Back to Journals » OncoTargets and Therapy » Volume 11
DcR3 induces proliferation, migration, invasion, and EMT in gastric cancer cells via the PI3K/AKT/GSK-3β/β-catenin signaling pathway
Authors Ge H, Liang C, Li Z, An D, Ren S, Yue C, Wu J
Received 30 April 2018
Accepted for publication 7 June 2018
Published 19 July 2018 Volume 2018:11 Pages 4177—4187
DOI https://doi.org/10.2147/OTT.S172713
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Dr William C. Cho
Hua Ge, Chaojie Liang, Zhixia Li, Dali An, Shulin Ren, Chaosen Yue, Jixiang Wu
Department of General Surgery, Beijing Tongren Hospital, Capital Medical University, Beijing, China
Background: Decoy receptor 3 (DcR3) has been reported to be overexpressed in a wide variety of malignancies and is correlated with tumorigenesis and progression. In gastric cancer (GC), DcR3 overexpression is associated with lymph node and distant metastasis, as well as poor prognosis. However, the functional role of DcR3 expression in GC remains elusive.
Purpose: The aim of this study is to elucidate the direct role of DcR3 in regulating GC progression and metastasis and identify the potential mechanism.
Methods: DcR3 expression was stably knocked down in HGC27 and MKN28 cells by transfecting the cells with DcR3 shRNA using lentiviral vector system. After the knockdown of DcR3 was confirmed, cell proliferation, colony formation, cell cycle distribution, apoptosis, cell invasion and migration were assessed in vitro. In addition, Western blot analysis was performed to evaluate the expression of downstream mediators of DcR3. Comparisons between multiple groups were performed using one-way analysis of variance (ANOVA) or unpaired Student’s t-test. Differences were considered significant at P<0.05.
Results: Our findings demonstrate that DcR3 induces proliferation, migration, invasion, and promotes epithelial-mesenchymal transition (EMT) of GC cells. In addition, DcR3 increases the expression levels of several components of the PI3K/AKT/GSK-3β/β-catenin signaling pathway, such as p-AKT, GSK-3β, p-GSK-3β and β-catenin. Additionally, DcR3 also enhances the expression of N-cadherin and Vimentin and decreases the expression of E-cadherin.
Conclusion: In summary, the findings of this study indicate that during GC progression, DcR3 plays a key role in cell proliferation and invasion via the PI3K/AKT/GSK-3β/β-catenin signaling pathway. Thus, targeting DcR3 might be a potential therapeutic approach for the treatment of GC.
Keywords: DcR3, gastric cancer, epithelial–mesenchymal transition, metastasis
Introduction
Gastric cancer (GC) is one of the most common malignant neoplasms of the digestive system and fourth most common cancer worldwide.1 The fatality rate of GC is 75%, accounting for 8.8% of the total deaths from cancer in the world, especially in developing countries.1,2 Although progress has been made in the diagnosis, and treatment of GC by surgical techniques and chemotherapeutic regimens, the prognosis of patients with GC remains poor. In China, due to the nonspecific symptoms of early GC, numerous GC patients are diagnosed with lymph node or distant metastasis when they initially seek medical care. GC cells have a high potential for invasion and metastasis, which present a serious challenge to patients. Therefore, a better insight into the mechanisms of GC invasion and metastasis is essential.
Metastasis is responsible for as much as 90% of cancer-related deaths, and it remains the most poorly understood component of cancer pathogenesis.3 Epithelial–mesenchymal transition (EMT) is recognized as an important mechanism for cancer metastasis, whereby the epithelial cell morphology changes from an epithelial cobblestone phenotype to an elongated fibroblastic phenotype.4 The process of EMT involves the disassembly of cell–cell junctions, actin cytoskeleton reorganization and enhancement of cell motility and invasion.5 EMT is characterized by the loss of the cell–cell adhesion molecule E-cadherin, and the E-cadherin–catenin complex plays a key role in cellular adhesion.6,7 Therefore, decreasing the depletion of E-cadherin will be of great help to reverse the development of malignant tumors.
The soluble decoy receptor 3 (DcR3), also known as TR6, M68, or TNFRSF6B, is a member of the tumor necrosis factor receptor (TNFR) superfamily. DcR3 lacks the transmembrane domain and is believed to be a secreted protein.8 There are 3 known ligands of DcR3, namely, the Fas ligand (FasL), lymphotoxin analogs (LIGHT), and tumor necrosis factor-like ligand 1A (TL1A), and it is capable of neutralizing FasL-mediated apoptosis, LIGHT-mediated immunomodulation, and TL1A-induced antiangiogenesis.8–10 Although DcR3 is almost undetectable in most normal individuals, upregulation of DcR3 has been reported in a variety of cancer cells and tumor tissues.11 Recent studies reported that DcR3 is closely related with metastasis of various cancers.12,13 However, the specific mechanisms whereby DcR3 participates in the EMT process remain unclear. Therefore, in this study, we explored the role of DcR3 in tumorigenesis and EMT and established a theoretical basis for DcR3-targeted therapy for GC.
Materials and methods
Cell culture
The human GC cell lines AGS, MKN45, HGC27, and SGC7901 were purchased from the Cancer Hospital of the Chinese Academy of Medical Sciences (Beijing, China). The MKN28, SW480, and HT29 cell lines were purchased from the Cell Bank of Shanghai Institute of Biochemistry and Cell Biology (Shanghai, China). Cells were cultured in Roswell Park Memorial Institute (RPMI)-1640 containing 10% fetal bovine serum (FBS) (Gibco, Crand Island, NY, USA), 100 U/mL penicillin, and 100 μg/mL streptomycin (Invitrogen, Carlsbad, CA, USA) in a humidified atmosphere of 5% CO2 at 37°C.
Antibodies and reagents
Rat monoclonal antibodies against DcR3 and E-cadherin; rabbit monoclonal antibodies against GSK3β, p-GSK3β, β-catenin, Vimentin, and N-cadherin were purchased from Abcam (Cambridge, UK). Rat monoclonal antibodies against glyceraldehyde 3-phosphate dehydrogenase was purchased from Abways Biotechnology Co., Ltd. (Shanghai, China). Rabbit monoclonal antibodies against AKT and p-AKT were purchased from Affinity Biosciences (Cincinnati, OH, USA). LY294002 and insulin-like growth factor 1 (IGF-1) were purchased from Sigma-Aldrich (Saint Louis, MO, USA).
Lentiviral shRNA-mediated DcR3 silencing
DcR3 cDNA sequence was retrieved from GenBank, and 3 siRNA sequences were designed, namely, siRNA-1 (5′-GCGTGCCGTCATCGACTTTGT-3′), siRNA-2 (5′-GCAGTTCTGGAACTACCTAGA-3′), and siRNA-3 (5′-GCTGCCGCACCGGCTTCTTCG-3′). A negative control sequence, si-NC (5′-TTCTCCGAACGTGTCACGT-3′) was also designed and synthesized. Target shDcR3 was cloned into the pGMLV-SC5 vector. Transfection was carried out with Lipofectamine LTX reagent (Invitrogen) and Opti-MEM media, according to the manufacturer’s instructions. Successfully transfected cells were then selected with puromycin (1 mg/L) for 2 weeks. Stable knockdown of DcR3 in the selected cells was confirmed using the reverse transcriptase polymerase chain reaction (RT-PCR) and Western blotting. siRNA with the highest interference efficiency was used in the subsequent experiments.
Enzyme-linked immunosorbent assay (ELISA) and RT-PCR analysis
The supernatants derived from cell culture media were analyzed by sandwich ELISA using a DcR3 ELISA Kit (R&D Systems, Minneapolis, MN, USA) according to the manufacturer’s instructions. Total RNA was extracted from cells using Trizol reagent (Takara, Tokyo, Japan), according to the manufacturer’s instructions; cDNA was synthesized from total RNAs using Prime Script RT Reagent Kit (Takara). RT-PCR analysis was performed using the Eppendorf PCR system (Eppendorf AG Hamburg, Germany). Relative quantification was determined by normalization to the amount of β-actin. The DcR3 primers were as follows: 5′-CTCAATGTGCCAGGCTCTTC-3′ and 3′-GAAAGCCACAAAGTCGATGA-5′. The β-actin primers were 5′-GATGAGATTGGCATGGCTTT-3′ and 3′-GTCACCTTCACCGTTCCAGT-5′. Expression levels of target-of-interest were calculated as relative expression using the 2−ΔΔCT method.
Cell viability assay and colony formation assay
Cells from 3 groups were seeded in 96-well plates (1×103/well) with 100 μL medium. Cell proliferation was assessed using the Cell Counting Kit-8 (CCK-8) (Dojindo Laboratories, Kumanoto, Japan) assay, according to the manufacturer’s protocols. A total of 10 μL of CCK-8 solution was added to each well and incubated for 2 hours in 5% CO2 at 37°C. Then, the absorbance at 450 nm was measured. The cell proliferation assay was performed on days 1, 2, 3, 4, and 5. DcR3 siRNA, si-NC, and control cells were seeded into 6-well plates at a cell density of 500 cells/well, and cells were allowed to grow for 14 days in RPMI-1640 supplemented with 10% FBS. PBS washed cells were fixed with 4% paraformaldehyde and then stained with 1% crystal violet. Colonies containing over 50 cells were counted. All experiments were performed in triplicate.
Transwell assay and wound healing assay
Cell invasion was measured using Transwell chambers (8 μm pore size; Costar) with Matrigel (BD Biosciences, San Jose, CA, USA). Briefly, cell suspensions (1×105 cells) prepared in serum-free medium were added into the upper chambers of the transwells. The matched lower chamber contained 600 μL of 10% FBS medium, which served as a chemo-attractant. After incubation for 24 hours, the cells remaining on the upper chamber were removed with cotton tips, while those that migrated onto the lower surface of the membrane were fixed with formaldehyde and stained with hematoxylin for 30 minutes. Then, the cells that migrated to the basal portion of the membrane in the lower compartment of the chamber were counted under the microscope in 5 randomly selected fields at 200× magnification. For the wound healing assay, cells were seeded into 6-well plates at a density of 1×105/mL and cultured. Once confluence was reached, a wound was then made along the center of each well by scratching the cell layer with the tip of a 200-μL pipette. After washing away the detached cells with PBS, the remaining cells were cultured in medium under standard condition. Images were taken at 0 and 24 hours to assess cell migration into the wound. Each experiment was performed in triplicate.
Apoptosis and cell cycle analysis
Apoptotic changes were detected by fluorescein isothiocyanate (FITC)-Annexin V staining. Briefly, cells were double-stained with Annexin V-FITC and propidium iodide (PI) and incubated at room temperature for 15 minutes in the dark. Then, the stained cells were analyzed by flow cytometer using a BD Accuri C6 flow cytometer system (BD Biosciences). The percentage of Annexin V-positive and PI-positive cells was compared among the different groups. The data were processed using the CellQuest software (BD Biosciences). For cell cycle analysis, cells were harvested and fixed using 75% ethanol overnight at 4°C, then cells were incubated with RNase A for 30 minutes at 37°C and stained with PI. Cell cycle analysis was performed by flow cytometry.
Western blotting
Whole-cell extract was prepared by scraping and suspending the cells in radioimmunoprecipitation assay buffer (Beyotime Institute of Biotechnology, Haimen, China) and the protein concentrations was quantified using the BCA Kit (Cwbiotech, Beijing, China). Equal amounts of protein were separated by 10% sodium dodecyl sulfate-polyacrylamide gel electrophoresis and then transferred to polyvinylidene fluoride membranes (EMD Millipore, Billerica, MA, USA). Non-specific protein interactions were blocked by incubation with 5% non-fat milk in tris-buffered saline with Tween-20 (TBST) for 1 hour. Then, the membranes were incubated with the appropriate individual primary antibody overnight at 4°C. After washing 3 times with 1× TBST, the membranes were incubated at room temperature for 50 minutes with the appropriate secondary antibody to detect binding of the primary antibody. Eventually, the expression signal was detected using an enhanced chemiluminescence system (EMD Millipore).
Statistical analysis
Statistical analyses were performed using the GraphPad Prism 6 software (GraphPad Software Inc., La Jolla, CA, USA) and SPSS 22.0 software (IBM, Armonk, NY, USA). All experiments were performed in triplicate, the data is represented as the mean ± SD. The one-way analysis of variance test or unpaired Student’s t-test was used to perform comparison among all groups. Differences were considered significant at P<0.05.
Results
DcR3 expression in various GC cell lines
Before we studied the functional role of DcR3 in GC cells, we first measured the expression of DcR3 in 5 different GC cell lines, namely, AGS, HGC27, MKN28, MKN45, and SGC7901. As shown in Figure 1A, the ELISA and RT-PCR analysis results revealed that the DcR3 expression was increased to a different extent in the 5 GC cell lines. The human colon cancer cell lines HT29 and SW480 were used as the negative control and the positive control for DcR3 expression, respectively. Compared with the negative control cell line HT29, MKN28 cells had the highest DcR3 expression level, while AGS cells had the lowest. Based on these findings, we chose the HGC27 and MKN28 cell lines for further investigation as they were the top DcR3 producers.
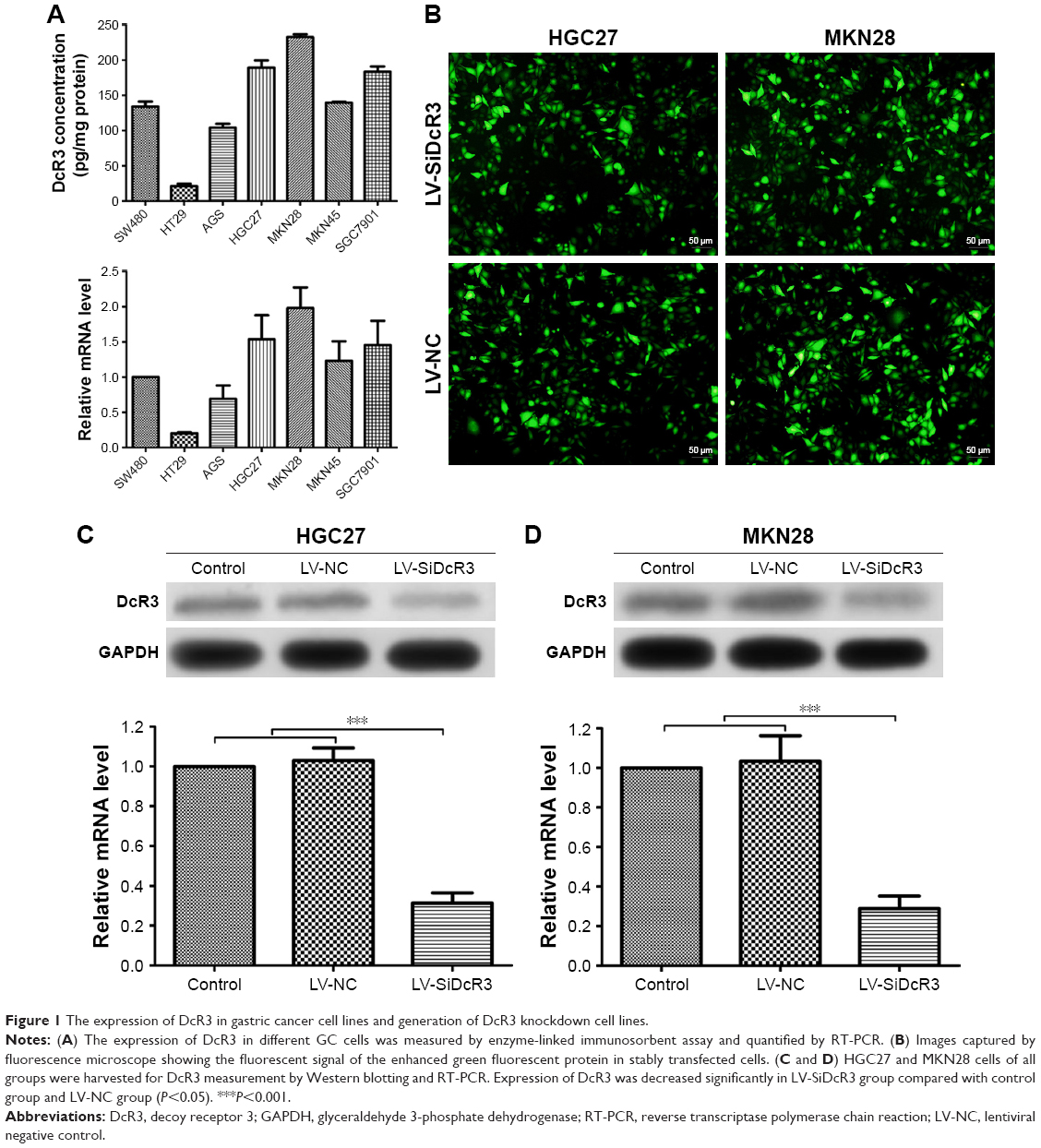 | Figure 1 The expression of DcR3 in gastric cancer cell lines and generation of DcR3 knockdown cell lines. |
Generation of DcR3 knockdown cell lines
We stably knocked down DcR3 expression in HGC27 and MKN28 cells by transfecting the cells with DcR3 shRNA using lentiviral vector system. The plasmid contained a reporter-enhanced green fluorescent protein (EGFP) so that the transfection efficiency could be evaluated using fluorescent microscopy. As shown in Figure 1B, positive EGFP signal could be detected in the LV-NC and LV-SiDcR3 groups, indicating the successful transfection of the plasmids into the target cells by the lentivirus. Western blotting and RT-PCR analysis further confirmed that both the DcR3 protein and gene expression were significantly downregulated in the LV-SiDcR3 group compared to the control and LV-NC groups, and whose differences were statistically significant (P<0.05) (Figure 1C and D).
Effects of DcR3 knockdown on the proliferation and colony formation of HGC27 and MKN28 cells
As shown in Figure 2A, according to the growth curves, the cell proliferation was significantly inhibited in the DcR3 knockdown groups compared with the control and LV-NC groups (P<0.05). In addition, the colony formation assays indicate that cells in the LV-SiDcR3 groups formed fewer colonies compared with other groups (P<0.05). These findings suggest that inhibition of DcR3 expression can restrain GC cell proliferation.
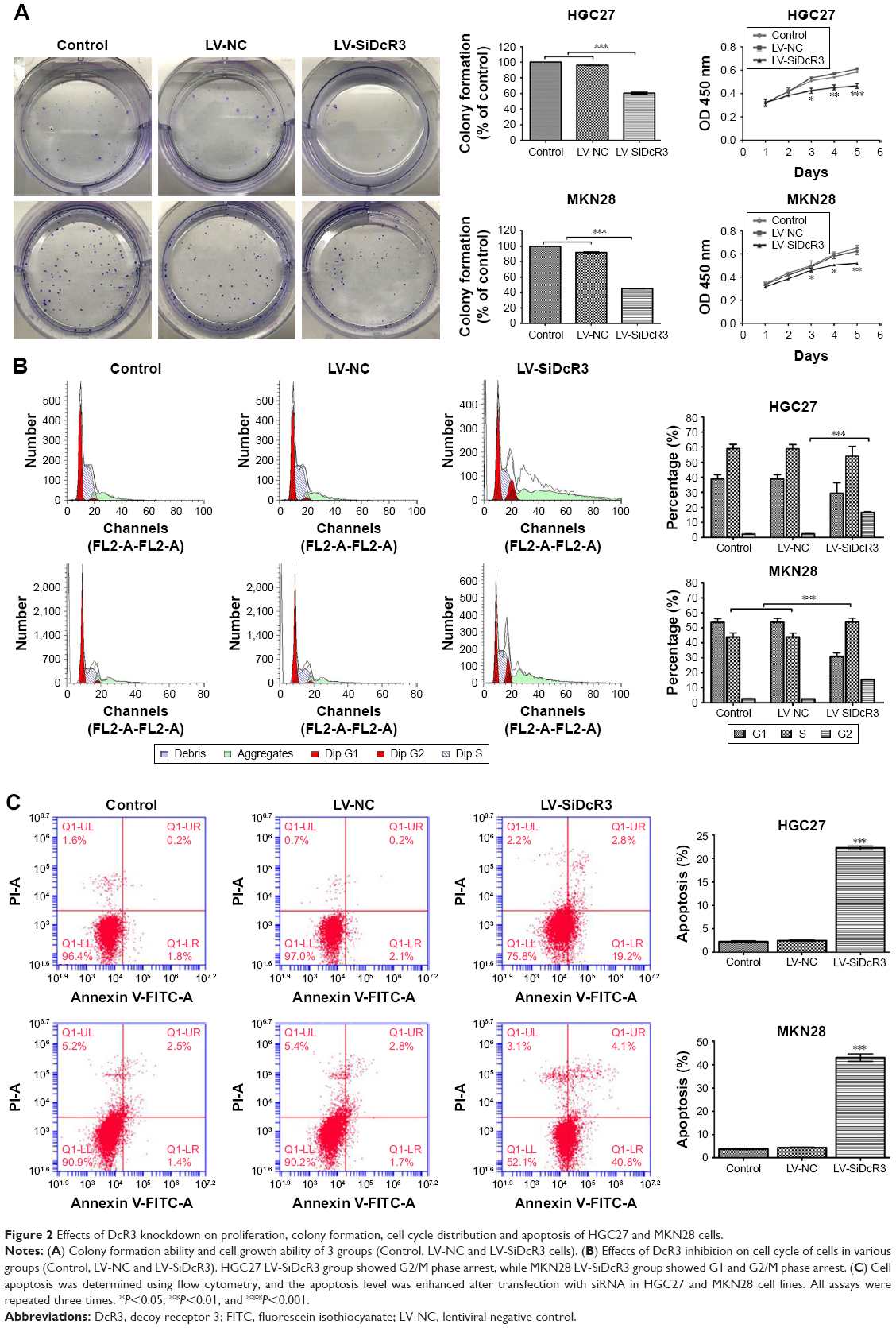 | Figure 2 Effects of DcR3 knockdown on proliferation, colony formation, cell cycle distribution and apoptosis of HGC27 and MKN28 cells. |
Effects of DcR3 knockdown on the cell cycle distribution and apoptosis of HGC27 and MKN28 cells
The results reveal that inhibiting DcR3 impairs the cell cycle and apoptosis in HGC27 and MKN28 cells compared with control cells. For instance, as shown in Figure 2B, the proportion of G2/M phase cells was increased in the HGC27 LV-SiDcR3 groups, indicating a possible G2/M phase arrest (P<0.05); in addition, increased proportion of G1 and G2/M phase cells were observed in the MKN28 LV-SiDcR3 groups (P<0.05) (Figure 2B). Also, as shown in Figure 2C, the downregulation of the DcR3 expression significantly increased cell apoptosis in HGC27 and MKN28 cells compared with the negative control cells (P<0.05). Taken together, these results demonstrate that the loss of DcR3 induced cell cycle distribution and apoptosis in GC cells.
Effects of DcR3 on cell viability
To further investigate the effects of DcR3 on cell viability, the wound healing assay and Transwell assay were conducted. The results revealed that cell migration was significantly inhibited when the cells were treated with siRNA compared with the control and LV-NC in HGC27 and MKN28 cells (P<0.05) (Figure 3A and C). Also, as shown in Figure 3B and D, cell invasion and migration were significantly inhibited in the LV-SiDcR3 groups (P<0.05).
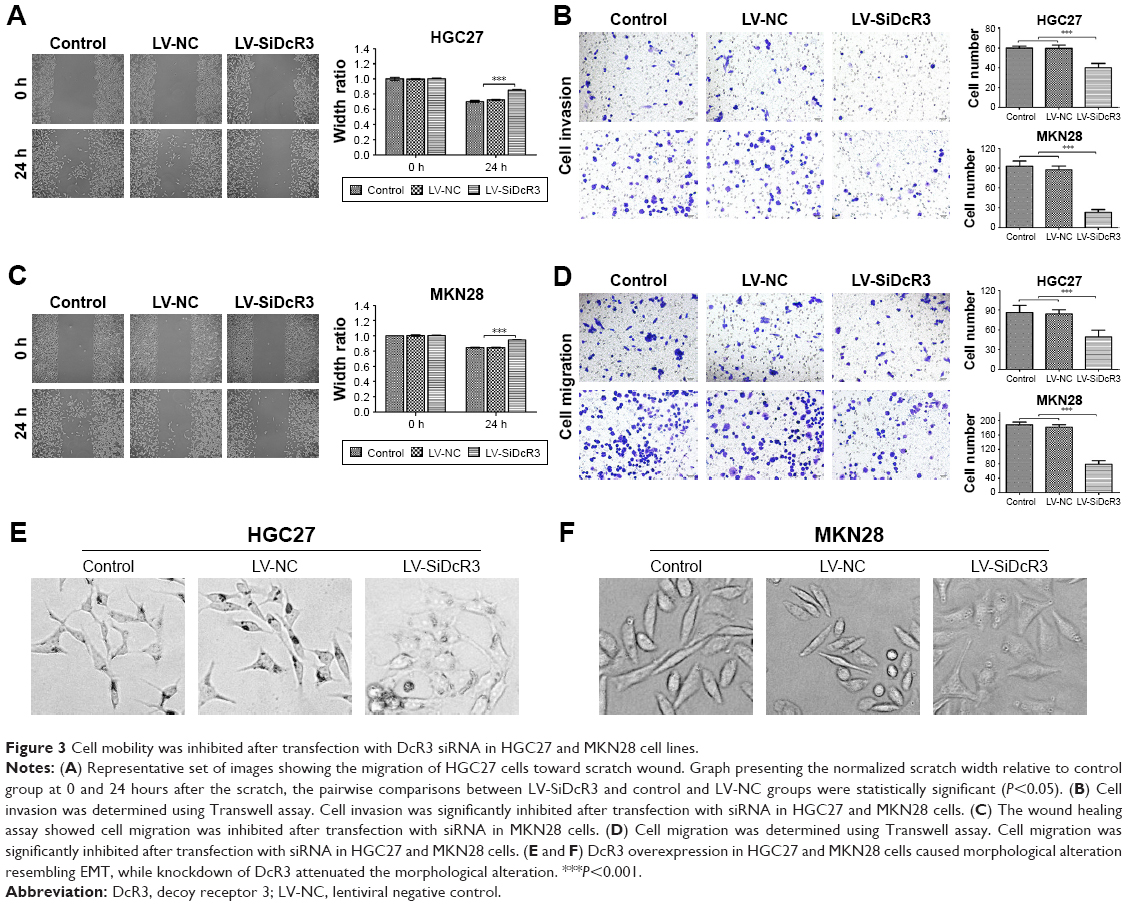 | Figure 3 Cell mobility was inhibited after transfection with DcR3 siRNA in HGC27 and MKN28 cell lines. |
DcR3 facilitated EMT of GC cells via the PI3K/AKT/GSK-3β/β-catenin signaling pathway
As shown in Figure 3E and F, DcR3 overexpression altered the HGC27 and MKN28 cell morphology, such that both cell lines exhibited a spindle-like, fibroblastic cell morphology, whereas DcR3 inhibition partially reversed the morphological changes. We also examined changes of EMT markers after DcR3 inhibition in GC cells. The result showed that the expression level of the epithelial-like cell marker E-cadherin was upregulated, and the expression level of the interstitial-like cell molecular markers N-cadherin and Vimentin were downregulated (Figure 4). To elucidate the possible mechanism whereby the knockdown of DcR3 suppresses GC cell proliferation, migration, and invasion, we analyzed the molecular expression of the PI3K/AKT/GSK-3β/β-catenin signaling pathway in DcR3 knockdown and control GC cells. The results revealed that the knockdown of DcR3 decreased the protein levels of p-AKT, GSK-3β, p-GSK-3β, and β-catenin in HGC27 and MKN28 cells (Figure 4).
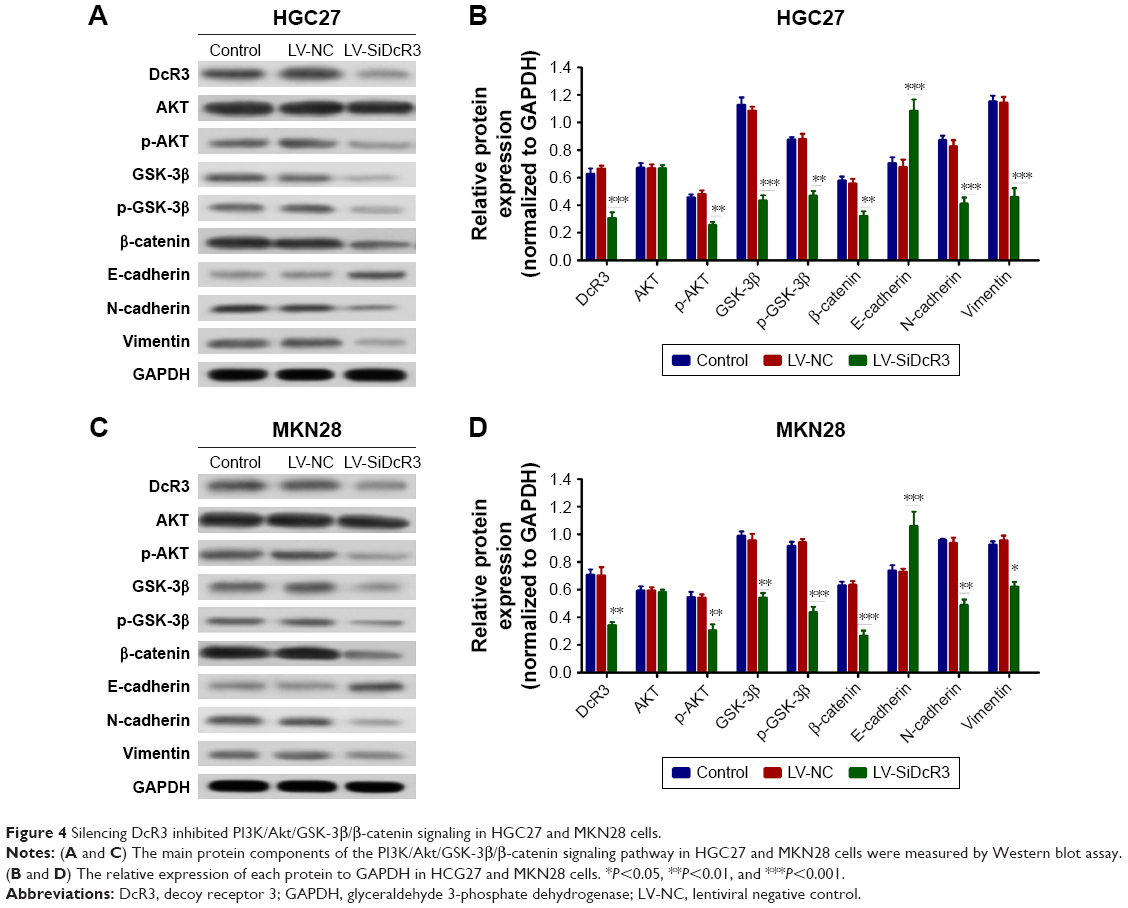 | Figure 4 Silencing DcR3 inhibited PI3K/Akt/GSK-3β/β-catenin signaling in HGC27 and MKN28 cells. |
To confirm the importance of DcR3 on PI3K/AKT/GSK-3β/β-catenin signaling, the HGC27 and MKN28 cells were treated with PI3K inhibitor LY294002 (50 μM) for 24 hours, and dimethyl sulfoxide was used as control. As shown in Figure 5A and C, inhibition of PI3K signaling restored E-cadherin expression and attenuated N-cadherin, p-GSK-3β, and β-catenin expression in both the HGC27 and MKN28 cells, which is similar to the effects of DcR3 knockdown. In contrast, treatment of the HGC27 and MKN28 DcR3 knockdown cells with the PI3K signaling activator IGF-1 (100 ng/mL) for 24 hours, the results showed that IGF-1 reversed the inhibitory effects of DcR3 silencing (Figure 5B and D).
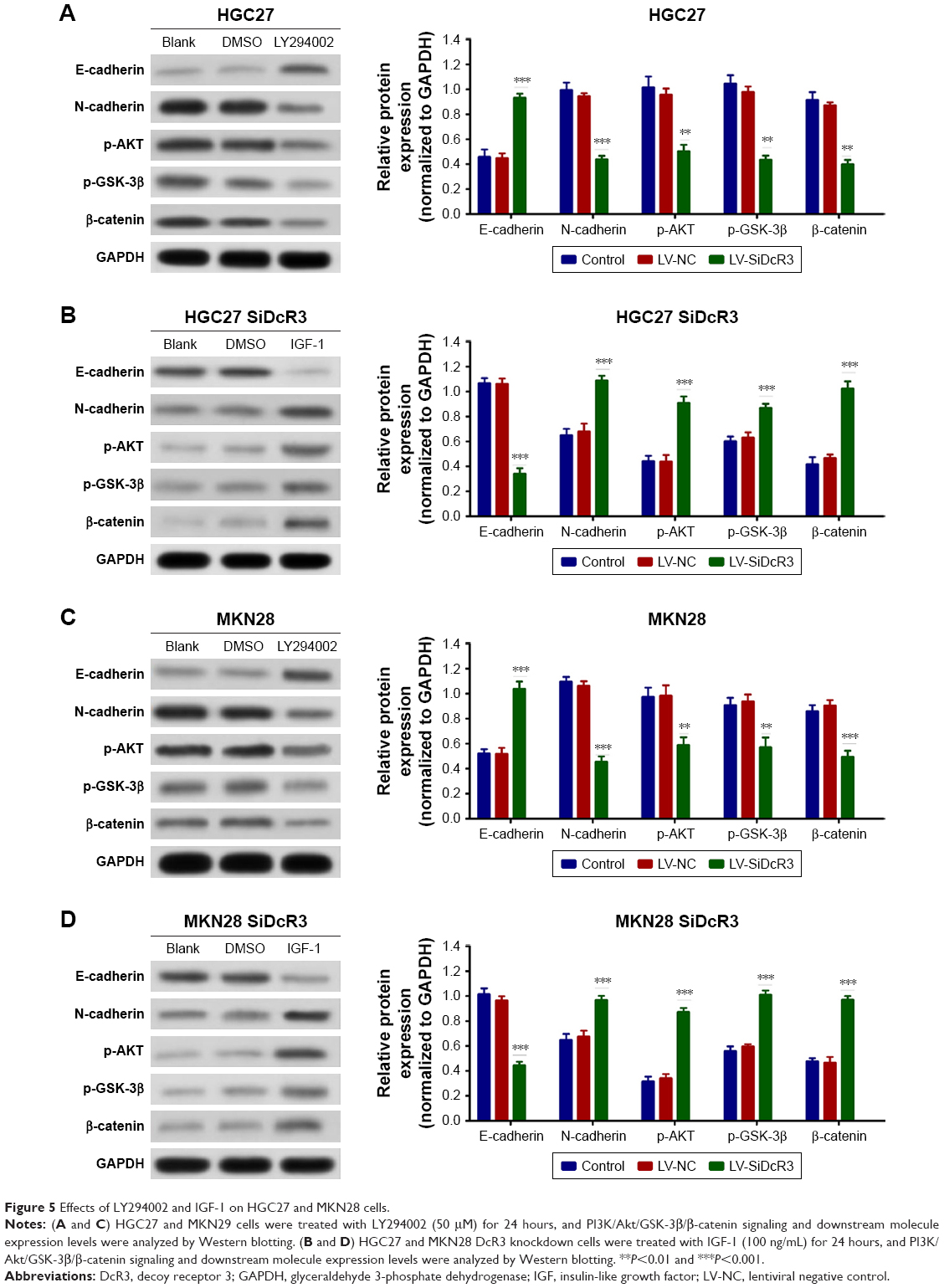 | Figure 5 Effects of LY294002 and IGF-1 on HGC27 and MKN28 cells. |
Taken together, the aforementioned findings demonstrate that DcR3 indeed reduces GC cell proliferation, invasion, and EMT by targeting the PI3K/AKT/GSK-3β/β-catenin signaling pathway.
Discussion
DcR3 was first discovered in 1998 in human lung and colon tumors as a decoy receptor, which binds to FasL and inhibits FasL-mediated apoptosis.8 Accumulating evidence suggests that DcR3 is overexpressed in human malignant tumors, such as stomach, colon, liver, breast, and bladder cancers.14–18 In addition, it has been reported that a high level of DcR3 was correlated with cancer risk, tumor differentiation, lymph node metastasis, advanced TNM stage, and poor survival.19–21 Our previous studies found that the expression of DcR3 in colon cancer and hepatocellular carcinoma (HCC) cell lines was significantly higher than in normal cells, while downregulation of DcR3 inhibited the malignant property of tumor cells and enhanced TRAIL-mediated apoptosis in HCC.22–24 However, the effect of DcR3 on GC cells has not yet been fully elucidated. In this study, we showed that DcR3 silencing significantly attenuates GC cell proliferation, migration and invasion capability. In addition, loss of DcR3 disrupts the cell cycle and induces apoptosis in these cells. As mentioned previously, DcR3 overexpression has been observed in various cancers, indicating that DcR3 upregulation might be a common tumor characteristic. Therefore, treatments targeting DcR3 may have the potential to serve as an anticancer therapy for GC.
Though the roles of DcR3 in GC cell proliferation, cell cycle distribution, and apoptosis have been investigated in the present study, the roles of this protein in other aspects relevant to GC are relatively unknown. Tumor metastasis is facilitated by various mechanisms. For instance, EMT is associated with invasion and metastasis of cancer cells.25,26 Indeed, we found that inhibition of DcR3 attenuates the morphological alteration of EMT in HGC27 and MKN28 cells. Moreover, DcR3 knockdown increases E-cadherin expression and decreases N-cadherin expression. It has also been reported that DcR3 regulates tumor metastasis by mediating the EMT of colorectal cancer and HCC cells, which is consistent with our findings.12,13 Together with our results, these observations indicate that DcR3 facilitates the occurrence of EMT.
The P13K/AKT signaling pathway is one of the most important signal transduction pathways in cells. It plays an important role in the regulation of biological processes, such as inhibition of apoptosis and induction of cell proliferation by activating downstream molecules. GSK-3β plays an important role in β-catenin phosphorylation and degradation and thus prevents β-catenin from translocating into the nucleus, in which it forms a complex with the TCF transcription factor family members to enhance β-catenin-responsive gene expression, such as E-cadherin.27 Furthermore, GSK-3β activity is inhibited by AKT phosphorylation at Ser9. In recent years, it has been documented that PI3K/AKT signaling components are frequently altered in human cancers.28 To further investigate the possible mechanisms whereby DcR3 participates in GC, we evaluated the relationship between DcR3 and the P13K/AKT/GSK-3β/β-catenin signaling pathway in HGC27 and MKN28 cells. As previously reported, DcR3 expression enhances the activity of P13K/AKT/GSK-3β/β-catenin signaling, whereas DcR3 inhibition has the opposite effect. To confirm the role of DcR3 in the PI3K/AKT/GSK-3β/β-catenin signaling pathway, we treated GC cells with the PI3K inhibitor LY294002, while the DcR3 knockdown cells were treated with the PI3K activator IGF-1. The result demonstrates that PI3K/AKT/GSK-3β/β-catenin signaling is involved in a DcR3-dependent mechanism.
Consistent with our findings, it has been reported that a DcR3-specifc siRNA efficiently inhibited cell growth and induced cell apoptosis of cells via the PI3K/AKT pathway in glioma and malignant fibrous histiocytoma cells.29,30 However, in pancreatic adenocarcinoma, Chen and Yang reported that the PI3K/AKT signaling was involved in the modulation of endogenous DcR3 expression.31 Another study linked the Epstein–Barr virus transcription activator Rta to PI3K/AKT signaling and increased DcR3 expression.32 Apparently, our findings are contrary to these findings. However, the discrepancy among different studies can be explained in the following aspects. A possible reason is that the differences in the findings are associated with the different cancer types. In addition, DcR3 also has “non-decoy” functions and acts as an effector molecule to modulate the activities of many cell types directly.11 Moreover, there might be a mechanism for feedback regulation between DcR3 and PI3K/AKT signaling. Without doubt, this needs further validation.
Conclusion
In summary, our study demonstrates that DcR3 enhances the malignant phenotypes of GC cells and promotes EMT and invasion in different GC cell lines through the PI3K/AKT/GSK-3β/β-catenin signaling pathway. These results provide insights into the mechanism of action of DcR3 in GC and suggest that DcR3 can be useful as a potential therapeutic target in GC.
Acknowledgment
This study was funded by the Beijing Municipal Administration of Hospital Clinical Medicine Development of Special Funding Support (ZYLX201612).
Disclosure
The authors report no conflicts of interest in this work.
References
Jemal A, Bray F, Center MM, Ferlay J, Ward E, Forman D. Global cancer statistics. CA Cancer J Clin. 2011;61(2):69–90. | ||
Fock KM. Review article: the epidemiology and prevention of gastric cancer. Aliment Pharmacol Ther. 2014;40(3):250–260. | ||
Chaffer CL, Weinberg RA. A perspective on cancer cell metastasis. Science. 2011;331(6024):1559–1564. | ||
Thiery JP, Sleeman JP. Complex networks orchestrate epithelial-mesenchymal transitions. Nat Rev Mol Cell Biol. 2006;7(2):131–142. | ||
Kong D, Li Y, Wang Z, Sarkar FH. Cancer stem cells and epithelial-to-mesenchymal transition (EMT)-phenotypic cells: are they cousins or twins? Cancers. 2011;3(1):716–729. | ||
Prieto-García E, Díaz-García CV, García-Ruiz I, Agulló-Ortuño MT. Epithelial-to-mesenchymal transition in tumor progression. Med Oncol. 2017;34:122. | ||
Benham-Pyle BW, Pruitt BL, Nelson WJ. Cell adhesion. Mechanical strain induces E-cadherin-dependent Yap1 and β-catenin activation to drive cell cycle entry. Science. 2015;348(6238):1024–1027. | ||
Pitti RM, Marsters SA, Lawrence DA, et al. Genomic amplification of a decoy receptor for Fas ligand in lung and colon cancer. Nature. 1998;396(6712):699–703. | ||
Yu KY, Kwon B, Ni J, Zhai Y, Ebner R, Kwon BS. A newly identified member of tumor necrosis factor receptor superfamily (TR6) suppresses LIGHT-mediated apoptosis. J Biol Chem. 1999;274(20):13733–13736. | ||
Migone TS, Zhang J, Luo X, et al. TL1A is a TNF-like ligand for DR3 and TR6/DcR3 and functions as a T cell costimulator. Immunity. 2002;16(3):479–492. | ||
Lin WW, Hsieh SL. Decoy receptor 3: a pleiotropic immunomodulator and biomarker for inflammatory diseases, autoimmune diseases and cancer. Biochem Pharmacol. 2011;81(7):838–847. | ||
Liu YP, Zhu HF, Liu DL, et al. DcR3 induces epithelial-mesenchymal transition through activation of the TGF-β3/SMAD signaling pathway in CRC. Oncotarget. 2016;7(47):77306–77318. | ||
Zhang H, Chen X, Li D, et al. DcR3 promotes hepatoma cell migration by downregulating E-cadherin expression. Oncol Rep. 2017;38(1):377–383. | ||
Wu Y, Guo E, Yu J, Xie Q. High DcR3 expression predicts stage pN2-3 in gastric cancer. Am J Clin Oncol. 2008;31(1):79–83. | ||
Liang QL, Wang BR, Li GH, Gh L. DcR3 and survivin are highly expressed in colorectal carcinoma and closely correlated to its clinicopathologic parameters. J Zhejiang Univ Sci B. 2009;10(9):675–682. | ||
Chen C, Zhang C, Zhuang G, et al. Decoy receptor 3 overexpression and immunologic tolerance in hepatocellular carcinoma (HCC) development. Cancer Invest. 2008;26(10):965–974. | ||
Wu Q, Zheng Y, Chen D, Li X, Lu C, Zhang Z. Aberrant expression of decoy receptor 3 in human breast cancer: relevance to lymphangiogenesis. J Surg Res. 2014;188(2):459–465. | ||
Yamana K, Bilim V, Hara N, et al. Prognostic impact of FAS/CD95/APO-1 in urothelial cancers: decreased expression of Fas is associated with disease progression. Br J Cancer. 2005;93(5):544–551. | ||
Tong J, Ao R, Wang Y, Chang B, Wang BY. Prognostic and clinicopathological differences of DcR3 in gastrointestinal cancer: evidence from meta-analysis. Int J Clin Exp Med. 2014;7(9):3096–3105. | ||
Jiang M, Lin X, He R, et al. Decoy receptor 3 (DcR3) as a biomarker of tumor deterioration in female reproductive cancers: a meta-analysis. Med Sci Monit. 2016;22:1850–1857. | ||
Ge H, Liang C, Ren S, Yue C, Wu J. Prognostic value of DcR3 in solid tumors: a meta-analysis. Clin Chim Acta. 2018;481:126–131. | ||
Yu W, Xu YC, Tao Y, et al. DcR3 regulates the growth and metastatic potential of SW480 colon cancer cells. Oncol Rep. 2013;30(6):2741–2748. | ||
Zhou XN, Li GM, Xu YC, Zhao TJ, Wu JX. Knockdown of decoy receptor 3 impairs growth and invasiveness of hepatocellular carcinoma cell line of HepG2. Chin Med J. 2016;129(21):2623–2629. | ||
Liang C, Xu Y, Li G, et al. Downregulation of DcR3 sensitizes hepatocellular carcinoma cells to TRAIL-induced apoptosis. Onco Targets Ther. 2017;10:417–428. | ||
Arvelo F, Sojo F, Cotte C. Tumour progression and metastasis. Ecancermedicalscience. 2016;10:617. | ||
Thompson EW, Newgreen DF, Tarin D. Carcinoma invasion and metastasis: a role for epithelial-mesenchymal transition? Cancer Res. 2005;65(14):5991–5995. | ||
Macdonald BT, Tamai K, He X. Wnt/beta-catenin signaling: components, mechanisms, and diseases. Dev Cell. 2009;17(1):9–26. | ||
Martini M, de Santis MC, Braccini L, Gulluni F, Hirsch E. PI3K/AKT signaling pathway and cancer: an updated review. Ann Med. 2014;46(6):372–383. | ||
Zhang Y, Huang S, Leng Y, et al. Effect of DcR3-specific siRNA on cell growth suppression and apoptosis induction in glioma cells via affecting ERK and AKT. Onco Targets Ther. 2016;9:5195–5202. | ||
Toda M, Kawamoto T, Ueha T, et al. ‘Decoy’ and ‘non-decoy’ functions of DcR3 promote malignant potential in human malignant fibrous histiocytoma cells. Int J Oncol. 2013;43(3):703–712. | ||
Chen PH, Yang CR. Decoy receptor 3 expression in AsPC-1 human pancreatic adenocarcinoma cells via the phosphatidylinositol 3-kinase-, Akt-, and NF-kappa B-dependent pathway. J Immunol. 2008;181(12):8441–8449. | ||
Ho CH, Hsu CF, Fong PF, et al. Epstein-Barr virus transcription activator Rta upregulates decoy receptor 3 expression by binding to its promoter. J Virol. 2007;81(9):4837–4847. |
 © 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.