Back to Journals » International Journal of Chronic Obstructive Pulmonary Disease » Volume 15
CYP1B1, VEGFA, BCL2, and CDKN1A Affect the Development of Chronic Obstructive Pulmonary Disease
Authors Yang D, Yan Y, Hu F, Wang T ![]()
Received 25 June 2019
Accepted for publication 30 December 2019
Published 23 January 2020 Volume 2020:15 Pages 167—175
DOI https://doi.org/10.2147/COPD.S220675
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Chunxue Bai
Danlei Yang, 1 Ying Yan, 2 Fen Hu, 1 Tao Wang 1
1Department of Respiratory and Critical Care Medicine, Tongji Hospital, Tongji Medical College, Huazhong University of Science and Technology, Wuhan 430030, People’s Republic of China; 2Department of Respiratory and Critical Care Medicine, Ningxia People’s Hospital, Yinchuan 750002, People’s Republic of China
Correspondence: Tao Wang
Department of Respiratory and Critical Care Medicine, Tongji Hospital, Tongji Medical College, Huazhong University of Science and Technology, 1095 Jiefang Avenue, Wuhan 430030, People’s Republic of China
Tel +86-13971477320
Email [email protected]
Purpose: Chronic obstructive pulmonary disease (COPD) is a progressive lung disease characterized by poor airflow. The purpose of this study was to explore the mechanisms involved in the development of COPD.
Patients and Methods: The mRNA expression profile GSE100281, consisting of 79 COPD and 16 healthy samples, was acquired from the Gene Expression Omnibus database. The differentially expressed genes (DEGs) between COPD samples and healthy samples were analyzed using the limma package. Functional enrichment analysis for the DEGs was carried out using the Database for Annotation, Visualization, and Integrated Discovery (DAVID) tool. Furthermore, DEG-compound pairs were predicted using the Comparative Toxicogenomics Database. The KEGG metabolite IDs corresponding to the compounds were also obtained through the MetaboAnalyst pipeline. Based on the diffusion algorithm, the metabolite network was constructed. Finally, the expression levels of key genes were determined using quantitative PCR (qPCR).
Results: There were 594 DEGs identified between the COPD and healthy samples, including 242 upregulated and 352 downregulated genes. A total of 696 DEG-compound pairs, such as BCL2-C00469 (ethanol) and BCL2-C00389 (quercetin) pairs, were predicted. CYP1B1, VEGFA, BCL2, and CDKN1A were included in the top 10 DEG-compound pairs. Additionally, 57 metabolites were obtained. In particular, hsa04750 (inflammatory mediator regulation of TRP channels)-C00469 (ethanol) and hsa04152 (AMPK signaling pathway)-C00389 (quercetin) pairs were found in the metabolite network. The results of qPCR showed that the expression of CYP1B1, VEGFA, BCL2, and CDKN1A was consistent with that predicted using bioinformatic analysis.
Conclusion: CYP1B1, VEGFA, BCL2, and CDKN1A may play important functions in the development and progression of COPD.
Keywords: chronic obstructive pulmonary disease, differentially expressed genes, enrichment analysis, disease metabolites, metabolite network
Introduction
Chronic obstructive pulmonary disease (COPD), which manifest as either chronic bronchitis or emphysema, is characterized by poor airflow.1 The primary symptoms of COPD are cough, shortness of breath, and expectoration.2,3 COPD is a progressive disease that may further develop into cor pulmonale and respiratory failure.4,5 Tobacco smoking and air pollution are the main risk factors of COPD, while metabolic disturbances have a critical influence on this disease.6 COPD is also associated with increased chronic inflammatory responses to toxic particles or gases in the airways and lungs.7 In 2015, COPD has impacted 174.5 million people that lead to 3.2 million deaths globally.8 Thus, the pathogenesis of COPD should be further investigated to improve COPD treatment strategies.
The expression of pleomorphic adenoma gene like-2 (PLAGL2) is significantly higher in COPD patients than that in healthy smokers, suggesting that PLAGL2 overexpression may be involved in the mechanisms underlying the development and progression of COPD.9 Further, overexpression of nuclear factor-κB2 (NF-κB2) is related to the expression of cyclooxygenase-2 (COX-2), interleukin-1 beta (IL-1β), and interleukin-8 (IL-8), which are also associated with the pathogenesis of COPD.10,11 Prostaglandin E2 (PGE2) is an important cyclic component that is responsible for fibroblast senescence and inflammation in COPD patients. Regulation of the PGE2 signaling pathway may suppress fibroblast senescence and inflammation during disease progression.12 Moreover, the interleukin-4/interleukin-13 (IL-4/IL-13) receptor complex induces the overproduction of mucus and enhances the viscosity of the airway surface liquid in patients with COPD and bronchial asthma by promoting pendrin expression.13 However, despite the previous reports, the mechanisms involved in the development of COPD have not been fully elucidated.
Willis-Owen et al generated the COPD-related dataset GSE100281 to analyze global gene transcription. In their study, the differential gene expression between healthy and COPD samples was analyzed using limma package, while the patterns of transcriptional coordination were analyzed using weighed gene co-expression network analysis (WGCNA). They found 1826 transcripts showing COPD-related variations, where 18 transcripts exhibited more than 2-fold change in expression.14 In the current study, the GSE100281 dataset was analyzed again. However, the study differed from their analyses in that enrichment analysis followed the screening for the differentially expressed genes (DEGs) between COPD and healthy samples. Additionally, prediction of disease metabolites and metabolite network analysis were also performed, which were not mentioned in the study by Willis-Owen et al The findings of our study might help establish the theoretical foundation for developing novel therapies for COPD.
Materials and Methods
Data Source
The mRNA expression profile GSE100281 (sequencing platform: GPL11532
[HuGene-1_1-st] Affymetrix Human Gene 1.1 ST Array [transcript (gene) version]) was extracted from Gene Expression Omnibus (GEO, http://www.ncbi.nlm.nih.gov/geo/) database. GSE100281 is composed of mRNA expression profiles from 79 COPD and 16 healthy samples.
Data Preprocessing and Differential Expression Analysis
The raw data expression matrix was obtained using the R Affy package (version 1.56.0, http://bioconductor.org/packages/release/bioc/html/affy.html).15 Data preprocessing, including background adjustment, quantile normalization, final summarization, and log base 2 scaling, was conducted using the robust multi-array average (RMA) method.16 Combined with the platform annotation files, probe IDs were mapped to the gene symbols. Probes without corresponding gene symbols were removed. For several probes that matched with only one gene symbol, the mean expression value of the probes was calculated and considered as the final gene expression value.
Using the R limma package (version 3.34.9, http://bioconductor.org/packages/release/bioc/html/limma.html), differential expression analysis was performed on COPD and healthy samples.17 The p-values obtained using t-test were adjusted according to the Benjamini & Hochberg method.18,19 Genes with adjusted p-values < 0.05 were considered as differentially expressed.
Functional Enrichment Analysis
To identify the Gene Ontology (GO) functional terms and Kyoto Encyclopedia of Genes and Genomes (KEGG) pathways involved with COPD, the DEGs were analyzed using Database for Annotation, Visualization, and Integrated Discovery (DAVID) tool (version 6.8, https://david.ncifcrf.gov/).20–22 GO terms and KEGG pathways with p-value < 0.05 and count > 1 were considered significant.
Prediction of Disease Metabolites
Using “Pulmonary Disease” and “Chronic Obstructive” as key words, the genes and compounds directly correlated with COPD were searched from the Comparative Toxicogenomics Database (http://ctd.mdibl.org/).23 The DEGs-associated compounds with Reference.Count > 1 were screened. The KEGG metabolite IDs corresponding to the compounds were also obtained using the Compound ID Conversion tool in the MetaboAnalyst database (http://www.metaboanalyst.ca).24
Metabolite Network Analysis
The R package was used for pathway enrichment analysis of the identified disease metabolites.25 Utilizing KEGG database as the background, a differential pathway network composed of pathways, compounds, reactions, enzymes, and KEGG modules was built according to the results of the functional enrichment analysis.22 Based on the diffusion algorithm, heat flows from a given metabolite node and diffuses to the differential pathway network.26 Heat diffusion caused the change of node scores in the differential pathway network. The p-values of the diffusion scores were obtained using the displacement test.27 The top 100 nodes with p-value < 0.05 were selected for constructing the gene-metabolite network.
The formula for calculating the diffusion scores was T = -KI - 1*G, where G is the inputted metabolite (ie if it is the inputted metabolite, G is 1; otherwise, G is 0) and KI is the matrix.
Validation with CTD Database
The genes directly associated with COPD were extracted from the CTD database. The parameter was set with an inference score > 5. The inference score reflects the degree of similarity between the CTD chemical–gene–disease networks and a similar scale-free random network. A high score means a high probability that the inference network has atypical connectivity. Furthermore, Venn diagram analysis was performed to identify the genes shared between the CTD databases and the identified DEGs.
Quantitative PCR (qPCR)
Based on the bioinformatics analysis results, the expression of key genes were verified in six CAD and five normal blood samples. Total RNA was extracted using TRIzol reagent (Invitrogen, Carlsbad, CA, USA) following the manufacturer’s instructions. The purity and quality of the extracted RNA were determined using the Infinite M100 PRO microplate reader ((Tecan, Männedorf, Switzerland). Complementary DNA (cDNA) was synthesized from high quality total RNA using PrimeScript™ RT Master Mix (No. RR036A, Takara Bio USA, Mountain View, CA, USA). Real-time qPCR was performed to validate gene expression using Power SYBR™ Green PCR Master Mix (No. A25742, Thermo Fisher Scientific, Waltham, MA, USA) on the 7900HT Fast Real-Time PCR System (Applied Biosystems, Foster City, CA, USA), with the following thermal cycling conditions: 50 °C for 2 min, 95 °C for 10 min, followed by 40 cycles at 95 °C for 10 s and 60 °C for 30 s. The melting curve was analyzed from 60 °C to 95 °C at an increment of 0.5 °C/10 s. The primer sequences for the genes were as follows: CYP1B1, forward 5ʹ-GGCAGAATTGGATCAGGTCG-3ʹ; CYP1B1, reverse 5ʹ-TGTGGTAGCCCAAGACAGAGG-3ʹ; VEGFA, forward 5ʹ-GAGGGCAGAATCATCACGAAG-3ʹ; VEGFA, reverse 5ʹ-CACCAGGGTCTCGATTGGAT-3ʹ; BCL2, forward 5ʹ-GACTTCGCCGAGATGTCCAG-3ʹ; BCL2, reverse 5ʹ-GGTGCCGGTTCAGGTACTCA-3ʹ; CDKN1A, forward 5ʹ-TGGCACCTCACCTGCTCTG-3ʹ; CDKN1A, reverse 5ʹ-CGGCGTTTGGAGTGGTAGA-3ʹ; GAPDH, forward 5ʹ-TGACAACTTTGGTATCGTGGAAGG-3ʹ; GAPDH, reverse 5ʹ-AGGCAGGGATGATGTTCTGGAGAG-3ʹ. The relative quantification of gene expression was based on the comparative CT (2−ΔΔCT) method. Statistical analysis was performed using GraphPad Prism 5 (GraphPad Software, San Diego, CA, USA). Data were presented as mean ± standard deviation (SD), with a statistical significance of P < 0.05.
Results
Differential Expression Analysis
Box plots showed that the medians of data after normalization were at the same level (Figure 1). Therefore, data preprocessing improved the quality of in silico data used for bioinformatics analysis. There were 594 DEGs, including 242 upregulated and 352 downregulated genes, between the COPD and healthy samples. The heatmap and volcano plot displaying the DEGs are presented in Figure 2.
 |
Figure 1 Box plots showing the medians of the mRNA expression data before and after normalization. The red and blue plots represent the chronic obstructive pulmonary disease (COPD) and healthy samples, respectively. |
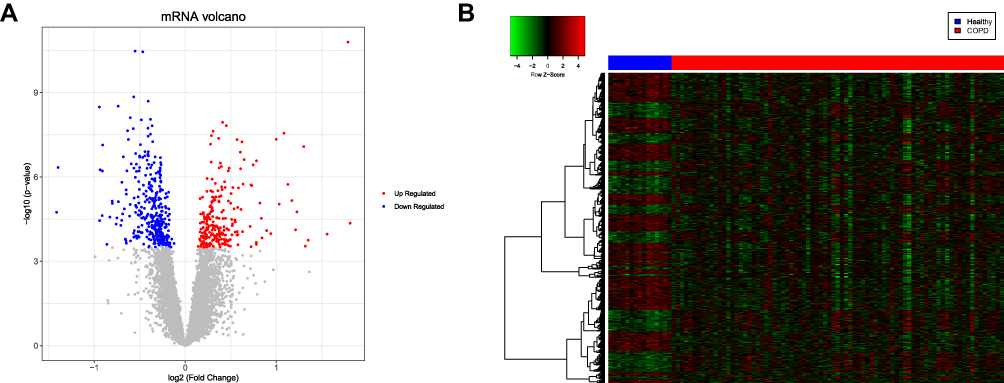 |
Figure 2 Volcano plot and heatmap displaying the differentially expressed genes (DEGs). (A) Volcano plot of the DEGs. The red and blue dots represent the upregulated and downregulated genes, respectively. (B) Heatmap of the DEGs. The red and blue sample strips represent the chronic obstructive pulmonary disease (COPD) and healthy samples, respectively. |
Functional Enrichment Analysis
The upregulated and downregulated genes were separately subjected to functional enrichment analysis. The top 5 results of GO terms and KEGG pathway analysis are shown in Figure 3. The upregulated genes were mainly implicated in the endocytosis pathway, with top GO terms being associated with signal transduction (GO_biological process, GO_BP), membrane (GO_cellular component, GO_CC), and protein binding (GO_molecular function, GO_MF). However, the downregulated genes were involved in the oxidative phosphorylation pathway, with top GO terms being associated with mitochondrial electron transport, NADH to ubiquinone (GO_BP), mitochondrion (GO_CC), and NADH dehydrogenase (ubiquinone) activity (GO_MF).
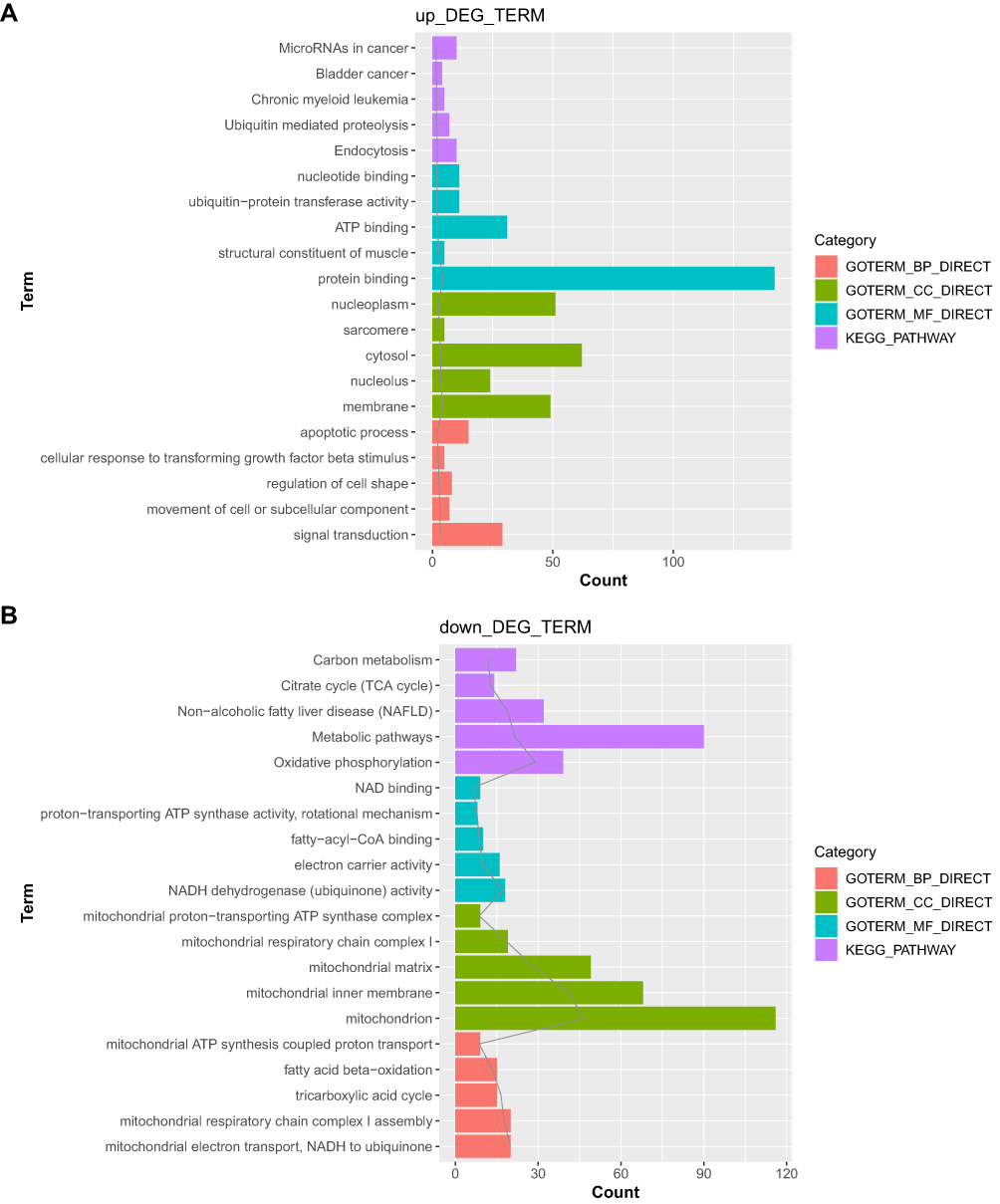 |
Figure 3 The Gene Ontology (GO)_biological process (BP), GO_cellular component (CC), and GO_molecular function (MF) functional terms and Kyoto Encyclopedia of Genes and Genomes (KEGG) pathways identified after functional enrichment analysis of the differentially expressed genes (DEGs). (A) Top 5 results for the upregulated genes. (B) Top 5 results for the downregulated genes. |
Prediction of Disease Metabolites
From the 21,555 genes and 4900 compounds directly related to COPD, a total of 123 compounds, 243 DEGs, and 696 DEG-compound pairs were screened. The top 10 DEG-compound pairs, including cytochrome P450 family 1 subfamily B member 1 (CYP1B1), vascular endothelial growth factor A (VEGFA), B cell leukemia/lymphoma 2 (BCL2), and cyclin dependent kinase inhibitor 1A (CDKN1A), are presented in Table 1. Furthermore, 57 compounds with corresponding KEGG metabolite IDs were obtained. In particular, both C00469 (ethanol) and C00389 (quercetin) were found to be related with BCL2.
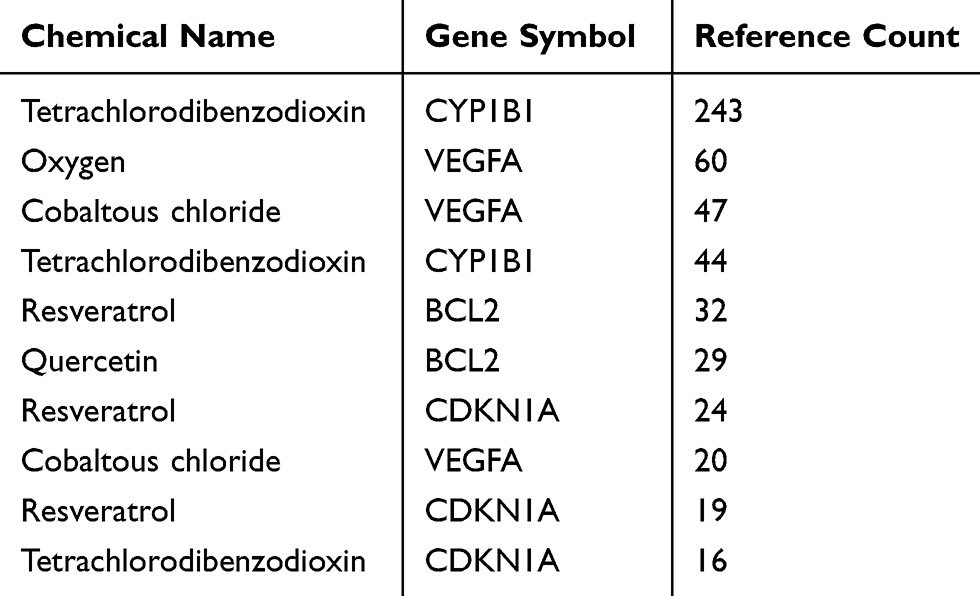 |
Table 1 The Top 10 Differentially Expressed Gene (DEG)-Compound Pairs |
Metabolite Network Analysis
Based on the differential pathway network, there were 9994 nodes and 33,828 edges identified. Among the 57 metabolites, 23 metabolites matched with nodes and edges in the differential pathway network. In addition, a gene-metabolite network composed of 77 nodes and 106 edges was constructed (Figure 4). Particularly, the hsa04750 (inflammatory mediator regulation of TRP channels)-C00469 (ethanol) and hsa04152 (AMPK signaling pathway)-C00389 (quercetin) pairs were discovered in the network.
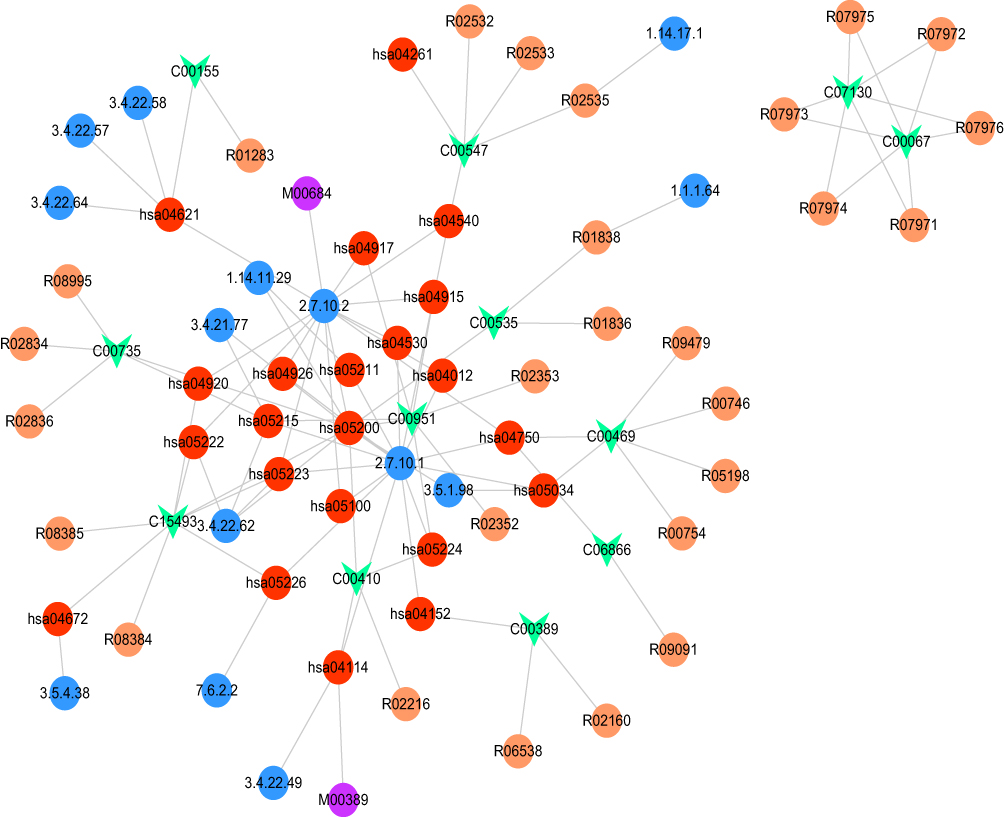 |
Figure 4 Constructed gene-metabolite network. The red, purple, blue, and orange circles and the green inverted triangle represent the pathway, module, enzyme, reaction, and disease metabolite, respectively. |
Validation with CTD Database
A total of 12,455 COPD-associated genes were obtained from the CTD database. The results of Venn diagram analysis are shown in Figure 5 and Supplemental Table 1. There were 221 shared genes, including CYP1B1, VEGFA, BCL2, and CDKN1A, between the COPD-associated genes and the 243 DEGs.
 |
Figure 5 Results of the Venn diagram analysis. |
Expression Levels of Key Genes
Based on in silico analysis, the key genes CYP1B1, VEGFA, BCL2, and CDKN1A were selected for validation using qPCR. As shown in Figure 6, the expression levels of CYP1B1 and CDKN1A were significantly upregulated (P < 0.05) in COPD samples than in the control. By contrast, the expression levels of BCL2 and VEGFA were significantly downregulated (P < 0.05) in COPD samples than in the control. These results were consistent with the results previously obtained from the bioinformatics analysis.
 |
Figure 6 Expression levels of CYP1B1, VEGFA, BCL2, and CDKN1A quantified using qPCR. *P<0.05. |
Discussion
In the present study, 594 DEGs consisting of 242 upregulated genes and 352 downregulated genes between COPD samples and healthy samples were screened and functionally analyzed. A total of 696 DEG-compound pairs, including CYP1B1, VEGFA, BCL2, and CDKN1A, and 57 metabolites were obtained. Moreover, both C00469 (ethanol) and C00389 (quercetin) were found to be associated with BCL2. In the gene-metabolite network, the hsa04750 (inflammatory mediator regulation of TRP channels)-C00469 (ethanol) and hsa04152 (AMPK signaling pathway)-C00389 (quercetin) pairs were also identified.
The levels of CYP1B1 mRNA are known to be upregulated in alveolar epithelial type II (ATII) cells relative to that in hepatocytes. In addition, increased CYP2C19 and reduced CYP2J2 levels have been detected in the ATII cells of COPD patients relative to that in smokers without COPD.28 CYP1A1, CYP1B1, CYP2J2, CYP2E1, CYP2B6, and CYP3A5 encode proteins that are localized in the lungs; thus, these proteins may activate the COPD-associated compounds.29 The transcription factor T-box (TBX) and its target CDKN2A are involved in the pathogenesis of COPD by affecting senescence. The expression of CDKN1A, CDKN2A, and caveolin 1 (CAV1) is known to be higher in the peripheral lungs of COPD patients.30 CDKN1A, a biomarker of cell cycle arrest and premature ageing, is upregulated in COPD patients, which is related to skeletal muscle wasting.31,32 Skeletal muscle dysfunction is common in COPD patients. A previous study identified 18 genes, including CDKN1A, that were upregulated in the quadriceps of COPD patients relative to that in healthy controls.14 These results suggest that CYP1B1 and CDKN1A might be implicated in the pathogenesis of COPD.
Decreased miR-503 function promotes the release of VEGF from lung fibroblasts, thereby mediating vascular homeostasis in patients with COPD.33 VEGF can be used to diagnose COPD in healthy donors (HD), with better overall accuracy and Youden’s index (YI), while IL-8 can be used to diagnose cancer in both HD and COPD patients.34 The serum levels of VEGF and its soluble receptor sVEGF R2 are higher in COPD patients than that in the control; therefore, VEGF and sVEGF R1 may be involved in aberrant pulmonary vascular remodeling in COPD patients.35 VEGF overexpression promotes the development of Th2 inflammatory disorders such as asthma, while VEGF downregulation can affect the mechanisms of viral disorders including COPD.36 Therefore, VEGFA may also be involved in the development of COPD.
BCL2 is implicated in apoptotic regulation and lung function, which may be correlated with the development and progression of COPD.37 The BCL2 family members mediate cell apoptosis through maintenance of mitochondrial membrane potential, which promotes the development of COPD and affects its severity.38 Through the AMPK/mTOR signaling pathway, β-arrestin2 reduces the expression of inflammatory cytokines in the BEAS-2B bronchial epithelial cells by suppressing autophagy.39 Thus, BCL2 may also be associated with the progression of COPD through the hsa04750 (inflammatory mediator regulation of TRP channels)-C00469 (ethanol)-BCL2 and hsa04152 (AMPK signaling pathway)-C00389 (quercetin)-BCL2 pairs.
Conclusion
A total of 594 DEGs between COPD and healthy samples were identified. The key genes CYP1B1, VEGFA, BCL2, and CDKN1A may affect the mechanisms underlying the development and progression of COPD. Specifically, BCL2 may be involved in the development of COPD via inflammation-mediated regulation of TRP channels and AMPK signaling pathway. However, in-depth experimental studies are still needed to confirm these results.
Funding
This study was supported by the National Natural Science Foundation of China (81470252, 81170049, 81570325).
Disclosure
The authors declare no conflicts of interest in this work.
References
1. Mirza S, Benzo R. Chronic obstructive pulmonary disease phenotypes: implications for care. Mayo Clin Proc. 2017;92(7):1104–1112. doi:10.1016/j.mayocp.2017.03.020
2. Bringsvor HB, Skaug K, Langeland E, et al. Symptom burden and self-management in persons with chronic obstructive pulmonary disease. Int J Chron Obstruct Pulmon Dis. 2018;13:365–373. doi:10.2147/COPD.S151428
3. Mohammed J, Derom E, De WI, Rambaut L, Calders P. Autonomic symptoms in patients with moderate and severe chronic obstructive pulmonary disease. Acta Clin Belg. 2018;73(3):182–190. doi:10.1080/17843286.2017.1379255
4. de Torres JP, Celli BR. Is chronic obstructive pulmonary disease really a progressive disease? Arch Bronconeumol. 2016;53(7):632–636.
5. Rutten FH, Hoes AW. Chronic obstructive pulmonary disease: a slowly progressive cardiovascular disease masked by its pulmonary effects? Eur J Heart Fail Suppl. 2014;14(4):348–350. doi:10.1093/eurjhf/hfs022
6. Wang X, Li W, Zhang Y, et al. Chronic obstructive pulmonary disease candidate gene prioritization based on metabolic networks and functional information. PLoS One. 2017;12(9):e0184299. doi:10.1371/journal.pone.0184299
7. Silva BSDA, Lira FS, Rossi FE, et al. Inflammatory and metabolic responses to different resistance training on chronic obstructive pulmonary disease: a randomized control trial. Front Physiol. 2018;9:262. doi:10.3389/fphys.2018.00262
8. Vos T, Allen C, Arora M, et al. Global, regional, and national incidence, prevalence, and years lived with disability for 310 diseases and injuries, 1990–2015: a systematic analysis for the global burden of disease study 2015. Lancet. 2016;388(10053):1545–1602. doi:10.1016/S0140-6736(16)31678-6
9. Ni K, Zhao ZH, Zhang JQ, et al. Study on relationship between pleomorphic adenoma gene-like 2 and chronic obstructive pulmonary disease. Nanosci Nanotechnol Lett. 2017;9(7):1009–1014. doi:10.1166/nnl.2017.2420
10. Zhou L, Ying L, Xin C, et al. Over-expression of nuclear factor-κb family genes and inflammatory molecules is related to chronic obstructive pulmonary disease. Int J Chron Obstruct Pulmon Dis. 2018;13:2131–2138. doi:10.2147/COPD.S164151
11. Gagliardo R, Chanez P, Profita M, et al. Iκb kinase–driven nuclear factor-κb activation in patients with asthma and chronic obstructive pulmonary disease. J Allergy Clin Immunol. 2011;128(3):635–645.e632. doi:10.1016/j.jaci.2011.03.045
12. Dagouassat M, Gagliolo JM, Chrusciel S, et al. The cyclooxygenase-2-prostaglandin e2 pathway maintains senescence of chronic obstructive pulmonary disease fibroblasts. Am J Respir Crit Care Med. 2013;187(7):703–714. doi:10.1164/rccm.201208-1361OC
13. Nofziger C, Vezzoli V, Dossena S, et al. Stat6 links il-4/il-13 stimulation with pendrin expression in asthma and chronic obstructive pulmonary disease. Clin Pharmacol Ther. 2011;90(3):399–405. doi:10.1038/clpt.2011.128
14. Willis-Owen SA, Thompson A, Kemp PR, et al. Copd is accompanied by co-ordinated transcriptional perturbation in the quadriceps affecting the mitochondria and extracellular matrix. Sci Rep. 2018;8(1):12165. doi:10.1038/s41598-018-29789-6
15. Gautier L, Cope LBolstad BM, Irizarry RA. Affy - analysis of affymetrix genechip data at the probe level. Bioinformatics. 2004;20:307–315. doi:10.1093/bioinformatics/btg405
16. Irizarry RA, Bridget H, Francois C, et al. Exploration, normalization, and summaries of high density oligonucleotide array probe level data. Biostatistics. 2003;4(2):249–264. doi:10.1093/biostatistics/4.2.249
17. Ritchie ME, Belinda P, Di W, et al. Limma powers differential expression analyses for rna-sequencing and microarray studies. Nucleic Acids Res. 2015;43(7):e47. doi:10.1093/nar/gkv007
18. Peng L, Tong T. A note on a two-sample test with one variance unknown. Stat Methodol. 2011;8(6):528–534. doi:10.1016/j.stamet.2011.07.001
19. Cai Q, Chan HP. A double application of the benjamini-hochberg procedure for testing batched hypotheses. Methodol Comput Appl Probab. 2016;19(2):1–15.
20. Da WH, Sherman BT, Lempicki RA. Systematic and integrative analysis of large gene lists using david bioinformatics resources. Nat Protoc. 2009;4(1):44–57. doi:10.1038/nprot.2008.211
21. Balakrishnan R, Harris MA, Huntley R, Auken KV, Cherry JM. A guide to best practices for gene ontology (go) manual annotation. Database. 2013;2013(1):2681–2694. doi:10.1093/database/bat054
22. Klukas C, Schreiber F. Dynamic exploration and editing of kegg pathway diagrams. Bioinformatics. 2007;23(3):344. doi:10.1093/bioinformatics/btl611
23. Allan Peter D, Grondin CJ, Kelley LH, et al. The comparative toxicogenomics database’s 10th year anniversary: update 2015. Nucleic Acids Res. 2015;43(Database issue):914–920. doi:10.1093/nar/gku935
24. Jianguo X, Sinelnikov IV, Beomsoo H, Wishart DS. Metaboanalyst 3.0–making metabolomics more meaningful. Nucleic Acids Res. 2015;43(W1):W251. doi:10.1093/nar/gkv380
25. Gaujoux R, Seoighe C. A flexible r package for nonnegative matrix factorization. BMC Bioinformatics. 2010;11(1):367. doi:10.1186/1471-2105-11-367
26. Ho JH, Shih HC, Liao BY, Chu SC. A ladder diffusion algorithm using ant colony optimization for wireless sensor networks. Inf Sci. 2012;192(6):204–212. doi:10.1016/j.ins.2011.03.013
27. Pan P, Nakashima M, Tomofuji H. Online test using displacement–force mixed control. Earthq Eng Struct Dyn. 2010;34(8):869–888. doi:10.1002/eqe.457
28. Kamata S, Fujino N, Yamada M, et al. Expression of cytochrome p450 mrnas in type ii alveolar cells from subjects with chronic obstructive pulmonary disease. Pharmacol Res Perspect. 2018;6(3):e00405. doi:10.1002/prp2.405
29. Hukkanen J, Pelkonen O, Hakkola J, Raunio H. Expression and regulation of xenobiotic-metabolizing cytochrome p450 (cyp) enzymes in human lung. Crit Rev Toxicol. 2002;32(5):391–411. doi:10.1080/20024091064273
30. Acquaahmensah GK, Malhotra D, Vulimiri M, Mcdermott JE, Biswal S. Suppressed expression of t-box transcription factors is involved in senescence in chronic obstructive pulmonary disease. PLoS Comput Biol. 2012;8(7):e1002597. doi:10.1371/journal.pcbi.1002597
31. Lakhdar R, Drost EM, Macnee W, Bastos R, Rabinovich RA. 2d-dige proteomic analysis of vastus lateralis from copd patients with low and normal fat free mass index and healthy controls. Respir Res. 2017;18(1):81. doi:10.1186/s12931-017-0525-x
32. Rabinovich RA, Drost E, Manning JR, et al. Genome-wide mrna expression profiling in vastus lateralis of copd patients with low and normal fat free mass index and healthy controls. Respir Res. 2015;16(1):1. doi:10.1186/s12931-014-0139-5
33. Ikari J, Nelson AJ, Obaid J, et al. Reduced microrna-503 expression augments lung fibroblast vegf production in chronic obstructive pulmonary disease. PLoS One. 2017;12(9):e0184039. doi:10.1371/journal.pone.0184039
34. Balla MM, Desai S, Purwar P, et al. Differential diagnosis of lung cancer, its metastasis and chronic obstructive pulmonary disease based on serum vegf, il-8 and mmp-9. Sci Rep. 2016;6(36065):36065. doi:10.1038/srep36065
35. Dorota KS, Tadeusz P, Pawel G, Henryk S. Serum vascular endothelial growth factor and its receptor level in patients with chronic obstructive pulmonary disease. Eur Cytokine Netw. 2006;17(1):75–79.
36. Chun Geun L, Bing M, Seyedtaghi T, et al. Studies of vascular endothelial growth factor in asthma and chronic obstructive pulmonary disease. Proc Am Thorac Soc. 2011;8(6):512–515. doi:10.1513/pats.201102-018MW
37. Makoto S, Noriaki T, Sumito I, et al. Intronic single-nucleotide polymorphisms in bcl-2 are associated with chronic obstructive pulmonary disease severity. Respirology. 2010;12(1):34–41.
38. Zeng H, Kong X, Peng H, et al. Apoptosis and bcl-2 family proteins, taken to chronic obstructive pulmonary disease. Eur Rev Med Pharmacol Sci. 2012;16(6):711–727.
39. Wu Y, Li Y, Wu B, et al. Β-arrestin2 inhibits expression of inflammatory cytokines in beas-2b lung epithelial cells treated with cigarette smoke condensate via inhibition of autophagy. Cell Physiol Biochem. 2018;50(4):1270–1285. doi:10.1159/000494586
 © 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.