Back to Journals » International Journal of General Medicine » Volume 13
Cushing’s Disease Caused by a Pituitary Microadenoma Coexistent with a Meningioma: A Case Report and Literature Review
Authors Wang Y ![]() , Sun Z, Jiang Z
, Sun Z, Jiang Z
Received 13 October 2020
Accepted for publication 16 November 2020
Published 24 November 2020 Volume 2020:13 Pages 1243—1248
DOI https://doi.org/10.2147/IJGM.S285833
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Dr Scott Fraser
Yu Wang, Zhixiang Sun, Zhiquan Jiang
Department of Neurosurgery, The First Affiliated Hospital of Bengbu Medical College, Bengbu, Anhui, People’s Republic of China
Correspondence: Zhiquan Jiang
Department of Neurosurgery, The First Affiliated Hospital of Bengbu Medical College, 287 Changhuai Road, Bengbu, Anhui 233004, People’s Republic of China
Tel +86-13966075971
Email [email protected]
Abstract: Cushing’s disease (CD), also known as adrenocorticotropic hormone (ACTH)-dependent pituitary Cushing’s syndrome, is a rare and serious chronic endocrine disease that is usually caused by a pituitary adenoma (especially a pituitary microadenoma). Meningioma is the most common type of primary intracranial tumor and is usually benign. The patient in this case report presented with CD coexisting with pituitary microadenoma and meningioma, which is an extremely rare comorbidity. The pathogenesis of CD associated with meningioma remains unclear. Here, we describe the case of bilateral lower extremity edema, lower limb pain, abdominal purplish striae, and abdominal distension for 9 months in a 47-year-old woman. Two years ago, the patient underwent a hysterectomy at a local hospital for hysteromyoma. She had no previous radiotherapeutic treatment or other medical history. Magnetic resonance imaging of her head revealed a sellar lesion (7.8 mm × 6.4 mm) and a spherical mass (3.0 cm × 3.0 cm) in the right frontal convexity. Her level of serum adrenocorticotropic hormone (ACTH) was 169 pg/mL, and her cortisol levels were 933 nmol/mL and 778 nmol/mL at 8 am and 4 pm, respectively. Preoperatively, she was diagnosed with ACTH-secreting pituitary microadenoma and meningioma. Excision of the meningioma was performed through a craniotomy, while an endoscopic endonasal transsphenoidal approach was used to remove the pituitary adenoma. Meningioma and pituitary adenoma were confirmed by postoperative pathology. On the basis of this unusual case, the relevant literature was reviewed to illustrate the diagnosis and treatment of Cushing’s disease and to explore the pathogenesis of pituitary adenoma associated with meningioma.
Keywords: Cushing’s disease, pituitary adenoma, meningioma
Introduction
Cushing’s disease (CD) is a severe condition caused by an adrenocorticotropic hormone (ACTH)-secreting pituitary tumor that accounts for approximately 70% of all cases of endogenous Cushing’s syndrome. It has a total incidence of 1–2 cases per million per year and a prevalence rate of approximately 30 patients per million per year, making it an uncommon disease.1 Meningiomas account for 15–25% of all intracranial tumors, with an annual incidence of 6 cases per 100,000 persons.2 CD combined with meningioma is a rare condition, and even rarer in patients who have no previously known risk factors for either tumor. To the best of our knowledge, its pathogenesis have not been clearly described to date.
Case Presentation
Clinical History and Laboratory Findings
A 47-year-old woman was admitted to the endocrinology department of our hospital with chief complaints of bilateral lower extremity edema, left lower limb pain, abdominal purplish striae, and abdominal distension for 9 months. Two years ago, the patient had a hysterectomy at a local hospital for hysteromyoma. She had no previous radiotherapeutic treatment or other medical history. She weighed 90 kg and was 165 cm tall with a body mass index (BMI) of 33. Physical examination showed typical features of Cushing’s syndrome, including centripetal obesity, moon face, pedal edema, and buffalo hump. Her skin was thin and dry, with acne and hirsutism. On admission, her blood pressure was 146/115 mmHg and routine biochemical blood tests confirmed comorbidity with diabetes mellitus, hyperlipidemia, and hypokalemia.
Endocrine measurements showed that her serum ACTH was 169 pg/mL (reference value: 5–50 pg/mL), cortisol (8 am) was 933 nmol/L (reference value: 138–690 nmol/L), and cortisol (4 pm) was 778 nmol/L (reference value: 69–345 nmol/L), indicating that her ACTH and cortisol levels were dramatically increased. Cortisol secretion was increased and had lost its circadian rhythm. The low-dose dexamethasone suppression test showed that cortisol suppression was < 50%, while a >50% suppression of cortisol was found in the high-dose dexamethasone suppression test. Serum prolactin, follicle-stimulating hormone, luteinizing hormone, testosterone, free thyroid hormone (FT3 and FT4), and thyrotropin values were normal. Endocrinological evaluation suspected that pituitary lesions caused Cushing syndrome.
Imaging Analysis
The patient underwent a magnetic resonance imaging (MRI) scan to image her head. T1-weighted MRI with contrast enhancement showed a spherical enhancing mass (3.0 cm × 3.0 cm) in the right frontal convexity and a dural tail sign (Figure 1A). In the sellar area, the enhancement degree of the lesion (7.8 mm × 6.4 mm) was significantly lower than that of the surrounding pituitary tissue, and the pituitary stalk was displaced to the right (Figure 1A and B). No abnormalities were found on plain or enhanced adrenal computed tomography scans.
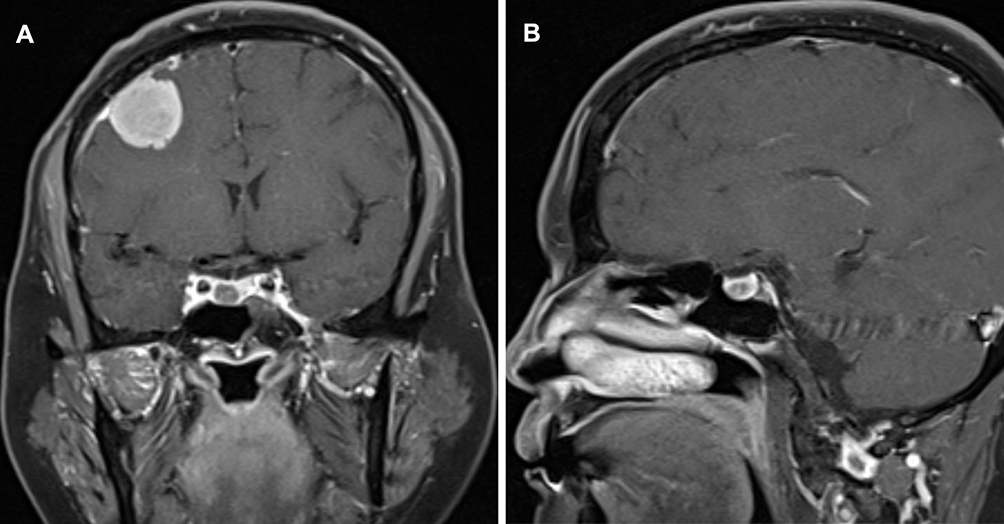 |
Figure 1 Enhanced magnetic resonance imaging (MRI) of the patient’s head: (A) Coronal view of the gadolinium-enhanced T1-weighted image showing a spherical enhancing mass in the right frontal convexity and a dural tail sign. A round low-intensity lesion can be seen on the right side of the pituitary gland, and the pituitary stalk is displaced to the right. (B) Sagittal T1-weighted sequence with contrast showing the degree of enhancement is lower than that of the pituitary in the sellar region. |
Treatment and Pathological Examination
Physical examination, endocrine examination, and head MRI successfully proved that pituitary microadenoma caused Cushing’s syndrome (specifically CD) comorbid with asymptomatic meningioma.
In order to receive surgical treatment, the patient was referred from the endocrinology department to neurosurgery. She underwent neuroendoscopic transsphenoidal surgery and the pituitary microadenoma was removed. The sellar floor was reconstructed with artificial dura mater, and after this reconstruction, no cerebrospinal fluid leakage was observed. The pathological specimen was examined and was determined to be consistent with a pituitary microadenoma (Figure 2A). One month later, excision of the meningioma was performed through a right frontal trephine craniotomy. Histological examination revealed a WHO grade I meningioma (Figure 2B).
 |
Figure 2 (A) Histopathologic examination revealed a pituitary adenoma (Hematoxylin and eosin staining, 100×). (B) Histopathologic examination revealed a meningioma (Hematoxylin and eosin staining, 100×). |
Outcome and Follow Up
On the second day after the operation, her cortisol level dropped below the normal range in the morning. Hydrocortisone replacement therapy was started on the same day. In addition, she had developed transient diabetes insipidus, which was treated with desmopressin. Three months postoperatively, after hydrocortisone replacement therapy, the symptoms of Cushing’s disease were alleviated, and the cortisol level returned to normal, which was 249nmol/L (reference value: 138~690nmol/L).
At the 1-year follow-up, no lesions were observed on the MRI scan and the symptoms of Cushing’s syndrome were in remission. The use of hydrocortisone supplements were discontinued and hormone levels remained normal, indicating recovery of the hypothalamic–pituitary–adrenal (HPA) axis. The patient had lost 30 kg and her BMI had dropped to 22, while her blood glucose, triglyceride level, and blood pressure had all returned to normal. Physical changes in the patient pre- and post-treatment are shown in Figure 3A and B.
 |
Figure 3 Abdominal appearance with striae (A) preoperation and (B) 4 months postoperation. |
Discussion
Cushing’s Disease
CD is a serious clinical condition caused by a pituitary adenoma secreting a high level of ACTH, leading to hypercortisolism. The proportion of ACTH-secreting pituitary adenomas (corresponding to CD) among hormone-secreting pituitary adenomas is 4.8%–10%, which affects women three times more frequently than men, mainly occurs in those 40–60 years old.3,4 Exposure to excessive cortisol can lead to various manifestations of Cushing’s syndrome and increases in morbidity and mortality.5 Therefore, early diagnosis and treatment of CD are very important.
The diagnosis and differential diagnosis of CD is very complicated, and these have always been challenging problems in clinical endocrinology. Once Cushing’s syndrome is diagnosed, its etiology should be determined. A diagnosis of Cushing’s disease is made based on a biochemical examination confirming the pituitary origin of the condition and exclude other sources (namely, ectopic ACTH secretion and adrenocortical tumors).3 High-dose dexamethasone suppression and corticotropin-releasing-hormone stimulation tests may be used to distinguish high-secretion sources of pituitary and ectopic ACTH. More than 90% of the pituitary adenomas that cause CD are microadenomas (≤10 mm in diameter), and 40% of the cases cannot be located by radiological examination.5 Examination with bilateral inferior petrosal sinus sampling (BIPSS) is necessary for CD patients in whom noninvasive biochemical and imaging examinations do not lead to a definitive diagnosis.6
The first-line treatment for CD is transsphenoidal selective tumor resection (TSS) with approximately 78% of the patients in remission after the operation, and 13% of patients relapse within 10 years after surgery. Therefore, there are a considerable number of patients who have experienced long-term surgical failure and require additional second-line treatment, such as radiotherapy, bilateral adrenalectomy, or medication.4
The pathogenesis of CD is unclear, but recent studies have confirmed that there are somatic activation mutations of multiple genes in adrenocorticotropin adenomas, while ubiquitin specific peptidase 8 (USP8) is the most common, accounting for about 50% of the mutations in these adenomas.7
Pituitary Adenoma Associated with Meningioma
Radiotherapy used to treat pituitary tumors is a well-known reason for the development of meningiomas. Gene mutations are a common molecular characteristic of meningiomas, with inactivation of the neurofibromatosis type 2 (NF2) tumor suppressor gene found in 55% of meningiomas, and a further 25% of meningiomas accounted for by recently described mutations in other genes.8
Simultaneous occurrence of pituitary adenoma and meningioma without a history of radiotherapy is a rare condition clinically, having only been described in 49 cases before 2019,9 while ACTH-secreting pituitary adenomas (CD) comorbid with meningioma have been reported even less frequently. In the reported cases, the most common site of meningioma is parasellar, accounting for 44.9%, while meningioma located in the distant part of the adenoma is rare.9,10
A number of clinicians have suggested that the coexistence of meningiomas and pituitary adenomas is incidental, with no relationship between the two diseases.2,11
Genetic imbalances have been found in pituitary adenomas, including in particular the chromosomal deletions of 1p, 2q, 4, 5, 6, 11q, 12q, 13q, and 18q, and the overexpression of 9q, 16p, 17p, 19, and 20q. Functional adenomas have more such imbalances than nonfunctional adenomas, corresponding in particular to deletions of chromosomes 4 and 18q, and the overexpression of chromosomes 17 and 19.12 Meanwhile, estrogen receptor positive de novo meningiomas significantly involve chromosomes 14 and 22.13
The study by Hwang et al14 reported that the expression levels of heterogeneous nuclear ribonucleoprotein (hnRNP) family proteins were significantly higher in pituitary adenomas and meningiomas than that in normal brain tissues. Leucine-rich repeat-containing G-protein coupled receptor 5 (LGR5) and its downstream signaling pathways play an pivotal role in pituitary tumor, meningioma, and other brain tumors. Zhu et al15 reported that multiple endocrine neoplasia type 1 (MEN1) plays an important role in pituitary adenoma associated with meningioma by upregulating the mammalian target of rapamycin signaling pathway. They found that rapamycin treatment promotes apoptosis in primary cells of the pituitary adenoma and meningioma in cases of pituitary adenoma associated with meningioma. Recurrence of pituitary adenoma, younger age, and larger size of meningioma have been shown to be significantly associated with MEN1 mutation.16
Mathuriya et al17 suggested that hormones may contribute to the occurrence of meningiomas.
de Vries et al9 reported that compared with other types of adenomas, the proportion of growth hormone adenomas is higher, accounting for about one third of cases. Meanwhile, Friend et al18 demonstrated that activation of GH/insulin-like growth factor-1 (IGF-1) axis clearly increased the growth rate of meningiomas. However, in the present case, we observed the coexistence of ACTH-secreting adenoma and meningioma. Further studies are required to understand whether ACTH or cortisol are related to the occurrence and development of meningioma.
In our case, pituitary microadenoma was the cause of Cushing’s syndrome, while the meningioma was an incidental imaging observation. With the popularity and technological progress of high-resolution imaging technology, the reported prevalence of intracranial lesions related to dominant pathology has increased.2 However, when imaging examinations are limited to specific regions, the diagnosis of lesions in other locations is likely to be omitted. For example, in our case, performing MRI of the sellar region alone may have meant that the meningioma was missed.
Conclusion
Cushing’s disease is the most common cause of endogenous Cushing’s syndrome and is caused by ACTH-secreting pituitary adenoma.It is associated with severe complications and reduced quality of life, so early diagnosis and treatment are critical. The coexistence of CD, pituitary adenoma, and meningioma is very rare, and the exact mechanisms underlying such comorbidity are currently unclear and need further study.
Data Sharing Statement
The data that support the findings of this study are available on request from the corresponding author, Zhiquan Jiang.
Ethics and Consent Statement
Based on the regulations of the department of research of the Bengbu Medical College, institutional review board approval is not required for case reports.
Consent for Publication
Written informed consent has been provided by the patient to have the case details and any accompanying images published.
Author Contributions
All authors made substantial contributions to conception and design, acquisition of data, or analysis and interpretation of data; took part in drafting the article or revising it critically for important intellectual content; agreed to submit to the current journal; gave final approval of the version to be published; and agree to be accountable for all aspects of the work.
Funding
The authors declared that this case has received no financial support.
Disclosure
The authors report no conflicts of interest in this work.
References
1. Lacroix A, Feelders RA, Stratakis CA, Nieman LK. Cushing’s syndrome. Lancet. 2015;386(9996):913–927. doi:10.1016/S0140-6736(14)61375-1
2. Curto L, Squadrito S, Almoto B, et al. MRI finding of simultaneous coexistence of growth hormone-secreting pituitary adenoma with intracranial meningioma and carotid artery aneurysms: report of a case. Pituitary. 2007;10(3):299–305. doi:10.1007/s11102-007-0011-4
3. Mehta GU, Lonser RR. Management of hormone-secreting pituitary adenomas. Neuro Oncol. 2017;19(6):762–773. doi:10.1093/neuonc/now130
4. Pivonello R, De Leo M, Cozzolino A, Colao A. The treatment of Cushing’s disease. Endocr Rev. 2015;36(4):385–486. doi:10.1210/er.2013-1048
5. Tritos NA, Biller BMK. Current management of Cushing’s disease. J Intern Med. 2019;286(5):526–541. doi:10.1111/joim.12975
6. Fan C, Zhang C, Shi X, et al. Assessing the value of bilateral inferior petrosal sinus sampling in the diagnosis and treatment of a complex case of Cushing’s disease. Intractable Rare Dis Res. 2013;2(1):24–29. doi:10.5582/irdr.2013.v2.1.24
7. Sbiera S, Kunz M, Weigand I, Deutschbein T, Dandekar T, Fassnacht M. The new genetic landscape of Cushing’s disease: deubiquitinases in the spotlight. Cancers. 2019;11(11):1761. doi:10.3390/cancers11111761
8. Apra C, Peyre M, Kalamarides M. Current treatment options for meningioma. Expert Rev Neurother. 2018;18(3):241–249. doi:10.1080/14737175.2018.1429920
9. de Vries F, Lobatto DJ, Zamanipoor Najafabadi AH, et al. Unexpected concomitant pituitary adenoma and suprasellar meningioma: a case report and review of the literature. Br J Neurosurg. 2019:1–5. doi:10.1080/02688697.2018.1556782.
10. Gosal JS, Shukla K, Praneeth K, et al. Coexistent pituitary adenoma and frontal convexity meningioma with frontal sinus invasion: a rare association. Surg Neurol Int. 2020;11:270. doi:10.25259/SNI_164_2020
11. Cannavo S, Curto L, Fazio R, et al. Coexistence of growth hormone-secreting pituitary adenoma and intracranial meningioma: a case report and review of the literature. J Endocrinol Invest. 1993;16(9):703–708. doi:10.1007/BF03348915
12. Szymas J, Schluens K, Liebert W, Petersen I. Genomic instability in pituitary adenomas. Pituitary. 2002;5(4):211–219. doi:10.1023/a:1025313214951
13. Pravdenkova S, Al-Mefty O, Sawyer J, Husain M. Progesterone and estrogen receptors: opposing prognostic indicators in meningiomas. J Neurosurg. 2006;105(2):163–173. doi:10.3171/jns.2006.105.2.163
14. Hwang M, Han MH, Park HH, et al. LGR5 and downstream intracellular signaling proteins play critical roles in the cell proliferation of neuroblastoma, meningioma and pituitary adenoma. Exp Neurobiol. 2019;28(5):628–641. doi:10.5607/en.2019.28.5.628
15. Zhu H, Miao Y, Shen Y, et al. The clinical characteristics and molecular mechanism of pituitary adenoma associated with meningioma. J Transl Med. 2019;17(1):354. doi:10.1186/s12967-019-2103-0
16. Zhu H, Miao Y, Shen Y, et al. Germline mutations in MEN1 are associated with the tumorigenesis of pituitary adenoma associated with meningioma. Oncol Lett. 2020;20(1):561–568. doi:10.3892/ol.2020.11601
17. Mathuriya SN, Vasishta RK, Dash RJ, Kak VK. Pituitary adenoma and parasagittal meningioma: an unusual association. Neurol India. 2000;48(1):72.
18. Friend KE, Radinsky R, McCutcheon IE. Growth hormone receptor expression and function in meningiomas: effect of a specific receptor antagonist. J Neurosurg. 1999;91(1):93–99. doi:10.3171/jns.1999.91.1.0093
 © 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.