Back to Journals » International Journal of Nanomedicine » Volume 12
Cyclic hexapeptide-conjugated nanoparticles enhance curcumin delivery to glioma tumor cells and tissue
Authors Zhang X, Li X, Hua H, Wang A, Liu W, Li Y, Fu F, Shi Y, Sun K
Received 31 March 2017
Accepted for publication 4 July 2017
Published 8 August 2017 Volume 2017:12 Pages 5717—5732
DOI https://doi.org/10.2147/IJN.S138501
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Professor Lei Yang
Xuemei Zhang,1–3 Xuejuan Li,1,4 Hongchen Hua,1 Aiping Wang,1 Wanhui Liu,1–3 Youxin Li,1–3 Fenghua Fu,1–3 Yanan Shi,5 Kaoxiang Sun1
1School of Pharmacy, Yantai University, Yantai, Shandong Province, People’s Republic of China; 2State Key Laboratory of Long-acting and Targeting Drug Delivery System, Yantai, Shandong Province, People’s Republic of China; 3Luye Pharmaceutical Co., Ltd., Shandong Province, People’s Republic of China; 4National Engineering and Technology Research Center of Chirality Pharmaceutical, Lunan Pharmaceutical Group Co., Ltd., Shandong Province, People’s Republic of China; 5School of Pharmacy, Binzhou Medical University, Shandong Province, People’s Republic of China
Abstract: Glioma has one of the highest mortality rates among primary brain tumors. The clinical treatment for glioma is very difficult due to its infiltration and specific growth locations. To achieve improved drug delivery to a brain tumor, we report the preparation and in vitro and in vivo evaluation of curcumin nanoparticles (Cur-NPs). The cyclic hexapeptide c(RGDf(N-me)VK)-C (cHP) has increased affinity for cells that overexpress integrins and was designed to target Cur-NPs to tumors. Functional polyethyleneglycol-modified poly(D,L-lactide-co-glycolide) (PEG-PLGA) conjugated to cHP was synthesized, and targeted Cur-NPs were prepared using a self-assembly nanoprecipitation process. The physicochemical properties and the in vitro cytotoxicity, accuracy, and penetration capabilities of Cur-NPs targeting cells with high levels of integrin expression were investigated. The in vivo targeting and penetration capabilities of the NPs were also evaluated against glioma in rats using in vivo imaging equipment. The results showed that the in vitro cytotoxicity of the targeted cHP-modified curcumin nanoparticles (cHP/Cur-NPs) was higher than that of either free curcumin or non-targeted Cur-NPs due to the superior ability of the cHP/Cur-NPs to target tumor cells. The targeted cHP/Cur-NPs, c(RGDf(N-me)VK)-C-modified Cur-NPs, exhibited improved binding, uptake, and penetration abilities than non-targeting NPs for glioma cells, cell spheres, and glioma tissue. In conclusion, c(RGDf(N-me)VK)-C can serve as an effective targeting ligand, and cHP/Cur-NPs can be exploited as a potential drug delivery system for targeting gliomas.
Keywords: glioma targeting, integrin targeting, c(RGDf(N-me)VK)-C peptide, curcumin nanoparticles, in vitro and in vivo evaluation
Introduction
Glioma has one of the highest mortality rates among primary brain tumors and accounts for ~50%–60% of all brain tumors, with a median survival time of ~14 months and a 5-year survival rate near zero.1 Glioma carries a poor prognosis and is histologically characterized by vascular proliferation and necrosis. Surgical excision is the primary clinical treatment option. However, it is very difficult to distinguish tumor tissue from normal brain tissue due to its infiltration and specific growth locations, and therefore, the tumor usually cannot be completely removed.2 Chemotherapeutic drugs rarely reach brain tumor cells due to the blood–brain barrier (BBB), and such drugs also have side effects on normal tissues and cells. All of these factors contribute to the poor efficacy of glioma treatment.3,4 As physicians and biologists have expanded the understanding of the physiological mechanisms of glioma formation and maintenance, adding molecularly targeted materials to common chemotherapies for the treatment of gliomas is a favorable approach and achieves good benefits. However, these therapeutic effects are limited by several factors, including molecular heterogeneity and the BBB.5 Therefore, overcoming these obstacles is an important research topic in glioma treatment. Some studies have suggested that gliomas are the most vascularized human tumors. The transition of gliomas from low to high grade is often accompanied by angiogenesis and changes in the microvascular network. The glioma microvasculature is heterogeneous, which can influence the distribution of chemotherapeutic agents to brain tumors or lead to the development of resistance to anti-angiogenic therapies.6 Concurrently, the angiogenesis process produces many invasion inhibitors, including vascular endothelial growth factor receptor (VEGFR), epidermal growth factor receptor (EGFR), hepatocyte growth factor, and integrins. Among these, integrin has been shown to be involved in promoting glioma adhesion, migration, and angiogenesis.7 Anti-angiogenic drugs have been applied in clinical studies for treatment of neoplastic and non-neoplastic diseases. Cilengitide c(RGDf(N-me)V) was the first anti-angiogenic small molecule discovered that targets αvβ3, αvβ5, and α5β1 integrins. Although the Phase III clinical study evaluating cilengitide for the treatment of glioblastomas failed, integrin targeting remains a potential treatment strategy for neoplastic diseases such as glioma.8
Invasive angiogenesis also disrupts the BBB in patients with glioma. For this reason, many researchers have been arguing for years that the BBB was not an important obstacle in influencing the delivery and efficacy of therapies targeting glioma.9 However, all glioma patients have some percentage of their BBB intact, and this region of the BBB can prevent the distribution of chemotherapy drugs into tumor cells. Scientists have been researching multiple approaches to penetrate the BBB, many of which have been reviewed by Oberoi et al.5 Earlier techniques to disrupt the BBB included osmotic pumps, ultrasound, and pharmacological agents; however, these approaches were limited by their toxic side effects, complexity, or poor clinical efficacy. The next issue is how to overcome the active efflux in the BBB.5 It is difficult to change the distribution of most molecularly targeted agents in tumors and tissues with changes in the expression of transporters. Recent studies have focused on nanoparticle drug carriers and peptide-based drug delivery to overcome the BBB. The method by which the drug is loaded or linked to the molecular targeting peptides or ligands affects targeting of the BBB. In most cases, these nanoparticles (NPs) or delivery systems penetrate the BBB via receptor-mediated endocytosis.10 Among these targeting peptides and ligands, the cyclic Arg-Gly-Asp (RGD) sequence peptide has been reported more frequently in the treatment of glioma.2,7 Moreover, some cyclic RGD sequence peptides such as iRGD(CRGDK/RGPD/EC), a tumor-homing peptide, can deliver the drug deep into the tumor parenchyma.11
Plant extracts such as curcumin, which is derived from turmeric, have been recently explored as candidates for cancer therapy due to their broad anti-angiogenic and pro-apoptotic properties. In addition, curcumin was reported to have the capacity to block the proliferation, migration, and invasion of glioma cells.12 However, the therapeutic applications of curcumin remain limited due to its low water solubility13 and poor systemic bioavailability, as patients would require 8–10 g of oral curcumin daily to achieve a therapeutic effect.14 One method of improving the bioavailability of curcumin is to increase its targeting capacity to tumors. NPs are believed to be suitable to deliver curcumin to tumors, and a series of curcumin nanoformulations have been studied over the past decade.13 However, the use of curcumin nanoparticles (Cur-NPs) for the treatment of glioma has not been studied due to the lack of an effective targeting ligand to deliver the drug through the BBB.
Materials and methods
Materials
Cyclic peptides (purity .95%) and fluorescein isothiocyanate (FITC)-labeled cyclic peptides (purity .95%) were synthesized by ChinaPeptides Co., Ltd. (Shanghai, China). FITC-labeled immunoglobulin G1 (IgG1) was purchased from Abcam Trading (Shanghai) Co., Ltd. (Shanghai, China). 1,1'-Dioctadecyl-3,3,3',3'-tetramethylindotricarbocyanine iodide (DiR iodide) and Hoechst33342 were purchased from Fanbo Biochemicals Co., Ltd. (Beijing, China). 3-(4,5-Dimethylthiazol-2-Yl)-2,5-diphenyltetrazolium bromide (MTT) was purchased from Aladdin Industrial Inc. (Shanghai, China). Maleimide-poly(ethylene glycol)-poly(lactic-co-glycolide) (mal-PEG-PLGA, 20,000:5,000 Da) was purchased from PolySciTech (West Lafayette, IN, USA). mPEG5000-PLGA20000 90/10 was purchased from Jinan Daigang Biomaterial Co., Ltd. (Jinan, China). Curcumin was purchased from Shanghai Zhanyun Biological Technology Co., Ltd. (Shanghai, China). Dulbecco’s Modified Eagle’s Medium (DMEM), Minimum Essential Medium (MEM), and RPMI 1640 cell culture media were purchased from Gibco® Roswell Park Memorial Institute (Grand Island, NA, USA). All other materials were reagent grade and obtained from commercial suppliers.
Human umbilical vein endothelial cells (HUVECs), HeLa human breast cancer cells, and C6 malignant glioma cells were purchased from Institute of Basic Medical Sciences, Chinese Academy of Medical Sciences (Peking, China), and kindly supplied by Shandong Luye Pharmaceutical Co., Ltd. BALB/c-nu nude mice and Sprague-Dawley (SD) rats were supplied by Beijing Vital River Laboratory Animal Technology Co., Ltd. (Beijing, China). The Experimental Animal Research Committee at Yantai University approved this study, and all animal studies were performed in accordance with the Regulations of Experimental Animal Administration (State Food and Drug Administration, 2003).
Screening of cyclic peptides
To obtain high-affinity peptides for integrins, a computer simulation molecular docking method was used to screen and evaluate peptides. We modified the structure of a widely known cyclic RGD peptide c(RGDf(N-Me)V) by introducing lysine and cysteine to yield the presence of active sulfhydryl to allow the peptide to easily couple with the polymer carrier. The new peptide structures were screened and evaluated using computer-aided drug discovery software (SYBYL-X 2.0, Tripos Inc., St Louis, MO, USA), and the preferred peptides were then further evaluated using in vitro cell assays.
The method of molecular docking is summarized as follows: the crystal structure data of the extracellular segment of integrin avβ3 in complex with c(RGDf(N-Me)V) ligand (1 L5G)15 was downloaded from the Protein Data Bank (PDB); the H2O and ligand were removed from the complex crystal structure, and the residual structure was subsequently used for molecular docking after hydrogenation.
Here, the docking functions of SYBYL-X 2.0 were utilized. Some previously reported RGD peptides were docked with the above residual structure, and the score was obtained depending on Hammerhead scoring function combined with electrostatic forces, van der Waals forces, hydrophobic interactions, and hydrogen bonding. The parameters used in the docking process were adjusted according to the score sequence and the reported binding affinity with integrin avβ3.16
New cyclic hexapeptides based on c(RGDf(N-Me)V) were designed considering previously reported structure–activity relationships,17 the required structure covalent coupled with the polymer carrier, and the synthesizability of the structure. These cyclic hexapeptides were docked with the residual integrin structure and they scored based on the above parameters. A cyclic hexapeptide c(RGDf(N-me)VK)–C was obtained for further study based on the docking score sequence.
Cell affinity evaluation of cyclic peptides
Cell culture
HUVECs and C6 cells were selected to explore the affinity of the cyclic hexapeptides with integrin αvβ3 under a high expression of integrin αvβ3.18 We cultured HUVECs in DMEM and C6 cells in MEM (both media were supplemented with 10% fetal bovine serum) at 37°C with 5% CO2. The medium was replaced every 1–2 d, and the growth state of the cells was observed daily through an inverted microscope.
Affinity evaluation of cyclic peptides
Flow cytometry was applied in the affinity studies. Two milliliters of cell suspension (1×105 cells/well) was inoculated in a 6-well plate and cultured for 24 h. The HUVECs and C6 cells were divided into two groups. The cells in group one were treated with 2 mL of 10 μg/mL FITC-labeled cyclic hexapeptide-containing medium for 15 min at 37°C. The cells in group two were pretreated with 2 mL of antibody-containing medium for 30 min at 37°C. The medium contained 2 μL of anti-integrin αvβ3 antibodies (Abcam; 27.1 [VNR-1]). Using this procedure, the cells in group two were treated with cyclic hexapeptide-containing medium consistent with the first group. The cells were treated with blank medium as a control. All cells were collected at the prescribed times after treating with the different media, washed twice with PBS, and re-suspended in 600 μL of PBS. Fluorescence data were collected using a FACS Jazz cell sorting system (BD Biosciences, San Jose, CA, USA), and the cell fluorescence and affinity were analyzed.
Covalent coupling of cyclic hexapeptide to mal-PEG-PLGA
The cyclic hexapeptide c(RGDf(N-me)VK)–C and mal-PEG-PLGA were covalently coupled using a previously reported method with modifications.19 In brief, mal-PEG-PLGA was dissolved in acetone, dried with nitrogen, and hydrated overnight in HEPES (pH =6.5). mal-PEG-PLGA was then mixed with cyclic hexapeptide at a 1:1 molar ratio in HEPES (pH =6.5). This mixture was gently stirred at 20°C for at least 48 h under nitrogen gas. The resulting reaction samples were purified from the unreacted cyclic hexapeptide by ultrafiltration (molecular weight cutoff =10 kDa). The products were collected and lyophilized to obtain cyclic hexapeptide-PEG-PLGA (cHP-PP).
The amount of cyclic hexapeptide conjugated to mal-PEG-PLGA was determined indirectly by measuring uncoupled cyclic hexapeptide in the ultrafiltrate after the ultrafiltration step. A reverse-phase high-performance liquid chromatography (RP-HPLC) (Agilent 1100 system, Agilent, Santa Clara, CA, USA) system with a UV detector at 220 nm was used to analyze uncoupled cyclic hexapeptide in the reaction system. The chromatographic column was a Phenomenex C18 (4.6×150 mm, 5 μm), and the mobile phase consisted of 0.1% trifluoroacetate (TFA) in acetonitrile and 0.1% TFA in water (10:90, v/v). The injection volume was 20 μL.
Characterization of synthetic functional polymers
The cHP-PP was dissolved in 0.5 mL of CDCl3, after which, nuclear magnetic resonance spectroscopy (Advance Bruker 400 M, Bruker Company, Fällanden, Switzerland) was used to identify the reaction product.
Preparation of NPs and cHP-modified nanoparticles (cHP-NPs)
NPs and cHP-NPs were prepared via one-step nanoprecipitation with different ratios of cHP-PP, methoxy-poly(ethylene glycol)-poly(lactic-co-glycolic acid) (mPEG-PLGA), curcumin, DiR, or coumarin (Cou). Here, curcumin acts as the drug entrapped in the NPs and as a fluorescent probe in the in vitro cell evaluation. DiR and coumarin were used as fluorescent indicators in the in vivo targeting evaluation of the NPs due to the weak fluorescence of curcumin in such conditions. The average molecular weights of PEG and PLGA in mPEG-PLGA were 5 and 20 kDa, respectively. According to the proposed formulation table (Table 1), mPEG-PLGA, cHP-PP, curcumin, DiR, or coumarin was added to 2 mL of acetone solution to prepare the oil phase. Under stirring, the oil phase was dropped into 20 mL of purified water. The suspension system was then stirred uncovered for at least 4 h at 20°C to evaporate the acetone. To determine the incorporation efficiency of curcumin in NPs or cHP-NPs, the NP suspensions were centrifuged (10 min, 10 krpm), and the supernatant was analyzed using an RP-HPLC (Agilent 1100 system, Agilent, Santa Clara, CA, USA) system with a UV detector at 425 nm. The chromatographic column was a Phenomenex C18 column (4.6×150 mm, 5 μm), and the mobile phase consisted of acetonitrile with 0.5% acetic acid in water (60:40, v/v). The injection volume was 20 μL, and the flow speed was 1.0 mL/min1. The final NPs were purified, collected by ultrafiltration (15 min, 3,000 rpm, MW 30 kDa), and lyophilized. The particle size and polydispersity of the resulting NPs were detected with a dynamic light scattering (DLS) particle size analyzer (Nicomp 380 ZLS; PSS, Port Richey, FL, USA). The surface morphology of the NPs was examined using a transmission electron microscope (TEM) (JEM 1400, JOEL, Tokyo, Japan).
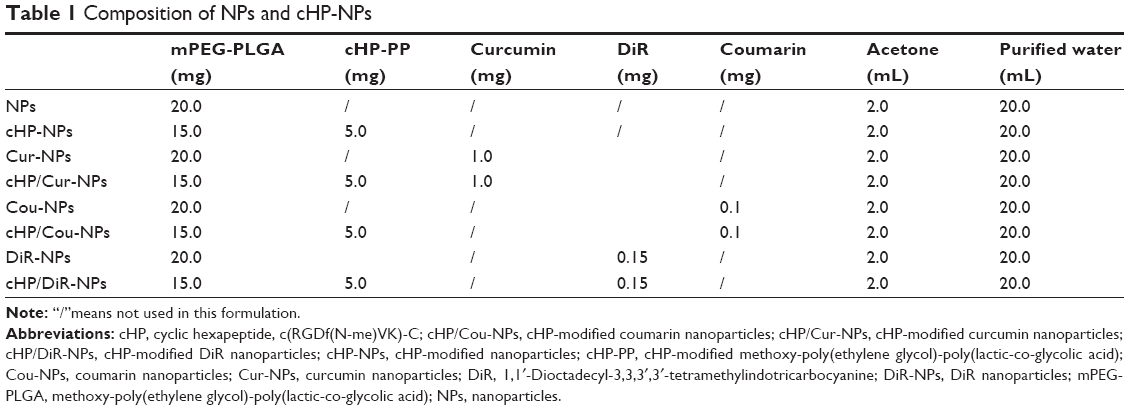 | Table 1 Composition of NPs and cHP-NPs |
In vitro cytotoxicity assay
The cytotoxicity of the prepared free curcumin and its NPs was determined using the MTT assay. C6 cells and HeLa cells were seeded in 96-well plates at 1×104 cells per well in 100 μL of corresponding media. The cells were incubated at 37°C in 5% CO2 for 24 h followed by treatment with increasing concentrations (1.25–40 μM) of either free curcumin or the Cur-NPs (Cur-NP, cHP-modified curcumin nanoparticles [cHP/Cur-NPs]) for 24, 48, and 72 h; the cells were then incubated in 5% CO2 at 37°C. Subsequently, 20 μL of MTT solution (5 mg/mL) was added per well and incubated with the cells for 4 h. Next, the culture medium was discarded, and 200 μL of DMSO was used to dissolve the formazan crystals. After the plate was gently shaken for 10 min, the absorbance at 570 nm of each well was measured using a microplate reader (SpectraMax M5, Molecular Devices Corporation, Sunnyvale, CA, USA). The data are shown as the percentage of viable cells per sample compared with the survival of the control group (untreated cells were used as the control group, and the cell viability of this group was set to 100%).
Targeting and penetration capabilities of the NPs
In vitro cellular targeting of C6 cells by NPs
The C6 cells were seeded in 6-well plates (1×105 cells in 2 mL per well) and incubated for 24 h at 37°C. The cultured medium was replaced with 2 mL of medium containing different NPs (Cur-NP, cHP/Cur-NP; the curcumin concentration in the NPs was 2–10 μM). The cells were harvested by trypsin and centrifuged after 30, 60, and 90 min of incubation at 37°C. The percentage of fluorescent cells in each NP group was subsequently measured by flow cytometry (FACS, BD FACSJazz, BD Biosciences, San Jose, CA, USA), and the cellular uptake efficiency was calculated. Cells not treated with NPs served as the control group.
To investigate the targeting mechanism of curcumin NPs, C6 cells were preincubated for 1 h with c(RGDf(N-me)VK)–C (40 μM), the competitive inhibitor of RGD with integrin αvβ3. The C6 cells were subsequently co-incubated with Cur-NPs or cHP/Cur-NPs at a curcumin concentration of 10 μM for 90 min at 37°C. The control group did not receive inhibitor treatment. The cellular uptake efficiency was determined by flow cytometry, and the data were compared between the test group and the control group.
In vitro cellular penetration of NPs to C6 spheroids
In vitro C6 spheroids were established using a previously reported method20 with a lipid overlay system. C6 cells (2×103 cells/well) were seeded in 200 μL of culture medium in a 96-well plate loaded with 2% (m/V) agarose in basal DMEM. After the cells were cultured for 5 d at 37°C in an atmosphere containing 5% CO2, they slowly aggregated to form a solid sphere. The tumor cell spheroids were then incubated with 200 μL of medium containing NPs, coumarin nanoparticles (Cou-NPs), or cHP-modified coumarin nanoparticles (cHP/Cou-NPs) (the concentration of coumarin was 2 μM) for 1, 2, or 4 h. After the plates were washed three times with PBS, the spheroids were observed via confocal laser scanning microscopy (CLSM, Olympus FV1000, Olympus Corporation, Tokyo, Japan) to investigate NP penetration.
In vivo evaluation
In vivo targeting evaluation of NPs in a rat glioma model
Adult male SD rats (180–220 g) were randomly divided into a sham operation group, a model group, and a blank group and provided free access to chow and water. The number of rats in the three groups was 6, 4, and 4, respectively. Before surgery, all of the rats were anesthetized with 10% chloral hydrate (4 mL/kg intraperitoneally). A small incision was made through the right hemisphere. A stereotactic apparatus (Neurostar, Tübingen, Germany) was used for head fixation and location. A high-speed drill was used to create a hole ~1.0 mm in diameter at 1.0 mm anterior of the bregma coronal suture and 3.0 mm right lateral of the sagittal suture. A total of 10 μL (1×106 cells) of C6 cells was transplanted into the caudate nucleus of the rats using a Hamilton syringe through the drilled holes. Ten minutes after injection, the needle was removed, and bone wax was used to fill the burr hole. The skin was sutured together, and the rats recovered. The sham operation group was injected with the same volume of PBS as described above, and the blank group was used as the control group.
One rat in each group was sacrificed with excessive 10% chloral hydrate (i.p.) on days 14 and 21 after tumor cell implantation. Brain tissues were collected for morphological observation to determine tumor establishment. After the final observation, 0.2 mL of DiR nanoparticles (DiR-NPs) or cHP-modified DiR nanoparticles (cHP/DiR-NPs) was injected into each group of rats (300 μg/kg intravenous [i.v.]). One rat in each group was euthanized 8 h after NP injection. The rat hearts were perfused with saline and then with 4% paraformaldehyde (PFA). The brains tissue and tumors were collected and fixed for 24 h in 3% PFA and then cryopreserved in 3% PFA/sucrose for 48 h. Afterward, the tumor tissues were sliced into 10-μm sections, which were fixed for 10 min with cold acetone and then washed with PBS. Then, 10 μg/mL Hochest33342 was added, and the sections were cultured at room temperature for 10 min. A fluorescence-quenching agent was added to seal the sections. The tumor cells and NPs that penetrated the tumor tissue were observed using CLSM.
At 1, 4, 8, 12, and 24 h after injection, the rats were anesthetized with 5% chloral hydrate. In vivo imaging (Care stream In Vivo FX, Bruker, Madison, WI, USA) was utilized to capture images of the fluorescence distribution and assess the in vivo targeting ability of the NPs. At 8 h post-injection, the rats were euthanized and the brain and other organs were harvested. The fluorescent images of the brains and other organs were recorded and analyzed with Kodak MI In Vivo Fx Pro software.
Results and discussion
Screening and affinity of the tested cyclic peptides
The results of the simulated SYBYL-X 2.0 score, which was a comprehensive result that integrated the electrostatic interactions, van der Waals forces, hydrophobic interactions, and hydrogen bonding of the cyclic RGD peptide to integrin αvβ3, are shown in Table 2. It is generally accepted that scores below 4 indicate poor affinity, and scores >7 represent strong affinity. Among all of the peptides tested, the top three scores were observed for c(RGDf(N-me)VK)-C, c(RGDfKV)-C, and c(RGDf(N-me)V); therefore, these three peptides were utilized in the affinity study of cells overexpressing integrin.
 | Table 2 Scores of the cyclic peptide according to computer-aided drug discovery software |
To evaluate the affinity of the screened cyclic peptides with integrins, a cellular uptake capability test on integrin-overexpressing cells was applied. It was previously demonstrated that HUVECs20 and C6 cells21 express integrin αvβ3 on their cell surface, an observation that was confirmed in our cell sorting experiment using αvβ3 antibodies. Therefore, HUVECs and C6 cells were selected to explore the specificity of cyclic RGD peptides in subsequent in vitro studies.
Figure 1 shows the cellular uptake efficiency results after a 15-min treatment with FITC-labeled cyclic peptide or integrin antibody plus FITC-labeled cyclic peptide in HUVECs and C6 cells. Prior to this experiment, we preliminarily studied the relationship between the cell uptake efficiency and treatment time of the FITC-labeled cyclic peptide medium and the concentration of FITC-labeled cyclic peptide. The specific experimental parameters were then determined as described in the methods of affinity evaluation of cyclic peptides.
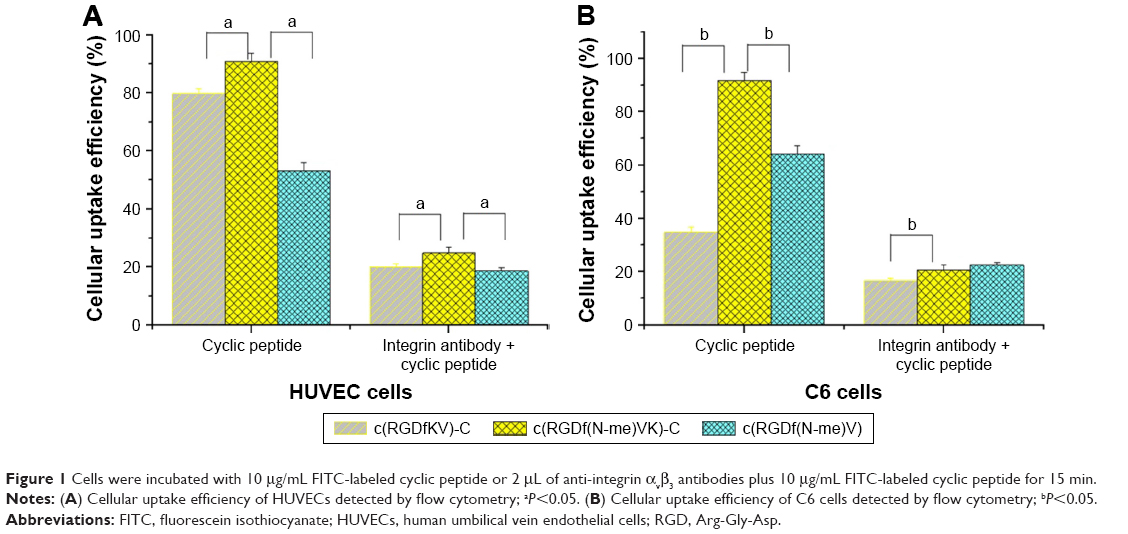 | Figure 1 Cells were incubated with 10 μg/mL FITC-labeled cyclic peptide or 2 μL of anti-integrin αvβ3 antibodies plus 10 μg/mL FITC-labeled cyclic peptide for 15 min. |
According to the results, for the cyclic peptide group, the average cellular uptake efficiency of FITC-labeled c(RGDf(N-me)VK)-C (90.7%) was significantly higher than those of FITC-labeled c(RGDfKV)-C (79.6%) and c(RGDf(N-me)V) (53.0%) (a, P<0.05, n=3) in HUVECs, and similar results for the cyclic peptide group were observed in C6 cells. For the second group, the average cellular uptake efficiency of FITC-labeled cyclic peptide decreased to <25% after the HUVECs and C6 cells were treated with integrin antibody for 30 min. Based on the uptake efficiency difference between groups 1 and 2 in HUVECs and C6 cells, we considered that the uptake efficiency of cyclic peptide was associated with the affinity between the overexpressed integrin in the cells and the cyclic peptides. FITC-labeled c(RGDf(N-me)VK)-C had a stronger ability to bind with the integrin and a stronger cellular uptake ability than the other two peptides. Therefore, c(RGDf(N-me)VK)-C was utilized as the preferred cyclic peptide in the following experiments.
Characterization of the synthetic functional polymers
Michael addition reaction occurs between c(RGDf(N-me)VK)-C with mal-PEG-PLGA in PBS (pH 6.5) yielding the product cHP-PP. Figure 2 shows the 1H-NMR spectrum of cHP-PP. In Figure 2B, the peak (2H) at δ 6.71 was the characteristic peak of MAL. However, the peak disappeared after binding with cyclic hexapeptide, as shown in Figure 2C, indicating that a new substance had been generated. In Figure 2A, the peaks at δ 1.42, 2.78, and 2.95 are the characteristic peaks of c(RGDf(N-me)VK)-C, but these are covered by the peak of mal-PEG-PLGA and therefore cannot be an indicator of cHP-PP. Content analysis via HPLC showed that the link efficiency of mal-PEG-PLGA with cyclic hexapeptide was ~90%.
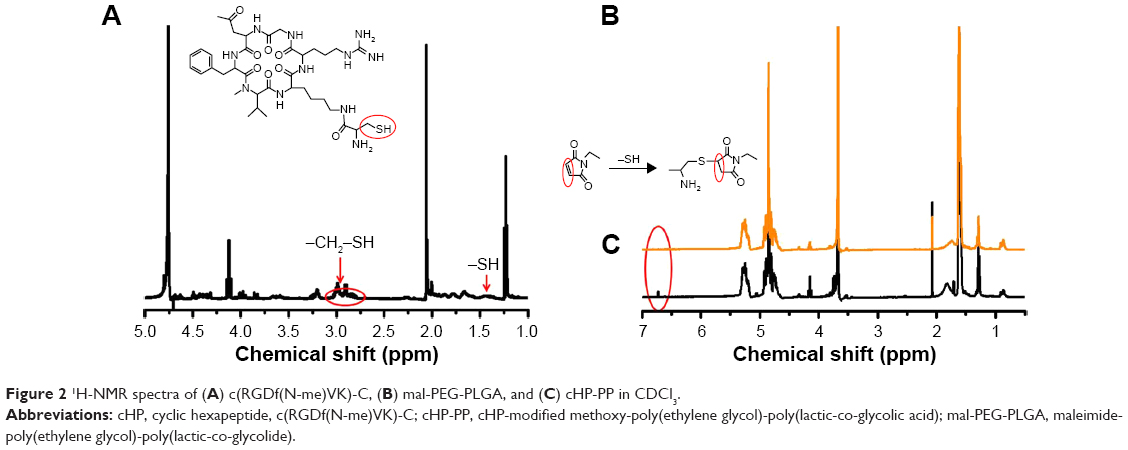 | Figure 2 1H-NMR spectra of (A) c(RGDf(N-me)VK)-C, (B) mal-PEG-PLGA, and (C) cHP-PP in CDCl3. |
Physicochemical characteristics of NPs and cHP-NPs
The cHP-PP synthesis and cHP-NP preparation processes are illustrated in Figure 3. We studied the physicochemical properties of different NPs with respect to particle size, polydispersity index, and encapsulation efficiency. The properties of the NPs and cHP-NPs are shown in Table 3 and Figure 4. The NPs are solid spheres, and compared with the diameter of the NPs, the mean diameter of the cHP-NPs increased as a result of coupling with cyclic hexapeptides. However, their average diameter remained <100 nm, which is an important factor to avoid uptake by the reticuloendothelial system.22 Similar results were noted for the NPs and cHP-NPs loaded with curcumin compared to the NPs and cHP-NPs without curcumin. In addition, this increase correlated with high drug loading. However, the particle size of the NPs and cHP-NPs was not affected by the loading of coumarin-6 for its low drug content. The polydispersity of all the NPs presented a narrow particle size distribution. Regarding the encapsulation efficiency of either drug or fluorescent agent, all the NPs showed high loads of >80%. After covalently binding the cyclic hexapeptide to PEG-PLGA-MAL, the encapsulation efficiency of the cHP-NPs was reduced, possibly due to the increasing internal osteoporosis observed as a result of macromolecular steric hindrance in the formation of the NPs.
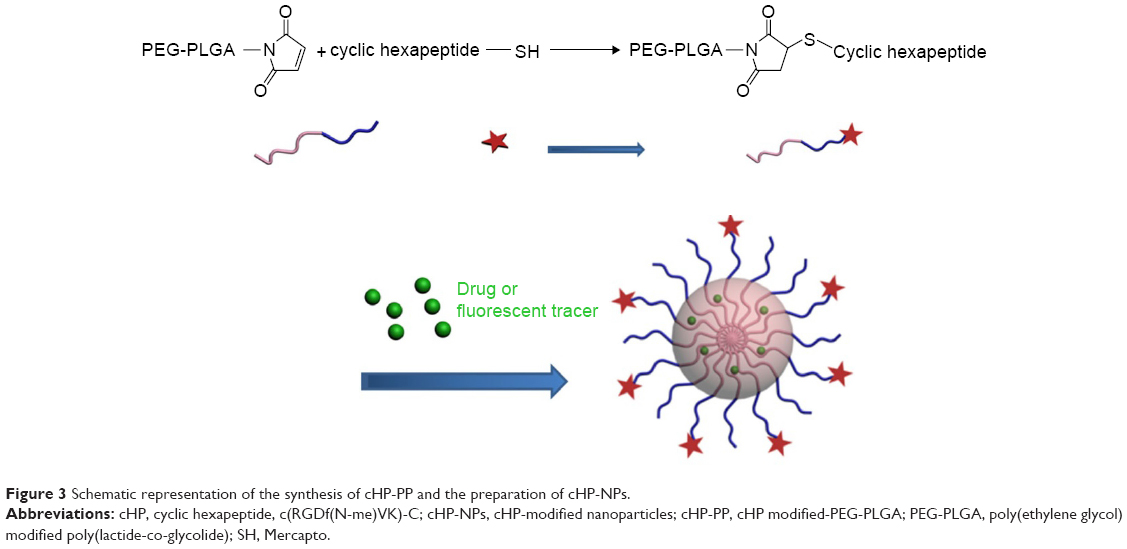 | Figure 3 Schematic representation of the synthesis of cHP-PP and the preparation of cHP-NPs. |
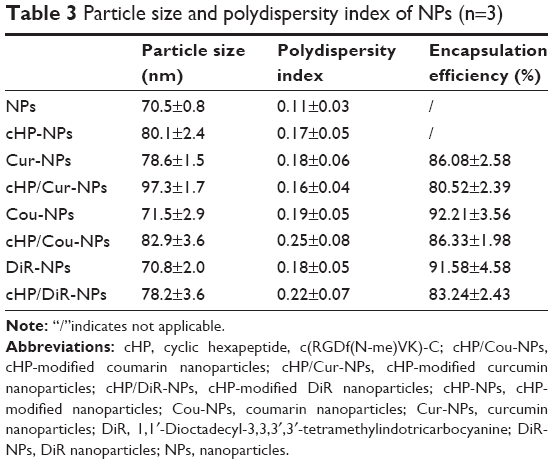 | Table 3 Particle size and polydispersity index of NPs (n=3) |
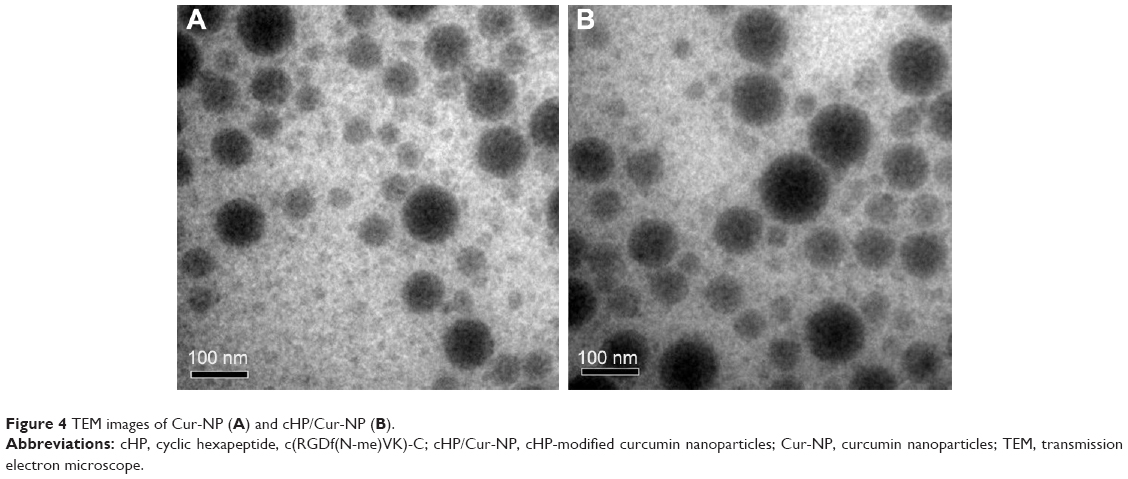 | Figure 4 TEM images of Cur-NP (A) and cHP/Cur-NP (B). |
Cytotoxicity of free curcumin, Cur-NPs, and cHP/Cur-NPs
C6 and HeLa cells were incubated with different concentrations of free curcumin and its NPs for 24, 48, and 72 h. The cell viabilities of the different cell types are shown in Figure 5.
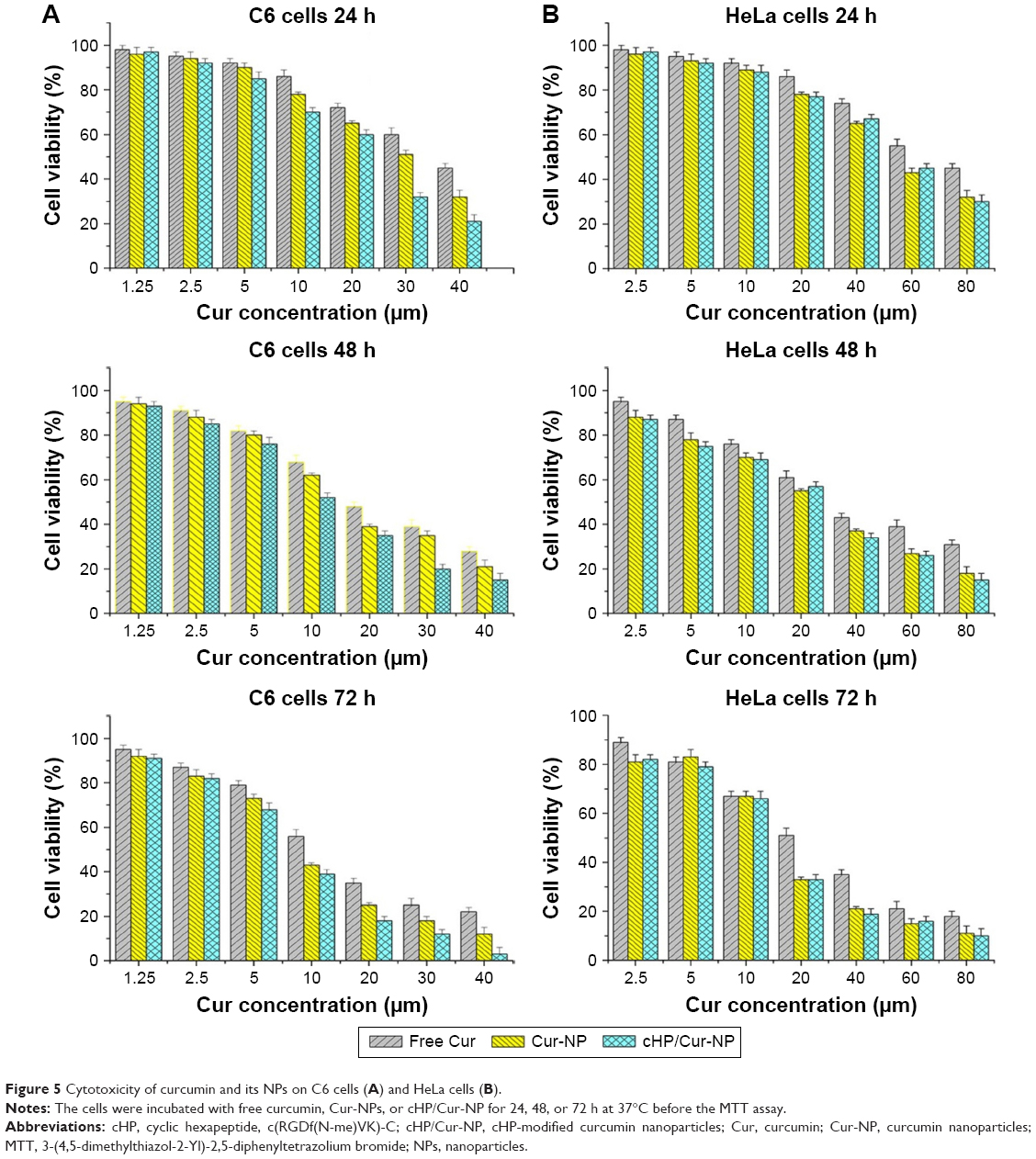 | Figure 5 Cytotoxicity of curcumin and its NPs on C6 cells (A) and HeLa cells (B). |
For C6 cells, high integrin αvβ3-expressing cells,21 there was no significant difference between free curcumin and its NPs at low concentrations (eg, 1.25 and 2.5 μM) after a 24-h incubation before the MTT assay. However, the C6 cell viability decreased with increasing curcumin concentrations and extended incubation times. When the concentrations exceeded 5 μM, the toxicity of Cur-NPs and cHP/Cur-NPs increased faster and was significantly higher than that of free curcumin (P<0.05). At the same curcumin concentration, such as 30 μM curcumin, the toxicity of the cHP/Cur-NPs (32.4%±2.3%) was significantly greater than that of the unmodified Cur-NPs (51.2%±3.1%) (P<0.01). Figure 5 shows that the cytotoxicity of curcumin and its NPs were positively correlated with the curcumin concentration and incubation time, and similar results were obtained for different incubation times.
To verify whether the increasing cytotoxicity of the curcumin NPs was caused by the toxicity of cyclic hexapeptide and nanomaterials, blank NPs and blank cHP-NPs without curcumin were prepared, and additional cytotoxicity experiments in C6 cells and HeLa cells were performed. Both types of blank NPs showed good cell compatibility and low toxicity (data not shown here). For the blank NPs, at the highest concentration of 1,600 μg/mL (equivalent to that in 80 μM of Cur-NPs), the cell viability was still >85% after a 72-h incubation. The cytotoxicity of blank NPs modified by cyclic hexapeptide slightly increased, which is likely due to cell growth inhibition by cyclic hexapeptide. This finding indicated that the material itself had low cytotoxicity, and, therefore, the observed cytotoxicity was caused by curcumin and its NPs.
For HeLa cells, integrin αvβ3-negative cells,23 no significant differences were identified between the free curcumin and its NPs at low curcumin concentrations for 24 h of incubation. The cytotoxicity increased with increasing concentrations. When the concentration in HeLa cells reached 20 μM after 24 h of incubation, the average cell viabilities for treatment with free curcumin, Cur-NPs, and cHP/Cur-NPs were 86.2%, 78.6%, and 77.1%, respectively. Curcumin NPs had a higher cytotoxicity than free curcumin, but the cell viability was similar between Cur-NPs and cHP/Cur-NPs. In addition, extended incubation times increased the cytotoxicity of equal curcumin concentrations. In these experiments, curcumin showed a lower cytotoxicity in C6 and HeLa cells than the corresponding NPs due to the improved biocompatibility and affinity to cells of the PEG-PLGA incorporated in the NPs.24
The cHP/Cur-NPs were more lethal to C6 cells than Cur-NPs in the low integrin-expressing HeLa cells. In HeLa cells, no significant difference was observed in the cytotoxicity between Cur-NPs and cHP/Cur-NPs in most cases, possibly because the RGD sequence peptides can bind to integrin-αvβ3 in C6 cells but not in HeLa cells,25,26 and the retention time and cellular uptake efficiency of the NPs can be greatly improved by receptor-mediated endocytosis of the existing targeted ligands.27 The experimental cytotoxicity data indicated that the effect of Cur-NPs coupled with cyclic hexapeptide on cells was concentration and time dependent. After a 24-h incubation with 10 μM cHP/Cur-NPs, 70% of C6 cells were alive; therefore, the cells were incubated with curcumin for less time in subsequent in vitro cellular targeting evaluations.
In vitro cellular targeting of NPs in C6 cells
To evaluate the ability of the cyclic hexapeptide-modified Cur-NPs to specifically recognize high integrin αvβ3-expressing cells, the cellular uptake efficiency of the Cur-NPs and cHP/Cur-NPs in C6 cells was determined by flow cytometry. The results are shown in Figure 6. In this experiment, curcumin was used as a fluorescent indicator for detecting the ratio of fluorescence in the cells.
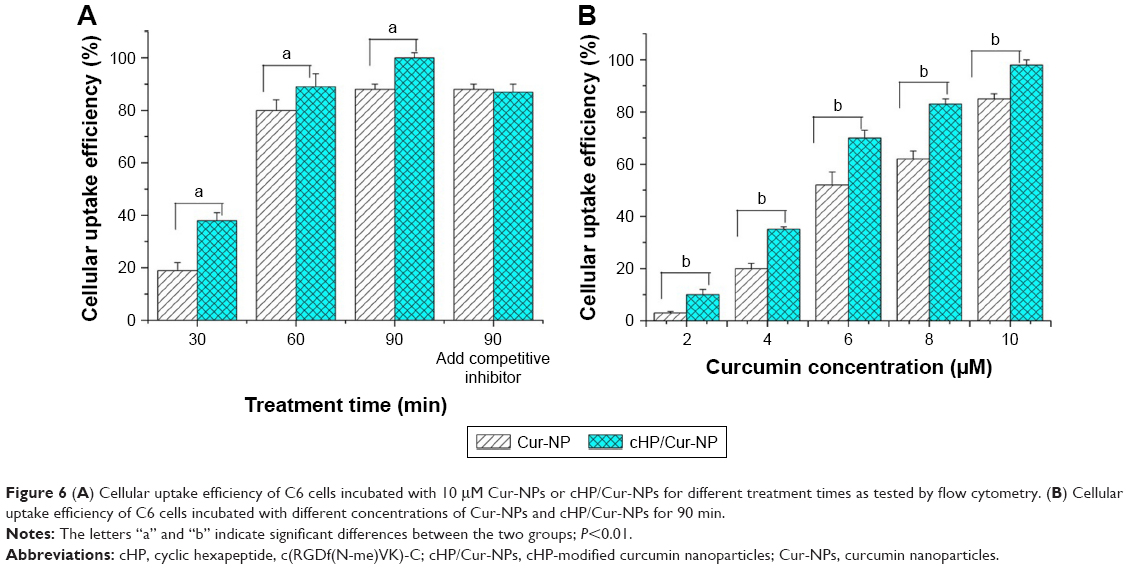 | Figure 6 (A) Cellular uptake efficiency of C6 cells incubated with 10 μM Cur-NPs or cHP/Cur-NPs for different treatment times as tested by flow cytometry. (B) Cellular uptake efficiency of C6 cells incubated with different concentrations of Cur-NPs and cHP/Cur-NPs for 90 min. |
For the cHP/Cur-NP and Cur-NP groups (n=3), the average cellular uptake efficiency of Cur-NPs was dependent on incubation time. At a concentration of 10 μM, the cellular uptake efficiency of the cHP/Cur-NP group reached 38.3% after 30 min of treatment. In addition, the average cellular uptake efficiency of Cur-NPs was 19.4% at 30 min, which was significantly decreased compared with that of the cHP/Cur-NP group (P<0.01). With increasing incubation time, the cellular uptake efficiency of the two groups increased correspondingly. For 90 min of culture time, the uptake efficiency of the cHP/Cur-NP group reached 99.5%. At all treatment times, the cHP/Cur-NP group had a better cell uptake efficiency than the Cur-NP group (P<0.01). After pre-incubation for 1 h with a competitive integrin αvβ3 inhibitor, the cell uptake efficiencies of the cHP/Cur-NP and Cur-NP groups were 87.6% and 88.3%, respectively, at 90 min of incubation, and no significant difference was observed between these two groups. These data presented that the different cell uptake efficiencies for cells treated with the cHP/Cur-NPs and Cur-NPs were related to the affinity of cyclic hexapeptide with the integrin on the surface of C6 cells. When the integrin was conjugated with free cyclic hexapeptide, the Cur-NPs modified by cyclic hexapeptide could not specifically recognize C6 cells. Therefore, the cell uptake efficiency of the cHP/Cur-NPs was similar to the Cur-NP group.
To further investigate the cellular uptake efficiency of cHP/Cur-NPs, we studied the dose-dependent relationship of the NPs in C6 cells via flow cytometry. The uptake efficiency of Cur-NPs and cHP/Cur-NPs showed a positive dose–efficiency relationship (Figure 6, right). The cell uptake efficiency of curcumin NPs increased with increasing concentrations of curcumin, and significant differences between the cHP/Cur-NP and Cur-NP groups (P<0.01) were observed at all concentrations.
The experimental results indicated that the cHP/Cur-NPs had a significant capacity to specifically recognize C6 cells because they had a better affinity for C6 cells (αvβ3+) than the Cur-NPs for promoting the cellular uptake of the NPs.
In vitro cellular penetration of NPs to C6 spheroids
In the above experiments, compared to Cur-NPs, the cHP/Cur-NPs showed better targeting ability for monolayer cancer cells that highly express integrin αvβ3. However, this benefit might not be a key to cancer treatment because of tumor drug resistance mechanisms.28 Cancer cell spheroids represent a better model of drug resistance than monolayer cells. Therefore, we established a tumor cell spheroid model for evaluating the penetration of Cur-NPs into tumor tissue in vitro.29 In this experiment, curcumin is not suitable for observing drug penetration into compact cell spherical entities because of its weak fluorescence. Therefore, coumarin was used as an indicator instead of curcumin in these experiments.
Agarose was added to 96-well plates, and C6 cells were then cultured in the wells. The cell aggregates grew and gradually formed tight tumor spheres. The growth states of the cells at 1, 2, and 5 d are shown in Figure 7A. The tight spheroids were used for the experiment.
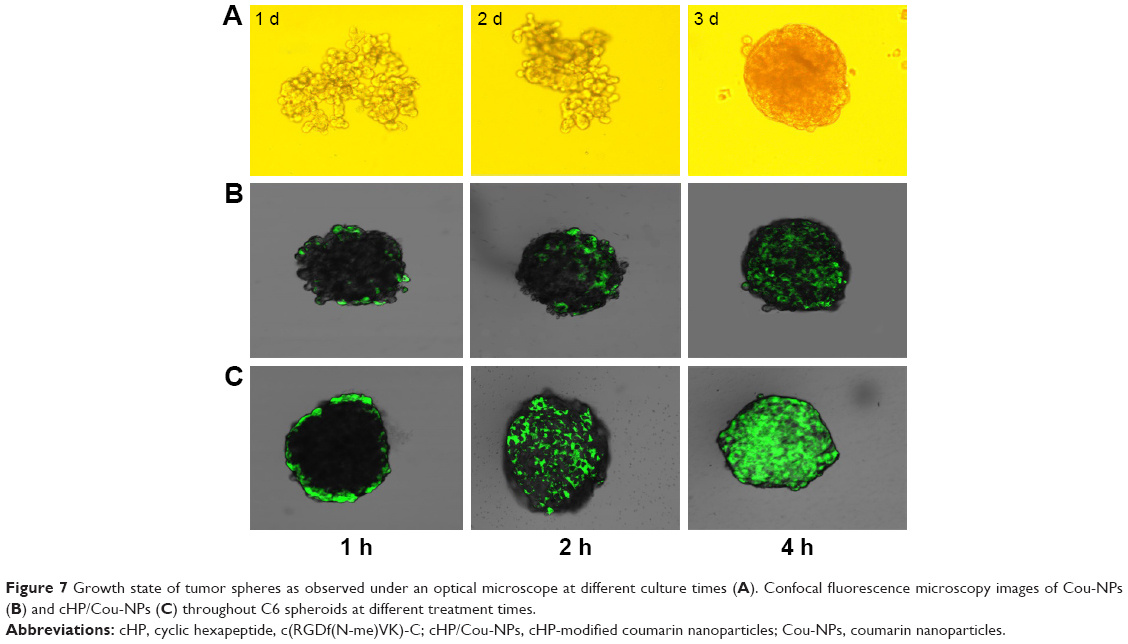 | Figure 7 Growth state of tumor spheres as observed under an optical microscope at different culture times (A). Confocal fluorescence microscopy images of Cou-NPs (B) and cHP/Cou-NPs (C) throughout C6 spheroids at different treatment times. |
Penetration of the cHP/Cou-NPs into the C6 spheroids was observed via confocal microscopy, and the results are presented in Figure 7B and C. The image shows that fluorescence was observed on the periphery of the cell spheroids after a short time of incubation with cHP/Cou-NPs that were modified by the cyclic hexapeptide. This result indicated that the cHP/Cou-NPs had gathered at the surface of the tumor spheroids. With increased incubation time, the fluorescence became uniformly distributed in the spheroids, and the intensity gradually increased. The Cou-NPs, which were not modified with cyclic hexapeptide, exhibited difficulty in penetrating the C6 tumor spheroids, and weak fluorescence was observed in each spheroid at various time points. These results indicated that the cHP/Cou-NPs had a stronger ability to penetrate the tight tumor tissue due to the presence of cyclic hexapeptide. The addition of the cyclic hexapeptide improved the cell specificity recognition and nanoparticle uptake, and these NPs showed similar results with the in vitro cellular targeting of NPs in C6 monolayer cells.
In vivo targeting of NPs in a rat glioma model
The infiltrative nature of glia and the existence of the BBB are responsible for the failure of surgery and chemotherapy to completely remove gliomas.30 Modeling this process in vivo will lead to a better simulation of the pathophysiology of gliomas and improved evaluations of the targeting ability of NPs to gliomas.
On the basis of our results, cyclic hexapeptide can allow NPs to target C6 cells overexpressing integrins. However, for gliomas, which grow at specific sites and are protected by the BBB, the targeting of the drug delivery system requires further studies for validation in in vivo gliomas. To investigate the targeting of NPs modified with a cyclic hexapeptide in a pathological condition, a glioma model was developed using C6 cells in rats. DiR was used as a tracing agent for in vivo imaging instead of curcumin and was encapsulated into NPs and NPs modified with cyclic hexapeptide to monitor their distributions in tumor-bearing rats.
We established a C6 glioma model in SD rats using a stereotaxic apparatus, and images of the brains and tumors for each group are presented in Figure 8A–C. Twenty-one days after the injection of PBS, the brains of rats in the sham surgery group exhibited no significant changes in appearance. In the surgery group, C6 cells were injected into the brains of test rats, and the brains gradually changed in appearance as time progressed. At 14 and 21 d after the injection of tumor cells, gliomas formed gradually, and significant midline shifts occurred due to tumor extrusion. Therefore, in subsequent experiments, rats were used at 21 d after injection of C6 cells.
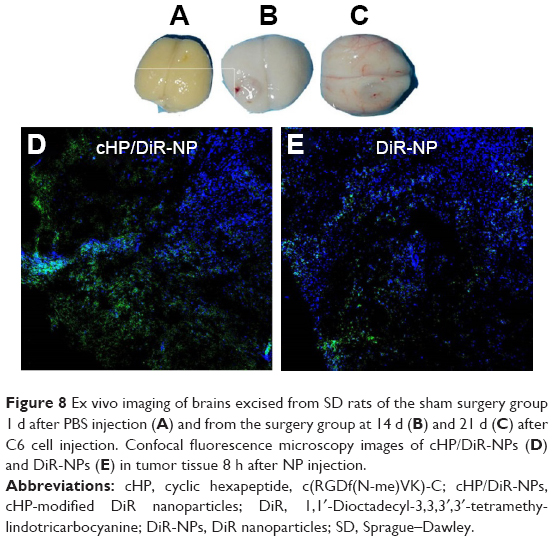 | Figure 8 Ex vivo imaging of brains excised from SD rats of the sham surgery group 1 d after PBS injection (A) and from the surgery group at 14 d (B) and 21 d (C) after C6 cell injection. Confocal fluorescence microscopy images of cHP/DiR-NPs (D) and DiR-NPs (E) in tumor tissue 8 h after NP injection. |
Confocal fluorescence images of cHP/DiR-NPs and DiR-NPs in tumor tissue after NP injection for 8 h in C6 glioma rats are shown in Figure 8D and E. The blue regions in the image are nuclei stained with Hochest33342, and the green fluorescence indicates DiR encapsulated in the NPs. The green fluorescence signal in the cHP/DiR-NP group was stronger than that in the DiR-NP group.
Figure 9 shows in vivo fluorescence images of C6 gliomas of SD rats at 1, 4, 8, 12, and 24 h after the tail vein injection of free DiR-NPs or cHP/DiR-NPs. For the cHP/DiR-NP group, a weak fluorescence signal was observed in the brain 1 h after injection, and the signal increased gradually as time progressed. At 12 h after injection, the signal reached a maximum and was still observed in the brain at 24 h. For the DiR-NP group, a fluorescent signal in the tumor appeared at 4 h post-injection and reached a maximum at 12 h. The rats in the cHP/DiR-NP group exhibited a stronger signal in the tumor compared with the rats in the DiR-NP group at all points. Due to the thickness of the rat’s body, it is difficult to observe the fluorescent signal distribution of NPs in the other organs within the animal. Fluorescence signals were observed only in the superficial brain and abdomen. Therefore, the fluorescence images of the excised tissues obtained after 24 h are shown in Figure 9 to further confirm the DiR-NP distribution. The image shows that the fluorescent signal of the brain in the cHP/DiR-NP group was significantly stronger than that in the DiR-NP group. The fluorescent cHP/DiR-NPs of the excised tissues were successively distributed in the lungs, spleen, liver, brain, heart, and kidneys. The signals in the brain of the cHP/DiR-NP group were stronger than that of the DiR-NP group. Similar to our earlier results in the C6 ectopic tumors (presented in the Supplementary materials), fewer cHP/DiR-NPs were observed in the lungs, spleens, and kidneys of the glioma rats than DiR-NPs in the respective groups.
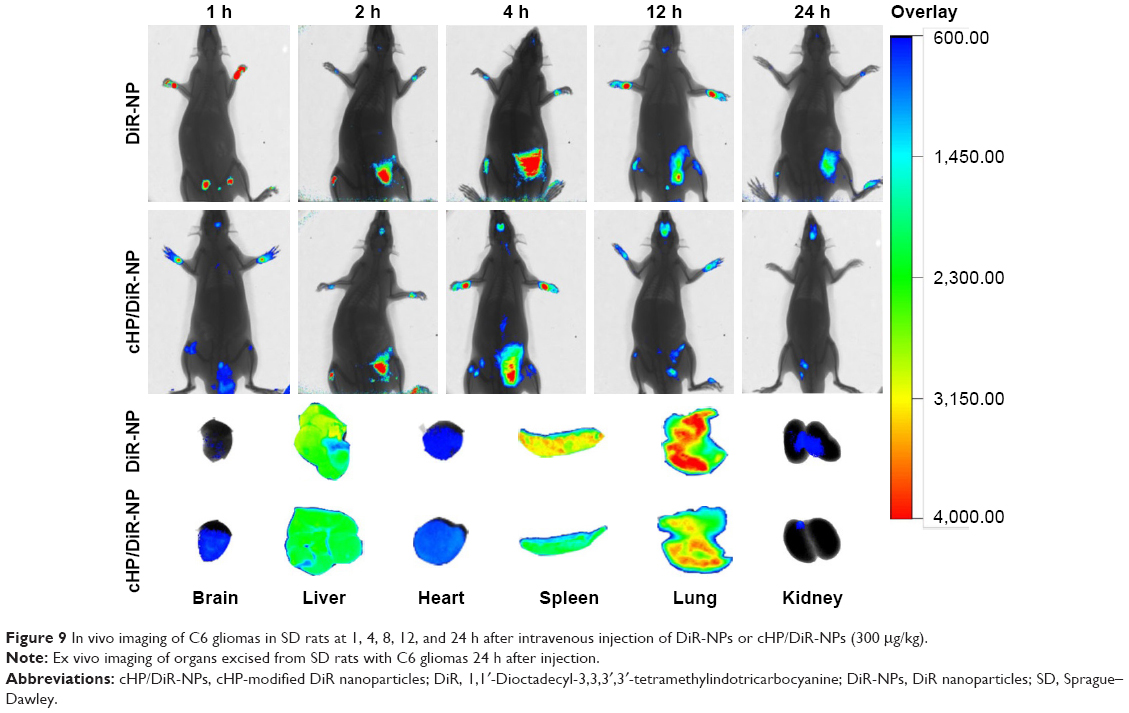 | Figure 9 In vivo imaging of C6 gliomas in SD rats at 1, 4, 8, 12, and 24 h after intravenous injection of DiR-NPs or cHP/DiR-NPs (300 μg/kg). |
Although in vivo targeting of NPs in the rat glioma model is only a qualitative experiment, this result further highlights that NPs modified with a cyclic hexapeptide can pass through the BBB and accumulate in a targeted manner in glioma tumor tissues.
Conclusion
In this study, various cyclic hexapeptides were screened via computer simulation docking and in vitro cell evaluations. The cyclic hexapeptide of c(RGDf(N-me)VK)–C, which exhibited strong affinity to integrin αvβ3-overexpressing cells, enhanced targeting specificity, as demonstrated through an in vitro cell evaluation. c(RGDf(N-me)VK)–C and PEG-PLGA conjugates were successfully synthesized. Cur-NPs modified with cyclic hexapeptides were prepared through the self-assembly method and they exhibited a spherical shape and a narrow size distribution. The NPs could specifically target monolayer cells and cell spheroids overexpressing integrin αvβ3. The in vitro cytotoxicity of the cyclic hexapeptide-modified cHP/Cur-NPs was higher than that of free curcumin and Cur-NP because of their superior ability to target tumor neovascularization and tumor cells. Compared with NPs not modified with cyclic hexapeptides, the NPs modified with cyclic hexapeptides show better penetration and aggregation ability in the brain of glioma rats and less distribution in the lungs and spleens. These results demonstrated that c(RGDf(N-me)VK)-C can serve as an effective targeting ligand, and the Cur-NPs modified with the cyclic hexapeptides can be exploited as a potential drug delivery system for targeting gliomas. In future studies, we will continue to evaluate their therapeutic efficacy in animal glioma models.
Acknowledgments
This work was supported by the Natural Science Foundation of Shandong (grant number ZR2014HM062) and the National Science and Technology Major Project (grant number 2013ZX09402201).
Disclosure
The authors report no conflicts of interest in this work.
References
Price RL, Chiocca EA. Evolution of malignant glioma treatment: from chemotherapy to vaccines to viruses. Neurosurgery. 2014;61(Suppl 1):74–83. | ||
Qin L, Wang CZ, Fan HJ, et al. A dual-targeting liposome conjugated with transferrin and arginine-glycine-aspartic acid peptide for glioma-targeting therapy. Oncol Lett. 2014;8(5):2000–2006. | ||
Pardridge WM. Brain drug development and brain drug targeting. Pharm Res. 2007;24(9):1729–1732. | ||
Alnaami IM, Al-Nuaimi SK, Senthilselvan A, et al. Effectiveness of adjuvant temozolomide treatment in patients with glioblastoma. Neurosciences (Riyadh). 2013;18(4):349–355. | ||
Oberoi RK, Parrish KE, Sio TT, Mittapalli RK, Elmquist WF, Sarkaria JN. Strategies to improve delivery of anticancer drugs across the blood-brain barrier to treat glioblastoma. Neuro Oncol. 2016;18(1):27–36. | ||
Vartanian A, Singh SK, Agnihotri S, et al. GBM’s multifaceted landscape: highlighting regional and microenvironmental heterogeneity. Neuro Oncol. 2014;16(9):1167–1175. | ||
Kurozumi K, Ichikawa T, Onishi M, Fujii K, Date I. Cilengitide treatment for malignant glioma: current status and future direction. Neurol Med Chir (Tokyo). 2012;52(8):539–547. | ||
Mas-Moruno C, Rechenmacher F, Kessler H. Cilengitide: the first anti-angiogenic small molecule drug candidate design, synthesis and clinical evaluation. Anticancer Agents Med Chem. 2010;10(10):753–768. | ||
Vick NA, Khandekar JD, Bigner DD. Chemotherapy of brain tumors. Arch Neurol. 1977;34(9):523–526. | ||
Herve F, Ghinea N, Scherrmann JM. CNS delivery via adsorptive transcytosis. AAPS J. 2008;10(3):455–472. | ||
Sugahara KN, Teesalu T, Karmali PP, et al. Tissue-penetrating delivery of compounds and nanoparticles into tumors. Cancer Cell. 2009;16(6):510–520. | ||
Senft C, Polacin M, Priester M, Seifert V, Kogel D, Weissenberger J. The nontoxic natural compound curcumin exerts anti-proliferative, anti-migratory, and anti-invasive properties against malignant gliomas. BMC Cancer. 2010;10:491. | ||
Yallapu MM, Jaggi M, Chauhan SC. Curcumin nanoformulations: a future nanomedicine for cancer. Drug Discov Today. 2012;17(1–2):71–80. | ||
Wadhwa J, Asthana A, Gupta S, Shilkari Asthana G, Singh R. Development and optimization of polymeric self-emulsifying nanocapsules for localized drug delivery: design of experiment approach. Scientific World Journal. 2014;2014:516069. | ||
Xiong JP, Stehle T, Diefenbach B, et al. Crystal structure of the extracellular segment of integrin αvβ3. Science. 2001;294(5541):339–345. | ||
Chen H, Niu G, Wu H, Chen X. Clinical application of radiolabeled RGD peptides for PET imaging of integrin αvβ3. Theranostics. 2016;6(1):78–92. | ||
Liu S. Radiolabeled cyclic RGD peptides as integrin αvβ3-targeted radiotracers: maximizing binding affinity via bivalency. Bioconjugate Chem. 2009;20(12):2199–2213. | ||
Liu X, Wang W, Samarsky D, et al. Tumor-targeted in vivo gene silencing via systemic delivery of cRGD-conjugated siRNA. Nucleic Acids Res. 2014;42(18):11805–11817. | ||
Yu KF, Zhang WQ, Luo LM, et al. The antitumor activity of a doxorubicin loaded, iRGD-modified sterically-stabilized liposome on B16-F10 melanoma cells: in vitro and in vivo evaluation. Int J Nanomedicine. 2013;8:2473–2485. | ||
Perche F, Torchilin VP. Cancer cell spheroids as a model to evaluate chemotherapy protocols. Cancer Biol Ther. 2012;13(12):1205–1213. | ||
Gao H, Hu Y, Xiong Y, et al. Glioma targeting and anti-glioma effect of interleukin 13 peptide and RGD peptide dual functionalized nanoparticles. Curr Pharm Biotechnol. 2014;14(13):1118–1126. | ||
Sawant RR, Torchilin VP. Challenges in development of targeted liposomal therapeutics. AAPS J. 2012;14(2):303–315. | ||
Borgne-Sanchez A, Dupont S, Langonne A, et al. Targeted Vpr-derived peptides reach mitochondria to induce apoptosis of αvβ3-expressing endothelial cells. Cell Death Differ. 2007;14(3):422–435. | ||
Fu YC, Fu TF, Wang HJ, et al. Aspartic acid-based modified PLGA-PEG nanoparticles for bone targeting: in vitro and in vivo evaluation. Acta Biomater. 2014;10(11):4583–4596. | ||
Trusolino L, Serini G, Cecchini G, et al. Growth factor-dependent activation of αVβ3 integrin in normal epithelial cells: implications for tumor invasion. J Cell Biol. 1998;142(4):1145–1156. | ||
Liu Z, Liu S, Wang F, Liu S, Chen X. Noninvasive imaging of tumor integrin expression using 18F-labeled RGD dimer peptide with PEG4 linkers. Eur J Nucl Med Mol Imaging. 2009;36(8):1296–1307. | ||
Shi J, Votruba AR, Farokhzad OC, Langer R. Nanotechnology in drug delivery and tissue engineering: from discovery to applications. Nano Lett. 2010;10(9):3223–3230. | ||
Tannock IF, Lee CM, Tunggal JK, Cowan DS, Egorin MJ. Limited penetration of anticancer drugs through tumor tissue: a potential cause of resistance of solid tumors to chemotherapy. Clin Cancer Res. 2002;8(3):878–884. | ||
Sethi P, Jyoti A, Swindell EP, et al. 3D tumor tissue analogs and their orthotopic implants for understanding tumor-targeting of microenvironment-responsive nanosized chemotherapy and radiation. Nanomedicine. 2015;11(8):2013–2023. | ||
DeArmond SJ, Stowring L, Amar A, et al. Development of a non-selecting, non-perturbing method to study human brain tumor cell invasion in murine brain. J Neurooncol. 1994;20(1):27–34. |
Supplementary materials
In vivo targeting evaluation in ectopic tumors of nude mice
Methods
C6 cells (5×106 per mice) were transplanted into the right forelimbs of nude mice (weighing 16–22 g), and the mice were used in subsequent experiments ~1 month later, when the tumors had grown to ~800 mm3.
Six tumor-bearing nude mice were randomly divided into three groups, and 0.2 mL of 1,1′-Dioctadecyl-3,3,3′,3′-tetramethylindotricarbocyanine (DiR) nanoparticles (DiR-NPs), cyclic hexapeptide, c(RGDf(N-me)VK)-C (cHP)-modified DiR nanoparticles (cHP/DiR-NPs), or DiR solution was injected (300 μg/kg i.v.) into the mice. At 1, 4, 6, 8, 10, 12, and 24 h after injection, the mice were anesthetized with 5% chloral hydrate. In vivo imaging (Care stream In Vivo FX, Bruker, Madison, WI, USA) was performed to screen the fluorescence distribution and assess the in vivo targeting ability of the NPs. At 8 h post-injection, the mice were euthanized, and the tumor and certain organs were harvested. The fluorescent images of the tumors and organs were recorded and analyzed using Kodak MI In Vivo Fx Pro software (Kodak, Rochester, NY, USA).
Results
To investigate the targeting of NPs modified with a cyclic hexapeptide in a pathological condition, an ectopic tumor model was developed using C6 cells in the forelimbs of nude mice. Instead of curcumin, DiR was used as a tracing agent for in vivo imaging and was encapsulated into NPs and NPs modified with a cyclic hexapeptide to monitor their distributions in tumor-bearing nude mice. Figure S1A shows in vivo fluorescence images of C6 tumor-bearing nude mice at 1, 4, 6, 8, 10, 12, and 24 h after tail vein injection of free DiR, DiR-NPs, or cHP/DiR-NPs. For the cHP/DiR-NP group, weak fluorescence was observed in the tumor 4 h after injection, and the signal gradually increased thereafter. At 8 h after injection, the signal reached a maximum value and was still detectable in the tumors at 24 h. For the DiR-NP group, a fluorescent signal appeared in the tumor at 6 h post-injection and reached a maximum at 8 h. However, the signal rapidly weakened thereafter, and very weak fluorescence was observed at the last time point. Free DiR can easily enter all tissues and tumors, but the distribution of free DiR in the tumors was low due to its broad tissue distribution. Therefore, the fluorescence signal in the free DiR group appeared earlier than in the other two groups, but its signal strength was weak at all time points. For the two NP groups, the mice in the cHP/DiR-NP group presented a stronger signal in the tumor tissue than the mice in the DiR-NP group at most time points. This result indicates that DiR-NPs modified with a cyclic hexapeptide have a better targeting effect than DiR-NPs that are not modified with a cyclic hexapeptide.
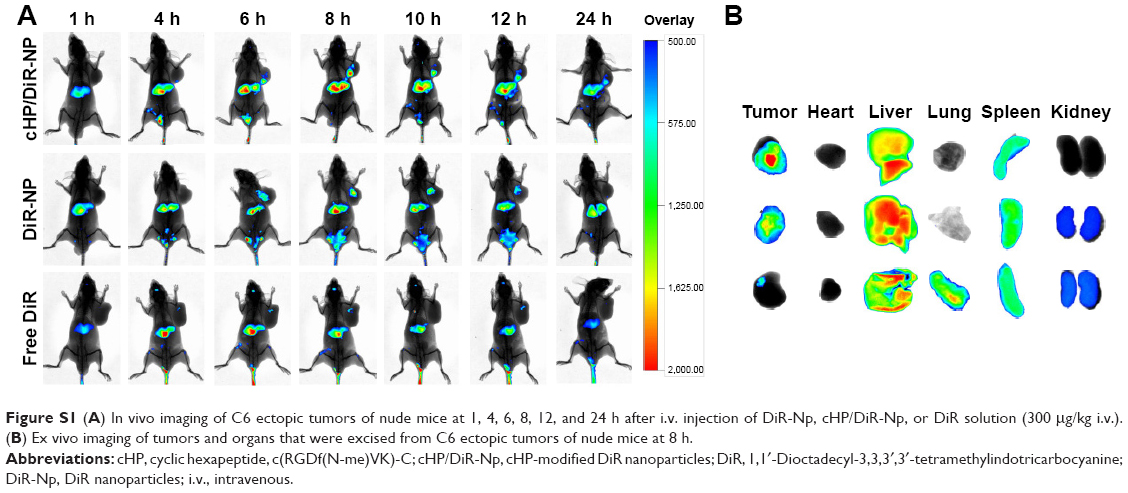 | Figure S1 (A) In vivo imaging of C6 ectopic tumors of nude mice at 1, 4, 6, 8, 12, and 24 h after i.v. injection of DiR-Np, cHP/DiR-Np, or DiR solution (300 μg/kg i.v.). (B) Ex vivo imaging of tumors and organs that were excised from C6 ectopic tumors of nude mice at 8 h. |
Figure S1B shows fluorescence images of the excised tissues acquired 8 h after treatment initiation, which further confirmed the distribution of free DiR and its NPs. As shown in the image, the fluorescent signal of the tumor in the cHP/DiR-NP group was stronger than that in the other groups. This finding further emphasized that cyclic hexapeptide-modified NPs aggregate better in tumors than NPs not modified with cyclic hexapeptide. In the free DiR group, the signal observed in the liver tissue was the strongest, followed by signals in the lungs, spleen, and tumor. The signal strength in tumor of the free DiR group was significantly weaker than the signals in the two NP groups. The NPs of the two NP groups were also primarily distributed in the liver for phagocytosis and ingestion by macrophages,1 followed by distribution in the tumor, spleen, and kidneys. As expected, the signal in the tumor tissue of the cHP/DiR-NP group was significantly stronger than that of the DiR-NP group because the enhanced permeability and retention effect at the tumor site increases NP accumulation. In addition, αvβ3 integrin-overexpressing C6 cells can also increase the specific binding to cyclic hexapeptide in the cHP/DiR-NP group. The aggregate fluorescence in the lungs and kidneys of the tumor-bearing nude mice was lower in the cHP/DiR-NP group than in the other two groups. This finding demonstrated that the cyclic hexapeptide could target integrin-overexpressing tumors.
Reference
Davis ME, Chen ZG, Shin DM. Nanoparticle therapeutics: an emerging treatment modality for cancer. Nat Rev Drug Discov. 2008;7(9):771–782. |
 © 2017 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2017 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.