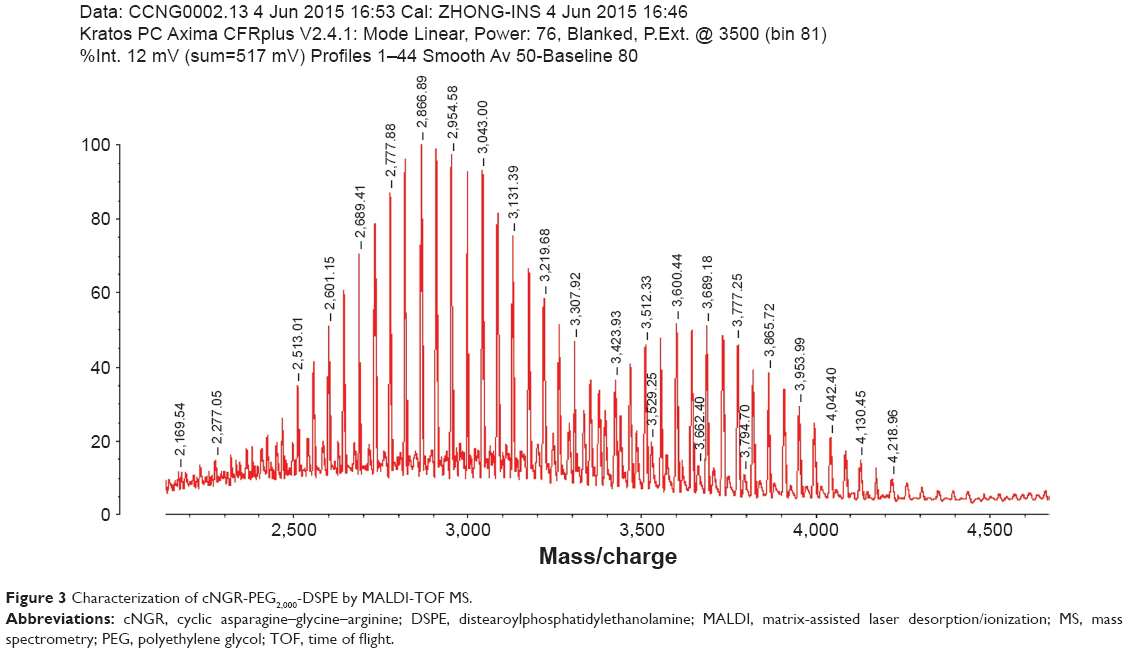 | Figure 3 Characterization of cNGR-PEG2,000-DSPE by MALDI-TOF MS. |
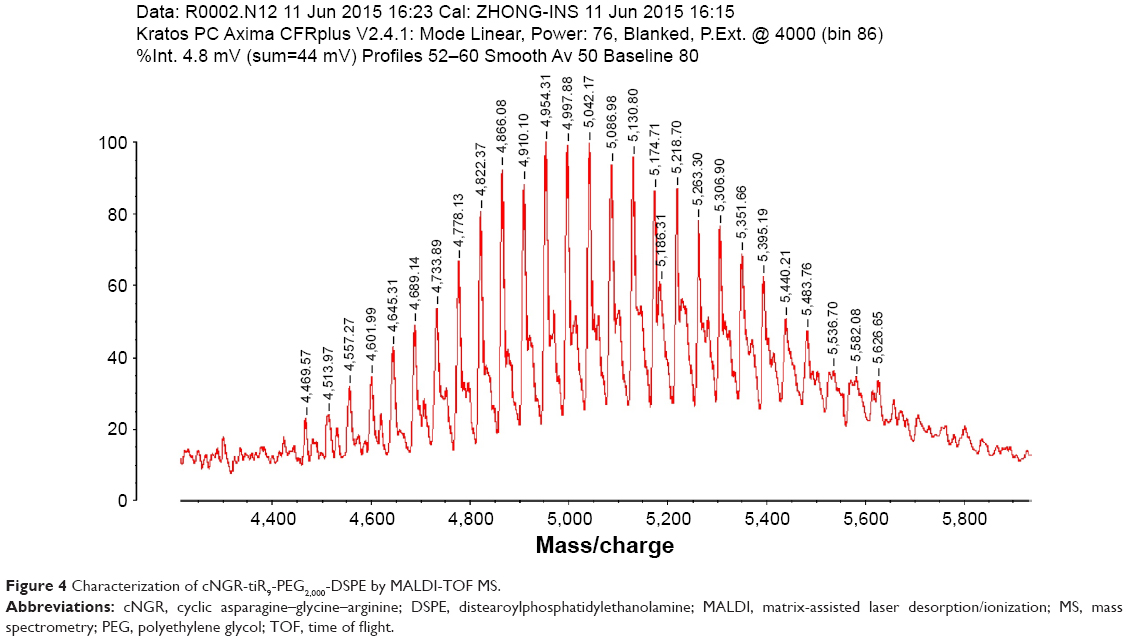 | Figure 4 Characterization of cNGR-tiR9-PEG2,000-DSPE by MALDI-TOF MS. |
Characteristics of functional liposomes
Characteristics of Lip-Dox, cNGR-Lip-Dox, and cNGR-tiR9-Lip-Dox are summarized in Table S1. It was shown that common liposomes were about 157 nm in diameter with a polydispersity-index value <0.12, while both cNGR- and cNGR-tiR9-modified liposomes were similar to common liposomes. All liposomal systems showed weak negative surface charges <−5 mV, average particle sizes below 160 nm, and polydispersity index <0.15. It was observed that >96% of Dox could be loaded into liposomes. Moreover, decoration of the liposomes with these ligands (cNGR and cNGR-tiR9) had no obvious impact on drug-encapsulation efficiency. Good particle-size uniformity was observed for Lip-Dox (Figure 5A), cNGR-Lip-Dox (Figure 5B), and cNGR-tiR9-Lip-Dox (Figure 5C), and this was important, since it would not lead to significant differences in receptor-mediated internalization during intracellular delivery for active targeting of nanocarriers.41–43 Transmission electron-microscopy images demonstrated spherical nanostructures for Lip-Dox (Figure 5D), cNGR-Lip-Dox (Figure 5E), and cNGR-tiR9-Lip-Dox (Figure 5F). Profiles of Dox release vs time in vitro are depicted in Figure 5G. After 24 hours, about 45% of Dox had been released from liposomes, suggesting a favorable drug-releasing property of PBS (pH 7.4). The relatively slow leakage of drugs from lipid nanocarriers was probably due to their encapsulation mechanism. However, there were no significant differences between cNGR-Lip-Dox and cNGR-tiR9-Lip-Dox with regard to drug release, suggesting little influence of the tiR9 modification. The similar physicochemical characteristics of nanocarriers allowed us specifically to compare the effects of CPP modification on cellular uptake and anticancer abilities of cNGR-mediated liposomes.43
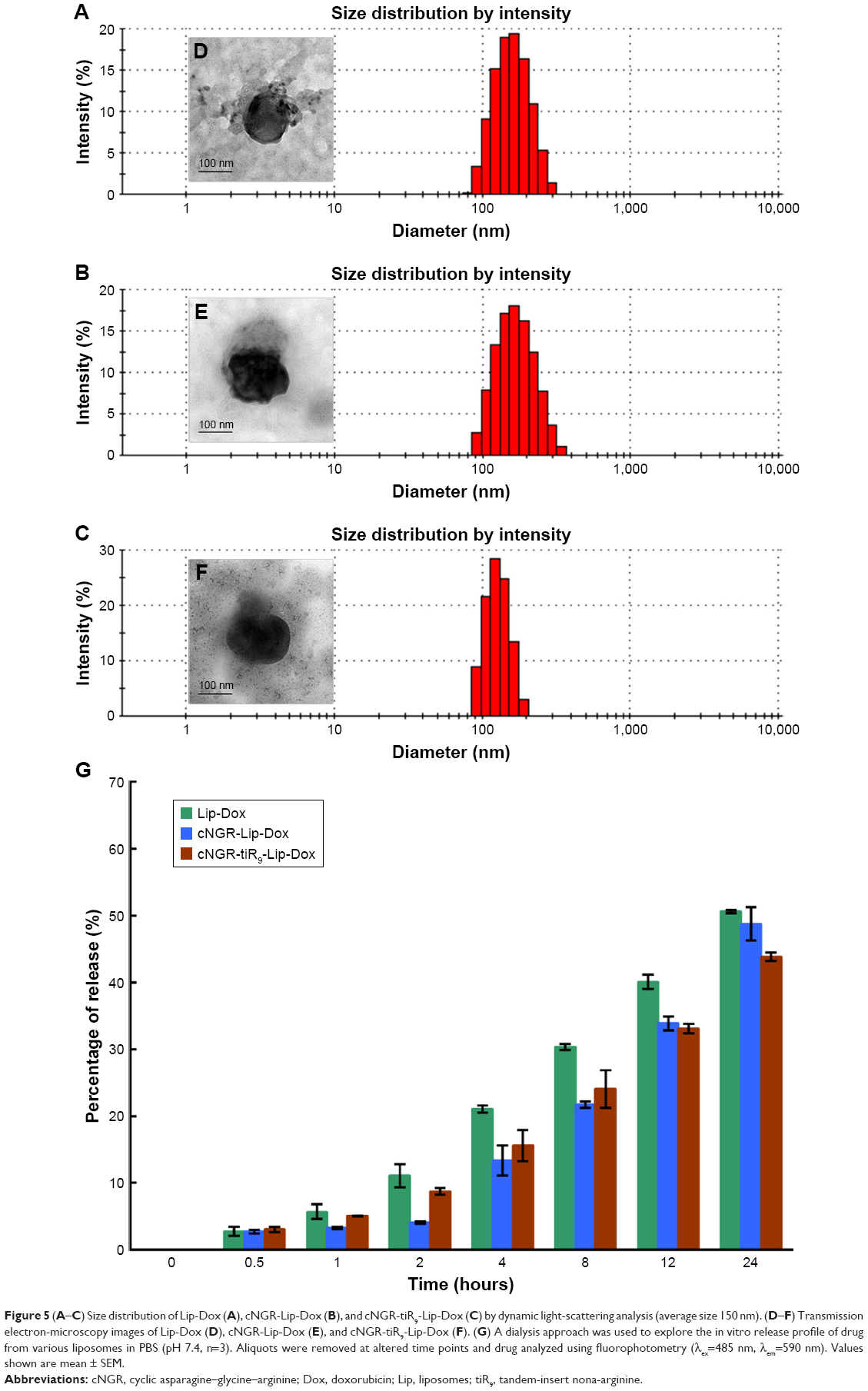 | Figure 5 (A–C) Size distribution of Lip-Dox (A), cNGR-Lip-Dox (B), and cNGR-tiR9-Lip-Dox (C) by dynamic light-scattering analysis (average size 150 nm). (D–F) Transmission electron-microscopy images of Lip-Dox (D), cNGR-Lip-Dox (E), and cNGR-tiR9-Lip-Dox (F). (G) A dialysis approach was used to explore the in vitro release profile of drug from various liposomes in PBS (pH 7.4, n=3). Aliquots were removed at altered time points and drug analyzed using fluorophotometry (λex=485 nm, λem=590 nm). Values shown are mean ± SEM. |
Total Dox uptake by HT1080 cells for various Dox formulations was quantified by flow cytometry. As shown in Figure 6, cellular Dox fluorescence intensity for cNGR-tiR9-Lip-Dox was about 1.3-fold higher than that of cNGR-Lip-Dox, while the strongest fluorescence was observed in the free-Dox group. This result revealed that tiR9 modification induced enhanced accumulation and absorption for improved active targeting by nanocarriers. Exploration of intracellular localization was next performed to understand further the effect of tandem polyarginine on internalization of cNGR-Lip-Dox, since previous studies have shown that CPPs can deliver cargoes into cytosol, nuclei, and other organelles.27,29,36
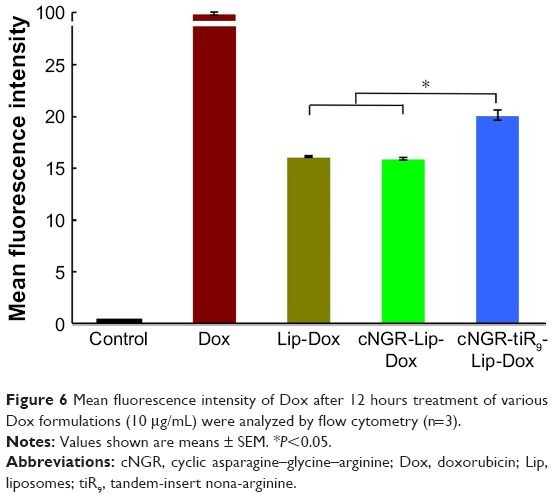 | Figure 6 Mean fluorescence intensity of Dox after 12 hours treatment of various Dox formulations (10 μg/mL) were analyzed by flow cytometry (n=3). |
Because single-layer scanning is not adequate for providing more detailed information on the intracellular distribution of nanocarriers, layer-by-layer scanning by confocal microscopy was used to determine the depth of drug penetration and drug localization from cellular center to cellular margin. There are eight layers of scanning. The thickness of cells was about 2 μm. The center region of cells was mainly between layers 4 and 5. Confocal microscopy images of HT1080 cells after 24 hours’ incubation with Dox formulations are shown in Figure 7. Free Dox localized mainly to the nucleus and exhibited the most intense intracellular fluorescence, due to its direct and rapid access into the membrane independently of nanocarriers, as well as its native high nucleophilic characteristics. Marginal layers (eg, 1, 2, 7, and 8) showed relatively stronger fluorescence for free Dox compared with other treatment groups, suggesting that there was greater distribution of Dox in the cytoplasm far from the cellular center. The apparent red images between layers 3 and 4 for Lip-Dox and cNGR-Lip-Dox suggest that an active targeting effect by cNGR resulted in increased delivery of drugs to cellular centers. cNGR-tiR9-Lip-Dox exhibited obviously enhanced accumulation in cellular centers (layers 4 and 5) relative to cNGR-Lip-Dox and Lip-Dox. It is noteworthy that cNGR-tiR9-Lip-Dox induced improved cellular uptake to cytosol, as indicated by observation from layers 1, 2, 7, and 8. In addition to cytosol accumulation, greater accumulation was observed in nuclei for cNGR-tiR9-Lip-Dox than for other liposomal formulations, based on observations of intensity in each layer. These results indicate that tiR9 helped nanocarriers to achieve deeper penetration into tumor cells than plain Lip-Dox. Judging from the red image in Figure S4, it is also apparent that cNGR-tiR9-Lip-Dox had increased fluorescence intensity in each layer over cNGR-Lip-Dox and Lip-Dox. Actually, poor penetration into cancer cells is a main shortcoming of commercial nanopreparations currently and may explain their modest clinical efficacy.24,25,44 In this study, cNGR-mediated active targeting aided by tiR9 presented enhanced penetration and greater accumulation in tumor cells, which is beneficial and critical to improving antitumor activity.
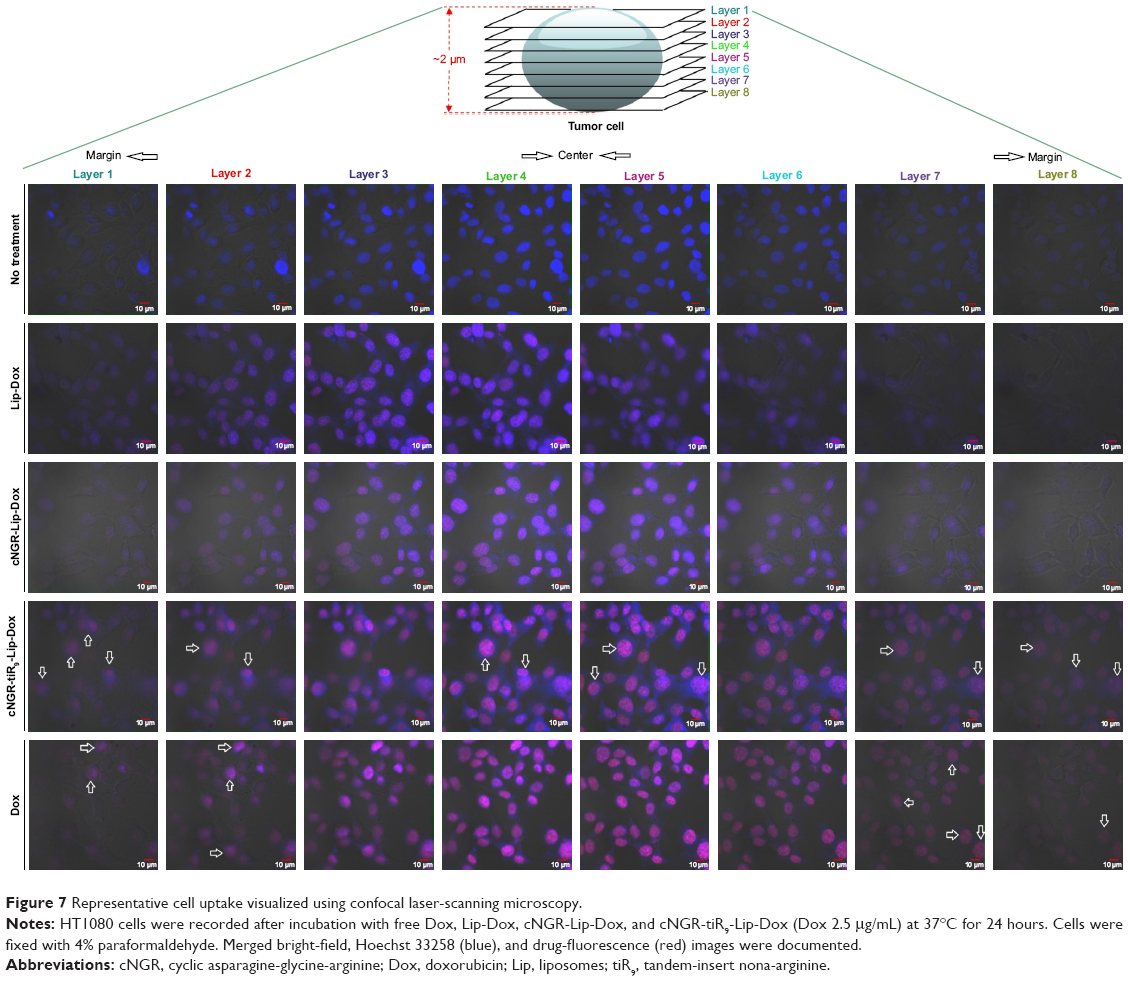 | Figure 7 Representative cell uptake visualized using confocal laser-scanning microscopy. |
Subcellular localization
Subcellular localization is shown in Figure 8 with confocal laser-scanning microscopy images after treatment of HT1080 cells with free Cou6 and Cou6-loaded liposomes. Free Cou6 and Cou6-loaded formulations are observed as green fluorescence, while red fluorescence is observed in lysosomes, mitochondria, endoplasmic reticulum, and Golgi apparatus cell staining with organelle-selective probes. Colocalization of formulations with organelle-selective probes is evidenced by yellow fluorescence, while no colocalization is indicated by red. The relatively stronger red image was detected for free Cou6 (Figure 8A1), Lip-Cou6 (Figure 8A2) and cNGR-Lip-Cou6 (Figure 8A3) compared to cNGR-tiR9-Lip-Cou6 (Figure 8A4) in lysosomes, suggesting that the acceleration of tiR9 enhanced the escape of active targeted liposomes from lysosomes. A similar lysosome-escape function aided by tiR9 has been documented.13 Of note, judging from their intensity of green fluorescence, cNGR-tiR9-Lip-Cou6 accumulated selectively in mitochondria (Figure 8B4), endoplasmic reticulum (Figure 8C4), and Golgi apparatus (Figure 8D4). In subcellular organelles (lysosomes, mitochondria, endoplasmic reticulum, and Golgi apparatus), as opposed to nuclei, lipophilic probe Cou6 exhibited greater dispersion. Moreover, free Cou6 exhibited greater distribution in the Golgi apparatus, resulting in the stronger green image. Generally, tiR9 enhanced penetration of cNGR-Lip-Cou6 through subcellular membrane barriers and caused its specific accumulation in mitochondria (Figure 7B4), endoplasmic reticulum (Figure 8C4), and Golgi apparatus (Figure 8D4), as visualized by augmented green fluorescence in those regions.
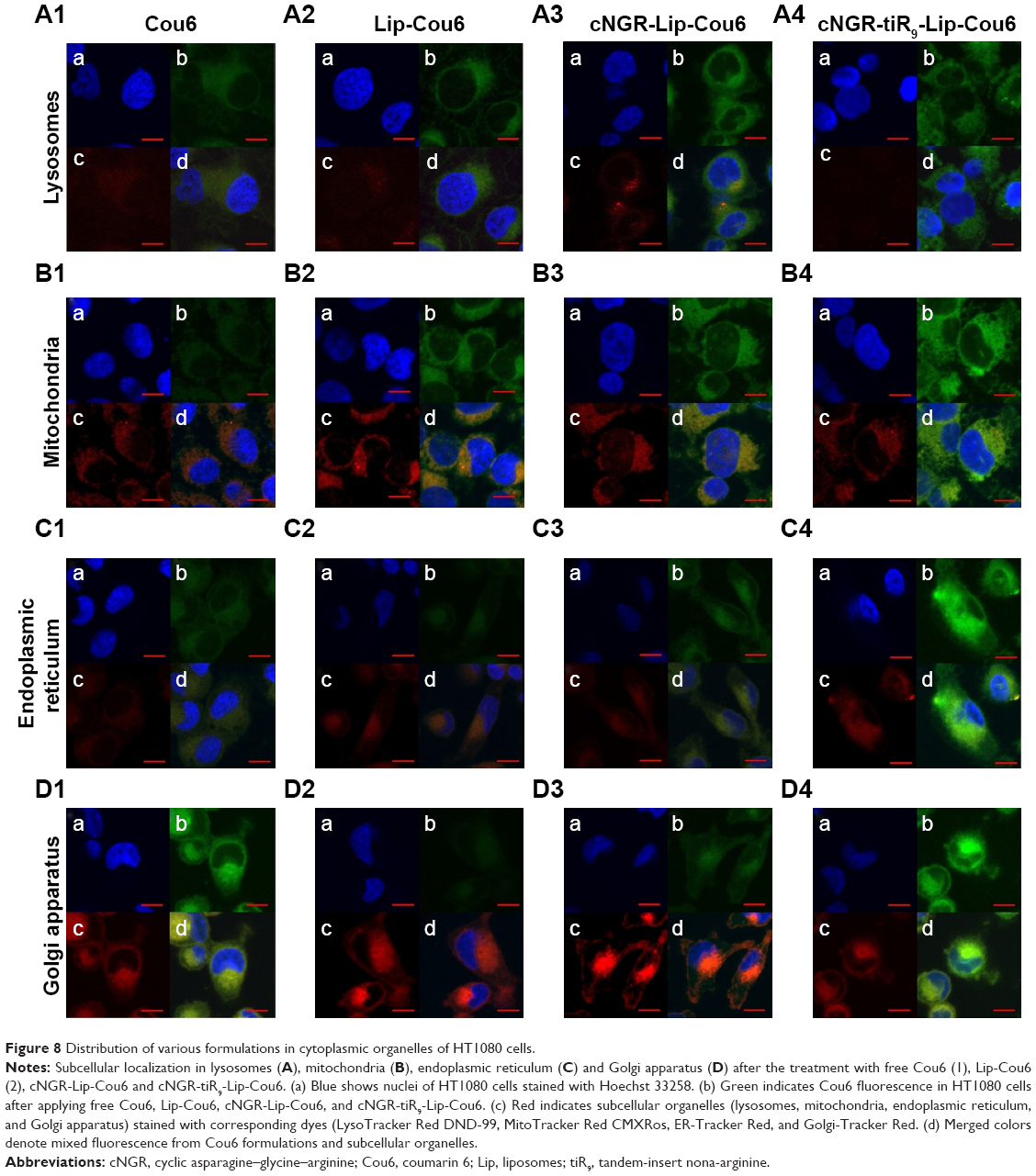 | Figure 8 Distribution of various formulations in cytoplasmic organelles of HT1080 cells. |
Apoptosis and caspase 3/7 activation
In vitro induced apoptosis results for HT1080 cells are shown in Figure 9 for blank culture medium, free Dox, Lip-Dox, cNGR-Lip-Dox, and cNGR-tiR9-Lip-Dox at various concentrations (0.5 μg/mL, 5 μg/mL, and 15 μg/mL). By measuring apoptotic percentage during the early period plus apoptotic percentage during the late period, the apoptosis-inducing effect was assessed (Figure 9A). At low Dox concentrations of 0.5 μg/mL and 5 μg/mL, apoptotic indices in all Dox formulations were similar (below 20%) and accounted for very small apoptosis percentages. With increased Dox concentrations, apoptosis-inducing effects appeared and were enhanced. After treatment with free Dox, Lip-Dox, cNGR-Lip-Dox, or cNGR-tiR9-Lip-Dox at the highest concentration (15 μg/mL), induced apoptosis proportions (Figure 9B) were 19.4%, 30.6%, 33.2%, and 33.5%, respectively. Modification of cNGR or cNGR-tiR9 induced greatly increased apoptosis of liposomes relative to values observed for common liposomes and parent drug. Taken together, these results demonstrate that tiR9 induced increased apoptosis of HT1080 cells when conjugated to cNGR-Lip-Dox.
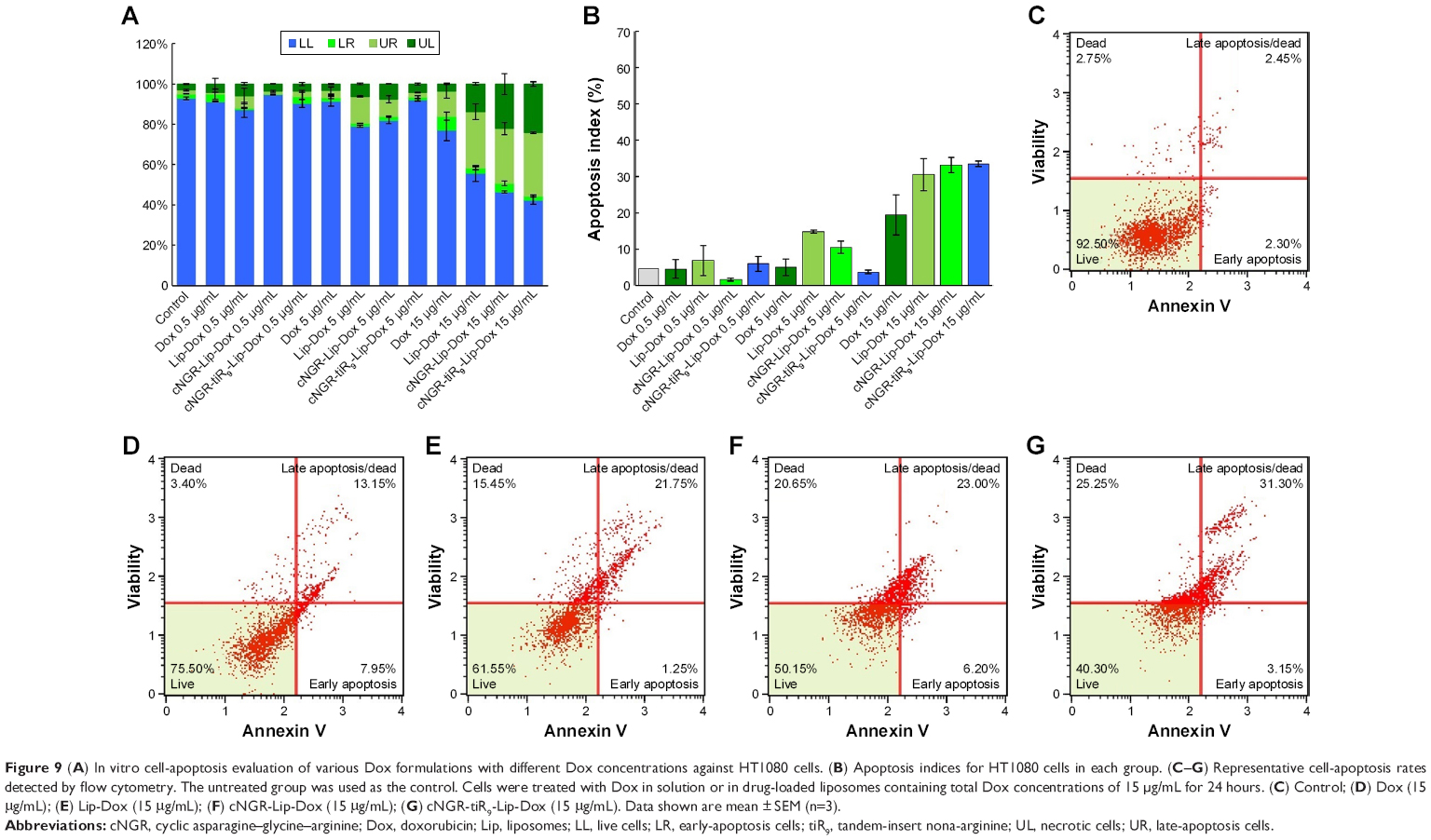 | Figure 9 (A) In vitro cell-apoptosis evaluation of various Dox formulations with different Dox concentrations against HT1080 cells. (B) Apoptosis indices for HT1080 cells in each group. (C–G) Representative cell-apoptosis rates detected by flow cytometry. The untreated group was used as the control. Cells were treated with Dox in solution or in drug-loaded liposomes containing total Dox concentrations of 15 μg/mL for 24 hours. (C) Control; (D) Dox (15 μg/mL); (E) Lip-Dox (15 μg/mL); (F) cNGR-Lip-Dox (15 μg/mL); (G) cNGR-tiR9-Lip-Dox (15 μg/mL). Data shown are mean ± SEM (n=3). |
Activation of caspase 3/7 during apoptosis induction is shown in Figure 10 in HT1080 cells. After application of free Dox, Lip-Dox, cNGR-Lip-Dox, and cNGR-tiR9-Lip-Dox, caspase-activation measurements indicated significantly greater activation of caspase 3/7 for cNGR-Lip-Dox and cNGR-tiR9-Lip-Dox treatments than free Dox or Lip-Dox at Dox concentrations of 5 μg/mL and 15 μg/mL. Proportions of caspase 3/7-activated cells were 27.0%, 31.7%, 60.8%, and 70.8% for 5 μg/mL concentration and 43.6%, 41.0%, 58.4%, and 79.6% for 15 μg/mL Dox concentration. Accordingly, at 15 μg/mL concentration, caspase 3/7-activity in HT1080 cells was about 3.2-, 3.0-, 4.3-, and 5.8-fold higher than the blank control. This result indicated that activities of caspases 3 and 7 were significantly increased after the addition of cNGR-tiR9 modified Dox-loaded liposome formulations.
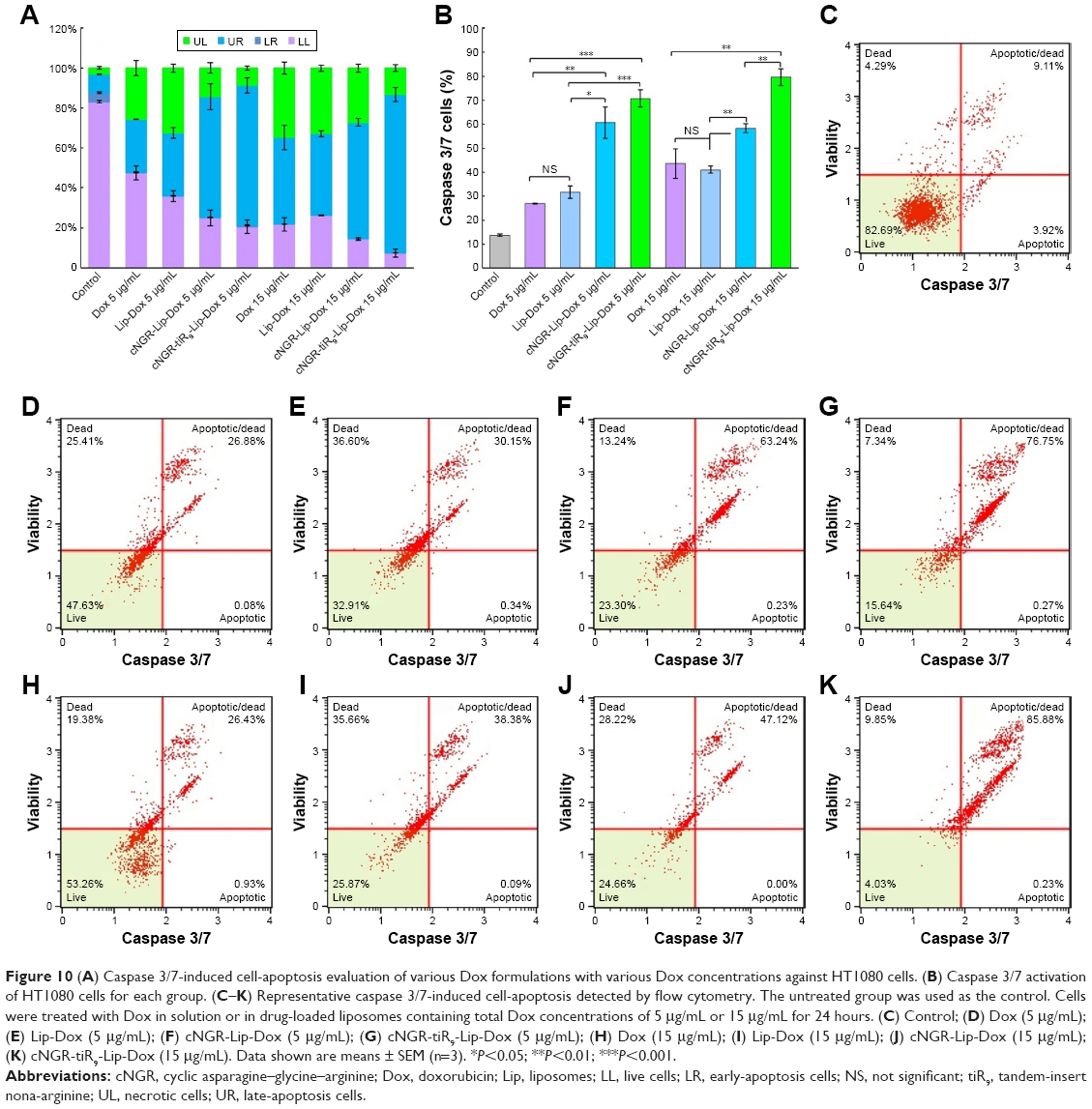 | Figure 10 (A) Caspase 3/7-induced cell-apoptosis evaluation of various Dox formulations with various Dox concentrations against HT1080 cells. (B) Caspase 3/7 activation of HT1080 cells for each group. (C–K) Representative caspase 3/7-induced cell-apoptosis detected by flow cytometry. The untreated group was used as the control. Cells were treated with Dox in solution or in drug-loaded liposomes containing total Dox concentrations of 5 μg/mL or 15 μg/mL for 24 hours. (C) Control; (D) Dox (5 μg/mL); (E) Lip-Dox (5 μg/mL); (F) cNGR-Lip-Dox (5 μg/mL); (G) cNGR-tiR9-Lip-Dox (5 μg/mL); (H) Dox (15 μg/mL); (I) Lip-Dox (15 μg/mL); (J) cNGR-Lip-Dox (15 μg/mL); (K) cNGR-tiR9-Lip-Dox (15 μg/mL). Data shown are means ± SEM (n=3). *P<0.05; **P<0.01; ***P<0.001. |
Internalization mechanisms
Effects of ATP depletion and various endocytosis inhibitors on cNGR-tiR9-Lip-Dox uptake were evaluated in HT1080 cells. Commonly, endocytosis is an energy-dependent route and generally inhibited at low temperatures.45 As shown in Figure 11A, decreasing the incubation temperature from 37°C to 4°C caused a marked decline in mean fluorescence levels (P<0.05), which suggests that the entry route of cNGR-tiR9-Lip-Dox into cells was dominated mainly by ATP-dependent access. A similar result was found for uptake of the CPP–avidin complex, as documented previously by our lab.27 Chloroquine, a potent lysosomotropic compound, can prevent endosomal acidification and consequently slow down endocytosis, thus leaving more time for endosomal escape.46 Conversely, the transmembrane mechanism of CPPs is often involved in a direct translocation that is independent of both energy and endocytosis. Meanwhile, attached cargoes of various sizes influence the resulting internalization pathways of CPPs. The results showed that chloroquine induced marked enhancement in cell uptake of cNGR-tiR9-Lip-Dox, indicating that chloroquine increased the retention of this CPP-containing nanocarrier in acidic compartments and providing further evidence for endocytosis of cNGR-tiR9-Lip-Dox. To examine the involvement of macropinocytosis in CPP-cargo transduction, we determined whether amiloride affected entry of cNGR-tiR9-Lip-Dox due to its action as an inhibitor of Na+−H+ exchange.47 In the presence of amiloride, there was no decrease in internalization compared to the control. Therefore, the macropinocytosis pathway did not play a crucial role in the entry of cNGR-tiR9-Lip-Dox into cells. Alternatively, clathrin-mediated endocytosis, considered the major and best-characterized endocytic route, has been proposed as the main mechanism for the uptake of arginine-rich transporters.48 Moreover, chlorpromazine also significantly intensified the cellular fluorescence of cNGR-tiR9-Lip-Dox to a higher extent (P<0.05) in tested cell lines. Taken together, these findings provide evidence that cNGR-tiR9-Lip-Dox did not require the clathrin-dependent endocytic pathway for intracellular access. Finally, β-cyclodextrin, a Chol-depletion agent, was used to explore whether lipid-raft-dependent endocytosis occurred.49 cNGR-tiR9-Lip-Dox entry displayed higher levels in the presence of β-cyclodextrin, which was approximately 1.2-fold higher than the control after incubation for 4 hours. Therefore, cNGR-tiR9-Lip-Dox tended to enter into cells by lipid-raft-independent endocytosis.
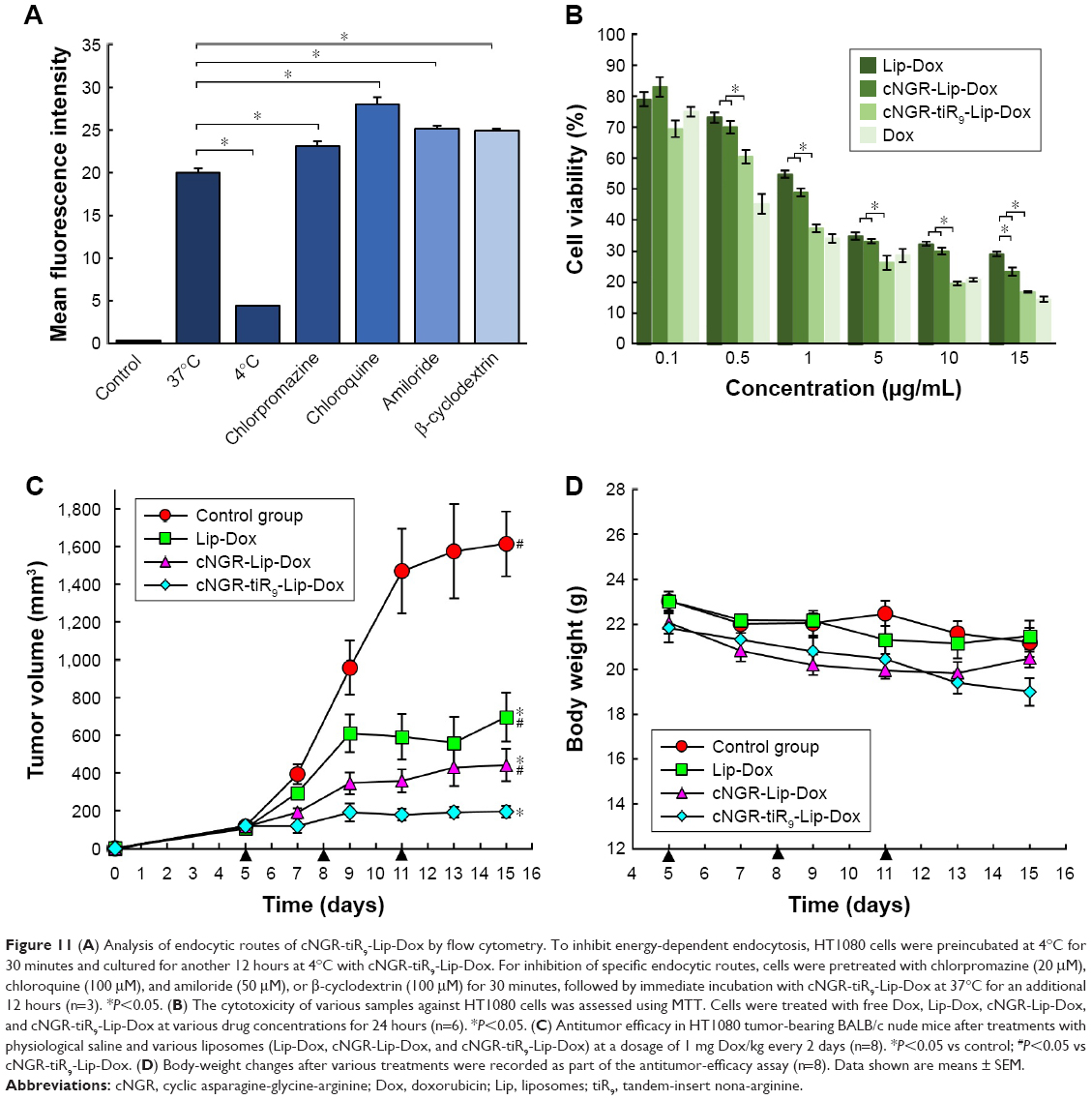 | Figure 11 (A) Analysis of endocytic routes of cNGR-tiR9-Lip-Dox by flow cytometry. To inhibit energy-dependent endocytosis, HT1080 cells were preincubated at 4°C for 30 minutes and cultured for another 12 hours at 4°C with cNGR-tiR9-Lip-Dox. For inhibition of specific endocytic routes, cells were pretreated with chlorpromazine (20 μM), chloroquine (100 μM), and amiloride (50 μM), or β-cyclodextrin (100 μM) for 30 minutes, followed by immediate incubation with cNGR-tiR9-Lip-Dox at 37°C for an additional 12 hours (n=3). *P<0.05. (B) The cytotoxicity of various samples against HT1080 cells was assessed using MTT. Cells were treated with free Dox, Lip-Dox, cNGR-Lip-Dox, and cNGR-tiR9-Lip-Dox at various drug concentrations for 24 hours (n=6). *P<0.05. (C) Antitumor efficacy in HT1080 tumor-bearing BALB/c nude mice after treatments with physiological saline and various liposomes (Lip-Dox, cNGR-Lip-Dox, and cNGR-tiR9-Lip-Dox) at a dosage of 1 mg Dox/kg every 2 days (n=8). *P<0.05 vs control; #P<0.05 vs cNGR-tiR9-Lip-Dox. (D) Body-weight changes after various treatments were recorded as part of the antitumor-efficacy assay (n=8). Data shown are means ± SEM. |
In vitro cytotoxicity assay
The effect of tiR9 conjugation on the antitumor efficacy of active targeted liposomes was investigated by in vitro cytotoxicity assays and in vivo antitumor efficacy studies. The in vitro cytotoxicity of free Dox, Lip-Dox, cNGR-Lip-Dox, and cNGR-tiR9-Lip-Dox was evaluated after incubation with HT1080 cells for 24 hours (Figure 11B). cNGR-tiR9-Lip-Dox and cNGR-Lip-Dox exhibited stronger growth inhibition of cells than Lip-Dox, while cNGR-tiR9-Lip-Dox produced significantly higher inhibition relative to all other treatment groups. Despite the overexpression of APN in HT1080 tumor cells vs normal cells, the absolute level of this biomarker is still low and not high enough to generate significant cellular inhibition at groups with low concentrations of drugs for cNGR-Lip-Dox. However, a significant antiproliferation effect of cNGR-Lip-Dox was observed in the high-concentration group (15 μg/mL), underscoring the low potency of this active targeted approach. Nevertheless, it is worth noting that cNGR-tiR9-Lip-Dox exhibited altered antiproliferation potency (Figure 11B) in the presence of tiR9. Specifically, treatment of HT1080 cells with almost all concentrations of cNGR-tiR9-Lip-Dox except 0.1 μg/mL led to remarkably enhanced cytotoxicity among all groups (P<0.05). According to cellular uptake analysis, it is reasonable to assume that the tiR9-CPP modification was responsible for the observed enhanced cytotoxicity of cNGR-Lip-Dox in vitro because of its penetration effect. In addition, several studies have demonstrated that tiR9-mediated cellular internalization played an important role in overcoming the multidrug resistance that is prevalent among various tumor cells by suppressing associated drug-efflux pumps.26,27,50–52 Oligoarginine CPPs have exhibited excellent ability to overcome this multidrug resistance,50–52 and this is probably another factor that contributes to the enhanced antiproliferation potency of cNGR-modified liposomes by tiR9 CPP.
In vivo antitumor efficacy
After verifying the enhanced penetration and antiproliferation potency of tiR9-conjugated cNGR-liposomes in vitro, we next used an animal-cancer model to detect therapeutic efficacy and evaluate the promotion of tiR9 on active targeted delivery in vivo. As shown in Figure 11C, the tumor volume of mice treated with 1 mg/kg Dox doses of Lip-Dox, cNGR-Lip-Dox and cNGR-tiR9-Lip-Dox were ~650 mm3, ~400 mm3, and ~200 mm3, respectively, significantly smaller than that after treatment with physiological saline (~1,600 mm3) by the end of the study. The short circulation time of common liposomes, as well as their poor ability to traverse cellular membranes, are both due to their sole reliance on the EPR effect. As such, Lip-Dox displayed only weak restraint of tumor growth. Conversely, the active targeting ability of the cNGR moiety11,15–18 enabled cNGR-Lip-Dox to exhibit stronger ability to suppress tumor growth than Lip-Dox, with enhanced antitumor efficacy at day 15 after tumor inoculation. Notably, among all treated groups, the smallest tumors were observed in the cNGR-tiR9-Lip-Dox group, revealing that the modification of tiR9 significantly enhanced and improved the anticancer activity of active targeted liposomes (cNGR-Lip-Dox). Moreover, this phenomenon was in agreement with a former investigation of cellular uptake and cytotoxicity assays of cNGR-tiR9-Lip-Dox. Furthermore, a synergistic effect of tiR9 and cNGR was evident in this study, indicating that the approach combining active targeting with tandem-insert CPPs was more effective in suppressing tumor growth than active targeting alone. Overall, these results collectively underscore the suitability of tiR9 to upgrade tumor therapy of active targeted nanocarriers.
With regard to drug safety, body-weight changes in animals were determined over the experimental period (Figure 11D). There was no significant variation in body weight for the physiological saline, Lip-Dox, cNGR-Lip-Dox, or cNGR-tiR9-Lip-Dox (1 mg/kg Dox equivalence) groups of mice throughout the experimental period. Moreover, no obvious weight loss was found in the cNGR-Lip-Dox or cNGR-tiR9-Lip-Dox groups when experiments were terminated, suggesting that cNGR-mediated or cNGR-tiR9-mediated delivery is safe. Finally, at the conclusion of antitumor assays, H&E tissue sections (Figure S5) showed that there was no noticeable pathological change after systematic administration of these liposomal formations, further underscoring the safety of this drug-delivery strategy.
Numerous studies have demonstrated that oligoarginine CPPs play an important role in overcoming both cell-membrane barriers27 and tissue barriers.54–57 Candan et al53 developed a system to deliver hydrophilic drugs and proteins across the skin by employing polyarginine (R11) and found that topical therapy using R11 enhanced the delivery of hydrophilic cargo by the transdermal route through directed conjugation with payloads. Taking this strategy further, Shah et al54 investigated the effect of polyarginine on topical delivery of surface-modified nanostructured lipid carriers containing spantide II/ketoprofen and demonstrated that the surface modification of these carriers with R11 improved transport of drugs across the deeper skin layers. In addition, penetration of other tissue barriers, such as the blood–brain barrier,55 inner ear membranes,56 and intestinal mucosa,57 have also been reported using polyarginine CPPs with directed linkage. However, this study revealed that undirected conjugation of tiR9 also exhibited enhanced antitumor activity not only for tumor cells in vitro (Figures 9–11) but also for tumor tissues in vivo (Figure 11C), mainly due to its deeply accelerating penetration effect across cellular (Figures 6, 7, and S4) or subcellular membrane barriers (Figure 8).
Conventional chemotherapy often fails due to poor drug penetration of solid tumors. Moreover, membrane permeability of nanotherapeutics is a related and critical intrinsic limitation of this newer approach.24,25,44 Nevertheless, while traditional nanocarriers decorated with active targeting ligands have achieved only limited success, they have demonstrated their effectiveness in tumor therapy to some extent.3,4 Their ineffectiveness may be due to their entry into cells mainly by ligand-receptor binding acting in concert with a passive-diffusion effect. Consequently, such a relatively slow absorption process diminishes their potency for tumor therapy and is largely responsible for their weak penetration.44 Through evaluations at cellular and organism levels using tiR9 as tandem-insert CPP and cNGR-Lip as active targeted delivery systems, the current work has demonstrated the effectiveness of tiR9 as an accelerating component to accelerate cellular/subcellular penetration and improve delivery of active targeted nanocarriers. In addition, in terms of the nonselectivity of CPPs for in vivo application,26,29,30 conjugation of the cNGR ligand adds a homing-target motif to reduce aspecific uptake by untargeted cells as well. Overall, this study provides a strategy to upgrade the antitumor efficacy of active targeted nanocarrier systems by combining an accelerating motif, tiR9, with cNGR liposomes for more effective targeted antitumor drug delivery. Generated accelerating active targeted nanotherapeutics in this work are also new to the development of multifunctional nanomedicine.
Conclusion
The concept that active targeted cNGR-mediated nanoliposomes containing tiR9 as an accelerating component can accelerate drug uptake by cells was successfully established and developed in this investigation. The nanocarriers fabricated here exhibited suitable physicochemical indicators, including small particles, narrow particle-size distribution, good ζ-potential/encapsulation efficiency, and ability to achieve drug release. Moreover, tiR9 led to enhanced cellular uptake of active targeted cNGR liposomes. Based on a cubic observation from layer-by-layer scanning using confocal microscopy, cNGR-tiR9-Lip-Dox was distributed deeply in both marginal and central regions compared to cNGR-Lip-Dox. Arginine-rich tiR9 enhanced penetration of cNGR-Lip-Cou6 through subcellular membrane barriers and caused its specific accumulation in mitochondria, endoplasmic reticulum, and Golgi apparatus. The outstanding merit of nanocarriers developed in this study derives from tandem incorporation of tiR9 as an accelerating motif that can potentially overcome membrane barriers or other biobarriers. It is also obvious that cNGR-tiR9-Lip-Dox induced enhanced apoptosis and activated caspase 3/7. Consequently, the tandem insertion of R9 improved antitumor efficacy of active tumor-targeting nanocarriers and promoted the enhanced cytotoxic effect observed from cNGR-tiR9-Lip-Dox. After intravenous administration, cNGR-tiR9-Lip-Dox exhibited improved antitumor efficacy and suppressed tumor growth at a higher level relative to other treatments. Therefore, the active targeted nanocarrier fabricated in this research based on the accelerating concept exhibited prominently superior efficacy and appears to be an upgraded platform for peptide-mediated active targeted delivery systems of therapeutic antitumor agents. In addition, cNGR-tiR9-Lip-Dox also showed promise as an effective accelerating active targeted nanomedicine by enhancing the penetration of biobarriers for specific tumor therapies.
Acknowledgments
This investigation was supported by the National Natural Science Foundation of China (81602654 and 82102953), Science and Technology Development Program of Jilin Province of China (201464053), State Key Laboratory of Medicinal Chemical Biology of Nankai University (201503001), China Postdoctoral Science Foundation (2015M571373), Science and Technology Development Program of Jilin Province of China (20160520046JH and 20140203012YY), and Scientific Research Foundation of the Education Department of Jilin Province of China (2015–401).
Disclosure
The authors report no conflicts of interest in this work.
References
Stewart BW, Kleihues P. World Cancer Report. Geneva: World Health Organization Press; 2003. | ||
Peer D, Karp JM, Hong S, et al. Nanocarriers as an emerging platform for cancer therapy. Nat Nanotechnol. 2007;2(12):751–760. | ||
Dawidczyk CM, Kim C, Park JH, et al. State-of-the-art in design rules for drug delivery platforms: lessons learned from FDA-approved nanomedicines. J Control Release. 2014;187:133–144. | ||
Dawidczyk CM, Russell LM, Searson PC. Nanomedicines for cancer therapy: state-of-the-art and limitations to pre-clinical studies that hinder future developments. Front Chem. 2014;2:69. | ||
Russell LM, Dawidczyk CM, Searson PC. Quantitative evaluation of the enhanced permeability and retention (EPR) effect. Methods Mol Biol. 2017;1530:247–254. | ||
Ngoune R, Peters A, von Elverfeldt D, Winkler K, Pütz G. Accumulating nanoparticles by EPR: A route of no return. J Control Release. 2016;238:58–70. | ||
Zhong Y, Meng F, Deng C, Zhong Z. Ligand-directed active tumor-targeting polymeric nanoparticles for cancer chemotherapy. Biomacromolecules. 2014;15(6):1955–1969. | ||
Wang X, Li S, Shi Y, et al. The development of site-specific drug delivery nanocarriers based on receptor mediation. J Control Release. 2014;193:139–153. | ||
Ruoslahti E. Peptides as targeting elements and tissue penetration devices for nanoparticles. Adv Mater. 2012;24(28):3747–3756. | ||
Accardo A, Morelli G. Review peptide-targeted liposomes for selective drug delivery: advantages and problematic issues. Biopolymers. 2015;104(5):462–479. | ||
Pasqualini R, Koivunen E, Kain R, et al. Aminopeptidase N is a receptor for tumor-homing peptides and a target for inhibiting angiogenesis. Cancer Res. 2000;60(3):722–727. | ||
Shim JS, Kim JH, Cho HY, et al. Irreversible inhibition of CD13/aminopeptidase N by the antiangiogenic agent curcumin. Chem Biol. 2003;10(8):695–704. | ||
Dunne M, Zheng J, Rosenblat J, Jaffray DA, Allen C. APN/CD13-targeting as a strategy to alter the tumor accumulation of liposomes. J Control Release. 2011;154(3):298–305. | ||
Luo LM, Huang Y, Zhao BX, et al. Anti-tumor and anti-angiogenic effect of metronomic cyclic NGR-modified liposomes containing paclitaxel. Biomaterials. 2013;34(4):1102–1114. | ||
Graziadio A, Zanda M, Frau S, et al. NGR tumor-homing peptides: Structural requirements for effective APN (CD13) targeting. Bioconjug Chem. 2016;27(5):1332–1340. | ||
Hendrikx G, de Saint-Hubert M, Dijkgraaf I, et al. Molecular imaging of angiogenesis after myocardial infarction by (111)In-DTPA-cNGR and (99m)Tc-sestamibi dual-isotope myocardial SPECT. EJNMMI Res. 2015;5:2. | ||
Soudy R, Ahmed S, Kaur K. NGR peptide ligands for targeting CD13/APN identified through peptide array screening resemble fibronectin sequences. ACS Comb Sci. 2012;14(11):590–599. | ||
Son S, Singha K, Kim WJ. Bioreducible BPEI-SS-PEG-cNGR polymer as a tumor targeted nonviral gene carrier. Biomaterials. 2010;31(24):6344–6354. | ||
Colombo M, Fiandra L, Alessio G, et al. Tumour homing and therapeutic effect of colloidal nanoparticles depend on the number of attached antibodies. Nat Commun. 2016;7:13818. | ||
Chaplin DJ, Olive PL, Durand RE. Intermittent blood flow in a murine tumor: radiobiological effects. Cancer Res. 1987;47(2):597–601. | ||
Padera TP, Stoll BR, Tooredman JB, et al. Pathology: cancer cells compress intratumour vessels. Nature. 2004;427(6976):695. | ||
Jain RK, Munn LL, Fukumura D. Dissecting tumour pathophysiology using intravital microscopy. Nat Rev Cancer. 2002;2(4):266–276. | ||
Heldin CH, Rubin K, Pietras K, Ostman A. High interstitial fluid pressure – an obstacle in cancer therapy. Nat Rev Cancer. 2004;4(10):806–813. | ||
Sriraman SK, Aryasomayajula B, Torchilin VP. Barriers to drug delivery in solid tumors. Tissue Barriers. 2014;2:e29528. | ||
Minchinton AI, Tannock IF. Drug penetration in solid tumours. Nat Rev Cancer. 2006;6(8):583–592. | ||
Shi NQ, Qi XR, Xiang B, Zhang Y. A survey on “Trojan Horse” peptides: opportunities, issues and controlled entry to “Troy”. J Control Release. 2014;194:53–70. | ||
Ma DX, Shi NQ, Qi XR. Distinct transduction modes of arginine-rich cell-penetrating peptides for cargo delivery into tumor cells. Int J Pharm. 2011;419(1–2):200–208. | ||
Xiang B, Dong DW, Shi NQ, et al. PSA-responsive and PSMA-mediated multifunctional liposomes for targeted therapy of prostate cancer. Biomaterials. 2013;34(28):6976–6991. | ||
Shi NQ, Gao W, Xiang B, Qi XR. Enhancing cellular uptake of activable cell-penetrating peptide-doxorubicin conjugate by enzymatic cleavage. Int J Nanomedicine. 2012;7:1613–1621. | ||
Shi NQ, Qi XR. Taming the wildness of “Trojan-Horse” peptides by charge-guided masking and protease-triggered demasking for the controlled delivery of antitumor agents. ACS Appl Mater Interfaces. 2017;9(12):10519–10529. | ||
Macewan SR, Chilkoti A. Harnessing the power of cell-penetrating peptides: activatable carriers for targeting systemic delivery of cancer therapeutics and imaging agents. Wiley Interdiscip Rev Nanomed Nanobiotechnol. 2013;5(1):31–48. | ||
Melikov K, Hara A, Yamoah K, et al. Efficient entry of cell-penetrating peptide nona-arginine into adherent cells involves a transient increase in intracellular calcium. Biochem J. 2015;471(2):221–230. | ||
Haran G, Cohen R, Bar LK, Barenholz Y. Transmembrane ammonium sulfate gradients in liposomes produce efficient and stable entrapment of amphipathic weak bases. Biochim Biophys Acta. 1993;1151(2):201–215. | ||
Wang X, Wang Y, Chen X, et al. NGR-modified micelles enhance their interaction with CD13-overexpressing tumor and endothelial cells. J Control Release. 2009;139(1):56–62. | ||
Sawant R, Torchilin V. Intracellular transduction using cell-penetrating peptides. Mol Biosyst. 2010;6(4):628–640. | ||
Futaki S. Membrane-permeable arginine-rich peptides and the translocation mechanisms. Adv Drug Deliv Rev. 2005;57(4):547–558. | ||
Sharmin S, Islam MZ, Karal MA, et al. Effects of lipid composition on the entry of cell-penetrating peptide oligoarginine into single vesicles. Biochemistry. 2016;55(30):4154–4165. | ||
Fischer R, Bächle D, Fotin-Mleczek M, et al. A targeted protease substrate for a quantitative determination of protease activities in the endolysosomal pathway. Chembiochem. 2006;7(9):1428–1434. | ||
Wender PA, Mitchell DJ, Pattabiraman K, et al. The design, synthesis, and evaluation of molecules that enable or enhance cellular uptake: peptoid molecular transporters. Proc Natl Acad Sci U S A. 2000;97(24):13003–13008. | ||
Cerrato CP, Künnapuu K, Langel Ü. Cell-penetrating peptides with intracellular organelle targeting. Expert Opin Drug Deliv. 2017;14(2):245–255. | ||
Riezman H, Woodman PG, van Meer G, Marsh M. Molecular mechanisms of endocytosis. Cell. 1997;91(6):731–738. | ||
Chen S, Tam YYC, Lin PJC, et al. Influence of particle size on the in vivo potency of lipid nanoparticle formulations of siRNA. J Control Release. 2016;235:236–244. | ||
Allen TM, Cheng WW, Hare JI, Laginha KM. Pharmacokinetics and pharmacodynamics of lipidic nano-particles in cancer. Anticancer Agents Med Chem. 2006;6(6):513–523. | ||
Wong C, Stylianopoulos T, Cui J, et al. Multistage nanoparticle delivery system for deep penetration into tumor tissue. Proc Natl Acad Sci U S A. 2011;108(6):2426–2431. | ||
Madani F, Lindberg S, Langel U, Futaki S, Gräslund A. Mechanisms of cellular uptake of cell-penetrating peptides. J Biophys. 2011;2011:414729. | ||
Zaro JL, Vekich JE, Tran T, Shen WC. Nuclear localization of cell-penetrating peptides is dependent on endocytosis rather than cytosolic delivery in CHO cells. Mol Pharm. 2009;6(2):337–344. | ||
Wadia JS, Stan RV, Dowdy SF. Transducible TAT-HA fusogenic peptide enhances escape of TAT-fusion proteins after lipid raft macropinocytosis. Nat Med. 2004;10(3):310–315. | ||
Richard JP, Melikov K, Brooks H, et al. Cellular uptake of unconjugated TAT peptide involves clathrin-dependent endocytosis and heparan sulfate receptors. J Biol Chem. 2005;280(15):15300–15306. | ||
Potocnik SJ, Jenkins N, Murphy TV, Hill MA. Membrane cholesterol depletion with beta-cyclodextrin impairs pressure-induced contraction and calcium signalling in isolated skeletal muscle arterioles. J Vasc Res. 2007;44(4):292–302. | ||
Dubikovskaya EA, Thorne SH, Pillow TH, Contag CH, Wender PA. Overcoming multidrug resistance of small-molecule therapeutics through conjugation with releasable octaarginine transporters. Proc Natl Acad Sci U S A. 2008;105(34):12128–12133. | ||
Wender PA, Galliher WC, Bhat NM, et al. Taxol-oligoarginine conjugates overcome drug resistance in-vitro in human ovarian carcinoma. Gynecol Oncol. 2012;126(1):118–123. | ||
Vargas JR, Stanzl EG, Teng NN, Wender PA. Cell-penetrating, guanidinium-rich molecular transporters for overcoming efflux-mediated multidrug resistance. Mol Pharm. 2014;11(8):2553–2565. | ||
Candan G, Michiue H, Ishikawa S, et al. Combining poly-arginine with the hydrophobic counter-anion 4-(1-pyrenyl)-butyric acid for protein transduction in transdermal delivery. Biomaterials. 2012;33(27):6468–6475. | ||
Shah PP, Desai PR, Channer D, Singh M. Enhanced skin permeation using polyarginine modified nanostructured lipid carriers. J Control Release. 2012;161(3):735–745. | ||
Pham W, Zhao BQ, Lo EH, Medarova Z, Rosen B, Moore A. Crossing the blood-brain barrier: a potential application of myristoylated polyarginine for in vivo neuroimaging. Neuroimage. 2005;28(1):287–292. | ||
Yoon JY, Yang KJ, Kim DE, et al. Intratympanic delivery of oligoarginine-conjugated nanoparticles as a gene (or drug) carrier to the inner ear. Biomaterials. 2015;73:243–253. | ||
Yamaki T, Uchida M, Kuwahara Y, et al. Effect of poly-L-arginine on intestinal absorption of hydrophilic macromolecules in rats. Biol Pharm Bull. 2013;36(3):496–500. |
Supplementary materials
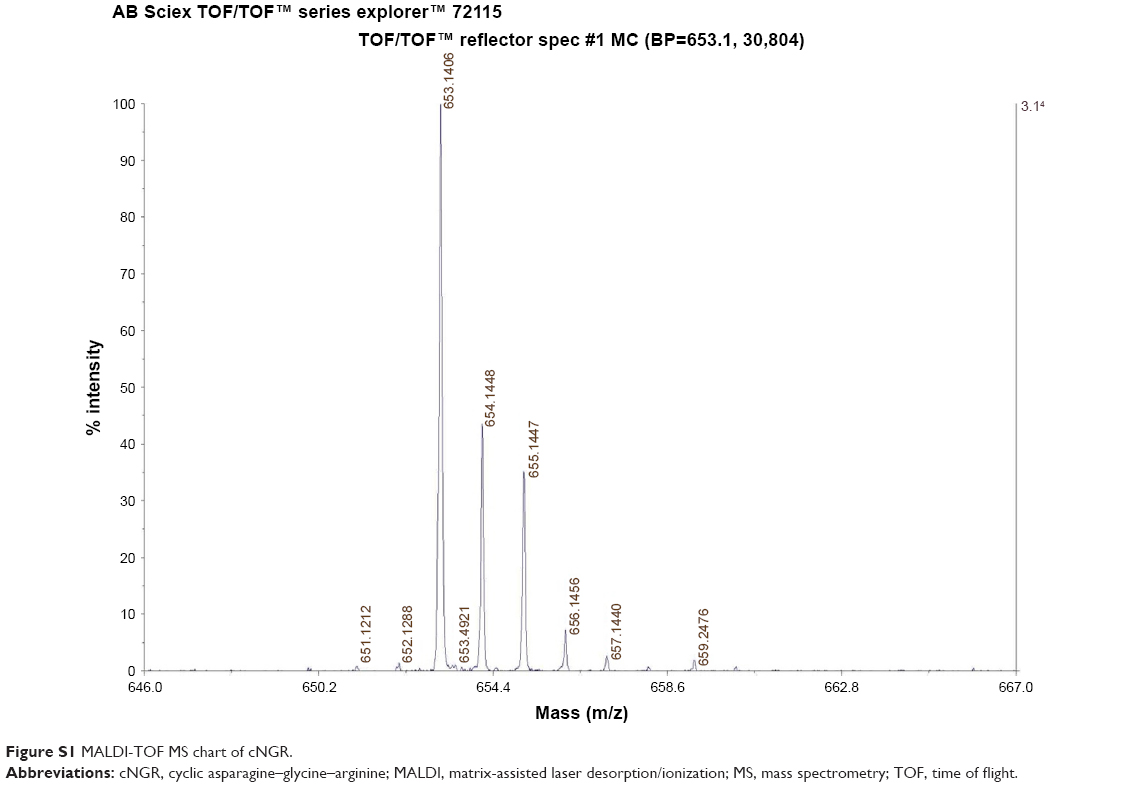 | Figure S1 MALDI-TOF MS chart of cNGR. |
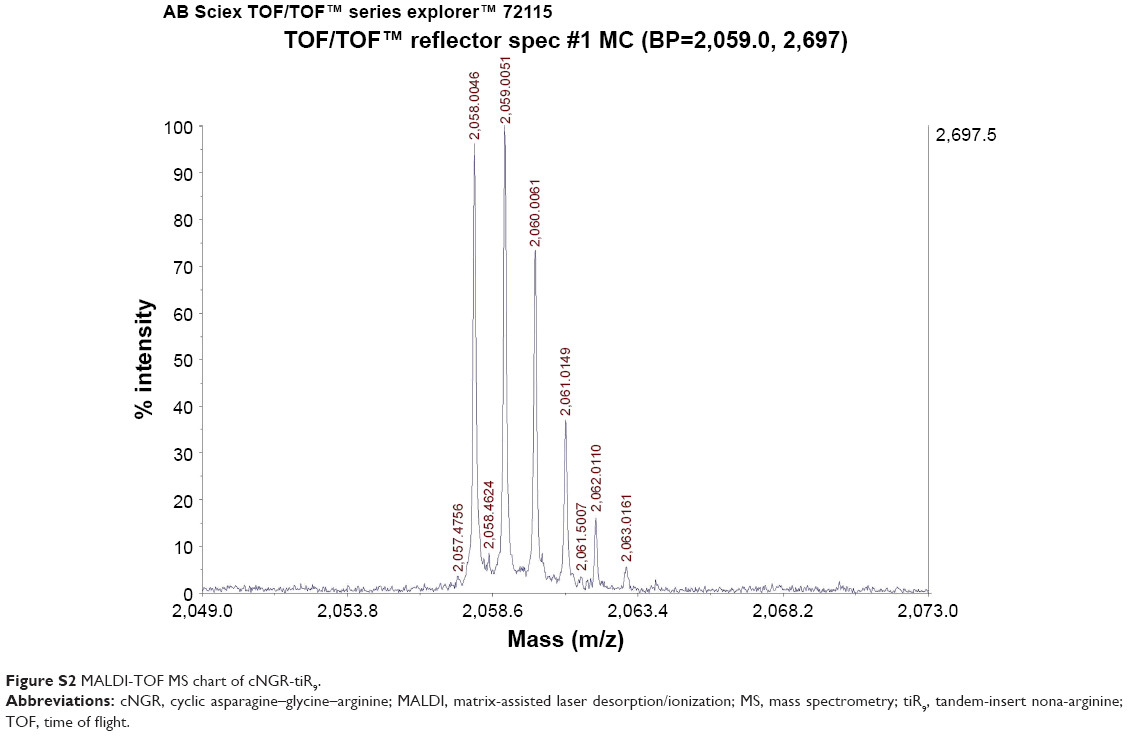 | Figure S2 MALDI-TOF MS chart of cNGR-tiR9. |
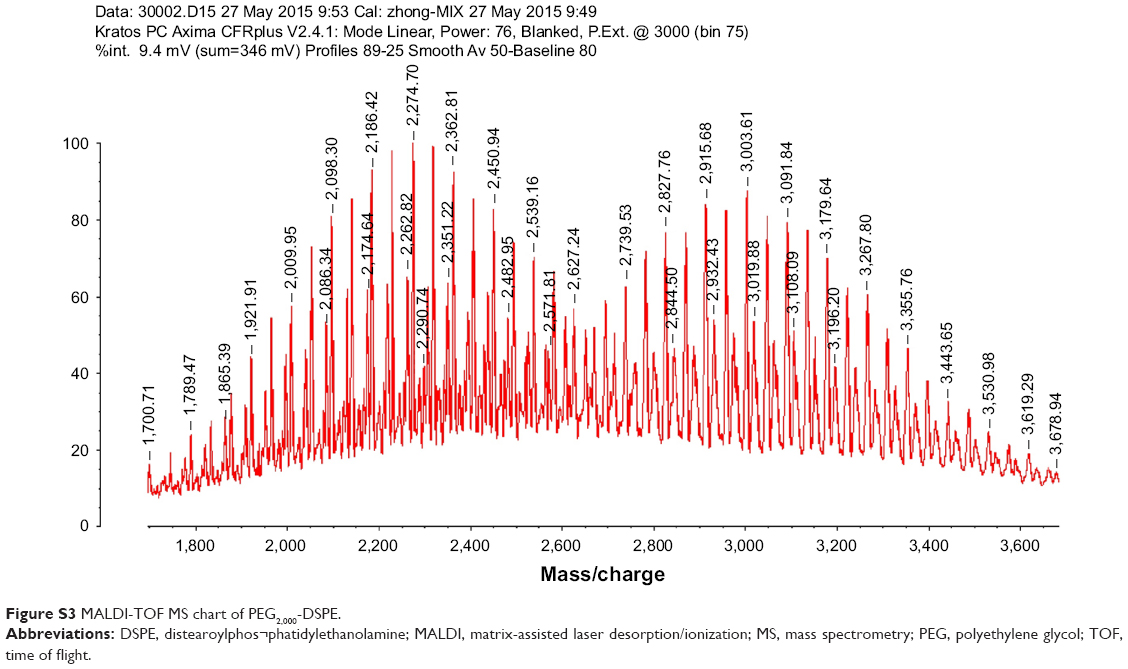 | Figure S3 MALDI-TOF MS chart of PEG2,000-DSPE. |
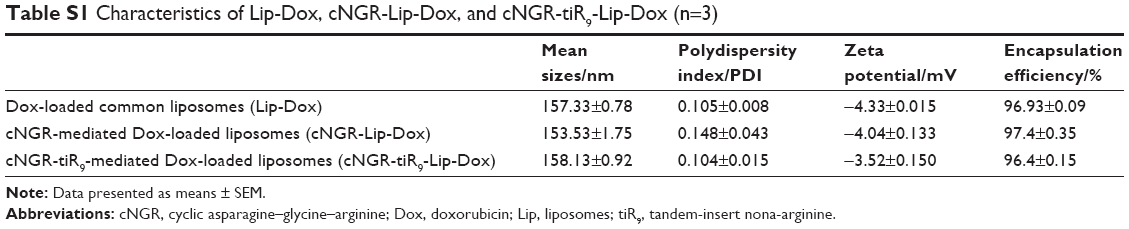 | Table S1 Characteristics of Lip-Dox, cNGR-Lip-Dox, and cNGR-tiR9-Lip-Dox (n=3) |
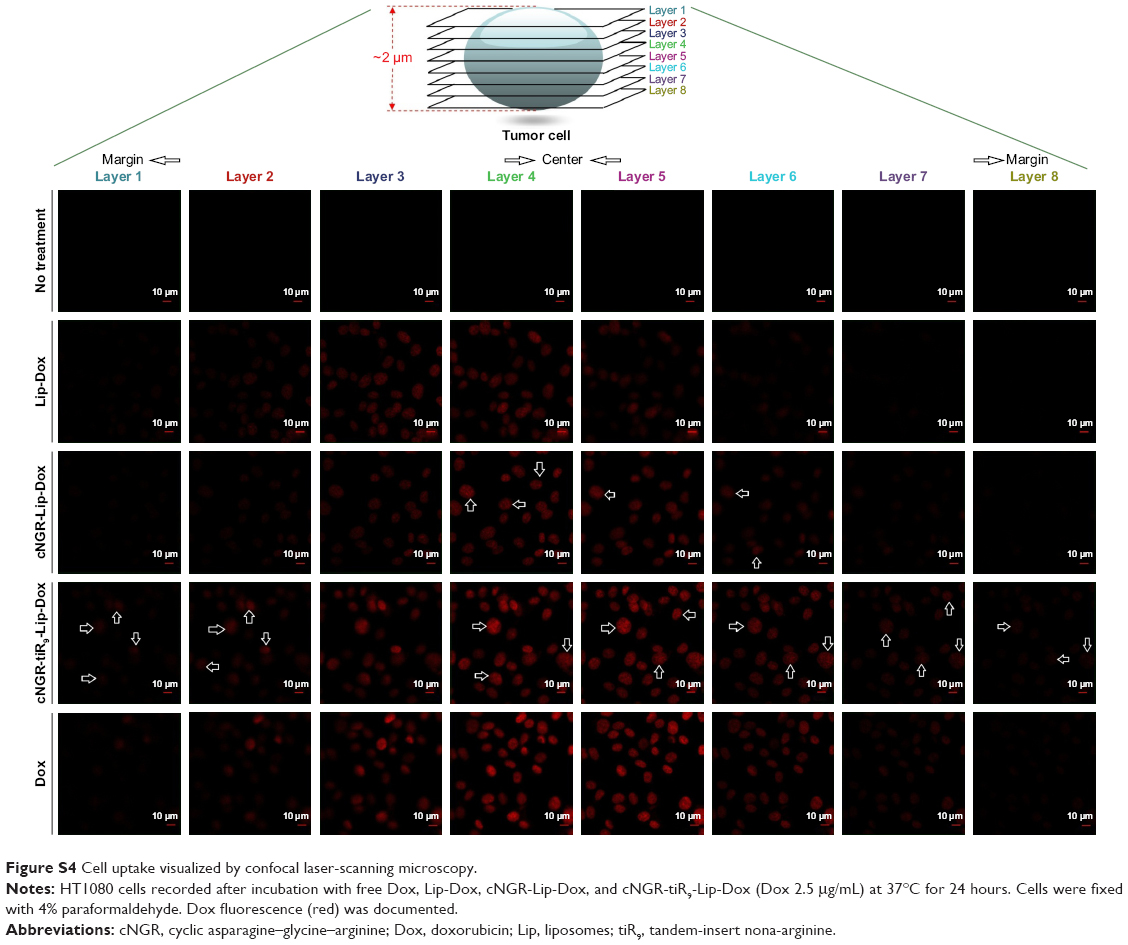 | Figure S4 Cell uptake visualized by confocal laser-scanning microscopy. |
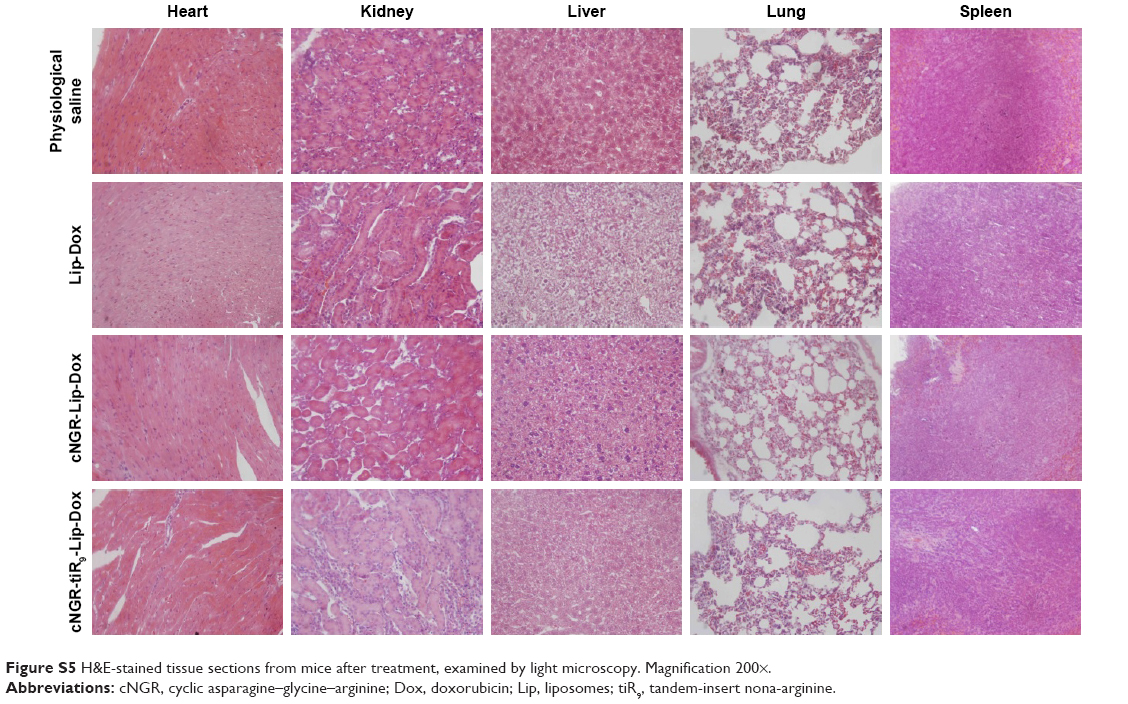 | Figure S5 H&E-stained tissue sections from mice after treatment, examined by light microscopy. Magnification 200×. |
 © 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.