Back to Journals » Cancer Management and Research » Volume 11
Current status of liquid biopsies for the detection and management of prostate cancer
Authors Lu YT ![]() , Delijani K, Mecum A
, Delijani K, Mecum A ![]() , Goldkorn A
, Goldkorn A
Received 3 January 2019
Accepted for publication 18 April 2019
Published 6 June 2019 Volume 2019:11 Pages 5271—5291
DOI https://doi.org/10.2147/CMAR.S170380
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 4
Editor who approved publication: Dr Ahmet Emre Eşkazan
Yi-Tsung Lu, Kevin Delijani, Andrew Mecum, Amir Goldkorn
Division of Medical Oncology, Department of Medicine, Keck School of Medicine and Translational and Clinical Science Program, USC Norris Comprehensive Cancer Center, University of Southern California, Los Angeles, CA, USA
Abstract: In recent years, new therapeutic options have become available for prostate cancer (PC) patients, generating an urgent need for better biomarkers to guide the choice of therapy and monitor treatment response. Liquid biopsies, including circulating tumor cells (CTCs), circulating nucleic acids, and exosomes, have been developed as minimally invasive assays allowing oncologists to monitor PC patients with real-time cellular or molecular information. While CTC counts remain the most extensively validated prognostic biomarker to monitor treatment response, recent advances demonstrate that CTC morphology and androgen receptor characterization can provide additional information to guide the choice of treatment. Characterization of cell-free DNA (cfDNA) is another rapidly emerging field with novel technologies capable of monitoring the evolution of treatment relevant alterations such as those in DNA damage repair genes for poly (ADP-ribose) polymerase (PARP) inhibition. In addition, several new liquid biopsy fields are emerging, including the characterization of heterogeneity, CTC RNA sequencing, the culture and xenografting of CTCs, and the characterization of extracellular vesicles (EVs) and circulating microRNAs. This review describes the clinical utilization of liquid biopsies in the management of PC patients and emerging liquid biopsy technologies with the potential to advance personalized cancer therapy.
Keywords: prostate cancer, biomarker, circulating tumor cell, circulating tumor DNA
Introduction
Liquid biopsy refers to the analysis of blood or other body fluids to obtain clinically or biologically relevant information about a solid malignancy, analogous to information obtained from a traditional tumor biopsy.1 Liquid biopsy encompasses a broad spectrum of approaches aimed at characterizing different components of body fluids, including circulating tumor cells (CTCs), cell-free DNA (cfDNA), circulating RNA, microRNAs, and extracellular vesicles (EVs). (Figure 1)
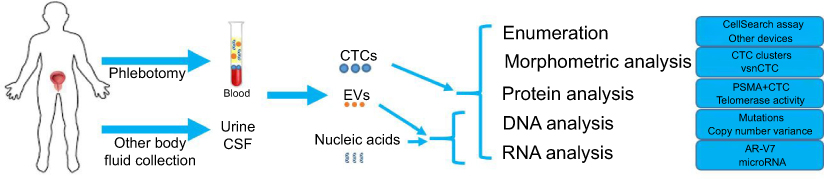 | Figure 1 Schematic overview of liquid biopsy analytes and profiling options in prostate cancer.Abbreviations: CTC, circulating tumor cell; EV, extracellular vesicle. |
There is an increasing interest in the use of liquid biopsies in the management of prostate cancer (PC), which remains the second leading cause of cancer death in men despite the development of many new therapies.2 From a clinical standpoint, liquid biopsies can be prognostic of PC outcome, predictive of response to treatment, or used to monitor disease. From a biological standpoint, a liquid biopsy serves as a surrogate source of tumor tissue that reflects the full molecular profile of the metastatic disease, thus revealing mechanisms of resistance and paving the way to the development of new therapies.
In this review, we discuss the recent advances and key technologies (Tables 1 and 2) in the field of liquid biopsy, focusing on their use as candidate clinical biomarkers in PC. Additionally, significant breakthrough discoveries and studies are summarized (Figure 2), as well as more recent emerging liquid biopsy fields and their potential impact on PC management.
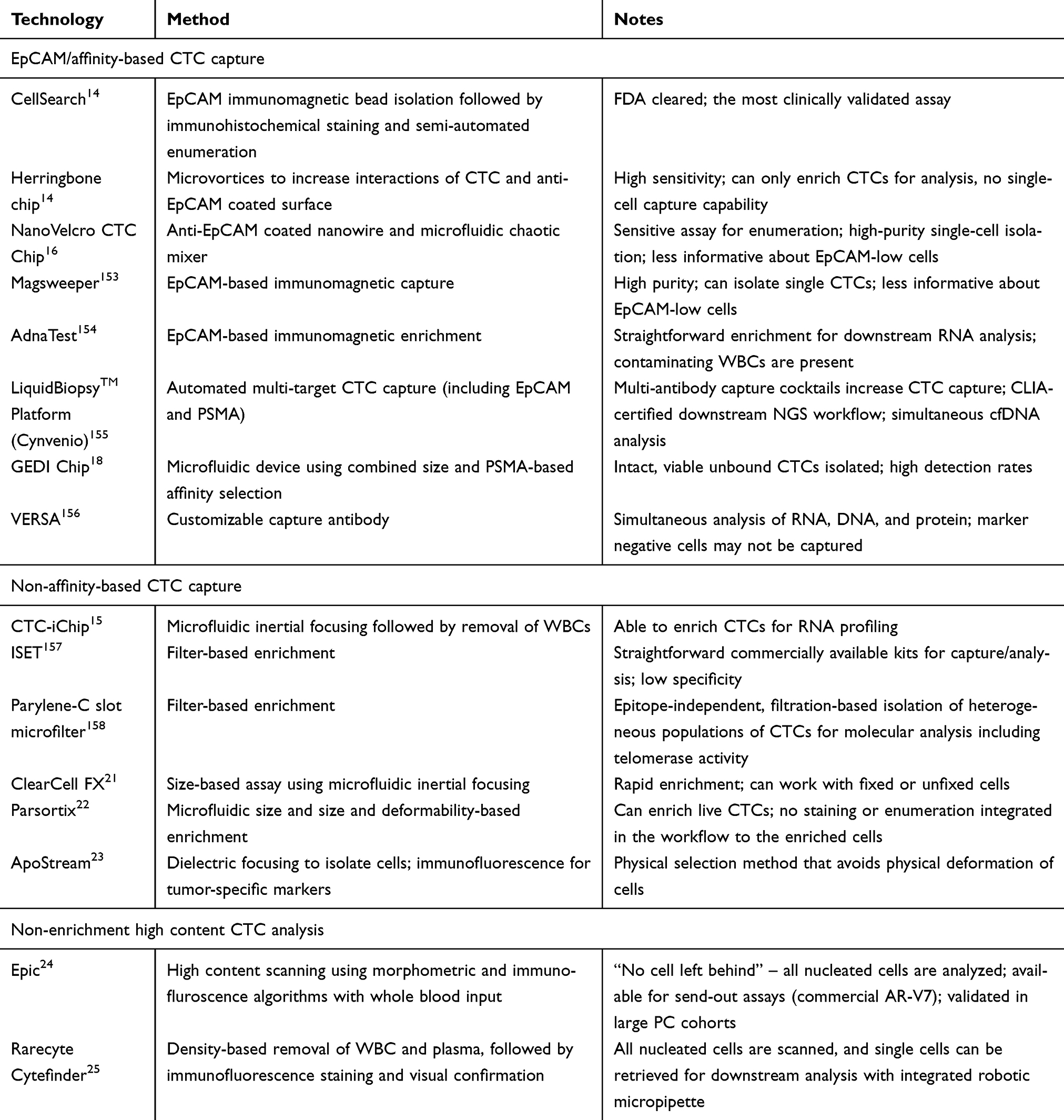 | Table 1 CTC capture methods |
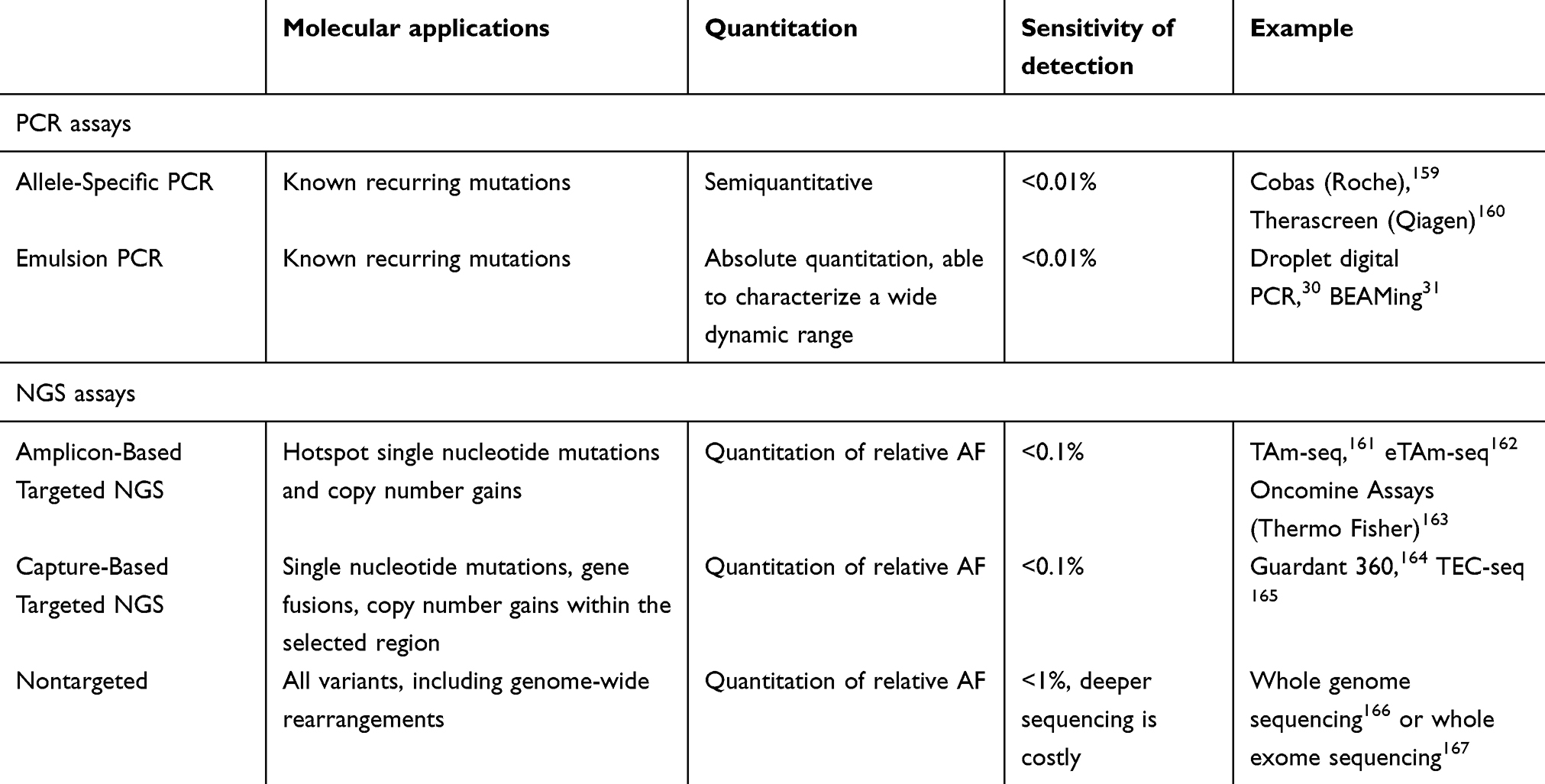 | Table 2 cfDNA technologies |
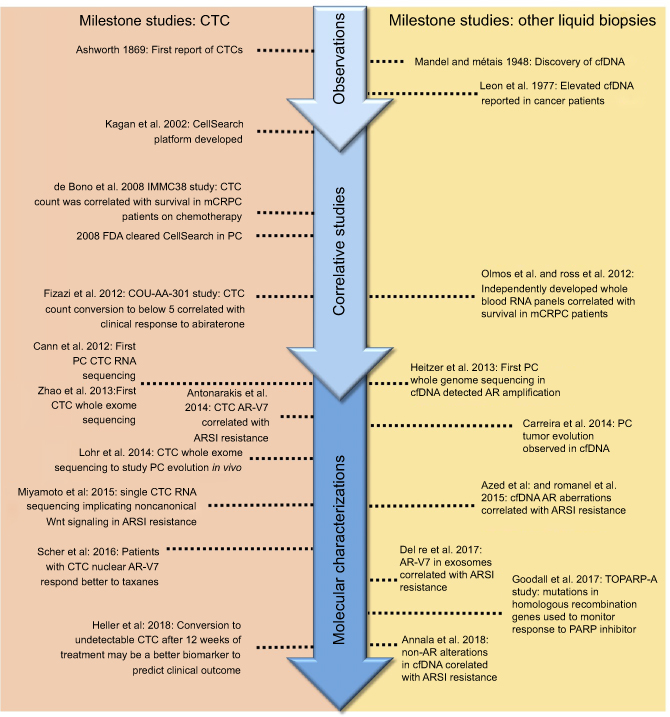 | Figure 2 Seminal liquid biopsy studies in prostate cancer.Abbreviations: CTC, circulating tumor cell; cfDNA, cell-free DNA; PC, prostate cancer; mCRPC, metastatic castration resistant prostate cancer; AR, androgen receptor; AR-V7, androgen receptor splice variant-7; ARSI, androgen receptor signaling inhibitor; PARP, poly (adenosine diphosphate [ADP]-ribose) polymerase. |
Technologies employed to characterize liquid biopsy samples
Circulating tumor cells (CTCs)
CTCs are cancer cells shed by primary or metastatic tumors into the bloodstream. CTC enumeration has been shown to correlate with prognosis and disease burden in patients with metastatic castration-resistant PC (mCRPC).3–7 More recent studies suggest that CTC molecular profiling also may be useful in predicting and monitoring the response of patients receiving specific therapies.8 A central barrier in studying CTCs is the rarity of the cells in the bloodstream, usually around one CTC per billion blood cells.9 This challenge has been addressed by a broad spectrum of new technology.
CellSearch (Menarini Silicon Biosystems Inc., Bologna, Italy) is the only FDA-cleared and clinically available assay for CTC enumeration.10 CellSearch is an affinity-based assay that uses immunomagnetic beads targeting EpCAM (epithelial cell adhesion molecule) to enrich CTCs.11 Most CTCs, which are epithelial in origin, express EpCAM, whereas the background white blood cells (WBCs) do not. The enumeration of CTCs is performed based on staining for DAPI (a nuclear marker), CK (cytokeratin, an epithelial marker), and CD45 (a WBC marker). DAPI+/CK+/CD45- cells are counted as CTCs.
The immunoaffinity method employed by CellSearch presents some limitations. Namely, due to its EpCAM-based enrichment, the assay cannot capture CTCs with down-regulated EpCAM. This can occur in cells undergoing epithelial–mesenchymal transition (EMT), an aggressive phenotypic shift observed in some tumor cells as they migrate and metastasize.12 Hence, an important population of CTCs undergoing EMT is under-represented in CellSearch.13 Similarly, EpCAM immunoaffinity assays cannot effectively capture CTCs shed by non-epithelial malignancies such as sarcomas and melanoma.
To improve upon affinity-based CTC capture, several platforms utilize microfluidic technology to enhance the capture of CTCs with lower EpCAM expression. For example, the herringbone chip utilizes a microfluidic architecture to generate microvortices and increase capture efficiency by creating more interactions between CTCs and the anti-EpCAM-coated surface.14 The same team subsequently developed CTC-iChip, a microfluidic device utilizing size-based separation based on inertial focusing, followed by surface antigen targeted magnetophoresis. CTC-iChip can capture CTCs via EpCAM-based enrichment but also is capable of EpCAM-independent CTC enrichment by depletion of CD45 and CD15 positive WBCs.15 The NanoVelcro CTC Chip is a device with improved CTC capture efficiency by utilizing anti-EpCAM antibody coated silicon nanowires to enhance the interactions with the microvilli on the CTCs and a microfluidic chaotic mixer to generate turbulent flow to enhance the substrate-CTC contact.16 Despite these advances, EpCAM-based strategies cannot isolate CTCs expressing very little or no EpCAM.17 To address this issue, additional capture targets, including PSMA (Prostate-Specific Membrane Antigen)18 and cadherin-1119 have been incorporated into affinity-based assays. Recently, a novel mechanism using rVAR2 (recombinant VAR2CSA) as a capture agent to interact with oncofetal chondroitin sulfate on tumor cells demonstrated efficacy for EpCAM-independent capture of CTCs in patients across different cancer types including PC.20
As an alternative strategy to affinity-based capture, CTC enrichment can be achieved based on the relatively large size and low deformability of CTCs. For example, ClearCell FX (Clearbridge BioMedics, Singapore) is a size-based instrument utilizing microfluidic inertial focusing to separate larger CTCs from smaller cells of hematogenous lineage.21 Parsortix (Angle PLC, Surrey, United Kingdom) is another microfluidic device that separates CTCs based on their size and compressibility.22 ApoStream (ApoCell Inc., Houston, TX, USA) is a microfluidic assay that captures CTCs based on dielectrophoresis, which separates cells according to their diameter, membrane area, density, and volume.23 Besides the capability to isolate CTCs without EpCAM surface expression, another advantage of these non-affinity-based CTC devices is their capacity to isolate live cells. Without the need to incubate cells with staining antibodies, these devices have a faster turnover time and preserve the viability of cells for downstream analysis, likely CTC culture and RNA analysis. One of the criticisms to these non-affinity-based CTC capture devices is the possibility of missing smaller CTCs or those with physical characteristics similar to WBCs.
Other, more recent technologies have taken a direct analysis approach wherein all the mononuclear cells in a blood sample are analyzed by automatic imaging. Such platforms include Epic (Epic Sciences, San Diego, USA)24 and Rarecyte CyteFinder (Rarecyte Inc., Seattle, WA, USA),25 both of which utilize high content immunofluorescence microscopy to scan slides with millions of cells and identify candidate CTCs using morphometric and immunofluorescence calling algorithms. Using this approach, early studies using the Epic platform revealed that CK negative CTCs can be identified and correlated with poor patient outcomes.26 Table 1 summarizes some representative technologies in these various categories. Figure 3 provides a graphic illustration of various CTC technologies.
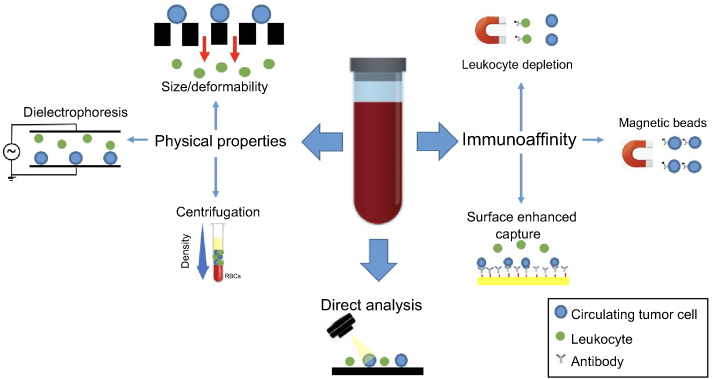 | Figure 3 CTC enrichment and detection strategies. CTCs are enriched from whole blood based on biological or physical properties or can be detected directly with high-content imaging. Immunoaffinity-based CTC enrichment is achieved via positive or negative selection for tumor or leukocyte-specific antigens, respectively. Capture efficiency can be enhanced by specifically engineered surfaces that maximize interaction in the microfluidic devices. Physical property-based enrichment exploits differential size, deformability, and dielectric properties to separate CTCs from background leukocytes. The direct analysis employs rapid, high content scanning of all blood cells after fixation in monolayer onto a slide. CTC candidates are identified using automated algorithms based on a combination of morphometric features and immunofluorescent staining for epithelial markers.Abbreviation: CTC, circulating tumor cell. |
To date, CellSearch remains the only FDA-cleared and most clinically validated CTC enumeration assay. It has been incorporated in numerous clinical trials and has repeatedly demonstrated its prognostic value in metastatic PC. Newer CTC assays demonstrated improved capture efficiency using various microfluidic designs. However, the association between patient outcome and CTCs enumerated by these novel devices has been less extensively validated. Furthermore, CellSearch developed optimized CTC calling algorithms that minimize inter-observer variability and maximize precision, a goal achieved with varying degrees of success by other platforms.
Beyond enumeration, we will discuss many reports of molecular analyses of CTCs in the review. These reports were all generated by novel CTC devices. While the reported data created great enthusiasm, we need to acknowledge that most of the CTC devices used have not undergone extensive prospective clinical validation in large patient cohorts, and in some cases, these devices may not even be available outside of the institutions where they were developed. Therefore, a systematic comparison of available platforms remains challenging, and many of these specialized biomarker assays must undergo further validation on their own merits with little head-to-head comparison to other approaches.
Cell-free DNA (cfDNA)
cfDNA refers to extracellular, short fragments of nucleic acids found in virtually all bodily fluids, including blood. cfDNA fragments are roughly 134–144 bp,27 suggesting that they are generated by the apoptotic degradation of cellular DNA.28 Circulating tumor DNA (ctDNA) is a component of cfDNA derived from tumor cells, and its ratio in cfDNA is largely dependent on overall tumor volume and the rate of turnover. Initially, detecting and analyzing somatic mutations in the relatively small portion of cfDNA comprised by ctDNA presented significant technical challenges.29 However, newer technologies can detect these alterations with increasing accuracy and are enabling the use of cfDNA as a biomarker for cancer diagnosis, prognosis, and therapeutic monitoring.
The first technology employed to characterize extremely low levels of mutated DNA was digital droplet PCR (ddPCR). Input DNA is diluted to the point at which one or less molecule is present per droplet containing fluorescent nucleotides for PCR amplification. The fluorescence of each droplet is subsequently measured by flow cytometry to quantify the number of fluorescent droplets with mutated copies of DNA.30 This technology is extremely sensitive for the detection of known mutations and remains widely utilized. Beads, emulsions, amplification, and magnetics (BEAM) is another technology that can identify and quantify rare mutations in plasma.31 Bead-containing emulsions are used instead of droplets to perform single-molecule PCR. Each DNA molecule is amplified on a bead, labeled with fluorescence, and the beads are later quantified by flow cytometry. Like ddPCR, BEAM is a sensitive assay capable of detecting mutations in the plasma at frequencies as low as 0.01%.32 Both assays have the same limitation in that a specific primer set needs to be designed to test each mutation. Therefore, they are relatively low throughput, and it is technically challenging to test more than 10 mutations simultaneously. Due to their high specificity, these PCR-based assays remain gold standards for the validation of a mutation of low allele frequency.
With the rapid advancement of next-generation sequencing (NGS) technologies, it is now possible to sequence multiple genes or even an entire genome in a single experiment. The general workflow of an NGS experiment includes a library preparation step, where the input genomic materials are converted into the library suitable for the sequencing platform, followed by the actual sequencing experiment and bioinformatic analysis. Whole genome sequencing (WGS) is performed when all the input materials are sequenced. The sequencing reads are distributed to a human genome of three giga base pairs. To achieve adequate coverage across the entire genome, the sequencing power needed for a WGS is very high, leading to a high cost. Alternatively, the library can be enriched to focus the sequencing power on the desired targets. The enrichment can be done by an amplicon-based approach or a hybridization capture approach. The amplicon-based approach uses pre-designed primers to amplify targeted sequences in the library, either before or at the time of library preparation. The amplicon approach is suitable for a small input of DNA but can only handle a relatively small panel of hotspot mutations due to the need for specific primer designs. In comparison, hybridization-based capture methods use designed oligonucleotides against complementary target regions as probes to bind the targeted DNA sequences, followed by the removal of background DNA.33 The capture approach has the advantage of enriching a large number of regions in a single assay and is the standard assay to perform whole exome sequencing (WES), but the required input DNA is higher.
Most of the recent cfDNA assays utilize deep sequencing with targeted enrichment for genes of interest to characterize mutations with low allele frequencies. To further increase the sensitivity and specificity, molecular barcoding technology was developed to discriminate the low-frequency mutations from sequencing errors.34 Many cfDNA assays are commercially available for routine clinical use. One example is the Guardant360 assay (Guardant Health, Redwood City, CA, USA). This CLIA-certified, commercially available assay performs capture-based enrichment and molecular barcoding, followed by the targeted sequencing of 73 clinically relevant genes.31 In a recent report, more than 10,000 samples were tested by the assay, and the authors reported a detection limit of 0.02% of allelic fraction, as well as a positive predictive value of 92–100% when primary tissue sequencing was used as the gold standard.35 The comparison of cfDNA technologies is summarized in Table 2.
Liquid biopsy as a biomarker
Biomarker development and qualification are comprised of three key phases: analytical validation, clinical validation, and assessment of clinical utility.36,37 Analytical validity describes the reproducibility and accuracy of the measurements. A thorough analytical validation requires the pre-analytical assessment of the specimen processing, the consistency of the measurement across different laboratories, and the post-analytical report of the results. Only after the analytical validity is confirmed and a laboratory workflow is established can a biomarker then be tested in specifically designed trials to assess its clinical validity, the degree to which it can be used as a surrogate for clinically relevant conditions or outcomes.37,38 A candidate biomarker’s sensitivity and specificity in discriminating a specific condition can be described as a binary outcome (ie, positive or negative) or plotted using a receiver operating characteristics (ROC) curve.39 Rigorous criteria, such as the Prentice criteria40 or the BEST resource,41 have been developed to guide the assessment of the adequacy of a candidate biomarker as a surrogate endpoint to patient outcome. The clinical utility of a biomarker is determined by how much additional information the biomarker provides and at what cost, thus measuring how useful a biomarker is in real-world clinical practice. Ideally, a biomarker should be less invasive, less expensive, widely available, and provide additional information that augments the utility of existing assays.42
Biomarkers can be further classified into three categories: a diagnostic biomarker that correlates with the presence of a medical condition; a prognostic biomarker that correlates with the likelihood of a future clinical event such as disease recurrence, progression, or death; and a predictive biomarker that correlates with the likelihood of response to a particular medical intervention.41 To date, several liquid biopsy assays have demonstrated prognostic value independent of treatment, but these seldom alter the choice of treatment and have limited utility in clinical practice. More recently, some assays have begun to yield predictive values that, with further rigorous validation, can significantly alter PC management.
CTC count as a prognostic biomarker in metastatic PC
The enumeration of CTCs by CellSearch has been clinically validated as a prognostic biomarker in metastatic PC. In the IMMC-38 study, 231 mCRPC patients starting first-line chemotherapy were categorized as having unfavorable CTC counts when greater than five CTCs per 7.5 ml of blood were enumerated by CellSearch.43 The median overall survival (OS) of the favorable group was 20.7 months, versus 9.5 months for the unfavorable group. Additionally, patients with unfavorable CTC counts at all time points had the lowest OS, while the patients who changed from unfavorable to favorable survived significantly longer. A subsequent reanalysis of the IMMC38 study showed that changes in CTC count outperformed PSA in prognostic value for OS at all time points post-treatment.4 Data from the IMMC-38 study led to the FDA clearance of the CellSearch assay, which remains the only CTC enumeration assay in the clinic.
The correlation between CTC counts and the response to hormonal therapy was characterized in the COU-AA‐301 trial assessing the efficacy of abiraterone in patients previously treated with docetaxel.44 In this study of 1195 mCRPC patients, CTC count by CellSearch was shown to be an independent predictor of survival in a multivariable analysis. Importantly, the study explored the utilization of CTC count four weeks after therapy as an intermediate endpoint of treatment. It demonstrated that a change of CTC count from favorable to unfavorable correlated with better survival in patients receiving abiraterone. Re-analysis of the data from the COU-AA‐301 trial showed that a biomarker panel using CTC count and lactate dehydrogenase (LDH) satisfy the four Prentice criteria for biomarker surrogacy and can be used as a surrogate biomarker for survival in patients treated with abiraterone, a potentially useful tool in future trial design.5
Additional trials have incorporated CTC count as a secondary endpoint. SWOG S0421 assessed the benefit of adding atrasentan, an endothelin receptor antagonist, to first-line docetaxel in mCRPC patients.45 Although atrasentan did not improve survival, CTC counts significantly correlated with PSA, baseline prognostic factors, and OS. Early changes in CTC count after the first 21-day cycle of chemotherapy were prognostic, and the increase in CTC counts was associated with reduced OS. Similar findings were observed in castrate sensitive patients in the SWOG S0925 trial. Cixutumumab, an antibody targeting insulin-like growth factor I receptors, did not significantly improve PSA response over ADT alone, but lower baseline CTC counts were associated with higher rates of PSA response regardless of the treatment the patient received.46
As a summary to all the CellSearch CTC enumeration studies, a recent report evaluated the utility of CTC counts to predict survival after the initiation of treatment using data pooled from five large randomized clinical trials. The CTC counts at baseline and 12 weeks after treatment were evaluated, and the two most significant predictors of survival were a conversion of CTC count to zero (CTC0) and a conversion of CTC count from more than five to less than five.7 Compared with the conventional biomarker of CTC conversion to less than five, CTC0 had similar prognostic value but was applicable to a greater number of PC patients with less disease burden and fewer CTCs. The change in CTC count also was a better marker than the change in PSA for predicting clinical response and patient survival. Therefore, a week 13 CTC conversion may be incorporated into clinical trials as an intermediate endpoint in mCRPC patients to offers earlier assessment of therapeutic efficacy.
While the prognostic value of CTC counts by CellSearch has been well validated, the predictive value of CTC count to guide therapy selection has not yet been established. The only study to utilize CTC count as a predictive biomarker was done in breast cancer. In SWOG S0500 trial, it was investigated whether an early switch of chemotherapy based on poor CTC response would lead to improved OS in metastatic breast cancer patients receiving first-line therapy.47 Patients with persistently elevated CTC counts (more than 5 per 7.5 mL of blood) after one cycle of cytotoxic chemotherapy were randomized to early switching therapy versus maintaining the same treatment. The results showed that while CTC counts remained a highly prognostic biomarker, the early switch of chemotherapy guided by CTC count did not lead to better OS. While disappointing, these results should be interpreted in the context of the trial design: the treatment arms were relatively small (60 patients per arm) and required a large treatment effect to reach statistical significance, something unlikely to occur given the poor response expected for second-line therapy in patients who demonstrated resistance to first-line therapy.
CTC AR-V7 assays in mCRPC patients on therapy
Androgen receptor (AR) is a nuclear receptor that, when bound by a ligand, translocates to the nucleus where it activates gene expression networks and plays a pivotal driver role in the pathophysiology of PC.48 Several splice variants of ARs have been described, most notably AR-V7 (Androgen Receptor Splice Variant-7),49 which encodes an AR lacking the ligand-binding domain. AR-V7, therefore, remains constitutively active as a transcription factor in a ligand-independent manner. Preclinical studies indicated that AR-V7 is associated with castration resistance and the AR encoded by AR-V7 cannot be targeted by direct (enzalutamide) or indirect (abiraterone) androgen receptor signal inhibitors (ARSIs).50
Due to the difficulty of obtaining repeat tissue biopsies in mCRPC patients, the association between AR-V7 expression and therapy response has been demonstrated using liquid biopsies. Antonarakis and colleagues first reported a correlation between AR-V7 mRNA and resistance to ARSIs, using qPCR for AR-V7 after CTC enriched by CTC AdnaTest (AdnaGen, Langenhagen, Germany), an EpCAM-based enrichment assay.8 Among the 62 patients in the study, no patients with positive CTC AR-V7 had a PSA response to ARSIs, and progression-free survival (PFS) and OS were shorter in patients with positive AR-V7. The same group later confirmed the association between CTC AR-V7 and survival in a larger cohort of 202 mCRPC patients starting ARSIs. CTCs were enumerated by CellSearch in parallel to differentiate patients with negative CTC and positive CTC with negative AR-V7. Patients with positive CTC and AR-V7 had lower PSA response to ARSIs and worse survival compared with patients with positive CTC but negative AR-V7. This study demonstrated that CTC AR-V7 was a prognostic biomarker independent of CTC count.51 Using a similar approach, another group independently reported that the presence of CTC AR-V7 mRNA was unrelated to the response to cabazitaxel,52 further indicating that CTC AR-V7 is a predictive biomarker to ARSI resistance rather than a general prognostic indicator.
Another way to characterize AR-V7 is to measure nuclear expression of AR-V7 protein, which is the sign of its persistent AR activation.53 Scher and colleagues investigated the utility of Epic platform to correlate the nuclear localization of AR-V7 in CTCs with the response to ARSI or taxane-based therapy.54 In a cohort of 161 patients, the nuclear localization of the AR-V7 protein in CTCs was correlated with poor PSA response to ARSI and shorter OS.54 In the study, the predictive value of the CTC nuclear AR-V7 assay was validated as patients with CTCs with AR-V7 nuclear localization had longer OS when treated with taxanes relative to ARSIs. Another observational study validated the conclusion that patients with positive CTC nuclear AR-V7 had observed better survival treated with taxanes compared with ARSIs.55
Recently, the PROPHECY study validated the association of CTC AR-V7 and resistance to ARSIs using both the Epic CTC nuclear AR-V7 protein assay and a CTC AR-V7 mRNA assay. It was concluded that AR-V7 detection by both assays was independently associated with shorter PFS and OS. Compared with the CTC mRNA assay, the commercially available Epic assay detected fewer patients with positive CTC AR-V7 (9% vs 24%) but with higher specificity. The patients with positive CTC AR-V7 mRNA had 6% to 11% chance to respond to ARSI, while no patient with positive Epic CTC nuclear AR-V7 had an objective response to ARSI.56
The importance of CTC AR-V7 was also reported using a different workflow. Another report investigated the utility of a ddPCR panel to characterize CTC RNA after CTC-iChip enrichment. CTCM score was developed based on an 8-gene RNA ddPCR panel to allow fast and quantitative detection of prostate CTCs. This score was shown to predict outcome and response to therapy in a prospective study of mCRPC patients receiving first-line abiraterone.57 It was shown that CTCM scores were more sensitive than the microscopic CTC enumeration in detecting CTCs in PC patients. AR-V7 was not among the panel of CTCM score, but the expression of CTC AR-V7 by ddPCR after CTC-iChip enrichment was associated with shorter OS.
Taken together, these studies provided solid evidence that CTC AR-V7 assays can serve as biomarkers to predict ARSI resistance. Ideally, a randomized prospective trial is needed to validate the clinical benefit of patients with positive CTC AR-V7 to receive taxanes instead of ARSIs. However, given the fact that the oncology community is already choosing therapies based on the results of CTC AR-V7 positivity,58 we envision that CTC AR-V7 assays will soon be incorporated into the management of PC to help select the best treatment options.
CTC phenotypes as prognostic biomarkers in metastatic PC
Beyond enumeration, CTC recovery offers the benefit of yielding tumor cells for morphometric and molecular analysis. CTC clusters are groups of more than 2–3 CTCs tightly adhered to each other. These clusters have been observed for more than 60 years, but their role in cancer dissemination has not been fully understood.59 In the initial study of the herringbone-chip, CTC clusters were observed in PC patients.14 In a later study, it was demonstrated that CTC clusters preserved cell–cell adhesion, reduced apoptosis, and had an increased metastatic potential.60 Similar observations were also made by groups using the Epic platform,6,26 which showed that CTC clusters were observed in PC patients, and neuroendocrine PC patients had more CTC clusters.
Very small nucleus CTCs (vsnCTCs) were shown to be significantly elevated in patients with visceral rather than non-visceral metastatic PC.61 Similar observations were also reported using the Epic platform, which showed that CTCs from patients with neuroendocrine PC had smaller sizes.6 These smaller CTCs or vsnCTCs may represent a clone with properties similar to small cell PC or the aggressive variant of PC.62
Telomerase is an enzyme that lengthens and protects telomeres, the tandem DNA repeats at the end of human chromosomes. Telomerase is crucial for cell proliferation and immortality,63 and increased telomerase expression has been observed in the majority of cancer types.64 CTC telomerase activity was measured in mCRPC patients in the SWOG S0421 trial. With 215 telomerase activity measurements, it was reported that low CTC telomerase activity was associated with a better OS in patients with intermediate range CTC counts (6–54 per 7.5 mL blood by CellSearch).65
Based on the knowledge that PC cells with high AR activity show a PSA+/PSMA- phenotype, the differential expression of PSA and PSMA on CTCs has been developed as a potential biomarker to monitor AR signaling activity. In a group of 18 patients, a persistently high percentage of PSA+/PSMA- CTCs was associated with worse responses when treated with abiraterone.66
CTCs from PC patients have also been profiled by NGS assays including WES,67,68 WGS,69 and RNA sequencing.70 These reports demonstrated the technological capability of performing comprehensive sequencing using single CTCs as input. In theory, all the mutations and alterations detected in CTCs have the potential to serve as biomarkers. However, these assays have only been tested in small cohorts of patients. While these reports demonstrated the technologies to study cancer biology, more studies are needed to use them as biomarkers.
In summary, these molecular and phenotypic characterizations of CTCs are promising but still relatively early in development, requiring more studies for validation as prognostic or predictive biomarkers.
CTC in the management of early stage PC
In the management of localized PC, due to the presence of PSA as a readily available sensitive biomarker, the utility of liquid biopsy has been less explored. There are several reports investigating the utility of CTC counts in predicting the risk for recurrence after prostatectomy. Pal and colleagues reported that peri-operational CTC counts by CellSearch were not associated with biochemical recurrence.71 However, the presence of EMT markers in CTCs was correlated with biochemical recurrence at 1 year. This finding was further supported by another report that CTC counts before surgery were not associated with the risk of recurrence.72
The utility of CTC counts in patients with biochemical recurrence was investigated in multiple reports.73,74 Using CellSearch, only 3–8% of the patients with biochemical recurrence had positive CTC counts. The small cohort size and low percentage of CTC positive patients limited the analysis of the correlation between CTC count and the risk of recurrence. It is possible that newer, more sensitive CTC or other liquid biopsy assays can be useful in the management of localized PC. However, more research is needed in the field.
Circulating nucleic acids in PC
In earlier studies, WGS was used to characterize cfDNA in PC patients. With a shallow sequencing depth, these reports demonstrated the detection of AR amplification as well as other driver alterations including TMPRSS2:ERG fusion, PTEN deletion, and MYC amplification.75,76 Building on these earlier observations, the association between AR aberration in cfDNA and clinical outcome was investigated in multiple studies in mCRPC patients.77,78 It was noted that AR amplification was more common in patients resistant to ARSI.79 Additionally, pre-treatment cfDNA AR amplifications and mutations were associated with poor clinical outcomes, including worse survival, lower rates of PSA response, and shorter time to radiographic/clinical progression.78 In another study, serial cfDNA samples were sequenced from 97 mCRPC patients treated with abiraterone. The emergence of AR point mutations (T878A or L702H) was observed in patients at progression on abiraterone.80 This suggests that although AR amplification is a possible mechanism of ARSI resistance, AR point mutations are more likely to represent the mechanism of acquired resistance to ARSI.
However, in a more recent report of 202 mCRPC patients starting enzalutamide or abiraterone, the presence of pre-treatment AR amplification was not significantly correlated with time to progression. Although AR aberrations were associated with ARSI response, the presence of these aberrations did not preclude the possibility of response to ARSI. On the other hand, alterations in homologous recombination repair genes (BRCA2 or ATM) and TP53 were observed to be significantly correlated with shorter time to progression.81 These promising results demonstrated an association between cfDNA alterations and ARSI resistance, and future studies will need to build upon this data to validate the predictive value of specific alterations for guiding choice of therapy and improve patient outcome.
Besides AR characterization in patients receiving ARSI, the clinical utility of ctDNA was also explored in the TOPARP-A trial studying olaparib, a poly (adenosine diphosphate [ADP]-ribose) polymerase (PARP) inhibitor in mCRPC patients. The TOPARP-A trial showed that mCRPC patients with defects in the DNA repair genes, including BRCA1/2, ATM, FANCA, and CHEK2 were associated with olaparib response.82 It was observed that the decrease of cfDNA quantity during treatment was independently associated with response to olaparib and improved OS. Furthermore, all the somatic DNA repair mutations associated with the response to olaparib were detectable in cfDNA. The decreased allele frequency of somatic mutations was noted in patients responding to olaparib, and the emergence of new sub-clonal aberrations was observed when patients progressed.83 Using ctDNA as a window to study clinical resistance to PARP inhibitors, it was demonstrated that the restoration of homologous recombination repair caused by BRCA2 reversion mutations is a resistance mechanism in PC patients previously responding to PARP inhibition.84
The utility of circulating RNA as a biomarker has also been investigated. De Bono and colleagues reported that microarray analysis of whole-blood RNA collected in PAXgene tubes (BD Biosciences, San Jose, CA, USA) can discriminate patients with castration-resistant PC and those on active surveillance. Based on the results, a 9-gene signature was designed using qPCR to identify patients with worse prognoses. This signature was validated in an independent cohort showing a median OS of 9.2 months with high-risk RNA signature and 21.6 months without high-risk RNA.85 A follow-up study from the same group demonstrated prostate-specific transcripts in the whole-blood RNA can be combined with CTC counts to improve the prognostic value.86 Another group reported a 6-gene whole-blood RNA panel separating patients with castration-resistant PC into high- and low-risk groups. A median OS of 7.8 months in the high-risk group and >34.9 months in the low-risk group was noted.87 As expected, the signatures derived in these whole blood RNA studies consisted of genes predominantly expressed in leukocytes rather than in cancer cells, given that white and red blood cells comprise >99% of the cellular content of whole blood. The measurement of AR-V7 in whole-blood RNA was also reported without the need for CTC enrichment. Around 10% of the mCRPC patients had detectable AR-V7 in whole-blood RNA, and the presence of AR-V7 was associated with poor response to abiraterone and shorter OS.88
In summary, cfDNA mutation assays analyzing DNA repair genes may be a predictive biomarker to the response to PARP inhibitors. At this time, many PARP inhibitors are actively tested in clinical trials, we envision subgroup analysis of these trials will help better evaluate the predictive value of cfDNA mutations in DNA repair genes to the response to various PARP inhibitors. AR mutations or amplification in cfDNA and whole-blood RNA assays looking at AR-V7 may predict the resistance to ARSIs. However, there was no reported comparison between these circulating nucleic acid AR assays and CTC AR-V7 assays, and the optimal biomarker to predict ARSI response has yet to be determined. Furthermore, we are awaiting a well-designed trial to demonstrate that patients with AR alterations can benefit from taxanes or other non-ARSI treatments. Finally, whole-blood RNA panels can be prognostic biomarkers for mCRPC patients, but the clinical utility of these biomarkers is limited as they do not alter our choice of treatment.
New frontiers in liquid biopsy
Liquid biopsies to characterize PC heterogeneity
A hallmark of cancer evolution is the generation of heterogeneity followed by the selection of clones suitable for the establishment of distant metastases and resistance to various therapies.89 In PC, heterogeneity has been recognized by pathologist since 1974, when it was observed that PC differed in nuclear morphology, cell proliferation, immune cell infiltration, differentiation status, and necrosis.90 Due to the heterogeneity in pathology within the prostate tumor tissue, the Gleason score was developed to characterize the two most prominent morphological features.91–93
Beyond morphological heterogeneity, heterogeneous copy number variations (CNVs) have also been reported in primary PC within an individual patient.94,95 Similarly, heterogeneity at somatic single nucleotide variance (SSNV) level was demonstrated among primary and metastatic tumor sites.96 The heterogeneity in mutational landscape has enabled re-tracing of the clonal evolution of individual patients’ disease using tissues from tumors and metastatic sites.97 As expected, this heterogeneity within primary and metastatic tumors has also been reflected in more recent studies utilizing liquid biopsies. Using WES, Lohr and colleagues compared CTCs and tumor tissues, and demonstrated that 30% of the exonic SSNVs in the CTCs were not present in the primary or metastatic tissues.67 Based on the comparison of the mutational landscape, CTCs were shown to be phylogenetically closer to a supraclavicular lymph node metastasis as compared with the primary prostate tissue. Another study using WGS to compare CTCs and tumor tissues demonstrated that 14% of the SSNVs from CTCs were not present in the tumor tissues. Besides SSNVs, heterogeneous rearrangements were found in important tumor suppressor genes in CTCs and tumor tissues.69
One challenge of studying heterogeneity using CTCs is the error rate intrinsic to single-cell sequencing. In order to characterize picogram levels of DNA input from single or small numbers of cells, most single-cell whole-genome or whole-exome analytic technologies require whole genome amplification (WGA), which has the potential to introduce false positives of SSNVs in PCR or non-PCR based amplification processes.98,99 WGA also introduces bias in the CNV analysis due to uneven amplification, limiting the detection of smaller CNVs. Currently, there is no perfect technology which can perform single-cell sequencing with the same level of reproducibility as standard sequencing. It is known that the amplification errors occur at a frequency of around one in a million base pairs or less. Using mathematical and experimental models, it was demonstrated that a detected SSNV was likely to be a true mutation rather than a false-positive error if it was observed in more than three independent single-cell sequencing results.100 However, this approach requires many single CTCs to be sequenced, which is costly and sometimes not possible in patients with few CTCs.
Compared to CTCs, the sequencing of cfDNA presents fewer false positives related to amplification, but significant heterogeneity is also observed. Two reports independently compared the mutations on the same patients reported by the FoundationOne assay (Foundation Medicine, Cambridge, USA), a widely used tissue-based cancer panel, and the Guardant360 assay, a commercially available cfDNA-based cancer panel. In the first report of 28 patients, the concordance of mutational landscape was 91.9%, but this calculation was driven mostly by the genes which were not mutated in both assays. When the reported alterations were compared, only 17.1% of the alterations reported were concordant in both platforms. As high as 33.7% of the alterations in the ctDNA were detected in the tumor tissue; likewise, 31.1% of the alterations in the tumor tissue were detected in ctDNA.101 The other smaller report of nine patients showed a similar finding, where only 22% of the alterations were reported by both platforms. It was observed that alterations with less than 1% allele frequency in cfDNA were more likely to be undetected in the tumor tissue,102 suggesting that some alterations detected in cfDNA may come from subclones not present in the tumor tissues.
The discordance in the mutational landscape was also observed when different cfDNA assays were compared head to head. In a recent report, plasma was collected simultaneously from 40 PC patients and was subjected to the Guardant360 and the PlasmaSELECT assays (Personal Genome Diagnostics, Baltimore, USA).103 A significant discordance in the reported mutations was observed, as only 30% of the patients had the same results reported from two assays. The analysis regarding the allele frequency of the discordant mutations was not available; however, it was likely that different cfDNA assays had different thresholds in reporting mutations with low allele frequency. These studies confirmed the well-recognized fact that tumor heterogeneity cannot be characterized by a single biopsy,104 and even liquid biopsies may need to be performed repeatedly. Furthermore, the characterization of subclonal alterations in ctDNA is even more technically challenging as these alterations are present in lower allele frequencies on the background of more abundant non-tumor derived cfDNA. Ideally, tissue sequencing and serial liquid biopsy should complement each other to capture the mutational landscape of patients with complex heterogeneity.
Whereas liquid biopsy analysis offers the capability of capturing alterations from disparate sites of disease as just discussed, it also offers the important benefit of allowing minimally invasive serial biopsies to study the dynamic change of molecular information and map temporal disease evolution. In PC, serial liquid biopsy analysis has led to the observation that patients develop an increased CTC AR-V7 mRNA expression after receiving ARSI treatment as a mechanism of resistance.105 Similarly, it was also observed that PC patients who developed resistance to PARP inhibition also developed mutations restoring the homologous recombination repair.84 Using serial liquid biopsy to study PC evolution, we will be able to better study the biology of drug resistance. We envision that liquid biopsies will be widely utilized in the near future to understand the mechanism of drug resistance and will eventually lead to the development of many new therapies to come.
CTC RNA: challenges and opportunities
Compared with DNA there are at least three major advantages in characterizing RNA for cancer research. First, RNA sequencing is a better tool to characterize gene fusion. While WGS can identify translocations leading to gene fusions, RNA sequencing identifies aberrant RNA species with a higher likelihood to cause biologic consequences, requires much less sequencing power and significantly reduces costs.106 Second, alternative RNA splicing is known to play key roles in the process. AR-V7 is one of the most characterized alternative splicing products, leading to the persistent activation of AR.48 Third, the biological importance of non-coding RNA, including microRNA and long non-coding RNA, are also well recognized.107 Therefore, the ability to accurately detect PC-relevant gene fusions and non-coding RNA, and to quantify the expression of disease driving genes, holds great promise for functional disease profiling in liquid biopsy.
Analysis of minute RNA quantities from rare cells like CTCs has been achieved with various techniques. Besides standard qPCR and ddPCR to quantify specific RNA targets in single cells, newer technologies are now available to perform whole transcriptome characterization, such as direct hybridization single RNA molecular imaging,108 single-cell whole transcriptomic RNA microarray,109 and single-cell RNA sequencing.110 In 2005, microarrays were first used to characterize RNA in CellSearch enriched CTCs in a PC patient to identify the presence of PC-specific transcripts. The finding was subsequently validated by qPCR of 31 additional PC patients. It was shown that the expression profile of CTCs obtained from PC patients was different from those obtained from breast and colorectal patients.111 This study set the cornerstone for all the following studies using qPCR to characterize CTC RNA in PC.
The first mRNA sequencing of CTCs in PC patients was reported by Cann and colleagues in 2012. After pure single-CTCs were isolated by MagSweeper, single-cell mRNA sequencing was performed using SMARTer Ultra Low Input RNA assay (Clontech, Mountain View, USA). The experiment demonstrated that CTC expression profiles were not contaminated by WBC transcripts. The PC-associated transcripts, including AR and TMPRSS2, were highly expressed in CTCs. At the same time, CTC RNA was significantly degraded, and CTCs also expressed transcripts associated with apoptosis.112 This pioneering report indicated that the challenge of analyzing the transcriptome of CTC lies not only in avoiding contamination with WBC transcripts, but also on the preservation of CTC viability and RNA quality, and successful sequencing on partially degraded RNA. After that, many more assays to perform single-cell sequencing became available, and single-CTC RNA sequencing has been utilized to study CTC biology in different cancer types. In pancreatic cancer, WNT2 was found to be highly expressed in CTCs, which has the function to enhance anchorage-independent sphere formation.113 In breast cancer, a group of CTCs were found to have dramatic enrichment for EMT-related transcripts, and their presence was associated with disease progression.17
In PC, Miyamoto and colleagues investigated the use of CTC mRNA sequencing to study the mechanism of enzalutamide resistance. Using CTC-iChip to enrich CTCs, followed by the isolation of single CTCs, RNA was extracted and amplified for standard sequencing. Unsupervised clustering of the sequencing results showed that CTCs from the same patient strongly clustered together. In patients with enzalutamide resistant disease, AR point mutations were only observed in one patient, while complex AR alternative splicing, including AR-V7, was observed in 8 out of 11 patients. When the whole transcriptome expression profile was compared between CTCs from CRPC and castrate sensitive PC patients, CRPC patients were noted to have a significant activation of noncanonical Wnt pathway. The association between enzalutamide resistance and noncanonical Wnt pathway activation was validated in a mouse model and multiple independent datasets.70
Besides demonstrating the feasibility of performing CTC RNA sequencing by different assays, these reports demonstrated that EMT and other biological processes can be reliably characterized in CTCs to study cancer biology in vivo. The use of liquid biopsies to observe tumor evolution at the RNA level may lead to a paradigm shift in the study of metastatic PC, as repeat biopsies have been very difficult in PC patients. This is particularly crucial in drug resistance studies, as the dynamic monitoring of expression profiles in CTCs can guide the development of novel therapies against resistant clones.
CTC culture and patient derived xenograft (PDX)
Besides directly analyzing DNA and RNA from rare cells, multiple groups aimed to assess cancer behavior at the cellular level using short-term cultures or patient-derived xenografts (PDXs) derived from CTCs. CTC cultures or PDXs not only expand the cells of interest to yield larger amounts of DNA and RNA, but also can be used in functional studies, such as drug susceptibility screening to predict the response in patients. The successful expansion of CTCs was first demonstrated in breast cancer patients.114,115 These reports demonstrated that these cultured CTCs harbored preexisting mutations from original cancer and can be used to test drug sensitivity.
PC has traditionally been difficult to expand into stable cell lines, all the more so from rare circulating cells. In one reported success, Gao and colleagues developed a 3D organoid system capable of long-term culture of PC from biopsy specimens and CTCs. These established organoids recapitulated the genomic and expression profiles of the matched tumor tissue, and the expression profiles of the organoids were highly correlated with those obtained from the tumor tissue.116 However, only one CTC-derived organoid was achieved in that study (the rest were derived from tissue biopsy), and the patient from whom it was established had a very high CTC count (more than 100 cells per 8 mL of blood), indicating that this technique may not be applicable to all PC patients.
In PDX models, freshly resected tumor pieces, or isolated CTCs, are subcutaneously or orthotopically implanted into immunocompromised mice. If established without prior expansion of CTCs ex vivo, PDXs offer the advantage of reduced bias created by selecting only subclones with the ability to survive in Petri dishes, thus conserving tumor heterogeneity. The successful generation of CTC-derived PDX was reported in small cell lung cancer117 and breast cancer.118 However, both reports suggested that a large number of CTCs were needed to generate a PDX, making most patients with less aggressive diseases unsuitable for this approach. In PC, a study reported the successful generation of PDX mice inoculating only 50 to 3000 CTCs. After a follow-up period of 10 months, microscopic diseases were found in all eight xenograft mice, when human cells were observed in the peripheral blood and bone marrow of grafted mice.119 These are promising results, and future studies will hopefully expand upon and refine such PDX protocols for use in functional studies. Currently, the technical challenges posed by the expansion of rare CTCs, in culture or in PDX models, continue to preclude the use of these approaches to analyze patient-derived cells in a clinical setting. Moreover, the clinical actionability of molecular data yielded by expanded cells will require further prospective validation, as such cells – by definition – represent a small subset of clones able to survive and proliferate under these artificial conditions.
Extracellular vesicles (EVs)
EVs are lipid-enclosed particles which contain DNA, RNA, or proteins released by their cell of origin.120 There are different populations of EVs which differ in size and mechanism of formation. The largest EVs, large oncosomes (LOs), range in size from 1 to 10 μm and are released preferentially from highly invasive tumor cells. Exosomes are the most commonly studied EV, ranging in size from 50 to 100 nm, and released by the fusion of multivesicular bodies with the plasma membrane.121
Many assays have been developed to enrich and profile EVs, but the relative absence of standardization has been a challenge in the field. Traditionally, differential centrifugation has been the most widely used assay for EV isolation.120 However, this methodology requires a large amount of starting material, and is labor intensive and not suitable for high-throughput assays. Alternatively, EV isolation can be achieved by ultrafiltration122 or affinity-based capture.123 NanoSight system (NanoSight, Amesbury, UK) is a commonly utilized device to measure the number and size distribution of EVs.124 After isolation, EV-derived proteins or nucleic acids can be analyzed by ELISA, Western blot, flow cytometry, qPCR, or sequencing. Such analyses yield intriguing new insights but must be interpreted cautiously, because the broad spectrum of isolation methods yield distinct populations of EVs, making it challenging to compare results across studies.
The advantage of analyzing EVs comes from the observation that tumor cells secrete more EVs compared with benign cells, leading to an enrichment of tumor DNA, RNA, and protein compared with other cell-free assays.125 In a recent study of 36 mCRPC patients receiving ARSI, exosomes were isolated for RNA extraction, and AR-V7 was tested using ddPCR. The detection of AR-V7 was associated with worse PFS and OS.126 Another study performed RNA sequencing on enriched exosomes and reported that higher levels of miR-1290 and -375 were associated with poor OS in PC patients.127 Another team reported that the P-glycoprotein expression on the circulating exosomes was higher in docetaxel-resistant patients than in treatment naïve patients.128 These early observations suggested that exosomal proteins and miRNAs have the potential to serve as prognostic or even predictive biomarkers in PC patients.
EVs can also be assayed for long non-coding RNAs and DNA. A group reported that SAP30L-AS1 and SChLAP1 can distinguish PC patients from those with benign prostate hypertrophy (BPH).129 Since there is a limited clinical utility for a new diagnostic biomarker in PC, exosomal long non-coding RNAs will need to find clinical niches for further development. At the DNA level, a recent report demonstrated that LOs isolated from the plasma of PC patients were enriched in chromosomal DNA with large fragments.130 Using WGS, the oncosomal DNA reflected the genomic make-up of the tumor of origin, indicating a potential utility of EV-derived DNA to study genomic alterations in PC patients. The mutation analysis of oncosomal DNA can be comparable to liquid biopsy from cfDNA and CTCs. An in-depth comparison will need to be demonstrated before we can better understand how to choose oncosomal DNA over other liquid biopsy assays for mutation analysis.
While our knowledge of plasma-derived EVs is advancing, a urine-based EV assay has already been clinically validated and is commercially available. ExoDx Prostate (IntelliScore) (EPI, Exosome Diagnostics Inc., Waltham, MA, USA) is a urine exosomal RNA assay analyzing the expression of three genes (ERG, PCA3, and SPDEF) to predict the risk of high-grade PC.131 The test is intended for men aged ≥50 with mildly elevated PSA (2–10 ng/mL), to determine the need for prostate biopsy. EPI was recently prospectively validated as having a negative predictive value of 89% for the absence of high-grade PC (Gleason Score ≥7).132 The authors reported that the incorporation of EPI can avoid 31% of all biopsies in the population, but will miss 11% of high-grade PC. A subsequent study is currently underway to utilize EPI scores as a risk stratification tool in the shared decision-making process in patients prior to prostate biopsy. EPI is endorsed by the NCCN (National Comprehensive Cancer Network) guideline to risk-stratify patients with low-risk of developing PC before diagnostic prostate biopsy.
These promising clinical results are evolving in the context of growing pre-clinical evidence of the functional importance of EVs, for PC cell motility and invasiveness, with payloads that modulate angiogenesis and promote pro-metastatic microenvironments.133,134 Taken together, these pre-clinical and clinical evidences strongly suggest that EVs will play a role in guiding PC management. New studies will seek to evaluate not only their analytical and clinical value of EVs, but also the clinical utility of analyzing the circulating tumor nucleic acids enriched in EVs, as some recent studies suggested that the majority of “cell-free” plasma nucleic acids in PC actually are EV-derived.130
Circulating microRNAs (miRNAs)
MicroRNAs (miRNAs) are small evolutionary conserved non-coding RNAs of 19–25 nucleotides capable of down-regulating gene expression by binding to the 3ʹ-untranslated region (3ʹ-UTR) of mRNAs and targeting them for degradation.135 miRNAs play a key regulatory role in cancer biology by interfering with the expression of tumor suppressors, oncogenes or other proteins associated with disease progression or drug resistance. Circulating miRNAs are the most abundant cell-free RNA in the blood,136 and they are stable in plasma and serum for quantification.137 This stability derives from the formation of complexes with proteins as well as the incorporation within EVs that protect miRNA from RNase digestion.138 Furthermore, the biological significance of miRNAs has been well studied in PC,139 which suggest the potential to develop miRNA assays as biomarkers.
Analysis of circulating miRNA can be accomplished with quantitative RT-PCR, microarray or deep sequencing after initial ultracentrifugation to remove cell debris, apoptotic bodies, and platelets containing their own background miRNA.140 In PC, a recent study demonstrated that a panel of four miRNAs (miR-4289, miR-326, miR-152-3p, and miR-98-5p) assayed from plasma could differentiate PC patients from healthy controls.141 Multiple other miRNAs have been reported to discriminate PC patients from controls.142–146 These miRNA-based biomarkers are early in their development, and further prospective validation is needed.
Several groups have focused their analysis on circulating microRNA from enriched EVs. One such study showed that exosomal miR-141 was progressively elevated in prostatic hypertrophy, localized PC, and metastatic disease, suggesting a potential diagnostic or prognostic role.147 Another report compared the performance of whole blood miRNA analysis with EV miRNA analysis. While miR-375 in whole plasma could differentiate PC from BPH patients, miR-200c-3p and miR-21-5p in EVs were better discriminators, and Let-7a-5p in EVs could distinguish PC patients with Gleason score ≥8 vs ≤6.148 Still other studies have evaluated miRNAs in urine for clinical utility, such as detection of local recurrence.149
More recently, the prognostic value of circulating miRNAs was evaluated in larger prospective clinical cohorts. In patients receiving first-line docetaxel for mCRPC, Lin and colleagues reported that elevated circulating miR-200b, miR-429, or miR-200a before chemotherapy were associated with worse OS; likewise, decreased or unchanged miR-20a, miR-20b, or miR-222 after chemotherapy was associated with worse OS.150 In another trial (SWOG S0925), Cheng and colleagues reported a correlation between baseline plasma miR‐141 and CellSearch CTC count, whereas baseline plasma miR-375 and miR-200b significantly correlated with week-28 PSA response.151
Taken together, these reports suggest that circulating miRNAs may serve as prognostic biomarkers in PC, perhaps not surprisingly given their recognized key regulatory function in this disease. As with most other candidate biomarkers in PC, additional prospective studies will be needed to validate the clinical value of specific miRNA expression cut points and assess their clinical utility individually and in combination with other liquid biopsy assays such as CellSearch CTC counts and serum PSA.
Future directions for liquid biopsy in PC
Liquid biopsy is a fast-evolving field with the potential to change patient management across a broad spectrum of cancer types, including PC. Until now, CTC enumerated by CellSearch has been by far the best clinically validated prognostic biomarker in metastatic PC. However, CTC count has limited utilization in the clinic, as it does not effectively guide the choice of therapy. A more predictive CTC-based phenotype has been AR-V7 in relation to ARSI response. This promising and now commercially available assay was recently validated in a prospective trial for its prognostic value.56 Despite the lack of a prospective study to demonstrate its predictive value in the selection of taxanes vs ARSIs, physicians are now starting to incorporate AR-V7 testing into clinical practice. As newer phenotyping techniques are applied to CTCs with greater efficiency and reduced costs, analyses such as the single-cell RNA sequencing will likely help identify new targets for therapy. For example, the patients who derived their ARSI resistance from the activation of noncanonical Wnt pathway70 can be enrolled in the trials targeting Wnt pathway.
CTC-based assays are likely to transition increasingly from enumeration and staining to molecular characterization, which until now has been limited by the cost and relatively low throughput of single-cell sequencing assays. With the rapid development of single-cell sequencing technology, assays such as the Chromium System (10x Genomics, Pleasanton, CA, USA) are now capable of simultaneously analyzing thousands of single cells in parallel.152 Such high-throughput single-cell platforms are not designed specifically for CTC research, as they cannot discriminate rare CTCs from background leukocytes. However, if successfully coupled with CTC identification, these powerful single-cell sequencing technologies can be leveraged to rapidly and efficiently characterize CTCs.
In parallel to CTC phenotyping, the analysis of plasma-based cell-free nucleic acids and EVs is advancing rapidly in PC, and promises to offer a robust and relatively simple means for monitoring the evolution of mutations in correlation to the emergence of therapy resistance. As with CTC-based AR-V7 assays, cfDNA assays, including those detecting mutations in AR and DNA damage repair genes, will require further prospective evaluation as a basis for therapy selection (ie, taxanes or PARP inhibitors) to improve outcome.
Ultimately, we envision the future of liquid biopsy is to analyze liquid biopsies in a multi-parametric approach. Mutations detected in CTCs can be cross-validated in cfDNA, and protein and RNA expression information from CTCs can provide additional information to the mutation profile. Likewise, the molecular information obtained from EVs and whole blood RNA should be compared with CTCs and ctDNA. By incorporating analyses from different components of liquid biopsy, we envision many predictive biomarkers will be developed to guide the clinicians about therapy selection. At the same time, utilizing these powerful tools, scientists can better understand the evolution of metastatic PC in vivo and develop new therapeutic strategies.
Conclusion
The liquid biopsy field continues to offer tantalizing opportunities to apply new technologies towards real-time tumor profiling. These assays have great potential to transform the clinical practice of PC by providing access to real-time molecular information and by reducing the need for costly and invasive biopsies. To be sure, it is a multidisciplinary field where physicians, scientists, and engineers with different expertise work closely to develop better assays, design clinical trials, and validate the clinical utility of these liquid biopsy assays. We are hopeful that liquid biopsies will continue to advance our understanding of cancer biology and ultimately improve patient outcomes in PC.
Disclosure
Dr. Amir Goldkorn reports personal fees from Acadia Woods (VC firm) outside the submitted work; In addition, Dr. Amir Goldkorn has a patent US8551425B2: Method for cancer detection, diagnosis, and prognosis licensed to Circulogix, Corestone. The authors report no other conflicts of interest in this work.
References
1. Merker JD, Oxnard GR, Compton C, et al. Circulating tumor DNA analysis in patients with cancer: american society of clinical oncology and college of american pathologists joint review. Arch Pathol Lab Med. 2018;142:1242–1253. doi:10.5858/arpa.2018-0901-SA
2. Siegel RL, Miller KD, Jemal A. Cancer statistics, 2019. CA Cancer J Clin. 2019;69(1):7–34. doi:10.3322/caac.21551
3. Danila DC, Heller G, Gignac GA, et al. Circulating tumor cell number and prognosis in progressive castration-resistant prostate cancer. Clin Cancer Res. 2007;13(23):7053–7058. doi:10.1158/1078-0432.CCR-07-1506
4. Scher HI, Jia X, de Bono JS, et al. Circulating tumour cells as prognostic markers in progressive, castration-resistant prostate cancer: a reanalysis of IMMC38 trial data. Lancet Oncol. 2009;10(3):233–239. doi:10.1016/S1470-2045(08)70340-1
5. Scher HI, Heller G, Molina A, et al. Circulating tumor cell biomarker panel as an individual-level surrogate for survival in metastatic castration-resistant prostate cancer. J Clin Oncol. 2015;33(12):1348–1355. doi:10.1200/JCO.2014.55.3487
6. Beltran H, Jendrisak A, Landers M, et al. The initial detection and partial characterization of circulating tumor cells in neuroendocrine prostate cancer. Clin Cancer Res. 2016;22(6):1510–1519. doi:10.1158/1078-0432.CCR-15-0137
7. Heller G, McCormack R, Kheoh T, et al. Circulating tumor cell number as a response measure of prolonged survival for metastatic castration-resistant prostate cancer: a comparison with prostate-specific antigen across five randomized phase III clinical trials. J Clin Oncol. 2018;36(6):572–580. doi:10.1200/JCO.2017.75.2998
8. Antonarakis ES, Lu C, Wang H, et al. AR-V7 and resistance to enzalutamide and abiraterone in prostate cancer. N Engl J Med. 2014;371(11):1028–1038. doi:10.1056/NEJMoa1315815
9. Yu M, Stott S, Toner M, Maheswaran S, Haber DA. Circulating tumor cells: approaches to isolation and characterization. J Cell Biol. 2011;192(3):373–382. doi:10.1083/jcb.201010021
10. Adams DL, Stefansson S, Haudenschild C, et al. Cytometric characterization of circulating tumor cells captured by microfiltration and their correlation to the cellsearch((R)) CTC test. Cytometry A. 2015;87(2):137–144. doi:10.1002/cyto.a.22613
11. Kagan M, Howard D, Bendele T, et al. A sample preparation and analysis system for identification of circulating tumor cells. J Clin Ligand Assay. 2002;25(1):104–110.
12. Kalluri R, Weinberg RA. The basics of epithelial-mesenchymal transition. J Clin Invest. 2009;119(6):1420–1428. doi:10.1172/JCI39104
13. Swennenhuis JF, van Dalum G, Zeune LL, Terstappen LW. Improving the cellsearch(R) system. Expert Rev Mol Diagn. 2016;16(12):1291–1305. doi:10.1080/14737159.2016.1255144
14. Stott SL, Hsu CH, Tsukrov DI, et al. Isolation of circulating tumor cells using a microvortex-generating herringbone-chip. Proc Natl Acad Sci U S A. 2010;107(43):18392–18397. doi:10.1073/pnas.1012539107
15. Ozkumur E, Shah AM, Ciciliano JC, et al. Inertial focusing for tumor antigen-dependent and -independent sorting of rare circulating tumor cells. Sci Transl Med. 2013;5(179):179ra147. doi:10.1126/scitranslmed.3005616
16. Lu YT, Zhao L, Shen Q, et al. NanoVelcro chip for CTC enumeration in prostate cancer patients. Methods. 2013;64(2):144–152. doi:10.1016/j.ymeth.2013.06.019
17. Yu M, Bardia A, Wittner BS, et al. Circulating breast tumor cells exhibit dynamic changes in epithelial and mesenchymal composition. Science. 2013;339(6119):580–584. doi:10.1126/science.1228522
18. Kirby BJ, Jodari M, Loftus MS, et al. Functional characterization of circulating tumor cells with a prostate-cancer-specific microfluidic device. PLoS One. 2012;7(4):e35976. doi:10.1371/journal.pone.0035976
19. Armstrong AJ, Marengo MS, Oltean S, et al. Circulating tumor cells from patients with advanced prostate and breast cancer display both epithelial and mesenchymal markers. Mol Cancer Res. 2011;9(8):997–1007. doi:10.1158/1541-7786.MCR-10-0490
20. Agerbaek MO, Bang-Christensen SR, Yang MH, et al. The VAR2CSA malaria protein efficiently retrieves circulating tumor cells in an EpCAM-independent manner. Nat Commun. 2018;9(1):3279. doi:10.1038/s41467-018-05793-2
21. Chudasama DY, Freydina DV, Freidin MB, et al. Inertia based microfluidic capture and characterisation of circulating tumour cells for the diagnosis of lung cancer. Ann Transl Med. 2016;4(24):480. doi:10.21037/atm.2016.04.05
22. Xu L, Mao X, Imrali A, et al. Optimization and evaluation of a novel size based circulating tumor cell isolation system. PLoS One. 2015;10(9):e0138032. doi:10.1371/journal.pone.0138032
23. Vishnoi M, Peddibhotla S, Yin W, et al. The isolation and characterization of CTC subsets related to breast cancer dormancy. Sci Rep. 2015;5:17533. doi:10.1038/srep17533
24. Werner SL, Graf RP, Landers M, et al. Analytical validation and capabilities of the epic CTC platform: enrichment-free circulating tumour cell detection and characterization. J Circ Biomark. 2015;4:3. doi:10.5772/60725
25. van der Toom EE, Groot VP, Glavaris SA, et al. Analogous detection of circulating tumor cells using the accucyte((R)) -cytefinder((R)) system and ISET system in patients with locally advanced and metastatic prostate cancer. Prostate. 2018;78(4):300–307. doi:10.1002/pros.23474
26. McDaniel AS, Ferraldeschi R, Krupa R, et al. Phenotypic diversity of circulating tumour cells in patients with metastatic castration-resistant prostate cancer. BJU Int. 2017;120(5B):E30–E44. doi:10.1111/bju.13631
27. Underhill HR, Kitzman JO, Hellwig S, et al. Fragment Length of Circulating Tumor DNA. PLoS Genet. 2016;12(7):e1006162. doi:10.1371/journal.pgen.1006162
28. Antonatos D, Patsilinakos S, Spanodimos S, Korkonikitas P, Tsigas D. Cell-free DNA levels as a prognostic marker in acute myocardial infarction. Ann N Y Acad Sci. 2006;1075:278–281. doi:10.1196/annals.1368.037
29. Butler TM, Spellman PT, Gray J. Circulating-tumor DNA as an early detection and diagnostic tool. Curr Opin Genet Dev. 2017;42:14–21. doi:10.1016/j.gde.2016.12.003
30. Hindson BJ, Ness KD, Masquelier DA, et al. High-throughput droplet digital PCR system for absolute quantitation of DNA copy number. Anal Chem. 2011;83(22):8604–8610. doi:10.1021/ac202028g
31. Diehl F, Li M, He Y, Kinzler KW, Vogelstein B, Dressman D. BEAMing: single-molecule PCR on microparticles in water-in-oil emulsions. Nat Methods. 2006;3(7):551–559. doi:10.1038/nmeth898
32. Dressman D, Yan H, Traverso G, Kinzler KW, Vogelstein B. Transforming single DNA molecules into fluorescent magnetic particles for detection and enumeration of genetic variations. Proc Natl Acad Sci U S A. 2003;100(15):8817–8822. doi:10.1073/pnas.1133470100
33. Mamanova L, Coffey AJ, Scott CE, et al. Target-enrichment strategies for next-generation sequencing. Nat Methods. 2010;7(2):111–118. doi:10.1038/nmeth.1419
34. Newman AM, Lovejoy AF, Klass DM, et al. Integrated digital error suppression for improved detection of circulating tumor DNA. Nat Biotechnol. 2016;34(5):547–555. doi:10.1038/nbt.3520
35. Odegaard JI, Vincent JJ, Mortimer S, et al. Validation of a plasma-based comprehensive cancer genotyping assay utilizing orthogonal tissue- and plasma-based methodologies. Clin Cancer Res. 2018;24(15):3539–3549. doi:10.1158/1078-0432.CCR-17-3831
36. Khleif SN, Doroshow JH, Hait WN. Collaborative A-F-NCB. AACR-FDA-NCI cancer biomarkers collaborative consensus report: advancing the use of biomarkers in cancer drug development. Clin Cancer Res. 2010;16(13):3299–3318. doi:10.1158/1078-0432.CCR-10-0880
37. Scher HI, Morris MJ, Larson S, Heller G. Validation and clinical utility of prostate cancer biomarkers. Nat Rev Clin Oncol. 2013;10(4):225–234. doi:10.1038/nrclinonc.2013.30
38. Bossuyt PM. Clinical validity: defining biomarker performance. Scand J Clin Lab Invest Suppl. 2010;242:46–52. doi:10.3109/00365513.2010.493383
39. Gutman S, Kessler LG. The US food and drug administration perspective on cancer biomarker development. Nat Rev Cancer. 2006;6(7):565–571. doi:10.1038/nrc1911
40. Prentice RL. Surrogate endpoints in clinical trials: definition and operational criteria. Stat Med. 1989;8(4):431–440.
41.
42. Parkinson DR, McCormack RT, Keating SM, et al. Evidence of clinical utility: an unmet need in molecular diagnostics for patients with cancer. Clin Cancer Res. 2014;20(6):1428–1444. doi:10.1158/1078-0432.CCR-13-2961
43. de Bono JS, Scher HI, Montgomery RB, et al. Circulating tumor cells predict survival benefit from treatment in metastatic castration-resistant prostate cancer. Clin Cancer Res. 2008;14(19):6302–6309. doi:10.1158/1078-0432.CCR-08-0872
44. Fizazi K, Scher HI, Molina A, et al. Abiraterone acetate for treatment of metastatic castration-resistant prostate cancer: final overall survival analysis of the COU-AA-301 randomised, double-blind, placebo-controlled phase 3 study. Lancet Oncol. 2012;13(10):983–992. doi:10.1016/S1470-2045(12)70379-0
45. Goldkorn A, Ely B, Quinn DI, et al. Circulating tumor cell counts are prognostic of overall survival in SWOG S0421: a phase III trial of docetaxel with or without atrasentan for metastatic castration-resistant prostate cancer. J Clin Oncol. 2014;32(11):1136–1142. doi:10.1200/JCO.2013.51.7417
46. Yu EY, Li H, Higano CS, et al. SWOG S0925: A randomized phase II study of androgen deprivation combined with cixutumumab versus androgen deprivation alone in patients with new metastatic hormone-sensitive prostate cancer. J Clin Oncol. 2015;33(14):1601–1608. doi:10.1200/JCO.2014.59.4127
47. Smerage JB, Barlow WE, Hortobagyi GN, et al. Circulating tumor cells and response to chemotherapy in metastatic breast cancer: SWOG S0500. J Clin Oncol. 2014;32(31):3483–3489. doi:10.1200/JCO.2014.56.2561
48. Watson PA, Arora VK, Sawyers CL. Emerging mechanisms of resistance to androgen receptor inhibitors in prostate cancer. Nat Rev Cancer. 2015;15(12):701–711. doi:10.1038/nrc4016
49.
50. Paschalis A, Sharp A, Welti JC, et al. Alternative splicing in prostate cancer. Nat Rev Clin Oncol. 2018;15(11):663–675. doi:10.1038/s41571-018-0085-0
51. Antonarakis ES, Lu C, Luber B, et al. Clinical significance of androgen receptor splice variant-7 mRNA detection in circulating tumor cells of men with metastatic castration-resistant prostate cancer treated with first- and second-line abiraterone and enzalutamide. J Clin Oncol. 2017;35(19):2149–2156. doi:10.1200/JCO.2016.70.1961
52. Onstenk W, Sieuwerts AM, Kraan J, et al. Efficacy of cabazitaxel in castration-resistant prostate cancer is independent of the presence of AR-V7 in circulating tumor cells. Eur Urol. 2015;68(6):939–945. doi:10.1016/j.eururo.2015.07.007
53. Watson PA, Chen YF, Balbas MD, et al. Constitutively active androgen receptor splice variants expressed in castration-resistant prostate cancer require full-length androgen receptor. Proc Natl Acad Sci U S A. 2010;107(39):16759–16765. doi:10.1073/pnas.1012443107
54. Scher HI, Lu D, Schreiber NA, et al. Association of AR-V7 on circulating tumor cells as a treatment-specific biomarker with outcomes and survival in castration-resistant prostate cancer. JAMA Oncol. 2016;2(11):1441–1449. doi:10.1001/jamaoncol.2016.1828
55. Scher HI, Graf RP, Schreiber NA, et al. Assessment of the validity of nuclear-localized androgen receptor splice variant 7 in circulating tumor cells as a predictive biomarker for castration-resistant prostate cancer. JAMA Oncol. 2018;4(9):1179–1186. doi:10.1001/jamaoncol.2018.1621
56. Armstrong AJ, Halabi S, Luo J. et al. Prospective multicenter validation of androgen receptor splice variant 7 and hormone therapy resistance in high-risk castration-resistant prostate cancer: the PROPHECY study. J Clin Oncol. 2019;JCO1801731. doi:10.1200/JCO.18.01731
57. Miyamoto DT, Lee RJ, Kalinich M, et al. An RNA-based digital circulating tumor cell signature is predictive of drug response and early dissemination in prostate cancer. Cancer Discov. 2018;8(3):288–303. doi:10.1158/2159-8290.CD-16-1406
58. Markowski MC, Silberstein JL, Eshleman JR, Eisenberger MA, Luo J, Antonarakis ES. Clinical utility of CLIA-grade AR-V7 testing in patients with metastatic castration-resistant prostate cancer. JCO Precis Oncol. 2017. doi:10.1200/PO.17.00127
59. Fabisiewicz A, Grzybowska E. CTC clusters in cancer progression and metastasis. Med Oncol. 2017;34(1):12. doi:10.1007/s12032-016-0875-0
60. Aceto N, Bardia A, Miyamoto DT, et al. Circulating tumor cell clusters are oligoclonal precursors of breast cancer metastasis. Cell. 2014;158(5):1110–1122. doi:10.1016/j.cell.2014.07.013
61. Chen JF, Ho H, Lichterman J, et al. Subclassification of prostate cancer circulating tumor cells by nuclear size reveals very small nuclear circulating tumor cells in patients with visceral metastases. Cancer. 2015;121(18):3240–3251. doi:10.1002/cncr.29455
62. Beltran H, Tomlins S, Aparicio A, et al. Aggressive variants of castration-resistant prostate cancer. Clin Cancer Res. 2014;20(11):2846–2850. doi:10.1158/1078-0432.CCR-13-3309
63. Shay JW. Role of telomeres and telomerase in aging and cancer. Cancer Discov. 2016;6(6):584–593. doi:10.1158/2159-8290.CD-16-0062
64. Shay JW, Bacchetti S. A survey of telomerase activity in human cancer. Eur J Cancer. 1997;33(5):787–791. doi:10.1016/S0959-8049(97)00062-2
65. Goldkorn A, Ely B, Tangen CM, et al. Circulating tumor cell telomerase activity as a prognostic marker for overall survival in SWOG 0421: a phase III metastatic castration resistant prostate cancer trial. Int J Cancer. 2015;136(8):1856–1862. doi:10.1002/ijc.29212
66. Miyamoto DT, Lee RJ, Stott SL, et al. Androgen receptor signaling in circulating tumor cells as a marker of hormonally responsive prostate cancer. Cancer Discov. 2012;2(11):995–1003. doi:10.1158/2159-8290.CD-12-0222
67. Lohr JG, Adalsteinsson VA, Cibulskis K, et al. Whole-exome sequencing of circulating tumor cells provides a window into metastatic prostate cancer. Nat Biotechnol. 2014;32(5):479–484. doi:10.1038/nbt.2892
68. Zhao L, Lu YT, Li F, et al. High-purity prostate circulating tumor cell isolation by a polymer nanofiber-embedded microchip for whole exome sequencing. Adv Mater. 2013;25(21):2897–2902. doi:10.1002/adma.201205237
69. Jiang R, Lu YT, Ho H, et al. A comparison of isolated circulating tumor cells and tissue biopsies using whole-genome sequencing in prostate cancer. Oncotarget. 2015;6(42):44781–44793. doi:10.18632/oncotarget.6330
70. Miyamoto DT, Zheng Y, Wittner BS, et al. RNA-Seq of single prostate CTCs implicates noncanonical Wnt signaling in antiandrogen resistance. Science. 2015;349(6254):1351–1356. doi:10.1126/science.aab0917
71. Pal SK, He M, Wilson T, et al. Detection and phenotyping of circulating tumor cells in high-risk localized prostate cancer. Clin Genitourin Cancer. 2015;13(2):130–136. doi:10.1016/j.clgc.2014.08.014
72. Meyer CP, Pantel K, Tennstedt P, et al. Limited prognostic value of preoperative circulating tumor cells for early biochemical recurrence in patients with localized prostate cancer. Urol Oncol. 2016;34(5):
73. Aragon-Ching JB, Siegel RS, Frazier H
74. Grisanti S, Antonelli A, Buglione M, et al. Analysis of circulating tumor cells in prostate cancer patients at PSA recurrence and review of the literature. Anticancer Res. 2016;36(6):2975–2981.
75. Heitzer E, Ulz P, Belic J, et al. Tumor-associated copy number changes in the circulation of patients with prostate cancer identified through whole-genome sequencing. Genome Med. 2013;5(4):30. doi:10.1186/gm493
76. Ulz P, Belic J, Graf R, et al. Whole-genome plasma sequencing reveals focal amplifications as a driving force in metastatic prostate cancer. Nat Commun. 2016;7:12008. doi:10.1038/ncomms12008
77. Carreira S, Romanel A, Goodall J, et al. Tumor clone dynamics in lethal prostate cancer. Sci Transl Med. 2014;6(254):254ra125. doi:10.1126/scitranslmed.3009448
78. Conteduca V, Wetterskog D, Sharabiani MTA, et al. Androgen receptor gene status in plasma DNA associates with worse outcome on enzalutamide or abiraterone for castration-resistant prostate cancer: a multi-institution correlative biomarker study. Ann Oncol. 2017;28(7):1508–1516. doi:10.1093/annonc/smdx155
79. Azad AA, Volik SV, Wyatt AW, et al. androgen receptor gene aberrations in circulating cell-free DNA: biomarkers of therapeutic resistance in castration-resistant prostate cancer. Clin Cancer Res. 2015;21(10):2315–2324. doi:10.1158/1078-0432.CCR-14-2666
80. Romanel A, Gasi Tandefelt D, Conteduca V, et al. Plasma AR and abiraterone-resistant prostate cancer. Sci Transl Med. 2015;7(312):312re310. doi:10.1126/scitranslmed.aad3106
81. Annala M, Vandekerkhove G, Khalaf D, et al. Circulating tumor DNA genomics correlate with resistance to abiraterone and enzalutamide in prostate cancer. Cancer Discov. 2018;8(4):444–457. doi:10.1158/2159-8290.CD-17-0937
82. Mateo J, Carreira S, Sandhu S, et al. DNA-repair defects and olaparib in metastatic prostate cancer. N Engl J Med. 2015;373(18):1697–1708. doi:10.1056/NEJMoa1506859
83. Goodall J, Mateo J, Yuan W, et al. Circulating cell-free DNA to guide prostate cancer treatment with PARP inhibition. Cancer Discov. 2017;7(9):1006–1017. doi:10.1158/2159-8290.CD-17-0261
84. Quigley D, Alumkal JJ, Wyatt AW, et al. Analysis of circulating cell-free DNA identifies multiclonal heterogeneity of BRCA2 reversion mutations associated with resistance to PARP inhibitors. Cancer Discov. 2017;7(9):999–1005. doi:10.1158/2159-8290.CD-17-0146
85. Olmos D, Brewer D, Clark J, et al. Prognostic value of blood mRNA expression signatures in castration-resistant prostate cancer: a prospective, two-stage study. Lancet Oncol. 2012;13(11):1114–1124. doi:10.1016/S1470-2045(12)70372-8
86. Danila DC, Anand A, Schultz N, et al. Analytic and clinical validation of a prostate cancer-enhanced messenger RNA detection assay in whole blood as a prognostic biomarker for survival. Eur Urol. 2014;65(6):1191–1197. doi:10.1016/j.eururo.2013.07.006
87. Ross RW, Galsky MD, Scher HI, et al. A whole-blood RNA transcript-based prognostic model in men with castration-resistant prostate cancer: a prospective study. Lancet Oncol. 2012;13(11):1105–1113. doi:10.1016/S1470-2045(12)70263-2
88. Todenhofer T, Azad A, Stewart C, et al. AR-V7 transcripts in whole blood RNA of patients with metastatic castration resistant prostate cancer correlate with response to abiraterone acetate. J Urol. 2017;197(1):135–142. doi:10.1016/j.juro.2016.06.094
89. Swanton C, McGranahan N, Starrett GJ, Harris RS. APOBEC enzymes: mutagenic fuel for cancer evolution and heterogeneity. Cancer Discov. 2015;5(7):704–712. doi:10.1158/2159-8290.CD-15-0344
90. Gleason DF, Mellinger GT. Prediction of prognosis for prostatic adenocarcinoma by combined histological grading and clinical staging. J Urol. 1974;111(1):58–64.
91. Miller GJ, Cygan JM. Morphology of prostate cancer: the effects of multifocality on histological grade, tumor volume and capsule penetration. J Urol. 1994;152(5 Pt 2):1709–1713.
92. Epstein JI, Allsbrook WC
93. Stephenson AJ, Scardino PT, Kattan MW, et al. Predicting the outcome of salvage radiation therapy for recurrent prostate cancer after radical prostatectomy. J Clin Oncol. 2007;25(15):2035–2041. doi:10.1200/JCO.2006.08.9607
94. Cheng L, Song SY, Pretlow TG, et al. Evidence of independent origin of multiple tumors from patients with prostate cancer. J Natl Cancer Inst. 1998;90(3):233–237.
95. Macintosh CA, Stower M, Reid N, Maitland NJ. Precise microdissection of human prostate cancers reveals genotypic heterogeneity. Cancer Res. 1998;58(1):23–28.
96. Lindberg J, Klevebring D, Liu W, et al. Exome sequencing of prostate cancer supports the hypothesis of independent tumour origins. Eur Urol. 2013;63(2):347–353. doi:10.1016/j.eururo.2012.03.050
97. Gundem G, Van Loo P, Kremeyer B, et al. The evolutionary history of lethal metastatic prostate cancer. Nature. 2015;520(7547):353–357. doi:10.1038/nature14347
98. Navin N, Kendall J, Troge J, et al. Tumour evolution inferred by single-cell sequencing. Nature. 2011;472(7341):90–94. doi:10.1038/nature09807
99. Xu X, Hou Y, Yin X, et al. Single-cell exome sequencing reveals single-nucleotide mutation characteristics of a kidney tumor. Cell. 2012;148(5):886–895. doi:10.1016/j.cell.2012.02.025
100. Hou Y, Song L, Zhu P, et al. Single-cell exome sequencing and monoclonal evolution of a JAK2-negative myeloproliferative neoplasm. Cell. 2012;148(5):873–885. doi:10.1016/j.cell.2012.02.028
101. Chae YK, Davis AA, Carneiro BA, et al. Concordance between genomic alterations assessed by next-generation sequencing in tumor tissue or circulating cell-free DNA. Oncotarget. 2016;7(40):65364–65373. doi:10.18632/oncotarget.11692
102. Kuderer NM, Burton KA, Blau S, et al. Comparison of 2 commercially available next-generation sequencing platforms in oncology. JAMA Oncol. 2017;3(7):996–998. doi:10.1001/jamaoncol.2016.4983
103. Torga G, Pienta KJ. Patient-paired sample congruence between 2 commercial liquid biopsy tests. JAMA Oncol. 2018;4(6):868–870. doi:10.1001/jamaoncol.2017.4027
104. Gerlinger M, Rowan AJ, Horswell S, et al. Intratumor heterogeneity and branched evolution revealed by multiregion sequencing. N Engl J Med. 2012;366(10):883–892. doi:10.1056/NEJMoa1113205
105. Nakazawa M, Lu C, Chen Y, et al. Serial blood-based analysis of AR-V7 in men with advanced prostate cancer. Ann Oncol. 2015;26(9):1859–1865. doi:10.1093/annonc/mdv282
106. Maher CA, Kumar-Sinha C, Cao X, et al. Transcriptome sequencing to detect gene fusions in cancer. Nature. 2009;458(7234):97–101. doi:10.1038/nature07638
107. Ozsolak F, Milos PM. RNA sequencing: advances, challenges and opportunities. Nat Rev Genet. 2011;12(2):87–98. doi:10.1038/nrg2934
108. Raj A, van Den Bogaard P, Rifkin SA, van Oudenaarden A, Tyagi S. Imaging individual mRNA molecules using multiple singly labeled probes. Nat Methods. 2008;5(10):877–879. doi:10.1038/nmeth.1253
109. Klein CA, Seidl S, Petat-Dutter K, et al. Combined transcriptome and genome analysis of single micrometastatic cells. Nat Biotechnol. 2002;20(4):387–392. doi:10.1038/nbt0402-387
110. Tang F, Barbacioru C, Wang Y, et al. mRNA-Seq whole-transcriptome analysis of a single cell. Nat Methods. 2009;6(5):377–382. doi:10.1038/nmeth.1315
111. Smirnov DA, Zweitzig DR, Foulk BW, et al. Global gene expression profiling of circulating tumor cells. Cancer Res. 2005;65(12):4993–4997. doi:10.1158/0008-5472.CAN-04-4330
112. Cann GM, Gulzar ZG, Cooper S, et al. mRNA-Seq of single prostate cancer circulating tumor cells reveals recapitulation of gene expression and pathways found in prostate cancer. PLoS One. 2012;7(11):e49144. doi:10.1371/journal.pone.0049144
113. Yu M, Ting DT, Stott SL, et al. RNA sequencing of pancreatic circulating tumour cells implicates WNT signalling in metastasis. Nature. 2012;487(7408):510–513. doi:10.1038/nature11217
114. Zhang L, Ridgway LD, Wetzel MD, et al. The identification and characterization of breast cancer CTCs competent for brain metastasis. Sci Transl Med. 2013;5(180):180ra148. doi:10.1126/scitranslmed.3005109
115. Yu M, Bardia A, Aceto N, et al. Cancer therapy. Ex vivo culture of circulating breast tumor cells for individualized testing of drug susceptibility. Science. 2014;345(6193):216–220. doi:10.1126/science.1253533
116. Gao D, Vela I, Sboner A, et al. Organoid cultures derived from patients with advanced prostate cancer. Cell. 2014;159(1):176–187. doi:10.1016/j.cell.2014.08.016
117. Hodgkinson CL, Morrow CJ, Li Y, et al. Tumorigenicity and genetic profiling of circulating tumor cells in small-cell lung cancer. Nat Med. 2014;20(8):897–903. doi:10.1038/nm.3600
118. Baccelli I, Schneeweiss A, Riethdorf S, et al. Identification of a population of blood circulating tumor cells from breast cancer patients that initiates metastasis in a xenograft assay. Nat Biotechnol. 2013;31(6):539–544. doi:10.1038/nbt.2576
119. Rossi E, Rugge M, Facchinetti A, et al. Retaining the long-survive capacity of Circulating Tumor Cells (CTCs) followed by xeno-transplantation: not only from metastatic cancer of the breast but also of prostate cancer patients. Oncoscience. 2014;1(1):49–56.
120. Minciacchi VR, Zijlstra A, Rubin MA, Di Vizio D. Extracellular vesicles for liquid biopsy in prostate cancer: where are we and where are we headed? Prostate Cancer Prostatic Dis. 2017;20(3):251–258. doi:10.1038/pcan.2017.7
121. Di Vizio D, Kim J, Hager MH, et al. Oncosome formation in prostate cancer: association with a region of frequent chromosomal deletion in metastatic disease. Cancer Res. 2009;69(13):5601–5609. doi:10.1158/0008-5472.CAN-08-3860
122. Cheruvanky A, Zhou H, Pisitkun T, et al. Rapid isolation of urinary exosomal biomarkers using a nanomembrane ultrafiltration concentrator. Am J Physiol Renal Physiol. 2007;292(5):F1657–F1661. doi:10.1152/ajprenal.00434.2006
123. Zeringer E, Barta T, Li M, Vlassov AV. Strategies for isolation of exosomes. Cold Spring Harb Protoc. 2015;2015(4):319–323. doi:10.1101/pdb.top074476
124. Lázaro-Ibáñez E, Sanz-Garcia A, Visakorpi T, et al. Different gDNA content in the subpopulations of prostate cancer extracellular vesicles: apoptotic bodies, microvesicles, and exosomes. Prostate. 2014;74(14):1379–1390. doi:10.1002/pros.22853
125. Soekmadji C, Corcoran NM, Oleinikova I, et al. Extracellular vesicles for personalized therapy decision support in advanced metastatic cancers and its potential impact for prostate cancer. Prostate. 2017;77(14):1416–1423. doi:10.1002/pros.23403
126. Del Re M, Biasco E, Crucitta S, et al. The detection of androgen receptor splice variant 7 in plasma-derived exosomal RNA strongly predicts resistance to hormonal therapy in metastatic prostate cancer patients. Eur Urol. 2017;71(4):680–687. doi:10.1016/j.eururo.2016.08.012
127. Huang X, Yuan T, Liang M, et al. Exosomal miR-1290 and miR-375 as prognostic markers in castration-resistant prostate cancer. Eur Urol. 2015;67(1):33–41. doi:10.1016/j.eururo.2014.07.035
128. Kato T, Mizutani K, Kameyama K, et al. Serum exosomal P-glycoprotein is a potential marker to diagnose docetaxel resistance and select a taxoid for patients with prostate cancer. Urol Oncol. 2015;33(9):
129. Wang YH, Ji J, Wang BC, et al. Tumor-derived exosomal long noncoding RNAs as promising diagnostic biomarkers for prostate cancer. Cell Physiol Biochem. 2018;46(2):532–545. doi:10.1159/000488620
130. Vagner T, Spinelli C, Minciacchi VR, et al. Large extracellular vesicles carry most of the tumour DNA circulating in prostate cancer patient plasma. J Extracell Vesicles. 2018;7(1):1505403. doi:10.1080/20013078.2018.1505403
131. McKiernan J, Donovan MJ, O‘Neill V, et al. A novel urine exosome gene expression assay to predict high-grade prostate cancer at initial biopsy. JAMA Oncol. 2016;2(7):882–889. doi:10.1001/jamaoncol.2016.0097
132. McKiernan J, Donovan MJ, Margolis E, et al. A prospective adaptive utility trial to validate performance of a novel urine exosome gene expression assay to predict high-grade prostate cancer in patients with prostate-specific antigen 2-10ng/ml at initial biopsy. Eur Urol. 2018;74(6):731–738. doi:10.1016/j.eururo.2018.08.019
133. Webber JP, Spary LK, Sanders AJ, et al. Differentiation of tumour-promoting stromal myofibroblasts by cancer exosomes. Oncogene. 2015;34(3):290–302. doi:10.1038/onc.2013.560
134. Minciacchi VR, Spinelli C, Reis-Sobreiro M, et al. MYC mediates large oncosome-induced fibroblast reprogramming in prostate cancer. Cancer Res. 2017;77(9):2306–2317. doi:10.1158/0008-5472.CAN-16-2942
135. Bartel DP. MicroRNAs: genomics, biogenesis, mechanism, and function. Cell. 2004;116(2):281–297.
136. Huang X, Yuan T, Tschannen M, et al. Characterization of human plasma-derived exosomal RNAs by deep sequencing. BMC Genomics. 2013;14:319. doi:10.1186/1471-2164-14-181
137. Mitchell PS, Parkin RK, Kroh EM, et al. Circulating microRNAs as stable blood-based markers for cancer detection. Proc Natl Acad Sci U S A. 2008;105(30):10513–10518. doi:10.1073/pnas.0804549105
138. Ahadi A, Brennan S, Kennedy PJ, Hutvagner G, Tran N. Long non-coding RNAs harboring miRNA seed regions are enriched in prostate cancer exosomes. Sci Rep. 2016;6:24922. doi:10.1038/srep24922
139. Formosa A, Markert EK, Lena AM, et al. MicroRNAs, miR-154, miR-299-5p, miR-376a, miR-376c, miR-377, miR-381, miR-487b, miR-485-3p, miR-495 and miR-654-3p, mapped to the 14q32.31 locus, regulate proliferation, apoptosis, migration and invasion in metastatic prostate cancer cells. Oncogene. 2014;33(44):5173–5182. doi:10.1038/onc.2013.451
140. Turchinovich A, Weiz L, Langheinz A, Burwinkel B. Characterization of extracellular circulating microRNA. Nucleic Acids Res. 2011;39(16):7223–7233. doi:10.1093/nar/gkr254
141. Matin F, Jeet V, Moya L, et al. A plasma biomarker panel of four microRNAs for the diagnosis of prostate cancer. Sci Rep. 2018;8(1):6653. doi:10.1038/s41598-018-24424-w
142. Bryant RJ, Pawlowski T, Catto JW, et al. Changes in circulating microRNA levels associated with prostate cancer. Br J Cancer. 2012;106(4):768–774. doi:10.1038/bjc.2011.595
143. Lodes MJ, Caraballo M, Suciu D, et al. Detection of cancer with serum miRNAs on an oligonucleotide microarray. PLoS One. 2009;4(7):e6229. doi:10.1371/journal.pone.0006229
144. Mahn R, Heukamp LC, Rogenhofer S, von Ruecker A, Muller SC, Ellinger J. Circulating microRNAs (miRNA) in serum of patients with prostate cancer. Urology. 2011;77(5):
145. Alhasan AH, Scott AW, Wu JJ, et al. Circulating microRNA signature for the diagnosis of very high-risk prostate cancer. Proc Natl Acad Sci U S A. 2016;113(38):10655–10660. doi:10.1073/pnas.1611596113
146. Kelly BD, Miller N, Sweeney KJ, et al. A circulating microRNA signature as a biomarker for prostate cancer in a high risk group. J Clin Med. 2015;4(7):1369–1379. doi:10.3390/jcm4071369
147. Li Z, Ma YY, Wang J, et al. Exosomal microRNA-141 is upregulated in the serum of prostate cancer patients. Onco Targets Ther. 2016;9:139–148. doi:10.2147/OTT.S95565
148. Endzelins E, Berger A, Melne V, et al. Detection of circulating miRNAs: comparative analysis of extracellular vesicle-incorporated miRNAs and cell-free miRNAs in whole plasma of prostate cancer patients. BMC Cancer. 2017;17(1):730. doi:10.1186/s12885-017-3737-z
149. Motamedinia P, Scott AN, Bate KL, et al. Urine exosomes for non-invasive assessment of gene expression and mutations of prostate cancer. PLoS One. 2016;11(5):e0154507. doi:10.1371/journal.pone.0154507
150. Lin HM, Castillo L, Mahon KL, et al. Circulating microRNAs are associated with docetaxel chemotherapy outcome in castration-resistant prostate cancer. Br J Cancer. 2014;110(10):2462–2471. doi:10.1038/bjc.2014.181
151. Cheng HH, Plets M, Li H, et al. Circulating microRNAs and treatment response in the phase II SWOG S0925 study for patients with new metastatic hormone-sensitive prostate cancer. Prostate. 2018;78(2):121–127. doi:10.1002/pros.23452
152. Zheng GX, Terry JM, Belgrader P, et al. Massively parallel digital transcriptional profiling of single cells. Nat Commun. 2017;8:14049. doi:10.1038/ncomms14049
153. Talasaz AH, Powell AA, Huber DE, et al. Isolating highly enriched populations of circulating epithelial cells and other rare cells from blood using a magnetic sweeper device. Proc Natl Acad Sci U S A. 2009;106(10):3970–3975. doi:10.1073/pnas.0813188106
154. Todenhofer T, Hennenlotter J, Feyerabend S, et al. Preliminary experience on the use of the adnatest(R) system for detection of circulating tumor cells in prostate cancer patients. Anticancer Res. 2012;32(8):3507–3513.
155. Winer-Jones JP, Vahidi B, Arquilevich N, et al. Circulating tumor cells: clinically relevant molecular access based on a novel CTC flow cell. PLoS One. 2014;9(1):e86717. doi:10.1371/journal.pone.0086717
156. Sperger JM, Strotman LN, Welsh A, et al. Integrated analysis of multiple biomarkers from circulating tumor cells enabled by exclusion-based analyte isolation. Clin Cancer Res. 2017;23(3):746–756. doi:10.1158/1078-0432.CCR-16-1021
157. Vona G, Sabile A, Louha M, et al. Isolation by size of epithelial tumor cells: a new method for the immunomorphological and molecular characterization of circulatingtumor cells. Am J Pathol. 2000;156(1):57–63. doi:10.1016/S0002-9440(10)64706-2
158. Xu T, Lu B, Tai YC, Goldkorn A. A cancer detection platform which measures telomerase activity from live circulating tumor cells captured on a microfilter. Cancer Res. 2010;70(16):6420–6426. doi:10.1158/0008-5472.CAN-10-0686
159. Kuang Y, Rogers A, Yeap BY, et al. Noninvasive detection of EGFR T790M in gefitinib or erlotinib resistant non-small cell lung cancer. Clin Cancer Res. 2009;15(8):2630–2636. doi:10.1158/1078-0432.CCR-08-2592
160. Alegre E, Fusco JP, Restituto P, et al. Total and mutated EGFR quantification in cell-free DNA from non-small cell lung cancer patients detects tumor heterogeneity and presents prognostic value. Tumour Biol. 2016;37(10):13687–13694.
161. Forshew T, Murtaza M, Parkinson C, et al. Noninvasive identification and monitoring of cancer mutations by targeted deep sequencing of plasma DNA. Sci Transl Med. 2012;4(136):136ra168. doi:10.1126/scitranslmed.3003726
162. Gale D, Lawson ARJ, Howarth K, et al. Development of a highly sensitive liquid biopsy platform to detect clinically-relevant cancer mutations at low allele fractions in cell-free DNA. PLoS One. 2018;13(3):e0194630. doi:10.1371/journal.pone.0194630
163. Bartels S, Persing S, Hasemeier B, Schipper E, Kreipe H, Lehmann U. Molecular analysis of circulating cell-free DNA from lung cancer patients in routine laboratory practice: a cross-platform comparison of three different molecular methods for mutation detection. J Mol Diagn. 2017;19(5):722–732. doi:10.1016/j.jmoldx.2017.05.008
164. Lanman RB, Mortimer SA, Zill OA, et al. Analytical and clinical validation of a digital sequencing panel for quantitative, highly accurate evaluation of cell-free circulating tumor DNA. PLoS One. 2015;10(10):e0140712. doi:10.1371/journal.pone.0140712
165. Phallen J, Sausen M, Adleff V, et al. Direct detection of early-stage cancers using circulating tumor DNA. Sci Transl Med. 2017;9(403). doi:10.1126/scitranslmed.aan2415.
166. Leary RJ, Sausen M, Kinde I, et al. Detection of chromosomal alterations in the circulation of cancer patients with whole-genome sequencing. Sci Transl Med. 2012;4(162):162ra154. doi:10.1126/scitranslmed.3004742
167. Adalsteinsson VA, Ha G, Freeman SS, et al. Scalable whole-exome sequencing of cell-free DNA reveals high concordance with metastatic tumors. Nat Commun. 2017;8(1):1324. doi:10.1038/s41467-017-00965-y
 © 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.