Back to Journals » Clinical, Cosmetic and Investigational Dermatology » Volume 16
Cytophagic Histiocytic Panniculitis Presenting as Subcutaneous Nodules and Generalized Edema – A Case Report
Authors Yang J, Chen L, Shi R, Zhao X, Pan M, Zheng J
Received 20 September 2023
Accepted for publication 28 November 2023
Published 12 December 2023 Volume 2023:16 Pages 3541—3545
DOI https://doi.org/10.2147/CCID.S437208
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Dr Anne-Claire Fougerousse
Jiayi Yang,* Lihong Chen,* Ruofei Shi, Xiaoqing Zhao, Meng Pan, Jie Zheng
Department of Dermatology, School of Medicine, Ruijin Hospital, Shanghai Jiao Tong University, Shanghai, People’s Republic of China
*These authors contributed equally to this work
Correspondence: Meng Pan, Email [email protected]
Background: Cytophagic histiocytic panniculitis (CHP) is a rare form of nodular panniculitis characterized by clinical manifestations such as skin erythema, nodules, fever, pancytopenia, liver failure, plasmacytosis, and hepatosplenomegaly. We report a case of CHP that was initially misdiagnosed as subcutaneous panniculitis-like T-cell lymphoma (SPTCL) but achieved complete remission with a favorable prognosis.
Methods: A 38-year-old female presented to the dermatology department with a 15-day history of subcutaneous nodules, generalized edema, and continuous fever.
Results: The patient was diagnosed as CHP combined with hemophagocytic syndrome by typical clinical manifestations, low value of SUVmax in positron emission tomography/computed tomography (PET/CT), benign differentiated T cells, negative TCR gene rearrangement, pancytopenia, abnormal coagulation, hypertriglyceridemia, decreased NK cell count, impaired liver function, and the presence of hemophagocytic cells observed in bone biopsy smears.
Conclusion: In our case, the patient presented with hemophagocytic syndrome with hemodynamic instability, indicating an intensive treatment is needed. The diagnosis of SPTCL necessitates a meticulous process of differential diagnosis, along with the cautious administration of an aggressive chemotherapy regimen. Extended follow-up is imperative to ascertain the long-term outcomes.
Keywords: cytophagic histiocytic panniculitis, SPTCL, HLH
Cytophagic histiocytic panniculitis (CHP) is a rare form of nodular panniculitis characterized by lobular panniculitis, which involves inflammation of the subcutaneous fat.1 Its clinical manifestations include skin erythema, nodules, fever, pancytopenia, liver failure, plasmacytosis, and hepatosplenomegaly. When combined with hemophagocytic syndrome, CHP can be fatal.2 The etiology of CHP remains unclear, although it has been associated with unknown T cell disorders, EBV infection, and connective tissue diseases. Here, we report a case of CHP that was initially suspected as subcutaneous panniculitis-like T-cell lymphoma (SPTCL) but achieved complete remission with a favorable prognosis.
A 38-year-old female presented to the dermatology department with a 15-day history of subcutaneous nodules, generalized edema, and continuous fever. The external examination revealed elevated levels of lactic dehydrogenase, reduced platelet count, abnormal disseminated intravascular coagulation (DIC), positive Epstein-Barr virus (EBV) IgM antibody, and a positron emission tomography/computed tomography (PET/CT) indicated widespread subcutaneous nodules with a maximum standardized uptake value(SUVmax) of 2.8. Additionally, multiple lymph nodes exhibited enlargement with SUVmax values ranging from 3.3 to 3.9, suggesting a potential diagnosis of subcutaneous panniculitis-like T-cell lymphoma. Despite treatment with dexamethasone (5mg) and methylprednisolone (80mg/day) for 5 days, along with antibiotics and antiviral therapy, her condition remained unchanged.
On admission, the patient presented with a fever of 39.3°C, mild tachycardia (108/min), and normal blood pressure (107/66 mmHg). Lymphadenopathy and hepatosplenomegaly were observed upon examination. Physical examination revealed generalized subcutaneous nodules and edema. Disseminated irregular painful nodules measuring 1 to 2cm were mainly found on the trunk and extremities (Figure 1). At her hospitalization, the patient experienced oliguria (600 mL/day) with a weight gain of 10 kg. Her laboratory results revealed progressive pancytopenia, abnormal DIC (elevated D-Dimer, APTT and PT, Decreased fibrinogen), hypertriglyceridemia, decreased NK cell count and impaired liver function. In addition, serum ferritin, lactate dehydrogenase, C-reactive protein, serum amyloid A and serum interleukin-2 receptor were all increased. Bacterial and fungal cultures of the blood and skin tissue were negative. Additional negative blood tests included EBV antibody, peripheral blood pathogens NGS. Other autoimmune antibody, such as immunoglobulin, complement, rheumatoid factor(RF), double-stranded deoxyribonucleic acid, antinuclear antibody(ANA) and extractable nuclear antigen(ENA), had normal results.
 |
Figure 1 Clinical presentation of cytophagic histiocytic panniculitis lesions. (A) Symmetric edematous erythematous plaque on cheeks; (B and C) Subcutaneous nodules and edema on abdomen and legs. |
Bone marrow smear showed hemophagocytic cell and giant lineage proliferation with impaired maturation. Flow cytometry, histopathology, and chromosome test of bone marrow were all normal. A biopsy of the nodules was performed, suggesting histiocytophagy (“beanbag-like” cells), degeneration of adipocytes within the fat lobules, infiltration with small mature lymphocytes and histiocytes. Immunohistochemistry showed CD3(+), CD8(+), GranzymeA(+), TIA-1(+), Ki67(<5%+), CD4(-), Perforin(-), EBER(-), CD56(-), and negative TCR gene rearrangement (Figure 2A and B).
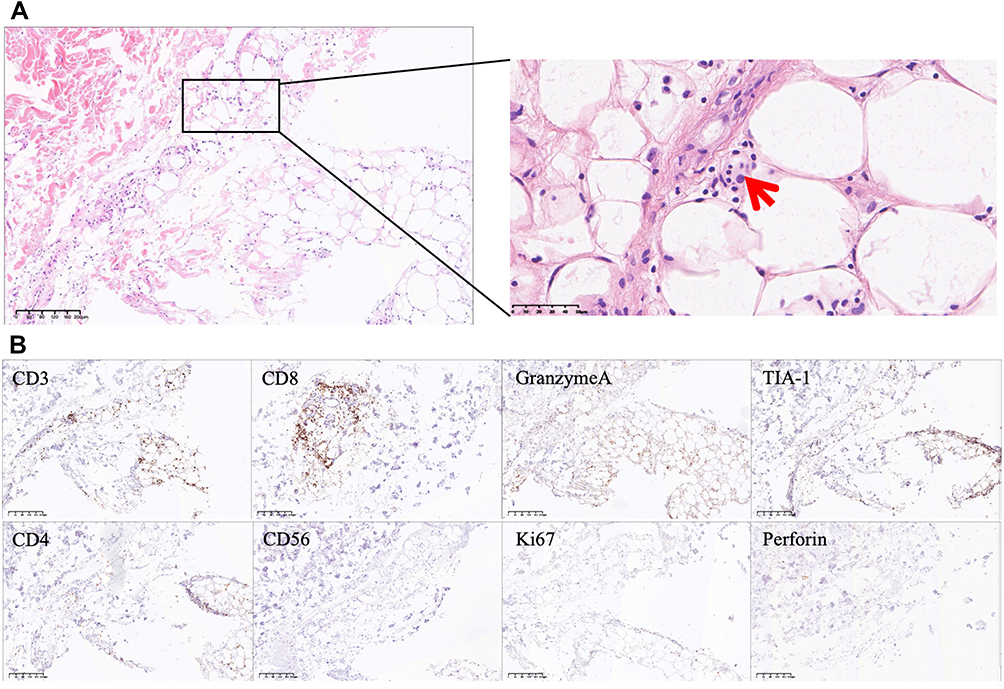 |
Figure 2 Hematoxylin-Eosin (HE) and immunohistochemical staining of skin biopsy. (A) HE staining showed histiocytophagy (“beanbag-like” cells, red arrow), degeneration of adipocytes within the fat lobules, infiltration with small mature lymphocytes and histiocytes. (B) Increased expression of CD3, CD8, GranzymeA and TIA-1, negative expression of CD4, CD56, Ki67 and Perforin in immunohistochemical staining. The scale bar is 200 μm for the 20X images. |
In summary, the patient’s condition can be characterized by several key factors: 1) Clinical manifestations: The patient presented with scattered painful erythema nodules throughout the body. PET/CT images revealed the presence of diffuse plaque-like deposits throughout the body with mild to moderate metabolic elevation. 2) Skin histopathology of erythema nodules displayed beanbag cells, panniculitis-like changes, and no lymphocyte atypia. 3) TCR gene rearrangement of erythema nodules in the skin showed negative results. 4) Bone marrow assessment, including flow cytometry, culture, and pathology, did not reveal any abnormalities. 5) Hemophagocytic syndrome (HLH) complication: the patient also presented with several HLH-associated features, including high fever, progressive cytopenia, abnormal DIC, hypertriglyceridemia, elevated ferritin levels, increased IL-2R, reduced NK cell count, abnormal liver function, splenomegaly, and the presence of hemophagocytic cells observed in bone biopsy smears. All the evidence leads to the diagnosis of CHP combined with HLH.
The patient underwent a less aggressive treatment regimen following the HLH-94 protocol, which involved daily administration of dexamethasone (10mg), VP16 (0.18g), and cyclosporine (100mg twice a day). Within two weeks, her systemic symptoms and hematologic profile showed significant improvement without any severe adverse effects. Subsequently, systemic glucocorticoids were gradually reduced, and the patient was maintained with a daily dose of 10mg prednisone after twelve months. Regular follow-up visits revealed no signs of relapse.
The term “CHP” was first proposed by Winkelmann et al in 1980.3 It is characterized by the presence of tender multiple subcutaneous nodules, high fever, hepatosplenomegaly, pancytopenia, bleeding, abnormal hemagglutination, and infiltration of histiocytes within the subcutaneous adipose tissue, resulting in a rare form of panniculitis. A distinctive feature observed in the histopathology of this disease is the phagocytosis of blood components by histiocytes, which leads to the formation of “bean bag” cells. The underlying cause of this condition involves infiltration of phagocytic cells within the subcutaneous adipocytes, often affecting multiple organs and resulting in symptoms such as subcutaneous nodules, fever, pancytopenia, bleeding, and liver and kidney failure. Interestingly, the subcutaneous lesions in histiocytophagocytic panniculitis contain significant numbers of benign T cells. This suggests a possible association with T-cell monoclonal proliferative disease as a potential pathogenic mechanism. However, it is important to note that there is currently no definitive evidence to support this speculation. Recently, there has been speculation that CHP may represent a natural progression of SPTCL.4,5
SPTCL is a form of cutaneous malignant lymphoma originating from cytotoxic T cells. This condition primarily affects subcutaneous adipose tissue, without involvement of the epidermis and dermis. Clinically, it typically presents as subcutaneous plaques, nodules, or skin ulcers, often accompanied by pain. These manifestations are most commonly observed in the limbs and trunk and are frequently associated with HLH.5,6 HLH is a frequently fatal complication that can be characterized by high fever, maculopapular eruption, growth retardation, CNS symptoms, hepatosplenomegaly, lymph node enlargement, hemocytopenia, clotting disorders, abnormal liver function tests, and significantly elevated levels of serum ferritin and soluble IL-2 receptor. Germline mutations disrupting the expression of the immune checkpoint molecule TIM3 are associated with both HLH and SPTCL.7 When SPTCL is associated with HLH, the prognosis is generally unfavorable. However, when CHP is combined with HLH, the prognosis tends to be more favorable and are less likely to experience the severe multi-organ involvement commonly observed in HLH cases. Besides, specific pathological characteristics of SPTCL consist of hyperchromatic atypical lymphocytes surrounding individual adipocytes in a nucleolar fashion. Immunohistochemical analysis reveals cytotoxic T cell immunophenotypes, with tumor cells predominantly expressing CD3, CD8, TCRβ, and cytotoxic granuloprotein, while being negative for CD4 and CD56. It also shows cytotoxic granuloplasmin granzyme B, T-cell intracellular antigen-1, and perforin. However, in 10%, 50%, and 44% of cases, CD2, CD5, and CD7 are absent, respectively.2,6 Confirming the diagnosis can be facilitated by detecting clonal TCR gene rearrangements. The key distinction lies in the histopathological presence of benign differentiated T cells in patients with CHP, compared to the malignant nature of SPTCL.
In addition to SPTCL, it is pertinent to consider the possibility of the following diseases in this patient: 1) Adult Still’s disease: the patient did not exhibit arthralgia or pharyngeal discomfort. The nodules presented atypically, leading to its exclusion from consideration. 2) Lupus panniculitis: this condition often accompanied by adipose tissue atrophy. The histopathological findings indicated lobular panniculitis-like alterations. Negativity of laboratory test in our patient helped to differentiate. 3) Infection-related disease: no evidence was found.
CHP currently lacks an effective treatment. However, the recommended approach involves using glucocorticoids in combination with immunosuppressants, such as cyclosporin A, azathioprine, etoposide, and others.2,8 Among the 23 reported cases of CHP, the fatality rate stands at 17.39%. Glucocorticoid therapy alone showed a remission rate of 20%. In comparison, glucocorticoids combined with immunosuppressants exhibited an 88.24% response rate. The CHOP-E regimen (consisting of cyclophosphamide, vincristine, doxorubicin, prednisone, and etoposide) achieved a 100% response rate, and the combination of glucocorticoids with cyclosporin A attained an 83% response rate. A maximum follow-up period of 35 months revealed no recurrence. Additionally, there have been reports of successful treatment of recurrent and refractory CHP using biologics and autologous peripheral blood stem cell transplantation.3 In this case, following a diagnosis of CHP complicated with HLH, the patient received a less aggressive treatment regimen following the HLH-94 therapy for 4 weeks. The therapeutic effect proved satisfactory with no recurrence observed during the 1-year follow-up. However, it is essential to remain vigilant for the development of SPTCL and continue careful clinical monitoring of the patient.
We present this case to add to current data regarding the patient presented CHP with HLH, indicating an intensive treatment is needed. The diagnosis of SPTCL necessitates a meticulous process of differential diagnosis, along with the cautious administration of an aggressive chemotherapy regimen, in order to minimize the risk of additional harm to patients. As no underlying primary disorder was found, particularly after excluding SPTCL, a less aggressive therapy was sufficient and was used to avoid fatal side effects. However, extended follow-up is imperative to ascertain the long-term outcomes.
Summary
- Dermatologists should be aware that the diagnosis of SPTCL necessitates a meticulous process of differential diagnosis, along with the cautious administration of an aggressive chemotherapy regimen.
- The key distinction lies in the histopathological presence of benign differentiated T cells with negative TCR gene rearrangement in patients with CHP, compared to the malignant nature of SPTCL.
- As no underlying primary disorder was found, particularly after excluding SPTCL, a less aggressive therapy was sufficient and was used to avoid fatal side effects. Extended follow-up is imperative to ascertain the long-term outcomes.
Ethics Statement
The written informed Consent has been provided by the patient to have the case details and any accompanying images published.
Disclosure
The authors declare that they do not have conflict of interest in this work.
References
1. Alegre VA, Winkelmann RK. Histiocytic cytophagic panniculitis. J Am Acad Dermatol. 1989;20(2 Pt 1):177–185. doi:10.1016/S0190-9622(89)70018-9
2. Aronson IK, Worobec SM. Cytophagic histiocytic panniculitis and hemophagocytic lymphohistiocytosis: an overview. Dermatol Ther. 2010;23(4):389–402. doi:10.1111/j.1529-8019.2010.01339.x
3. Winkelmann RK, Bowie EJ. Hemorrhagic diathesis associated with benign histiocytic, cytophagic panniculitis and systemic histiocytosis. Arch Intern Med. 1980;140(11):1460–1463. doi:10.1001/archinte.1980.00330220038015
4. Baxi KD, Rathod SP, Chaudhary RG, Jagati A. Subcutaneous panniculitis-like T-cell lymphoma. Indian J Dermatol Venereol Leprol. 2020;86(5):606. doi:10.4103/ijdvl.IJDVL_635_18
5. Gallardo F, Pujol RM. Subcutaneous panniculitic-like T-cell lymphoma and other primary cutaneous lymphomas with prominent subcutaneous tissue involvement. Dermatol Clin. 2008;26(4):529–540, viii. doi:10.1016/j.det.2008.05.008
6. Willemze R, Jansen PM, Cerroni L, et al. Subcutaneous panniculitis-like T-cell lymphoma: definition, classification, and prognostic factors: an EORTC Cutaneous Lymphoma Group Study of 83 cases. Blood. 2008;111(2):838–845. doi:10.1182/blood-2007-04-087288
7. Gayden T, Sepulveda FE, Khuong-Quang DA, et al. Germline HAVCR2 mutations altering TIM-3 characterize subcutaneous panniculitis-like T cell lymphomas with hemophagocytic lymphohistiocytic syndrome. Nat Genet. 2018;50(12):1650–1657. doi:10.1038/s41588-018-0251-4
8. Pasqualini C, Jorini M, Carloni I, et al. Cytophagic histiocytic panniculitis, hemophagocytic lymphohistiocytosis and undetermined autoimmune disorder: reconciling the puzzle. Ital J Pediatr. 2014;40(1):17. doi:10.1186/1824-7288-40-17
 © 2023 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2023 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.