Back to Journals » Drug Design, Development and Therapy » Volume 8
Cytoprotective effect of selective small-molecule caspase inhibitors against staurosporine-induced apoptosis
Authors Wu J ![]() , Wang Y, Liang S, Ma H
, Wang Y, Liang S, Ma H
Received 8 January 2014
Accepted for publication 1 April 2014
Published 24 May 2014 Volume 2014:8 Pages 583—600
DOI https://doi.org/10.2147/DDDT.S60283
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 9
Jianghong Wu, Yuren Wang, Shuguang Liang, Haiching Ma
Reaction Biology Corp, Malvern, PA, USA
Abstract: Caspases are currently known as the central executioners of the apoptotic pathways. Inhibition of apoptosis and promotion of normal cell survival by caspase inhibitors would be a tremendous benefit for reducing the side effects of cancer therapy and for control of neurodegenerative disorders such as Parkinson's, Alzheimer's, and Huntington's diseases. The objective of this study was to discover small-molecule caspase inhibitors with which to achieve cytoprotective effect. We completed the high-throughput screening of Bionet's 37,500-compound library (Key Organics Limited, Camelford, Cornwall, UK) against caspase-1, -3, and -9 and successfully identified 43 initial hit compounds. The 43 hit compounds were further tested for cytoprotective activity against staurosporine-induced cell death in NIH3T3 cells. Nineteen compounds were found to have significant cytoprotective effects in cell viability assays. One of the compounds, RBC1023, was demonstrated to protect NIH3T3 cells from staurosporine-induced caspase-3 cleavage and activation. RBC1023 was also shown to protect against staurosporine-induced impairment of mitochondrial membrane potential. DNA microarray analysis demonstrated that staurosporine treatment induced broad global gene expression alterations, and RBC1023 co-treatment significantly restored these changes, especially of the genes that are related to cell growth and survival signaling such as Egr1, Cdc25c, cdkn3, Rhob, Nek2, and Taok1. Collectively, RBC1023 protects NIH3T3 cells against staurosporine-induced apoptosis via inhibiting caspase activity, restoring mitochondrial membrane potential, and possibly upregulating some cell survival-related gene expressions and pathways.
Keywords: cell death, caspase inhibition, mitochondria, RBC1023
Introduction
Apoptosis is a genetically programmed, morphologically distinct form of cell death that can be triggered by many chemicals in a variety of cellular events, such as calcium influx, oxidative stress, cytoskeletal interference, inhibitors of protein synthesis, membrane disruption, and DNA disruption induced by radiation.1 Caspases, currently known as the central executioners of the apoptotic pathways, are cysteine proteases that cleave substrates after aspartate residues,2–4 and at least 14 different subtypes have been identified in mammalian cells. Two pathways of caspase activation for apoptosis induction have been identified. The first one starts at death receptors such as Fas.5 In the second and more common pathway, diverse proapoptotic signals converge at the mitochondrial level, inducing the translocation of cytochrome c from mitochondria to cytoplasm, thereby triggering the cascade of caspase activation.6,7 Caspase-3 is one of the most important and active executors in the apoptosis pathway.
Caspases are important targets of drug discovery8 because inhibition of apoptosis is a means of promoting cell survival. The significance of caspases in cancer therapy is implicated in two directions: while the activation of caspases leading to the cancer cell-specific apoptosis is critical to numerous and diverse cancer treatments, caspase inhibition in nonmalignant cells is a defined target for cytoprotection and reducing organ-specific toxicity during cancer therapy.9 The toxicity of normal cells exerted by chemotherapy and radiation therapy is often a limiting factor for optimal therapeutic dosage. Cytoprotection of nonmalignant cells by caspase inhibition would allow a higher upper limit of dose escalation of existing therapeutics to achieve maximal efficacy. Caspase inhibition has also been suggested for ultraviolet protection therapy to alleviate side effects such as ultraviolet-induced skin damage, inflammation, and aging.10–13 Furthermore, caspase inhibition may provide prevention or treatment against acute episodes such as stroke, traumatic brain injury, spinal cord injury, or myocardial infarction. Neuron-degenerative diseases, treatment of which may benefit from caspase inhibition, include amyotrophic lateral sclerosis, Parkinson’s, Alzheimer’s, and Huntington’s.14
To discover new caspase inhibitors, we screened Bionet’s 37,500-compound library against human caspase-1, -3, and -9 with the fluorogenic substrates Ac-YVAD-AMC, Ac-DEVD-AMC, and Ac-LEHD-AMC, and identified 43 initial hit compounds. Using staurosporine-induced apoptosis assay in NIH3T3 cell line, we discovered 19 small molecules with cell protective activities, in which compound RBC1023, a selective caspase-3 inhibitor, had shown a broad protection spectrum across many different cell types. Further mechanistic studies indicated that RBC1023 protected NIH3T3 cells from staurosporine-induced caspase cleavage and activation and restored staurosporine-induced loss of mitochondrial membrane potential. Moreover, we investigated the changes in gene expression in NIH3T3 cells treated with staurosporine in the presence or absence of RBC1023 using a DNA microarray analysis to understand the molecular mechanisms of the cytoprotective effect of RBC1023.
Materials and methods
Chemicals and reagents
Bionet’s 37,500-compound library was obtained from Key Organics Limited (Camelford, Cornwall, UK). Purified caspase-1, -3, and -9 enzymes; fluorogenic caspase substrates Ac-YVAD-AMC, Ac-DEVD-AMC, and Ac-LEHD-AMC; and control caspase inhibitor Ac-DEVD-CHO, Ac-LEHD-CHO, and Ac-YVAD-CHO were all obtained from Biomol International (Enzo Life Sciences, Inc., Farmingdale, NY, USA). Staurosporine was obtained from Sigma-Aldrich (St Louis, MO, USA). Cell Titer-Glo® Luminescent Cell Viability Assay Kit and Apo-ONE® Homogeneous Caspase-3/7 Assay Kit were obtained from Promega Corporation (Fitchburg, WI, USA). MTT (3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide) Cell Proliferation Assay Kit was obtained from Cayman Chemical (Ann Arbor, MI, USA). JC-10 Mitochondrial Membrane Potential Assay Kit was obtained from Abcam (Cambridge, UK). Anti-caspase-3 polyclonal antibody (catalog#9665, 1:2000 dilutions) was obtained from Cell Signaling Technology (Danvers, MA, USA). Anti-α-tubulin monoclonal antibody (catalog#T6074, 1:5000 dilutions) was obtained from Sigma-Aldrich. The secondary antibodies Alexa Fluor® 633 goat anti-rabbit immunoglobulin (Ig)G (heavy and light chains [H+L]) and Alexa Fluor® 532 goat anti-mouse IgG (H+L) were obtained from Thermo Fisher Scientific (Life Technologies, Waltham, MA, USA). Other chemicals were of reagent grade and were obtained from Sigma-Aldrich or VWR International (Radnor, PA, USA).
High-throughput screening (HTS) and hit confirmation
The primary HTS against caspase-1, -3, and -9 enzymes was performed using the Bionet compound library. In brief, 5 μL of reaction buffer (50 mM 4-(2-hydroxyethyl)piperazine-1-ethanesulfonic acid (HEPES) pH 7.4, 100 mM NaCl, 0.1% 3-[(3-cholamidopropyl) dimethylammonio]-1-propanesulfonate (CHAPS), 1 mM ethylenediaminetetraacetic acid (EDTA), and 3 mM dithiothreitol) with 1X enzyme was added to each well of Genetix 7020 low-volume 384-well plates (Molecular Devices LLC, Sunnyvale, CA, USA). Five nanoliters of 10 mM of each compound from the Bionet 37,500-compound library was delivered to each well of the Genetix plates (final compound concentration 10 μM) by acoustic droplet ejection liquid handler (Echo 550, Labcyte Inc., Sunnyvale, CA, USA). The plates were shaken to mix for 1 minute. One microliter of positive control inhibitors (Ac-YVAD-CHO, Ac-DEVD-CHO, and Ac-LEHD-CHO) was added to the control wells. Ten nanoliters of 5 mM of caspase substrates was delivered to the plates by Echo 550 to start the reaction. The fluorescent signal was measured by EnVision® 2104 Multilabel Reader (PerkinElmer Inc., Waltham, MA, USA) at an excitation wavelength of 355 nm and an emission wavelength of 460 nm at the 2-hour time point. The fluorescent signal generated was proportional to the caspase-1, -3, and -9 activities. The initial hits from the HTS were cherry-picked and further tested in a ten-dose half maximal inhibitory concentration (IC-50)-mode against caspase-1, -3, and -9.
Cells, cell culture, and compound treatment
NIH3T3 mouse fibroblast, PC12 rat pheochromocytoma cells, and U87MG human glioblastoma cells were obtained from American Type Culture Collection (ATCC) (Manassas, VA, USA). SH-SY5Y cell line was kindly provided by Dr Honglin Zhou at the University of Pennsylvania (Philadelphia, PA, USA). NIH3T3 cells were grown in Dulbecco’s Modified Eagle’s Medium, PC12 cells were grown in RPMI 1640 (Roswell Park Memorial Institute medium 1640), U87MG cells were grown in Modified Eagle’s Medium, and SH-SY5Y cells were grown in a 1:1 mixture of Modified Eagle’s Medium and F12 media. All culture media were supplemented with 10% heat-inactivated fetal bovine serum, 100 μg/mL penicillin, and 100 μg/mL streptomycin. Cultures were maintained at 37°C in a humidified atmosphere of 5% CO2 and 95% air. For most experiments, unless specified, 5,000/well of logarithmically growing cells were seeded in each of the 384-well plates containing proper medium plus 1% fetal bovine serum with or without compound. Dimethyl sulfoxide ([DMSO] ≤0.3%) was added to all the control cultures.
Cell Titer-Glo® Luminescent Cell Viability Assay and adenosine triphosphate (ATP) measurement
NIH3T3, PC12, U87MG, or SH-SY5Y cells were seeded overnight in triplicates in 384-well plates. The cells were incubated with compounds for 24 hours. A volume of Cell Titer-Glo® reagent equal to the volume of cell culture medium was added to each well. Contents were mixed for 2 minutes on an orbital shaker to induce cell lysis. The plates were incubated at room temperature for 10 minutes to stabilize luminescent signal. Luminescence was recorded by the EnVision® 2104 Multilabel Reader. The numbers of viable cells in culture were determined based on quantitation of the ATP present in each well. For quantifying cellular ATP contents in NIH3T3 cells, an ATP standard curve was generated starting at 1 μM in serial ten-fold dilutions in culture medium. The luminescence signals were converted into ATP concentration based on the ATP standard curve.
Apo-ONE® Homogeneous Caspase-3/7 Assay
NIH3T3 cells were seeded overnight in triplicates in 384-well plates. The cells were incubated with compounds for 6 hours. A volume of Apo-ONE® reagent equal to the volume of cell culture medium was added to each well. The contents were gently mixed using a plate shaker at 300 rpm for 30 seconds. The plates were incubated at room temperature for 30 minutes. Upon the cleavage of caspase substrate Z-DEVD-rhodamine 110, bis-(N-CBZ-L-aspartyl-L-glutamyl-L-valyl-L-aspartic acid amide) by caspase-3/7 activity, the fluorescence of each well was measured at an excitation wavelength of 485 nm and an emission wavelength of 535 nm using the EnVision® 2104 Multilabel Reader. The amount of fluorescence generated was proportional to the amount of caspase-3/7 cleavage activity present in the sample.
MTT cell proliferation assay
NIH3T3 cells were seeded in triplicates in 96-well plates at a density of 2×104 cells/well in 100 μL of culture medium with or without compounds to be tested. The cells were cultured in a CO2 incubator at 37°C for 24 hours. Ten microliters of MTT reagent was added to each well and mixed gently for 1 minute. The cells were incubated for 4 hours at 37°C. After incubation, the formazan produced in the cells appeared as dark crystals in the bottom of the wells. The culture medium from each well was aspirated carefully and 100 μL of crystal-dissolving solution was added to each well to dissolve the formazan crystals. The absorbance of each sample was quantified at 590 nm using the EnVision® 2104 microplate reader. The absorbance intensity was proportional to the amount of viable cells.
JC-10 mitochondrial membrane potential assay
Mitochondrial membrane potential (Δψm) was evaluated by cationic dye JC-10. In normal cells, JC-10 aggregates in mitochondria, fluorescing red. In apoptotic cells, JC-10 accrues in the cytosol, as a green fluorescing monomer. Briefly, NIH3T3 cells were plated in triplicates in growth medium in 384-well plates overnight. The cells were treated with or without compounds for 6 hours. JC-10 dye-loading solution was added and the plates were incubated in a 37°C, 5% CO2 incubator for 30 minutes. Assay buffer was added into the dye-loading plates and the fluorescence intensities were monitored at excitation/emission =485/535 nm and 485/590 nm for ratio analysis in the EnVision® 2104 Multilabel microplate reader.
Western blot analysis
NIH3T3 cells were seeded in six-well plates (0.8×106 cells/well) and treated with or without compounds for 6 hours. After the treatment, cells were washed with cold phosphate-buffered saline and lysed in cell lysis buffer (150 mM NaCl, 1% Triton X-100, 50 mM Tris pH 7.5, proteinase inhibitor cocktail, 0.1 mM phenylmethylsulfonyl fluoride). Protein concentrations of the cell lysates were measured using Bio-Rad protein assay dye reagent concentrate (Bio-Rad Laboratories Inc., Hercules, CA, USA). About 30 μg of total protein for each sample was separated under reducing conditions by sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE) and transferred to a nitrocellulose membrane (Life Technologies). The blots were blocked in 3% nonfat milk for 2 hours at room temperature with gentle shaking. This was followed by incubation with the respective antibody overnight at 4°C and then incubation with Alexa Fluor® 633 goat anti-rabbit IgG or Alexa Fluor® 532 goat anti-mouse IgG secondary antibody for 1 hour. Fluorescence signals of the immune complexes were scanned with a Typhoon 8610 variable mode imager (GE Healthcare Bio-Sciences Corp, Piscataway, NJ, USA).
DNA microarray analysis of gene expression profile
NIH3T3 cells were seeded in 10 cm dishes (2×106 cells/dish) and treated with DMSO or testing compounds. The cells were pelleted down by centrifuging and washed by cold phosphate-buffered saline. The cell pellets were frozen using dry ice, and submitted to the Penn Molecular Profiling Facility (University of Pennsylvania School of Medicine) for RNA extraction and transcript profiling. Microarray analysis was conducted in duplicate. RNA was extracted with TRIzol® by Life Technologies (Thermo Fisher Scientific) and purified using the Qiagen RNeasy Kit (Qiagen NV, Venlo, the Netherlands); total RNA yields ranged from 15.5 to 53.4 μg per sample with A260/280 of 2.07–2.18. Transcript profiling was conducted as described in the Affymetrix GeneChip Expression Analysis Technical Manual (Affymetrix Inc., Santa Clara, CA, USA).15 Briefly, 100 ng of total RNA was converted to first-strand complementary DNA (cDNA) using reverse transcriptase primed by poly (T) and random oligomers that incorporated the T7 promoter sequence. Second-strand cDNA synthesis was followed by in vitro transcription with T7 RNA polymerase for linear amplification of each transcript, and the resulting cRNA was converted to cDNA, fragmented, and biotinylated by terminal transferase end labeling. Labeled cDNA was added to Affymetrix hybridization cocktails, heated at 99°C for 5 minutes, and hybridized for 16 hours at 45°C to GeneChip Mouse Gene 1.0ST Arrays (Affymetrix Inc.). The microarrays were then washed at low (6× SSPE [sodium chloride, sodium phosphate monobasic, and ethylenediaminetetraacetic acid]) and high (100 mM MES [2-[N-morpholino]ethanesulfonic acid, tris base, ethylenediaminetetraacetic acid, and sodium dodecyl sulfate], 0.1 M NaCl) stringency, and stained with streptavidin/phycoerythrin. Fluorescence was amplified by adding biotinylated anti-streptavidin and an additional aliquot of streptavidin/phycoerythrin stain. A confocal scanner was used to collect the fluorescence signal after excitation at 570 nm. The intensity of the fluorescence signal was proportional to the levels of gene expression.
Statistical analysis
Statistical analysis in this study was performed using the unpaired Student’s t-test or one-way analysis of variance (ANOVA). The values are represented as mean ± standard deviation. A P-value <0.05 was considered to be statistically significant.
Results
HTS and hit confirmation
Using the Bionet 37,500-compound library, we successfully completed HTS against human caspase-1, -3, and -9. The screening was of high quality, with a robust Z-factor >0.6 (Table 1).16 By using simple signal-to-noise analysis, compounds with response signal >3 times the standard deviation of the noise were designated as “hits”. Totals of 453 compounds, 727 compounds, and 1,149 compounds were selected as initial hits for caspase-1, -3, and -9, respectively, which represent the hit rates of 1.20%, 1.94%, and 3.06%, respectively (Table 1). The hits were further confirmed in ten-dose IC50 mode in the presence of three different reducing agents (3 mM dithiothreitol, 5 mM β-mercaptoethanol, 5 mM cysteine) to eliminate any reactive electrophilic compounds.17 There were 143, 178, and 273 confirmed hits that inhibited caspase-1, -3, or -9, respectively. This provided overall hit rates of 0.38%, 0.47%, and 0.73%, respectively, which are typical for biochemical HTS against a diversity library. We further eliminated some compounds from these confirmed hits using three stringent selection processes: compounds with >20% inhibition on cytochrome P450 (Cyp) enzymes such as Cyp2C19, Cyp1A2, Cyp2A6, Cyp2C9, Cyp3A4, or Cyp2D6 at 10 μM (data not shown); compounds containing undesirable structures and metabolically fragile functionality (such as reactive chlorides, Michael acceptors, thiols, esters, quaternary salts, and detergents); and compounds showing significant toxicity against mouse embryonic fibroblast cells (data not shown). A final set of 43 compounds was selected for further testing in a cytoprotection assay (Table 2).
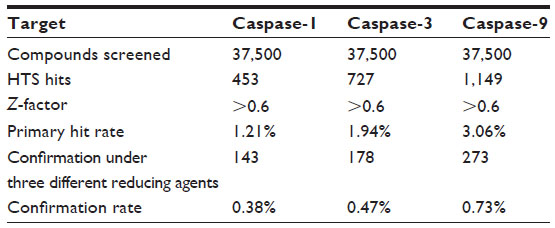 | Table 1 Primary high-throughput screening (HTS) of caspase-1, -3, and -9 |
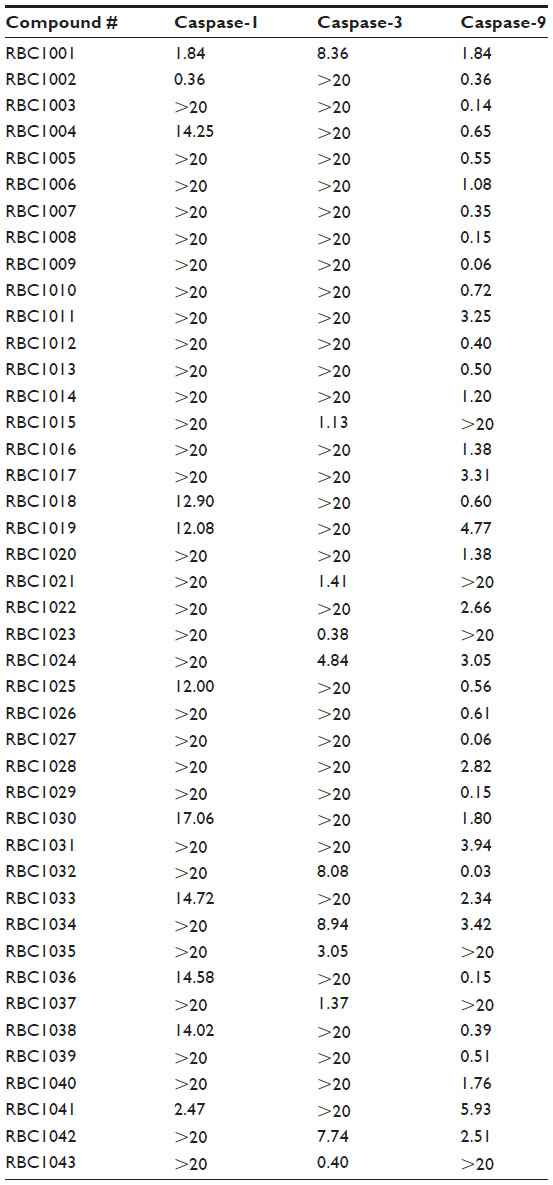 | Table 2 Caspase inhibition activity IC50 (μM) of 43 hit compounds from the in vitro caspase-1, -3, and -9 screening assays |
Cytoprotective activities of the caspase inhibitors
Staurosporine is a broad-spectrum inhibitor of protein kinases, and has been widely used in the induction of apoptosis in diverse cellular models.18–22 Staurosporine preferentially activates the mitochondrial apoptotic pathway relying on caspase activation and causes cell death. To confirm the caspase inhibition of the hit compounds in cell-based assay, NIH3T3 cells were treated with 20 μM of each of the 43 hit compounds in the presence or absence of 1 μM staurosporine for 24 hours. Then, cell viability was assessed by Cell Titer-Glo® Luminescent Cell Viability Assay. As shown in Figure 1, while staurosporine treatment alone caused over 90% cell death, 19 compounds showed significant protection against staurosporine-induced cell death in NIH3T3 cells (P<0.01). Several compounds that are toxic to cells alone were removed from the hit list (data not shown). Compound RBC1020 (a caspase-9 selective inhibitor) and RBC1023 (a caspase-3 selective inhibitor) with no cytotoxicity alone demonstrated 7.5-fold and 5.9-fold increases respectively of cell viability compared to cells treated with staurosporine alone, indicating a strong cytoprotective activity of these two compounds. Caspase inhibitors could have potential application in treating neurodegenerative diseases such as Parkinson’s and Alzheimer’s diseases, which are usually associated with neuron cell death. Therefore, we extended the study to further examine the cytoprotective activity of the leads RBC1020 and RBC1023 in several neural origin cell types. PC12, U87MG, and SH-SY5Y cells were treated with DMSO, staurosporine alone, RBC1020 or RBC1023 alone, or RBC1020 or RBC1023 plus staurosporine for 24 hours and then the cell viability was examined. As shown in Figure 2, quantitative measurement of cell viability showed that RBC1023 increased the percentage of staurosporine-treated viable cells from 4% to 66% in PC12 cells, from 8% to 71% in U87MG cells, and from 6% to 47% in SH-SY5Y cells. Interestingly, RBC1020 had no or minimum cytoprotection in these three cell lines. It is still unknown why RBC1020 and RBC1023 showed different effects in different cell lines. Additional studies are needed to investigate the mechanism that causes this difference. These results demonstrated that RBC1023, a selective caspase-3 inhibitor, exhibits strong cytoprotective effect against staurosporine-induced cell death in multiple cell lines.
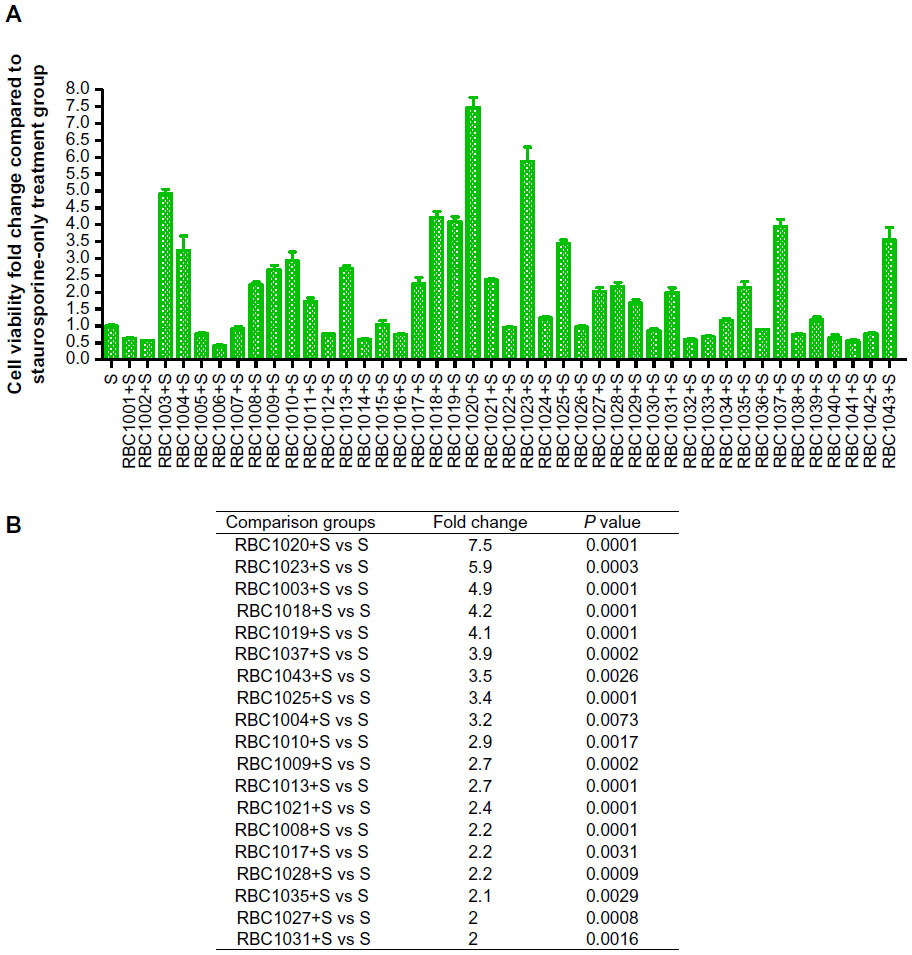 | Figure 1 Screening results for cytoprotection of 43 hit compounds against staurosporine (S)-induced cell death in NIH3T3 cells. |
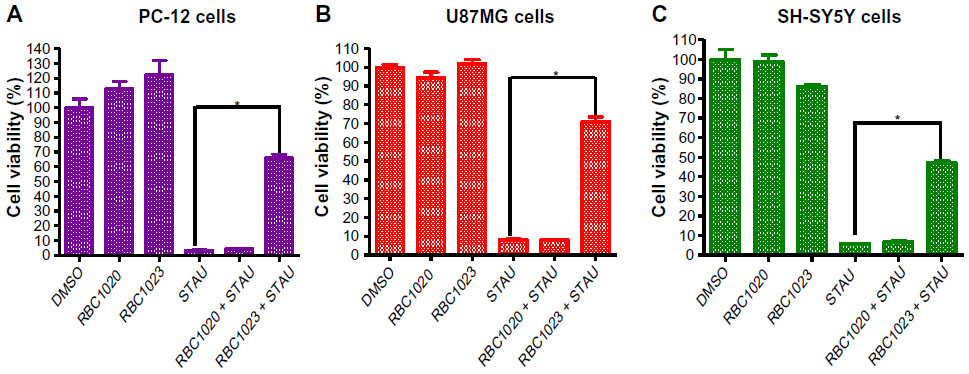 | Figure 2 RBC1023 protects STAU-induced cell death in PC-12, U87MG, and SH-SY5Y cells. |
Dose-dependent and time- dependent study of RBC1023 against staurosporine-induced cell death
Since the staurosporine/NIH3T3 apoptosis induction system is a very well-established model system for apoptosis-related mechanism studies, we next focused on the cytoprotection mechanism studies in the NIH3T3 cell line. We first investigated the protective effect of different concentrations of RBC1023 against different concentrations of staurosporine-induced cell death. NIH3T3 cells were treated with different dose combinations of RBC1023 (50 μM to 1 μM, three-fold dilution, ten doses) and staurosporine (0.768 μM or 2 μM) for 24 hours before the cell viability was measured. As shown in Figure 3A, RBC1023 started to show significant cytoprotection against the staurosporine-induced cell death at about 2 μM, when the concentration of staurosporine was at 0.768 μM. The cytoprotective effect by RBC1023 increased in a dose-dependent manner. To understand if the pretreatment or posttreatment of the caspase inhibitor would affect its cytoprotective effect, RBC1023 was added to cells at 1 hour before staurosporine treatment or 1 hour post-staurosporine treatment. The cell viability was measured after a total of 24 hours’ incubation. Interestingly, there were no significant differences of cytoprotective activity by RBC1023 between the 1-hour pretreatment and and 1-hour posttreatment group with staurosporine (Figure 3B).
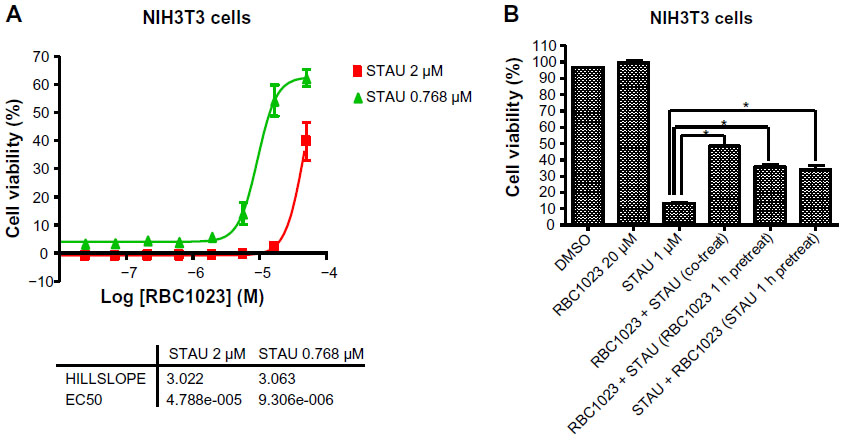 | Figure 3 Dose-response protection and treatment timing study of RBC1023 against Stau-induced cell death in NIH3T3 cells. |
RBC1023 blocks staurosporine-induced caspase activation in cell-based assays
To confirm RBC1023 as a caspase inhibitor in cell-based assays, we examined the caspase activity/activation-inducing effect in NIH3T3 cells that were treated with staurosporine in the presence or absence of RBC1023. As shown in Figure 4A, 6-hour staurosporine treatment induced significant caspase activation in NIH3T3 cells, as evaluated by Apo-ONE® Homogeneous Caspase-3/7 Assay using Z-DEVD-rhodamine 110 as a caspase substrate. Treatment of NIH3T3 cells with staurosporine for 6 hours also resulted in cleavage/activation of caspase-3, as was evident by the appearance of a 17 kD proteolytic product of caspase-3 as determined by Western blot analysis (Figure 4B). RBC1023 dramatically inhibited staurosporine-induced caspase-3 cleavage and activation (Figure 4B). These results confirmed that RBC1023 is a caspase inhibitor in cells.
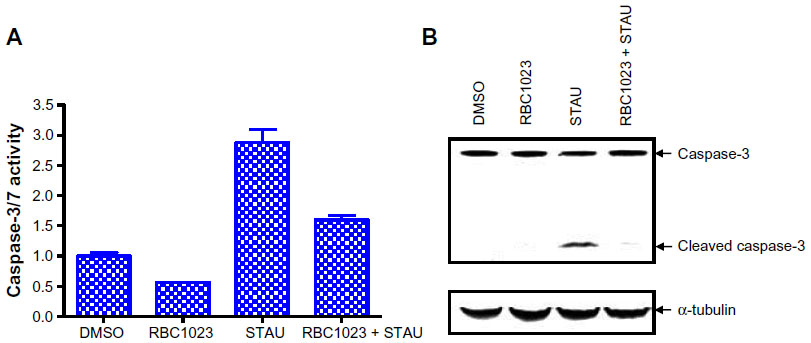 | Figure 4 RBC1023 inhibits Stau-induced caspase-3 cleavage and activation in NIH3T3 cells. |
RBC1023 restores NIH3T3 cells from the staurosporine-induced loss of cell viability and cellular ATP contents
To investigate if RBC1023 can rescue staurosporine-induced decrease of cell viability, we performed the MTT assay, in which the tetrazolium dye, MTT, is reduced to formazan by intracellular nicotinamide adenine dinucleotide phosphate (NADPH) oxidoreductases, which correlates with cellular metabolic activity and the number of viable cells present. As shown in Figure 5A, staurosporine treatment for 24 hours reduced the cell viability dramatically. RBC1023 co-treatment significantly rescued the loss of cell viability induced by staurosporine in NIH3T3 cells. We also performed ATP measurement by generating an ATP standard curve using Cell Titer-Glo® luminescent assay. As shown in Figure 5B, RBC1023 clearly restored the loss of ATP contents in cells treated with staurosporine. One-way ANOVA of the MTT and ATP assay data suggested that RBC1023 induces normalization or partial recovery of cell viability and ATP contents. Our results here further confirmed the cytoprotective effect of RBC1023 demonstrated by Cell Titer-Glo® assay.
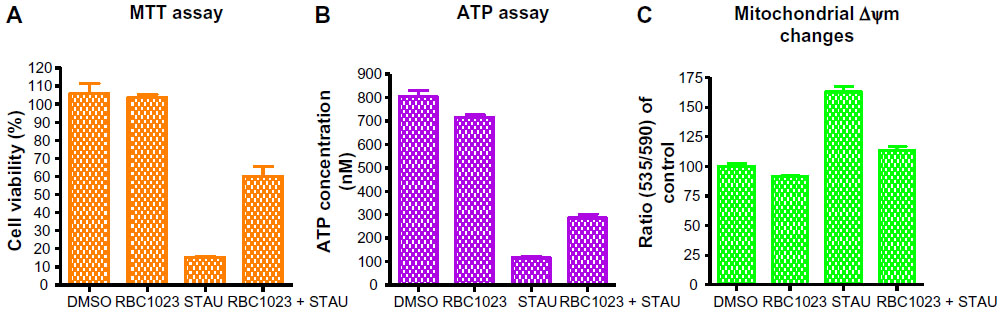 | Figure 5 RBC1023 restores NIH3T3 cells from Stau-induced loss of cell viability, ATP contents, and disruption of mitochondrial membrane potential. |
RBC1023 restores NIH3T3 cells from staurosporine-induced disruption of mitochondrial membrane potential
Disruption of mitochondrial membrane potential (Δψm), which is observed at an early stage of apoptosis in many cells, results in opening of the permeability transition pores (PTPs), causing a local disruption of the outer mitochondrial membrane.23,24 As a consequence, the soluble inter-membrane proteins including cytochrome c are released, and these proteins contribute to the activation of caspases for chromatin condensation and DNA fragmentation.7 Based on this theory, we next assessed if RBC1023 restores NIH3T3 cells from staurosporine-induced loss of mitochondrial membrane potential. The mitochondrial membrane potential changes were measured by the cationic, lipophilic JC-10 dye. As shown in Figure 5C, NIH3T3 cells that were treated with 1 μM of staurosporine for 6 hours significantly lost mitochondrial membrane potential, whereas co-incubating the cells with RBC1023 and staurosporine apparently prevented staurosporine-induced mitochondrial membrane impairment. One-way ANOVA of the data also supports this statement. Our findings suggest that RBC1023 mediates mitochondrial protection in addition to its direct caspase inhibitory effect.
Investigation of gene profile for the cytoprotective effect of RBC1023 by DNA microarray analysis
To further explore the potential molecular mechanisms of the cytoprotection effect by RBC1023, we performed DNA microarray analysis using Affymetrix GeneChip Mouse Gene 1.0ST, which covers 26,166 coding transcripts. NIH3T3 cells were treated by one of four conditions as follows: DMSO, RBC1023, staurosporine, or RBC1023 plus staurosporine. For each condition, the NIH3T3 cells were exposed to the indicated treatment for 20 hours. RNA was harvested from the cells treated under each condition and then converted to cDNA, which was used to query the genes on the Affymetrix GeneChip Mouse Gene 1.0ST Array. The general microarray results showed that staurosporine treatment dramatically changes the global gene expression profiling, and the changes caused by treatment of staurosporine were significantly restored by the co-treatment of RBC1023 in NIH3T3 cells. The gene expressions between each group, analyzed using supervised clustering of the 200 upregulated and downregulated messenger (m)RNAs that exhibited the most significant differences in all groups, were determined by one-way ANOVA. As shown in the heat map (Figure 6), the control and RBC1023 alone treatment samples showed similar clustering of these specific gene expressions. However, there was a dramatic increase or decrease in the expression of many genes in the staurosporine treatment samples. Importantly, the expression levels of these downregulated or upregulated genes caused by treatment of staurosporine were significantly restored by the co-treatment of RBC1023 in NIH3T3 cells. Molecular functions of the top 200 up- and downregulated gene transcripts are listed in Table S1. The gene list includes some apoptosis and cellular signaling-related genes such as Htra2 (Omi), caspase-12, Etv1, Egr1, Nek7, Upk1a, Skap2, and Tuba1b (Table S1). Furthermore, we summarized the top 50 genes upregulated (ranked by fold-change) in the RBC1023-plus-staurosporine treatment group in comparison with the staurosporine-alone treatment group, in which displays ~2.5- to ~4.9-fold increases can be seen in Table 3. Interestingly, no known proapoptotic genes can be seen in this list (Table 3). We hypothesize that the upregulation of some cell survival genes may be partially responsible for the protective effect of RBC1023 against staurosporine-induced cell death.
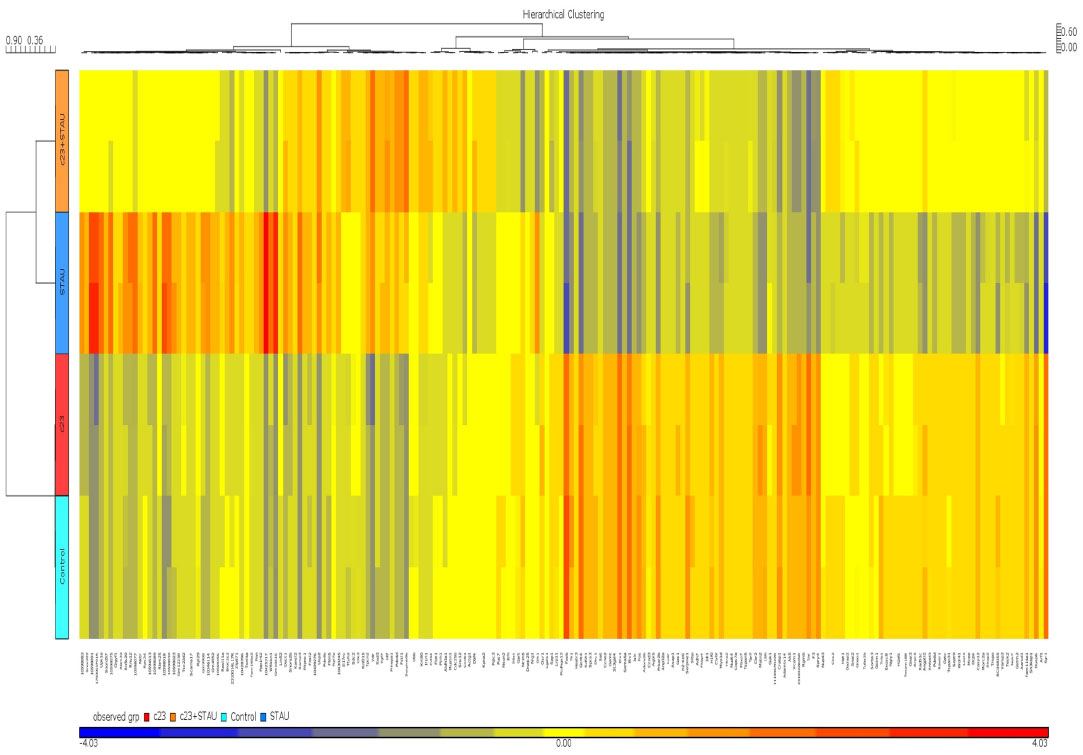 | Figure 6 Hierarchical clustering of 200 upregulated and downregulated genes from NIH3T3 samples treated with DMSO, RBC1023, STAU, or RBC1023 plus STAU. |
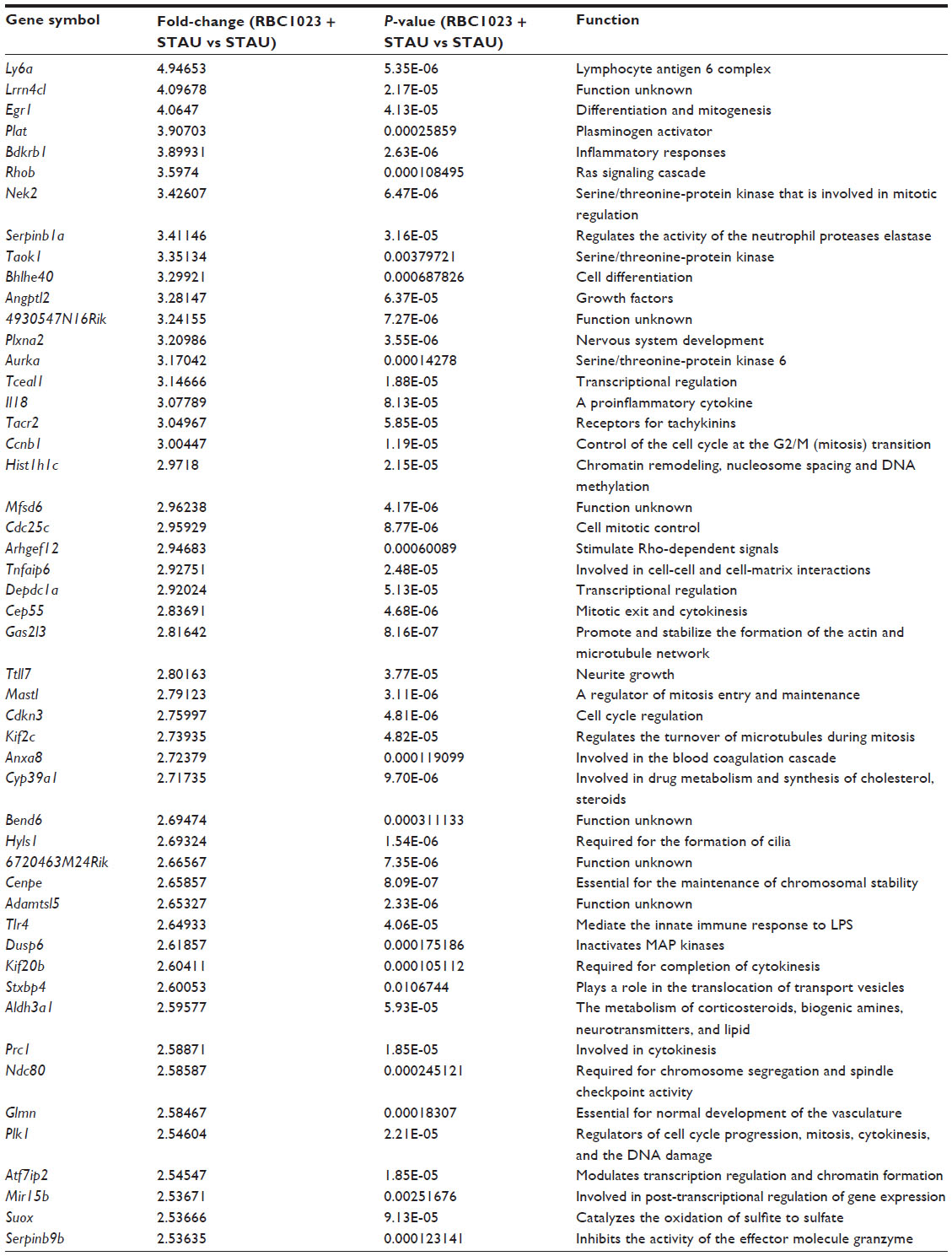 | Table 3 Top 50 genes upregulated (RBC1023 + STAU group versus STAU group) |
Discussion
In this study, we identified 19 caspase inhibitors that showed cytoprotection against staurosporine-induced cell death by screening Bionet’s 37,500-compound library against caspase-1, -3, and -9, and then through multiselective processes. RBC1023, a selective caspase-3 inhibitor, showed dose-dependent cytoprotection against staurosporine-induced cell death in different types of cell lines. We also confirmed that RBC1023 protects NIH3T3 cells from the staurosporine-induced caspase-3 cleavage and activation. These results indicate that reduced apoptotic cell death and increased cell proliferation are attributed to the inhibition of caspase activation by RBC1023. Interestingly, RBC1023 protected against cell death even up to 1 hour after staurosporine treatment.
Mitochondria play a central role in apoptosis,25 and there are reports that demonstrate the critical role of mitochondria in cytoprotection.26,27 We evaluated the possible correlation between the protection by RBC1023 and the mitochondrial function. First, our MTT assay results demonstrated that RBC1023 co-treatment was able to rescue the staurosporine-triggered loss of cell viability, suggesting RBC1023 restored the loss of the enzyme activity in mitochondria that reduces MTT during staurosporine treatment. Secondly, we found that co-treatment with RBC1023 and staurosporine resulted in a significant increase of cellular ATP content in comparison with the staurosporine treatment group. Our results suggest that RBC1023 restored the loss of ATP production during the staurosporine treatment. Furthermore, our results indicated that RBC1023 restored staurosporine-induced disruption of mitochondrial membrane potential. It is well known that mitochondrial dysfunction is the primary cause of staurosporine-induced apoptosis. A critical factor mediating mitochondrial dysfunction is the opening of mitochondrial PTP (mPTP). The opening of the mPTP can lead to a bioenergetic, biosynthetic, and redox crisis in a cell that can directly threaten the survival of the cell.28 When the mPTP is open, the mitochondrial inner membrane becomes permeable to protons, which then lead to the uncoupling of the electron respiratory chain and the collapse of membrane potential, which in turn leads to a cessation of ATP generation in mitochondria.28,29 In the RBC1023-pretreated NIH3T3 cells, the staurosporine-induced loss of mitochondrial membrane potential and decline in ATP levels was alleviated, supporting the notion that the cytoprotection of RBC1023 is, in part, due to the prevention of mitochondrial dysfunction. Upon activation of mPTP, functional breakdown and morphological disintegration of mitochondria occur, thus initiating cell death.30 Another threatening consequence of the altered mitochondrial permeability is the release of apoptogenic proteins from the mitochondrial inter-membrane space into the cytosol.28,29 Cytochrome c is associated with the inner mitochondrial membrane and serves as an essential component of the electron transfer chain. With opening of the mPTP and translocation of cytochrome c into the cytosol, mitochondrial function is compromised. However, in this study, the release of cytochrome c from the mitochondrial matrix into the cytosol by staurosporine was not significantly inhibited by RBC1023 (data not shown). This was possibly because caspase-3 is activated downstream of cytochrome c release and apoptosome formation in the mitochondrial pathway. It has been reported that B-cell lymphoma 2 protects endothelial cells against gamma radiation-induced caspase activation via a Raf-MEK-ERK cell survival signaling pathway that is independent of cytochrome c release.31 It is possible that RBC1023 also exerts its cytoprotective effect via some cell survival signaling pathways that are independent of the mechanism of the blockage of cytochrome c release. Taken together, we conclude that RBC1023, in addition to its direct caspase inhibitory effect, has some mitochondrial protective effect, which is at least partially responsible for its cytoprotection. For our next study, the mitochondrial oxygen consumption will be measured to directly evaluate mitochondrial function and cellular respiratory activity. Whether RBC1023’s mitochondrial protective effect is related to its caspase inhibitory effect could be an interesting subject for future study.
The DNA microarray analysis in the present study demonstrates that RBC1023 can recover the loss of expression of many genes induced by staurosporine treatment, correlating with its cytoprotection. The top 50 genes that were upregulated in the RBC1023-plus-staurosporine treatment group compared to the staurosporine-alone treatment group include several functional categories, such as cell differentiation and growth genes (Egr1, Bhlhe40, and Angptl2), cell cycle regulation genes (Ccnb1, Cdc25c, Cep55, Mastl, Cdkn3, Kif2c, and Plk1), cellular signaling genes (Rhob, Nek2, Taok1, Aurka, and Arhgef12), and transcription regulation genes (Tceal1, Depdc1a, and Atf7ip2), all of which are mainly responsible for cell growth and differentiation. While further studies need to be done to verify the roles of these genes by real time-quantitative polymerase chain reaction and/or Western blot, our current findings clearly suggest that upregulation of these pro-growth and pro-survival genes by RBC1023 plays a critical role in its cytoprotective effect.
RBC1023 is a selective, reversible, small-molecule caspase-3 inhibitor, which has multiple advantages compared to the known caspase inhibitors that are being developed. For example, the caspase inhibitors from Pfizer’s IDN6556 series (Pfizer, Inc., New York, NY, USA)32 and Vertex’s 166 series (Vertex Pharmaceuticals, Boston, MA USA)33 are irreversible, peptide-mimic and Pan-caspase inhibitors. Merck’s caspase-3 inhibitor M867 (Merck & Co., Inc., White House Station, NJ, USA) is selective and reversible, but a peptide mimic.34,35 Modified peptides generally do not make good therapeutic reagents, due to problems such as proteolysis and poor cellular permeability;36 therefore, selective small-molecule caspase inhibitors offer significant potential for protection of numerous cell types. Indeed, our preliminary results have shown that RBC1023 protects primary normal human oral keratinocytes from radiation-induced cell death using radiation colony formation assay (data not shown). During head and neck cancer therapy, oral mucositis caused by normal (nonmalignant) cell apoptosis is recognized as a major side effect of the therapy. Therefore, the potential use of cytoprotectants, such as the caspase inhibitors, for prevention of severe mucositis during head and neck cancer therapy could present a unique opportunity for effective therapy. In addition, caspases have also been demonstrated to be involved in some non-apoptotic functions, such as the regulatory events that are important for neural functions including axon pruning and synapse elimination, which are necessary to refine mature neuronal circuits.37 Small-molecule caspase inhibitor drugs may therefore have potential therapeutic or preventive efficacies against neurodegenerative diseases.
Conclusion
Taken together, our experimental study results indicate that RBC1023 protects NIH3T3 cells against staurosporine-induced apoptosis via inhibiting caspase activity, restoring mitochondrial membrane potential, and possibly upregulating some cell survival-related gene expressions.
Acknowledgments
We gratefully acknowledge Drs Kurumi Horiuchi and Yuan Wang for their early works in this project; Mr John Tobias from the Molecular Profiling Facility at University of Pennsylvania School of Medicine for performing the DNA microarray analysis; and Dr Honglin Zhou at University of Pennsylvania School of Medicine for providing the SHSY-5Y cell line and conducting the cell viability assay of SHSY-5Y cells in his lab. This work was partially supported by NIH Small Business Innovation Research grants 5R44DE017485 and 5R44CA140166 to HM.
Disclosure
The authors report no conflicts of interest in this work.
References
Pop C, Salvesen GS. Human caspases: activation, specificity, and regulation. J Biol Chem. 2009;284(33):21777–21781. | |
Cohen GM. Caspases: the executioners of apoptosis. Biochem J. 1997;326(Pt 1):1–16. | |
Nicholson DW, Thornberry NA. Caspases: killer proteases. Trends Biochem Sci. 1997;22(8):299–306. | |
Salvesen GS, Dixit VM. Caspases: intracellular signaling by proteolysis. Cell. 1997;91(4):443–446. | |
Nagata S. Apoptosis by death factor. Cell. 1997;88(3):355–365. | |
Green DR, Reed JC. Mitochondria and apoptosis. Science. 1998;281(5381):1309–1312. | |
Susin SA, Lorenzo HK, Zamzami N, et al. Molecular characterization of mitochondrial apoptosis-inducing factor. Nature. 1999;397(6718):441–446. | |
McBride CB, McPhail LT, Vanderluit JL, Tetzlaff W, Steeves JD. Caspase inhibition attenuates transection-induced oligodendrocyte apoptosis in the developing chick spinal cord. Mol Cell Neurosci. 2003;23(3):383–397. | |
Papaconstantinou HT, Xie C, Zhang W, et al. The role of caspases in methotrexate-induced gastrointestinal toxicity. Surgery. 2001;130(5):859–865. | |
Lee ER, Kim JH, Choi HY, Jeon K, Cho SG. Cytoprotective effect of eriodictyol in UV-irradiated keratinocytes via phosphatase-dependent modulation of both the p38 MAPK and Akt signaling pathways. Cell Physiol Biochem. 2011;27(5):513–524. | |
Park K, Lee JH. Protective effects of resveratrol on UVB-irradiated HaCaT cells through attenuation of the caspase pathway. Oncol Rep. 2008;19(2):413–417. | |
Pauloin T, Dutot M, Joly F, Warnet JM, Rat P. High molecular weight hyaluronan decreases UVB-induced apoptosis and inflammation in human epithelial corneal cells. Mol Vis. 2009;15:577–583. | |
Rass K, Reichrath J. UV damage and DNA repair in malignant melanoma and nonmelanoma skin cancer. Adv Exp Med Biol. 2008;624:162–178. | |
Smaili S, Hirata H, Ureshino R, et al. Calcium and cell death signaling in neurodegeneration and aging. An Acad Bras Cienc. 2009;81(3):467–475. | |
GeneChip® expression analysis: data analysis fundamentals [technical manual]. Sanra Clara, CA: Affymetrix Inc; 2004. | |
Zhang JH, Chung TD, Oldenburg KR. A simple statistical parameter for use in evaluation and validation of high throughput screening assays. J Biomol Screen. 1999;4(2):67–73. | |
Okun I, Malarchuk S, Dubrovskaya E, et al. Screening for caspase-3 inhibitors: effect of a reducing agent on identified hit chemotypes. J Biomol Screen. 2006;11(6):694–703. | |
Tamaoki T, Nomoto H, Takahashi I, Kato Y, Morimoto M, Tomita F. Staurosporine, a potent inhibitor of phospholipid/Ca++ dependent protein kinase. Biochem Biophys Res Commun. 1986;135(2):397–402. | |
Rüegg UT, Burgess GM. Staurosporine, K-252 and UCN-01: potent but nonspecific inhibitors of protein kinases. Trends Pharmacol Sci. 1989;10(6):218–220. | |
Belmokhtar CA, Torriglia A, Counis MF, Courtois Y, Jacquemin-Sablon A, Ségal-Bendirdjian E. Nuclear translocation of a leukocyte elastase Inhibitor/Elastase complex during staurosporine-induced apoptosis: role in the generation of nuclear L-DNase II activity. Exp Cell Res. 2000;254(1):99–109. | |
Yuste VJ, Sánchez-López I, Solé C, et al. The contribution of apoptosis-inducing factor, caspase-activated DNase, and inhibitor of caspase-activated DNase to the nuclear phenotype and DNA degradation during apoptosis. J Biol Chem. 2005;280(42):35670–35683. | |
Smaili SS, Hsu YT, Sanders KM, Russell JT, Youle RJ. Bax translocation to mitochondria subsequent to a rapid loss of mitochondrial membrane potential. Cell Death Differ. 2001;8(9):909–920. | |
Petit PX, Lecoeur H, Zorn E, Dauguet C, Mignotte B, Gougeon ML. Alterations in mitochondrial structure and function are early events of dexamethasone-induced thymocyte apoptosis. J Cell Biol. 1995;130(1):157–167. | |
Zamzami N, Marchetti P, Castedo M, et al. Sequential reduction of mitochondrial transmembrane potential and generation of reactive oxygen species in early programmed cell death. J Exp Med. 1995;182(2):367–377. | |
Wang X. The expanding role of mitochondria in apoptosis. Genes Dev. 2001;15(22):2922–2933. | |
Karachitos A, García Del Pozo JS, de Groot PW, Kmita H, Jordán J. Minocycline mediated mitochondrial cytoprotection: premises for therapy of cerebrovascular and neurodegenerative diseases. Curr Drug Targets. 2013;14(1):47–55. | |
Brenner C, Ventura-Clapier R, Jacotot E. Mitochondria and cytoprotection. Biochem Res Int. 2012;2012:351264. | |
Armstrong JS. The role of the mitochondrial permeability transition in cell death. Mitochondrion. 2006;6(5):225–234. | |
Galluzzi L, Morselli E, Kepp O, Kroemer G. Targeting post-mitochondrial effectors of apoptosis for neuroprotection. Biochim Biophys Acta. 2009;1787(5):402–413. | |
Kristián T. Metabolic stages, mitochondria and calcium in hypoxic/ischemic brain damage. Cell Calcium. 2004;36(3–4):221–233. | |
Kumar P, Coltas IK, Kumar B, Chepeha DB, Bradford CR, Polverini PJ. Bcl-2 protects endothelial cells against gamma-radiation via a Raf-MEK-ERK-survivin signaling pathway that is independent of cytochrome c release. Cancer Res. 2007;67(3):1193–1202. | |
Hoglen NC, Chen LS, Fisher CD, Hirakawa BP, Groessl T, Contreras PC. Characterization of IDN-6556 (3-[2-(2-tert-butyl-phenylaminooxalyl)-amino]-propionylamino]-4-oxo-5-(2,3,5,6-tetrafluoro-phenoxy)-pentanoic acid): a liver-targeted caspase inhibitor. J Pharmacol Exp Ther. 2004;309(2):634–640. | |
Weber P, Wang P, Maddens S, et al. VX-166: a novel potent small molecule caspase inhibitor as a potential therapy for sepsis. Crit Care. 2009;13(5):R146. | |
Méthot N, Huang J, Coulombe N, et al. Differential efficacy of caspase inhibitors on apoptosis markers during sepsis in rats and implication for fractional inhibition requirements for therapeutics. J Exp Med. 2004;199(2):199–207. | |
Kim KW, Moretti L, Lu B. M867, a novel selective inhibitor of caspase-3 enhances cell death and extends tumor growth delay in irradiated lung cancer models. PLoS One. 2008;3(5):e2275. | |
Callus BA, Vaux DL. Caspase inhibitors: viral, cellular and chemical. Cell Death Differ. 2007;14(1):73–78. | |
Hyman BT, Yuan J. Apoptotic and non-apoptotic roles of caspases in neuronal physiology and pathophysiology. Nat Rev Neurosci. 2012;13(6):395–406. |
Supplementary material
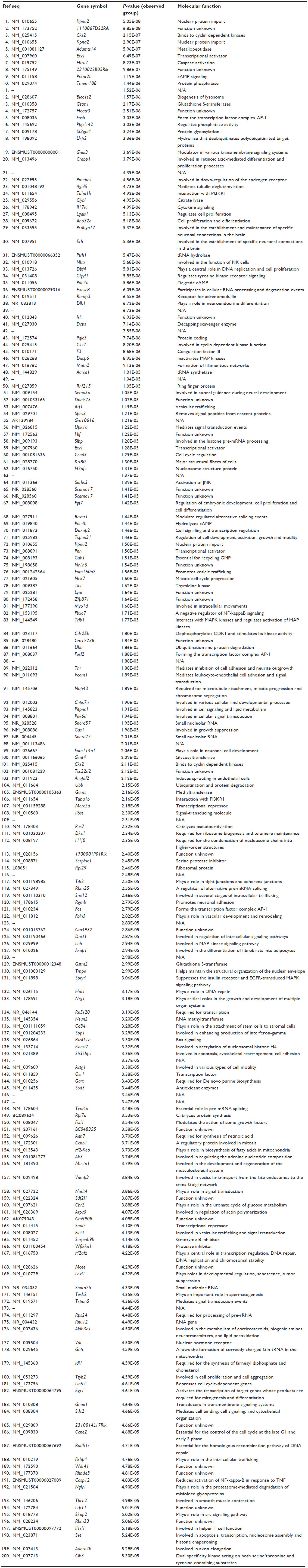 | Table S1 Molecular functions of the top 200 upregulated and downregulated clustering genes in the heat map |
 © 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.