Back to Journals » Research Reports in Clinical Cardiology » Volume 12
Dasatinib-Induced Pulmonary Arterial Hypertension: A Case Report
Authors Liu R, Tang Y, Fu T, Zhou J, Ma L, Yuan J ![]() , Xu O
, Xu O
Received 22 April 2021
Accepted for publication 14 July 2021
Published 10 August 2021 Volume 2021:12 Pages 33—39
DOI https://doi.org/10.2147/RRCC.S316980
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Richard Kones
Ru Liu,1,2 Yinjiang Tang,2 Ting Fu,2 Jianli Zhou,2 Lijiao Ma,2 Jinqing Yuan,1 Ou Xu2
1Department of Cardiology, Fuwai Hospital, Chinese Academy of Medical Sciences, Beijing, People’s Republic of China; 2Department of Pulmonary Vascular and General Medicine, Fuwai Yunnan Cardiovascular Hospital, Yunnan Provincial Cardiovascular Disease Clinical Medical Center, Kunming, Yunnan Province, People’s Republic of China
Correspondence: Ou Xu
Fuwai Yunnan Cardiovascular Hospital, Yunnan Provincial Cardiovascular Disease Clinical Medical Center, No. 528 North Shahe Road, Wuhua District, Kunming, 650102, People’s Republic of China
Tel +86-10-15901098523
Email [email protected]
Jinqing Yuan
Fuwai Hospital, Chinese Academy of Medical Sciences, No. 167 North Lishi Road, Xicheng District, Beijing, 100037, People’s Republic of China
Tel +86-10-88322457
Email [email protected]
Abstract: Dasatinib was identified to be associated with pulmonary arterial hypertension, also called dasatinib-induced pulmonary arterial hypertension. There was still little data on clinical characteristics of this rare but severe complication during hematological therapy in China. A 40-year-old female with worsening dyspnoea was diagnosed with severe pulmonary arterial hypertension and progressive right heart failure, and stabilized by standardized comprehensive management, including replacement of other tyrosine kinase inhibitor, administration of pulmonary vasodilators according to the right-heart catheterization-based risk stratification and traditional anti-heart failure drug therapy. Our case suggests that dasatinib-induced pulmonary arterial hypertension may be partially reversible, with a relatively good prognosis under formal target therapy of pulmonary arterial hypertension, providing new insight into its treatment algorithm.
Keywords: dasatinib-induced pulmonary arterial hypertension, right heart failure, pulmonary vasodilator, tyrosine kinase inhibitor
Background
Dasatinib, a second generation tyrosine kinase inhibitor (TKI), was approved as a first-line therapy for chronic myelocytic leukemia (CML). Since the introduction of dasatinib earlier this decade, more than 100 cases of dasatinib-induced pulmonary arterial hypertension (DASA-PAH) have been reported in Europe.1 Some of these reported cases were presented as a reversible course, while others not.2–4 The underlying pathogenesis is still unclear, as well as the predisposing factors.5 Herein, we present a case of severe PAH that developed during dasatinib treatment. The patient was successfully managed with stopping dasatinib (replacement of other TKI), administration of target combination therapy for PAH and comprehensive anti-heart failure therapy and management.
Case Presentation
A 40-year-old woman presented to our department with exertional dyspnea, accompanied by intermittent cough, cough sputum, edema, fatigue, abdominal distension, poor appetite. She had a 10.5-month history of exertional dyspnea that had worsened over the previous 20 days. Her shortness of breath was obvious at rest. She had been diagnosed with CML at the age of 29, for which a first-generation TKI, imatinib (400mg daily), was prescribed as her first-line therapy for 5 years. However, since thrombocytopenia was found, it was replaced by nilotinib (400 mg b.i.d.) for 2 years. And then nilotinib was also withdrawn due to thrombocytopenia and hyperbilirubinemia. A second-generation TKI, dasatinib (60 mg daily), was chosen 4 years before her initial presentation (Table 1). Concomitant pleural effusion was thought to have caused the exertional dyspnea. Subsequently, her symptoms were partially relieved by diuretics, repeated pleural puncture and corticosteroids in basic-level hospitals. However, probably due to the lack of understanding of DASA-PAH in primary hospitals, dasatinib was replaced by nilotinib (300 mg qd8, 150 mg qd16) 5 months after the initial presentation. She went to the clinic of our center 10 months after the initial presentation. At that time, transthoracic echocardiography (TTE) showed a very severe pulmonary hypertension, with estimated pulmonary arterial systolic pressure (PASP) as high as 116 mmHg and a marked enlargement of right ventricle, accordingly a compression of left ventricle. Bosentan (125 mg b.i.d.) initiated at the clinic. In the following 2 weeks, she reduced the drug by herself, and symptoms did not improve significantly. She was admitted 10.5 months after the initial presentation.
 |
Table 1 Timeline of History of Tyrosine Kinase Inhibitor Application and Pulmonary Arterial Hypertension |
Electrocardiography (Figure 1) and TTE (Figure 2A, Table 2) on admission indicated severe right ventricular pressure overload. Physical examination showed jugular vein dilatation, yellowing sclera and lip cyanosis. Right lung percussion dulled. Left lung respiratory sound decreased and right lung respiratory sound disappeared. No obvious dry or wet rales were heard. Cardiac auscultation revealed P2 hyperactivity and a systolic murmur on the left sternum 4–5 intercostal, grading 3/6 level. Liver could be reached, and severe pitting edema of both lower extremities was found. Laboratory data showed markedly elevated N-terminal of the prohormone brain natriuretic peptide (NT-proBNP) (10648.0 pg/mL; normal reference value, <125 pg/mL), and anti-nuclear and anti-centromere antibody negative. X-ray and chest computed tomography (CT) scan showed large pleural effusion on the right chest and small on the left (Figure 3). No evidence of pulmonary embolism showed by contrast enhanced CT scan for pulmonary arteries and perfusion scintigraphy. Abdominal ultrasonography showed no evidence of portal hypertension.
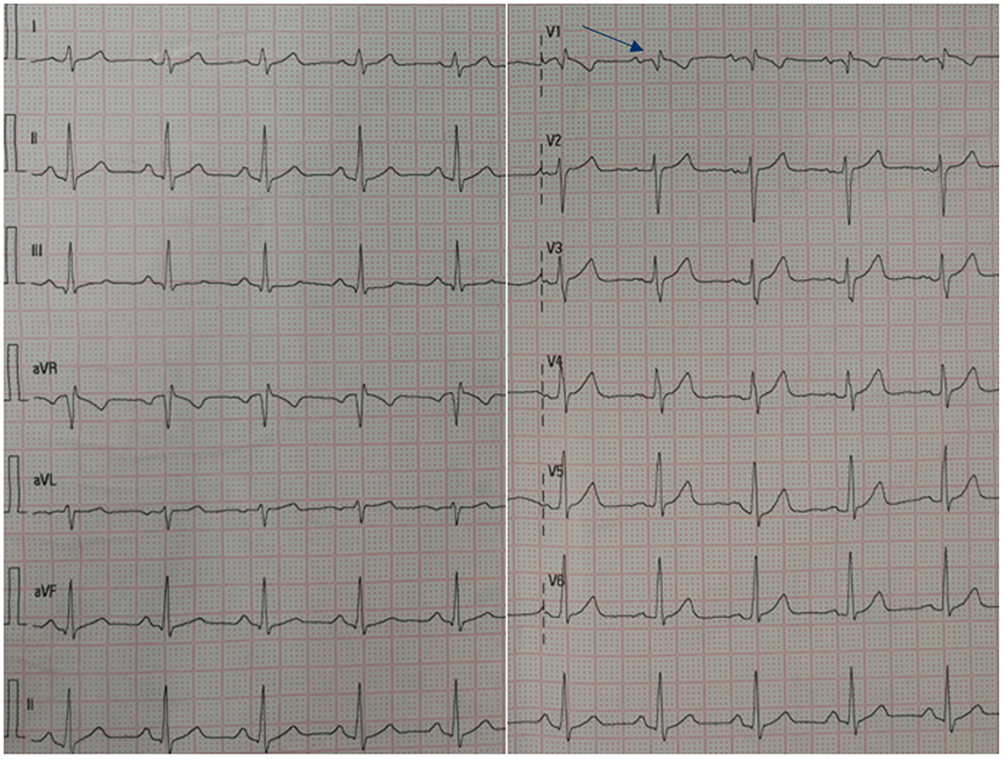 |
Figure 1 Electrocardiogram findings at the first admission. The arrow on electrocardiogram revealed an incomplete right bundle branch block (an R/S ratio >1 in lead V1). |
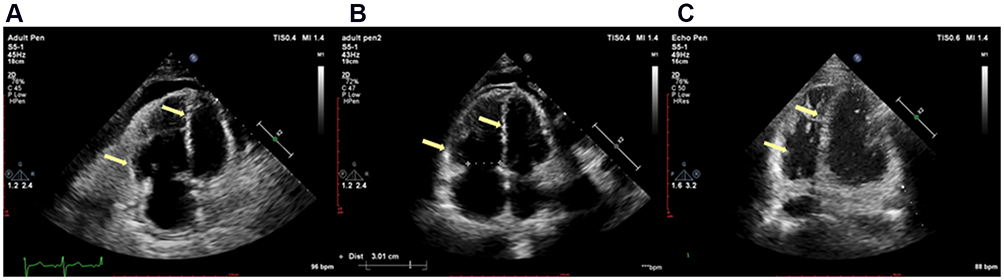 |
Figure 2 Transthoracic echocardiography. The four cavity heart section views are shown at (A) the first admission, (B) 5 months after discharge (10 months after dasatinib withdrawal) and (C) 12 months after discharge (17 months after dasatinib withdrawal). The arrows showed that enlargement of right ventricle and atrium was gradually restored, as well as the left transposition of the septum and compression of the left ventricle. |
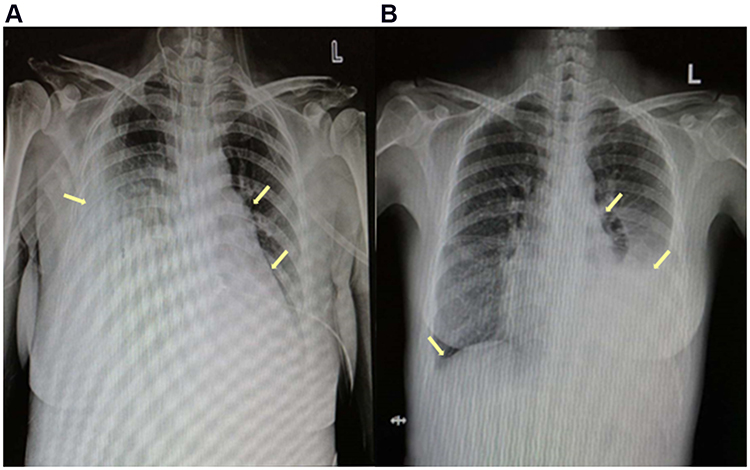 |
Figure 3 X-rays: (A) at the first admission: the arrows showed large pleural effusion on the right chest and small on the left, enlargement of right heart and a protruded pulmonary artery segment; (B) 5 months after discharge: the arrows showed significantly reduced pleural effusion, right heart enlargement and protruded pulmonary artery segment all improved. |
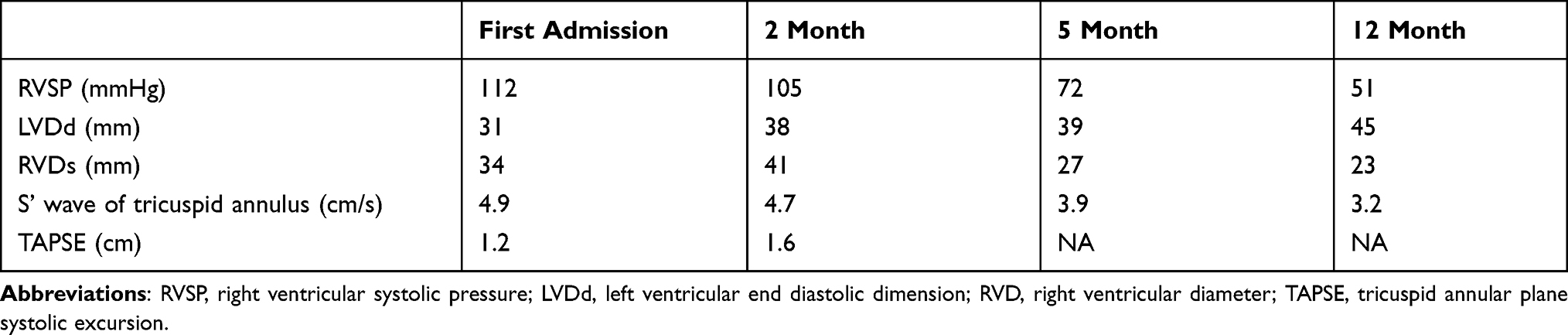 |
Table 2 Transthoracic Echocardiography Indexes During the Course of Disease |
Right-heart catheterization (RHC) confirmed markedly increased mean pulmonary artery pressure (MPAP; 61mmHg; normal reference value, ≤25 mmHg) and pulmonary vascular resistance [PVR; 6.98 wood units (WU); normal reference value, ≤3 WU3], as well as pulmonary arterial wedge pressure (PAWP; 21 mmHg; normal reference value, ≤15 mmHg) (Table 3). The acute pulmonary vascular dilatation test was negative. Mixed pulmonary hypertension was diagnosed and risk stratification was conducted. Tadalafil (10 mg o.d.), ambrisentan (5 mg o.d.), and treprostinil initiated. She was also received sustained oxygen intake, pleural puncture and traditional medication including diuretics, digitalis, dopamine, potassium supplement and albumin infusion. Nilotinib was maintained without adjustment (300 mg qd8, 150 mg qd16). The patient’s symptoms were significantly relieved and activity tolerance restored to two stories at discharge.
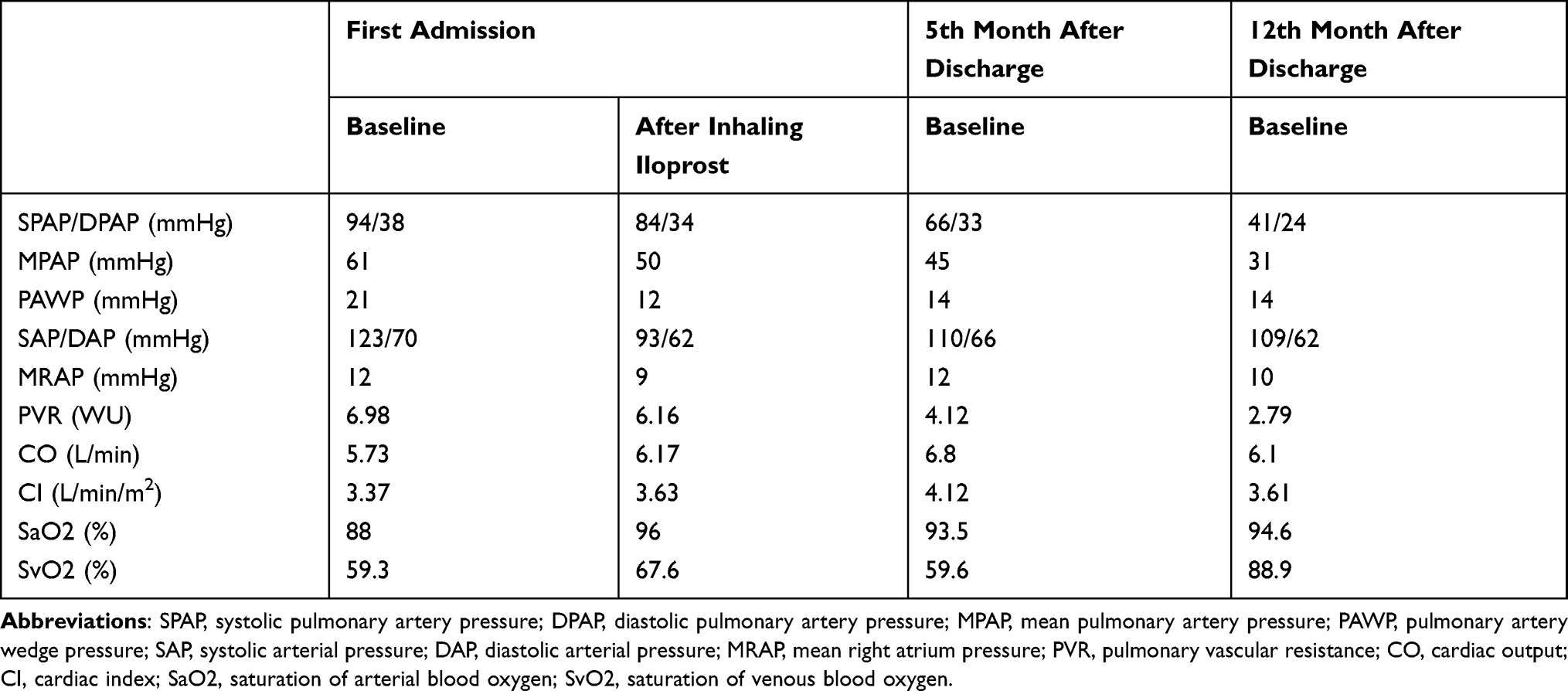 |
Table 3 Haemodynamical Indexes Measured by Right-Heart Catheter During the Course of Disease |
NT-proBNP reduced to 1993.0 pg/mL a month after discharge, 696.2 pg/mL on the 5th month after discharge. The amount of tadalafil, ambrisentan, and treprostinil was gradually increased during follow-up (Table 1). Her risk stratification reduced to low risk based on RHC on the 5th month after discharge. The diagnosis altered capillary anterior pulmonary hypertension. Her pulmonary circulation pressure and resistance were markedly reduced (Tables 2 and 3, Figure 2B). Then, treprostinil gradually reduced to stop, while tadalafil (20 mg o.d.) and ambrisentan (10 mg o.d.) were unchanged. Nilotinib (300 mg qd8, 150 mg qd16) was maintained after discharge. Fusion gene quantification 5th month after discharge suggests major molecular biological remission (MMR). The patient’s activity tolerance gradually restored and increased to seven stories during follow-up. On the 11th month after discharge, nilotinib was replaced by flumatinib (600 mg o.d.). Her risk stratification maintained low risk at 1 year after discharge. Her pulmonary circulation pressure and resistance were further reduced (Tables 2 and 3, Figure 2C). Fusion gene quantification showed sustained MMR at 1 year after discharge. The patient was followed up every three months in our Pulmonary Vascular Outpatient Department, and rechecked for RHC about half a year if necessary.
Discussion and Conclusion
Differences in the pharmacological mechanisms between dasatinib and other TKIs still do not explain the differences in pulmonary angiotoxicity between dasatinib and other TKIs. And researchers believe that the pathophysiology of DASA-PAH in human may reveal the role of tyrosine kinase pathway in pulmonary artery proliferative lesions.2 Guignabert et al found that exposing rats with dasatinib, before the administration of monocrotaline or exposure to hypoxia, resulted in markedly higher pulmonary artery pressure (PAP) and increased pulmonary vascular remodeling, compared with rats that were not pre-treated with dasatinib before monocrotaline or hypoxia exposure. Meanwhile, rats treated with dasatinib alone demonstrated no such increase in PAPs, supporting a two-hit theory of the pathophysiology of DASA-PAH in human.6 Takumi Toya et al experienced a case of DASA-PAH complicated with scleroderma that provides novel support for the two-hit hypothesis of DASA-PAH pathophysiology.5 Furthermore, in a report of 41 patients with RHC confirmed DASA-PAH, 94% showed improvement, but only 58% achieved complete resolution. These data indicate the underlying predisposing factors for DASA-PAH other than dasatinib drug toxicity, supporting the two-hit theory.7 However, the underlying predisposing factors remain unknown till now. Because of the limited number of reported events, formal statistical analyses could not be performed.1,7
This case provided no further support for the 2-hits hypothesis of DASA-PAH pathophysiology. However, this case has typical clinical manifestations of the disease. The diagnosis of DASA-PAH depends on dasatinib use history and hemodynamic diagnostic criteria for PAH, the first major category of pulmonary hypertension (according to the 6th World Symposium on Pulmonary Hypertension), as well as exclusion of all kinds of factors that could lead to PAH.8 Perhaps because the patient did not stop dasatinib and receive target treatment in time after the initial onset of PAH, the condition was further aggravated, and the cardiac function gradually deteriorated to New York Heart Association (NYHA) functional class (FC) IV. We suggest that the discontinuation of dasatinib, RHC-based hazard stratification, regular pulmonary vasodilators, combined with traditional anti-heart failure therapy, should become the routine treatment algorithm of DASA-PAH. And the preferred targeted drugs are still chosen based on current recommendations. Other scholars have also reported that many cases with severe symptoms (NYHA FC III or IV) or those with severe haemodynamic compromise [eg, cardiac index (CI) <3 L/min/m2] have been successfully treated by the similar treatment algorithm. Symptoms were controlled, hemodynamics improved, and PAP returned completely or near normal.3,9 However, it is still controversial on duration of pulmonary vasodilators. Some researchers think that if normal hemodynamics persists one year, de-escalation of pulmonary vasodilators can be considered.5 It is speculated that pulmonary vasodilator withdrawal is probably safe in reversible cases, while more clinical experience is needed to confirm whether pulmonary vasodilator maintenance is required for partially reversible cases.
Repeated massive pleural effusion is also characteristic of DASA-PAH. As reported in the literature, pleural effusion was the most common non-hematologic adverse reaction caused by dasatinib. The incidence of pleural effusion ranges from 14% to 60%, with 90% occurring within 1 year after dasatinib treatment, and 79% are bilateral.10,11 It had been reported that pleural effusion and pericardial effusion in most patients could disappear after dasatinib withdrawal and short-term symptomatic treatment, while PAH still existed, suggesting that pleural effusion might be not caused by right heart failure due to PAH, but the direct adverse reaction of dasatinib.12 The mechanism of pleural effusion associated with PAH may be similar to that directly caused by dasatinib, but the exact mechanism is still unclear.9
With regard to the management of dasatinib, DASA-PAH, a rare adverse reaction, should not negate the advantage of this drug in hematological field. However, as awareness of DASA-PAH increases, it is necessary to take some steps that may reduce its occurrence. Before use, it is suggested to evaluate for existing comorbidities that may lead to pulmonary hypertension to avoid “a possible second hit”. And, cardiopulmonary function should be routinely assessed before and during dasatinib treatment. Electrocardiography and TTE are the preferred non-invasive tests for screening PAH. If a hint of PAH occurs, dasatinib should be stopped immediately and for life, and target treatment for PAH should be considered according to the individualized situation.
There were some limitations associated with this case report. Due to the limitations of the overall level of specialized diagnosis and treatment of pulmonary vascular diseases in Yunnan Province, an economically backward mountainous province in the southwest of China, the diagnosis of this case was firstly made 10 months after the initial typical presentation. The delay of formal target therapy may influence the prognosis to some extent. Even so, the patient’s disease was reversed, confirming the conclusion that the disease had a relatively favorable prognosis. In addition, the measurement of echocardiography after each admission was performed by different doctors, and the measurement indexes were not identical each time.
In conclusion, we experienced a case of DASA-PAH with typical clinical characteristics and a partially reversible course, which was well controlled by a comprehensive strategy with combination targeted therapy guided by risk stratification as a core. However, further research is necessary to identify its predisposing factors, as well as the role of the tyrosine kinase pathway within the pulmonary vasculature, helping its early detection and future therapy.
Abbreviations
CML, chronic myeloid leukaemia; CI, cardiac index; DASA-PAH, dasatinib-induced pulmonary arterial hypertension; ECG, electrocardiogram; MMR, molecular biological remission; NT-proBNP, N-terminal of the prohormone brain natriuretic peptide; NYHA FC, New York Heart Association functional class; TKI, tyrosine kinase inhibitor; PAP, pulmonary artery pressure; PAH, pulmonary arterial hypertension; PASP, pulmonary arterial systolic pressure; PAWP, pulmonary arterial wedge pressure; RHC, right-heart catheterization; TTE, transthoracic echocardiography.
Ethics Approval and Consent to Participate
Written informed consent was obtained from the patient for publication of this case report and any accompanying images. The authors confirm that written consent for submission and publication of this case report including image(s) and associated text has been obtained from the patient in line with COPE guidance. Patients or the public were not involved in the design, or conduct, or reporting, or dissemination plans of our research. Ethical approvals were obtained from the Fuwai Hospital Research Ethics Committees (No. 2013‑449 and No. 2020-1310).
Author Contributions
All authors made a significant contribution to the work reported, whether that is in the conception, study design, execution, acquisition of data, analysis and interpretation, or in all these areas; took part in drafting, revising or critically reviewing the article; gave final approval of the version to be published; have agreed on the journal to which the article has been submitted; and agree to be accountable for all aspects of the work.
Funding
The National Natural Science Foundation of China (No. 81770365); National Key Research and Development Program of China (No. 2016YFC1301301); Beijing United Heart Foundation [No. BJUHFCSOARF201901-19]; Fuwai Yunnan Cardiovascular Hospital, Yunnan Provincial Cardiovascular Disease Clinical Medical Center Project (FZX2019-06-01).
Disclosure
None of the article contents are under consideration for publication in any other journal or have been published in any journal. We have no conflict of interest to disclose.
References
1. Ryan JJ. Tyrosine kinase inhibitors in pulmonary vascular disease. JACC: Basic Transl Sci. 2016;1(7):684–686.
2. Yurttaş NÖ, Eşkazan AE. Dasatinib‐induced pulmonary arterial hypertension. Br J Clin Pharmacol. 2018;84(5):835–845. doi:10.1111/bcp.13508
3. Weatherald J, Chaumais MC, Savale L, et al. Long-term outcomes of dasatinib-induced pulmonary arterial hypertension: a population-based study. Eur Respir J. 2017;50(1):1700217. doi:10.1183/13993003.00217-2017
4. Mattei D, Feola M, Orzan F, Mordini N, Rapezzi D, Gallamini A. Reversible dasatinib-induced pulmonary arterial hypertension and right ventricle failure in a previously allografted CML patient. Bone Marrow Transplant. 2009;43(12):967–968. doi:10.1038/bmt.2008.415
5. Toya T, Nagatomo Y, Kagami K, Adachi T. Dasatinib-induced pulmonary arterial hypertension complicated with scleroderma: a case report. Eur Heart J Case Rep. 2019;3(1):ytz025. doi:10.1093/ehjcr/ytz025
6. Guignabert C, Phan C, Seferian A, et al. Dasatinib induces lung vascular toxicity and predisposes to pulmonary hypertension. J Clin Invest. 2016;126(9):3207–3218. doi:10.1172/JCI86249
7. Shah NP, Wallis N, Farber HW, et al. Clinical features of pulmonary arterial hypertension in patients receiving dasatinib. Am J Hematol. 2015;90(11):1060–1064. doi:10.1002/ajh.24174
8. Xu XQ, Jing ZC. The 6th world symposium on pulmonary hypertension: focus on updates on definition and clinical classification of pulmonary hypertension. Med J PUMCH. 2018;9(3):197–201.
9. Hong JH, Lee SE, Choi SY, et al. Reversible pulmonary arterial hypertension associated with dasatinib for chronic myeloid leukemia. Cancer Res Treat. 2015;47(4):937–942. doi:10.4143/crt.2013.155
10. Paydas S. Dasatinib, large granular lymphocytosis, and pleural effusion: useful or adverse effect? Crit Rev Oncol Hematol. 2014;89(2):242–247. doi:10.1016/j.critrevonc.2013.10.005
11. Ouintas-Cardama A, Kantarjian H, O’Brien S, et al. Pleural effusion in patients with chronic myelogenous leukemia treated with dasatinib after imatinib failure. J Clin Oncol. 2007;25(25):3908–3914. doi:10.1200/JCO.2007.12.0329
12. Liu BC, Wang Y, Mi YC, Wang JX.. Reversible pulmonary arterial hypertension related to dasatinib in the treatment for chronic myelogenous leukemia: a case report and literature review. Chin J Hematol. 2014;35(7):581–586. doi:10.3760/cma.j.issn.0253-2727.2014.07.002
 © 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.