Back to Journals » The Application of Clinical Genetics » Volume 7
Current status of gene therapy for breast cancer: progress and challenges
Authors McCrudden CM, McCarthy H
Received 16 June 2014
Accepted for publication 9 August 2014
Published 10 November 2014 Volume 2014:7 Pages 209—220
DOI https://doi.org/10.2147/TACG.S54992
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Prof. Dr. Martin Maurer
Cian M McCrudden, Helen O McCarthy
School of Pharmacy, Queen’s University Belfast, Belfast, UK
Abstract: Breast cancer is characterized by a series of genetic mutations and is therefore ideally placed for gene therapy intervention. The aim of gene therapy is to deliver a nucleic acid-based drug to either correct or destroy the cells harboring the genetic aberration. More recently, cancer gene therapy has evolved to also encompass delivery of RNA interference technologies, as well as cancer DNA vaccines. However, the bottleneck in creating such nucleic acid pharmaceuticals lies in the delivery. Deliverability of DNA is limited as it is prone to circulating nucleases; therefore, numerous strategies have been employed to aid with biological transport. This review will discuss some of the viral and nonviral approaches to breast cancer gene therapy, and present the findings of clinical trials of these therapies in breast cancer patients. Also detailed are some of the most recent developments in nonviral approaches to targeting in breast cancer gene therapy, including transcriptional control, and the development of recombinant, multifunctional bio-inspired systems. Lastly, DNA vaccines for breast cancer are documented, with comment on requirements for successful pharmaceutical product development.
Keywords: breast cancer, gene therapy, nonviral, clinical trial
Introduction
There are in theory a plethora of targets available for breast cancer gene therapy, yet at the time of writing this review article, there are still no approved gene therapy products for breast cancer. Over 63% of all gene therapy clinical trials are in the field of cancer, so it could be extrapolated that a clinical product should ensue in due course. Yet the more that is known about the genetic dysregulation of the various subtypes of breast cancer, the more difficult it becomes to select a genetic therapy. The European Commission (EC) 2003/63 directive states that a gene therapy medicinal product is essentially a piece of nucleic acid that is delivered either in vivo or ex vivo that can be prophylactic, diagnostic, or therapeutic.1 This then opens the field up to encompass traditional corrective genes, cytotoxic genes, small interfering RNA (siRNA) or short hairpin RNA (shRNA) interference therapies, microRNA up- or downregulation, and DNA vaccination against overexpressed tumor antigens. Virtually all of these genetic therapies require assistance to overcome the many extra- and intracellular biological barriers. Therefore, for successful nucleic acid transfer, a delivery system must be considered that is typically either viral or nonviral. Taking all of these factors on board, it becomes apparent that the potential options for the “gene therapist” are actually limitless and with no clear “frontrunner”, it is not surprising that the progress of a therapy is moderate at best. The scope of this breast cancer review is to consider the progress that has been made with nucleic acid therapies, to examine possible future targets, and to identify some of the key challenges that are preventing product approval.
Viral breast cancer gene therapy
Viruses are highly efficient at transducing cells with their genetic material and so provide excellent delivery systems for the purposes of gene therapy. Recombinant viruses are nonreplicative, require a packaging cell line for particle production, and have been genetically engineered to carry the gene of interest. Increasingly, viral characteristics must be paired with the disease target (Table 1).
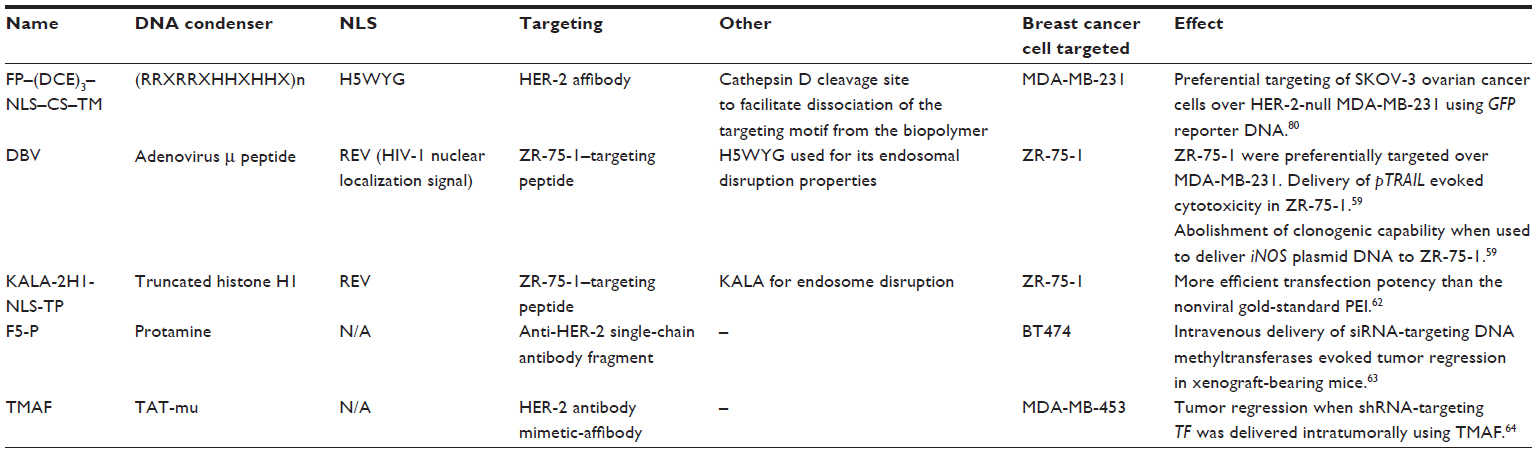 | Table 1 Summary of recombinant biopolymers and associated targeting strategies that have been used to target breast cancer in vitro and in vivo |
Retroviral viruses and breast cancer
Retroviral vectors are characterized by integration into the host genome and although this evokes long-term transgene expression, there are also some safety concerns with insertional mutagenesis and activation of oncogenes in normal tissues. However, if gene expression can be targeted only to tumors then recombinant retroviral vectors can be successfully employed. Expression may be controlled by direct intratumoral injection, although a significant limitation is that this strategy is only viable for accessible tumors. For example, MetXia-P450 is a recombinant retroviral vector that delivers cytochrome P450 2B6 in a gene-directed enzyme prodrug therapy (GDEPT) strategy to improve metabolism of cyclophosphamide (CPA). Initial preclinical studies determined that MetXia-P450 sensitized T47D breast cancer cells to CPA, produced a bystander effect, and significantly reduced growth of MDA-MB-231 breast tumors in vivo.2 MetXia-P450 entered Phase I clinical trials for nine breast cancer and three melanoma patients with cutaneous tumors to evaluate safety and then clinical response.3 After two intratumoral injections of MetXia-P450 and treatment with CPA, one breast cancer patient had a partial response and four had stable disease over the 12-week period. Although less than 1% of tumor cells were transduced, in this trial, 16/24 biopsies were positive for the β-galactosidase expression. Even with such a low transduction, the observed antitumor activity has led to further clinical trials for patients with pancreatic cancer.
Alternatively, the recombinant retrovirus may have a targeting motif for tumors, thus enabling systemic administration to less accessible metastases. Rexin-G is a highly engineered retroviral nanoparticle that achieves targeting to cancerous lesions through the attachment of a collagen motif that binds to “newly exposed” extracellular matrix, which is typically associated with tumor tissue. The transgene employed is a cytotoxic “dominant negative” cyclin G1 gene that results in tumor apoptosis and disrupts proliferation of the tumor vasculature.4 This therapy has entered several clinical trials with Phase I/II completed for metastatic breast cancer, and the authors have highlighted several noteworthy solid tumor cases.5 One such example was that of a 74-year-old woman who was diagnosed in 2001 with infiltrating ductal carcinoma of breast, stage T3N2. The patient went through a series of treatment regimens where following initial response, chemoresistant disease resulted, which was confirmed in 2006. Tumors were evident in the lymph node and anterior chest wall. In 2007, Rexin-G was administered three times a week for 3 weeks at a dose of 1×1011 particles. Regression of the tumors enabled surgical resection of a solitary residual tumor, which was a fibrotic mass with evidence of apoptosis and tumor-infiltrating CD8+ T-cells, again indicative of a good prognosis. This was further evidenced by patient survival in excess of 3 years at the time the authors published. Gordon and Hall give several other remarkable examples of the success of their therapy, which has been attributed to having a stealth nanoparticle that does not evoke an immune response, can be repeatedly administered, exploits retroviral characteristics with a tropism for only dividing cells thus sparing normal tissue damage, and confers widespread anticancer activity by blocking the cell cycle in G1.5
Oncolytic herpes simplex virus and breast cancer
The therapeutic potential of utilizing viruses that selectively replicate and destroy cancer cells is gaining momentum. Apart from having a tropism for cancer cells, oncolytic viruses can also be designed to deliver a transgene in addition to their cytotoxic capacity.6 The herpes simplex virus-1 (HSV-1) was genetically engineered to create the attenuated HF10 clone.7 Early preclinical tests with the MM102-TC breast cancer tumors in vivo were very promising, with HF10-treated mice surviving up to 120 days, >40 days longer than control mice after intratumoral injection.8,9 These promising mouse assays led to clinical trialing of the therapy, in which HF10 was injected into the cutaneous or subcutaneous nodules of six patients with recurrent breast cancer.9 For each patient, two nodules were selected, one of which received a saline control, the other an injection of HF10 (dose ranged from 104–5×105 pfu/0.5 mL). The injections were given daily for 3 days at various sites to ensure good distribution. After 2 weeks, the tumors were excised for histopathological examination. There were no local or systemic adverse effects following treatment and tumor regression of 30%–100% was observed.9 Furthermore, mean counts of CD8-infiltrating lymphocytes were significantly higher.10
Although initial clinical trials have been encouraging, delivery of the HF10 virus has been local-regional, and as yet, the systemic effects of this therapy have not been evaluated for breast cancer patients. Concerns over widespread delivery of HF10 may be alleviated by the fact that the HSV virus does not integrate into the host genome, thus avoiding unwanted mutations; should normal tissue replication occur, retroviral drugs could be employed to control the virus. Of course, extensive preclinical testing would be required before considering systemic delivery, but given how difficult distant metastases are to treat, a targeted oncolytic virus could potentially have very high rewards for breast cancer, in a similar manner to myeloma patients with disseminated disease.11
Oncolytic viruses can also have a dual function through delivery of an immune activating transgene, as is the case with talimogene laherparepvec (T-VEC [OncoVEXGM-CSF]).12 This oncolytic HSV expresses granulocyte-macrophage colony-stimulating factor (GM-CSF), which leads to an antitumor immune response.13 As with most HSVs, the deletion of the ICP34.5 gene confers tumor selectivity. The second generation T-VEC is designed to be more potent but still have the safety profile of the previous HSVs, which have been well-tolerated in the clinic.14–16 The main difference in the second generation was deletion of the ICP47 gene in order to enable antigen presentation on HSV-infected cells.12 Thirty patients with either cutaneous or subcutaneous metastases from a range of primary sites were selected, including those with breast cancer. Thirteen patients received a single dose and 17 received three doses. The main side effects were mild flu-like symptoms and local reactions at the injection site, which could be linked to the expression of GM-CSF. This local inflammation was dose-limiting and the study was amended in HSV-seronegative patients to a maximum of 107 pfu/mL. Of the 26 tumor biopsies, 14 stained for tumor necrosis, which correlated with HSV expression. The authors conclude that T-VEC is well-tolerated and can be safely administered with evidence of an antitumor effect.12 The question then still remains as to whether T-VEC could be administered systemically to target and destroy disseminated disease.
There are several oncolytic viruses in the pipeline; for example, one has been designed that targets breast cancer brain metastases, again with an immunotherapy approach.17 The oncolytic HSV (M002) has deletions in both copies of the y134.5 gene, thus conferring specificity by only replicating in tumor cells. The M002 virus has also been engineered to express interleukin (IL)-12, a known proinflammatory cytokine that confers an antitumor effect.18 The tumor microenvironment is heterogenous and so by delivering IL-12, the authors hope to eradicate uninfected tumor cells through immunotherapy. Results indicated that the levels of IL-12 produced following infection with M002 in vitro were at desired physiological levels. Perhaps more importantly though, analysis in vivo demonstrated that M002 significantly improved survival and reduced tumor burden over the R3659 virus, which did not express IL-12.17 This supports the dual functionality of combining lytic activity with immunotherapy.
Oncolytic parvovirus and breast cancer
The rodent parvovirus-H1 (H1-PV) selectively replicates in tumor cells and is not known to evoke pathogenicity in humans.19 Although the H1-PV has been shown to enter both normal and cancerous cells, it can only fully replicate its life cycle in the latter.20 In addition, the H1-PV can also activate the immune system via toll-like receptors 3 and 9 and cause NF-κB-dependent activation of the adaptive immune response.21 To evaluate the tropism of the H1-PV virus further, Muharram et al evaluated the cytotoxicity in 96 tumors taken from breast cancer patients with normal cells taken from the patients’ peritumoral tissue as a negative control.22 The clinical samples were taken prior to any other anticancer therapy. Of the 69 samples that gave cultures, sensitivity to the H1-PV was strongly correlated with high-grade tumors (48%). The authors also note the higher multiplicity of infection (MOI) that is required compared to routine in vitro cell lines.22 This study clearly highlights the benefits of using clinical breast tumors when evaluating the infectivity of the H1-PV. The lack of activation in the normal peritumoral tissue further confirms the selectivity of the H1-PV for higher-grade breast cancer tumors (>2). The tropism for aggressive breast cancer tissue should open up a plethora of therapeutic options, especially for those patients with limited treatment options. Parvoviruses are also attractive because they have been utilized as potent chemosensitizers, for example, with gemcitabine for pancreatic cancer, and could be employed with conventional therapies.23 In addition, the H1-PV has been genetically engineered to deliver complementary DNA (cDNA) immunotherapy such as TNFα and IFN-γ, thereby improving therapeutic outcomes.24
Challenges
The development of viral gene therapies have been somewhat limited for those patients with breast cancer, with the vast majority of trials to date focused on proof of principle in highly accessible cutaneous tumors. While these studies need to be completed, there is a dearth in the literature of experiments that go beyond intratumoral injection. Disseminated breast cancer is far from accessible, and therefore, studies with systemic administration of viral gene therapies are critical if progress is to be realized. The two major hurdles that need to be overcome are: 1) ensuring that viruses can be targeted by providing evidence in vivo and clinically in patients, and 2) that when the virus gets to its target site, the infectivity rate will be high enough to generate a significant therapeutic effect, while lacking immunogenicity.
Reovirus therapy has provided impressive therapeutic results in mouse models of experimental breast cancer. The progress of the reovirus arose from exciting preclinical data that demonstrated a therapy that could target distant tumors. Delivered systemically, replication of the virus is limited to cancer cells, and provoked regression of tumors at sites distant to the site of delivery in an MDA-MB-468 bilateral tumor model. Moreover, the reovirus was capable of replication in surgical breast tumor explants.25 The virus is only known to replicate in cancer cells that have an activated Ras pathway. Another reovirus, Reolysin® (Oncolytics Biotech, Inc., Calgary, AB, Canada) was administered for the first time in the US intravenously to 18 patients with advanced solid tumors, two of whom had breast cancer. This was a multiple-dosing experiment every 28 days to determine the maximally tolerated dose. The virus was relatively well-tolerated, with the majority having grade 1 toxicities. Only two patients had grade 2 toxicities, one of whom following seven administrations of 1×1010 viral particles.26 From trials such as this, it is invaluable to know that all toxicities were resolved with time and were mild compared to conventional chemotherapy. Unfortunately, all patients developed neutralizing antibodies that may restrict long-term administration of this oncolytic virus, but at the same time, this information points researchers in the right direction to further improve the therapy. Although clinical outcome was not the endpoint, one patient with chemoresistant metastatic breast cancer had a partial response with a 34% reduction in tumor burden. This patient also had a Ras mutation, supporting the premise of reovirus selective targeting.26 The constant shrinkage in tumor volume was maintained for 9 weeks, which provides important information when scheduling the dose and timing of such novel nucleic acid therapeutics. Similar results were also reported from systemic delivery of the reovirus in two trials in the UK.27,28 This gives researchers in the field confidence and the necessary clinical data to drive development of this therapy forward. Subsequently, at the time of writing this review, Oncolytics Biotech, Inc. have 23 clinical trials ongoing for Reolysin®. IND213 is one such trial, which combines intravenous administration of Reolysin® in combination with paclitaxel for patients with advanced or metastatic breast cancer.
With respect to breast cancer, preclinical in vivo experiments with viruses must be designed to demonstrate maximal targeting of the tumor in order for the therapy to progress into the clinic. For example, Nanni et al delivered a human epidermal growth factor receptor 2 (HER-2) targeted oncolytic virus for breast cancer and demonstrated efficacy in vivo following intraperitoneal administration.29 Taking on board new results is also critical to success. Oncolytic HSVs are hampered by poor potency in the clinic; studies by Cody et al that highlight the value of including histone deacetylase inhibitors to improve viral transcription could have key knock-on effects to those researching advanced development of HSVs.30
Development of a therapeutic product will only happen through the generation of translational data that at the earliest stages is designed for the end target, namely patients. Once in the clinic, data that demonstrates successful systemic administration coupled with a therapeutic response in patients will undoubtedly have the biggest impact in the drive toward a licensed viral therapy.
Nonviral breast cancer gene therapy
Typically, the delivery of genetic material involves either physical methods such as injection or gene gun, or chemical methods of delivery. Some physical methods of gene transfer have been assessed at clinical trial for breast cancer. Naked reporter plasmid DNA (pDNA) (LacZ) was successfully delivered by a needle-free jet injection to skin metastases from primary breast cancer in three patients, and also to melanoma lesions in 14 patients; the transgene was detectable at messenger RNA (mRNA) and protein levels in all patients.31 Electrotransfection was used to assist in the delivery of naked pDNA to BJMC3879 mouse mammary carcinoma; such a method used to deliver IL-12 pDNA significantly delayed tumor development in mice, reduced the incidence of lymph node metastases by over 50% compared with vector control, and resulted in smaller lung metastases.32 Despite impressive results that have been generated with electrotransfection, the practicality and acceptability issues involved in such techniques may lead to patient reluctance. Furthermore, the use of naked DNA and techniques such as jet injection are limited to disease conditions where tumors are superficial and easy to access. In the majority of disease states, systemic therapy delivery is required, necessitating use of a vehicle to protect nucleic acids from nuclease attack. Safety and immunogenicity issues associated with viral gene therapy delivery and the limitations of physical delivery methods have led to the development of a range of nonviral delivery vehicles, including polymers, liposomes, or peptides, or combination thereof, to protect the DNA from degradation and ensure that it reaches the nucleus. Of the multitude of nonviral delivery options published, it has become clear that although the safety issues are much less significant than is the case with their viral counterparts, efficacy remains the biggest stumbling block.
Nonviral gene therapy strategies are compelled to innovate to improve their nucleic acid cargo delivery in order to compete with the unrivalled performance of viral counterparts.33 Figure 1 details some of the agents that have been employed as vehicles for nonviral nucleic acid cargo delivery and some of the strategies that have been developed to improve delivery. Despite improvements in nonviral gene therapy strategies, there has been limited translation of impressive laboratory-generated results to the clinical setting for the targeting of breast cancer.
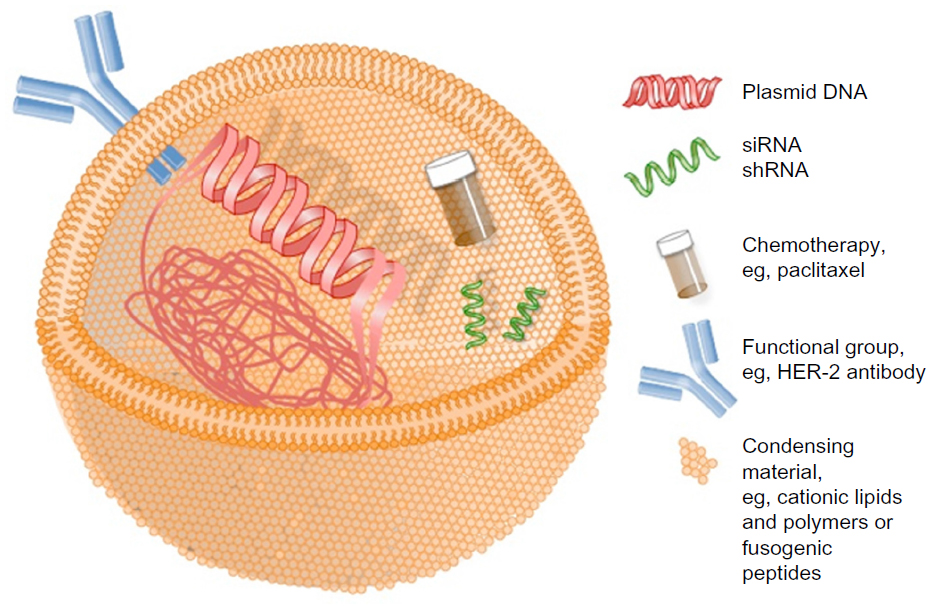 | Figure 1 Simplified schematic of nonviral gene therapy delivery vehicle. |
tgDCC-E1A is a nonviral therapy that was assessed for tolerability and clinical efficacy in nine patients with metastatic breast cancer. The therapeutic nucleic acid, pE1A-K2, which encodes E1A, a protein with known antitumor activity, is complexed using 3β[N-(N′, N′-dimethylaminoethane)-carbamoyl] cholesterol (DC-Chol) and dioleoylphosphatidylethanolamine (DOPE). E1A DNA was detectable in 14 of 15 samples tested following direct intratumoral injection of the therapy, although the authors were unable to assess mRNA or protein levels of E1A. There was some limited evidence of therapeutic potential following this initial trial.34 In a separate trial, the same therapy was delivered to the pleural cavity of six breast cancer patients. The therapy appeared to downregulate HER-2 expression in all of the breast specimens tested, and increased numbers of apoptotic cells were detected in ascites or pleural fluids, compared to those detected prior to gene therapy.35
ZYC300 is a pDNA encoding an inactivated form of the carcinogen activator cytochrome P450 1B1 (CYP1B1) that is complexed in poly-DL-lactide-coglycolide microparticles. The nonviral therapy is aimed at vaccinating patients against CYP1B1, which is expressed on most transformed tissues, but is absent from most normal tissues. A Phase I clinical trial undertaken to assess the safety, tolerability, and feasibility, as well as immunologic response and clinical activity of ZYC300 treatment comprised 17 patients with advanced cancers, one of whom had metastatic breast cancer. The disease of the breast cancer patient, whose disease had metastasized to the bone and liver, stabilized after ZYC300 treatment, before eventually progressing. ZYC300 proved extremely beneficial for a single patient in the trial, whose renal disease was regressing at the time of publication of the report.36
Nucleic acid cargo is generally condensed into nanoparticulate formulation using a cationic lipid such as N-[1-(2,3-dioleyloxy)propyl]-N,N,N-trimethylammonium chloride (DOTMA), dioctadecylamidoglycylspermine (DOGS), 1,2-bis(oleoyloxy)-3-(trimethylammonio)propane (DOTAP), DC-Chol, and DOPE, or cationic polymers, which include poly(L-lysine) (PLL), polyethylenimine (PEI), chitosan, and polyamidoamine (PAMAM). Fusogenic peptides capable of delivery of pDNA include KALA.37
Transcriptional control of genetic material
A common side effect of most, if not all, cancer therapies is damage to tissue that neighbors the diseased tissue. While it is usually imperative that maximal achievable delivery of therapeutic to the tumor is achieved, therapeutic efficacy is often limited by toxicities. It is well-understood that cancerous tissue has a genetic signature that differs from neighboring, nondiseased tissue. An attractive option for the gene therapist is to target these genetic differences in order to limit transgene expression to cells that aberrantly express certain genes.38 In breast cancer, the HER-2 receptor is a commonly targeted genetic aberration, although it is only targetable in 20% of breast cancers.39 Identification of suitable genes that are aberrantly expressed in breast cancer could aid in transcriptional targeting of gene expression in the remaining 80% of cases. In this manner, we the authors have used the osteocalcin promoter to drive the expression of our therapeutic transgene of interest, inducible nitric oxide synthase (iNOS), using nonviral means. Using this promoter to confine transgene expression to the tumor, we have demonstrated exquisite tumor abrogation in human prostate cancer models.40,41 Identification of further tumor-specific markers for promotion of transgene expression may lead to improved gene therapy potential and limited toxicities.
One of the earliest targeted nonviral gene therapy clinical trials was for patients that overexpress the HER-2 (erbB-2) receptor. Targeting was achieved with a HER-2 promoter containing a binding site for AP-2, which is an upregulated transcription factor in HER-2-positive tumors. The pERCY plasmid utilized this promoter to drive expression of cytosine deaminase, the enzyme that metabolizes 5-fluorocytosine (5-FC) to 5-fluorouracil (5-FU). Twelve patients were selected with skin metastases, analyzed for HER-2 expression, and the nodules were injected with up to 400 μg of naked pERCY plasmid followed by systemic administration of 200 mg/kg/24 hours of 5-FC. Four of the patients showed tumor reduction, two of whom had received only nucleic acid treatment with no prodrug. Cytosine deaminase gene expression was confirmed only in tumor tissue with no evidence of off-site targeting. However, the authors report that it took more than 18 months to recruit only 12 patients with nodular cutaneous HER-2 expressing lesions, and furthermore there was a high variability in cytosine deaminase expression, with the majority of patients having low levels at best.42
The survivin promoter was used to promote bax (both wild-type and S187del mutant, which has increased activity) expression in a range of breast cancer cell lines. Cytotoxicity elicited by the survivin promoter-driven bax expression was not as potent as that elicited by cytomegalovirus (CMV)-driven expression in SKBR3, MDA-MB-231, and MCF7 breast cancer cells. However, bax expression and related cytotoxicity was absent in normal cells when the tumor-specific promoter was used, but present in the case of CMV-driven expression. Intratumoral delivery of the survivin-driven bax S187del complexed with in vivo-jetPEI resulted in a tumor growth delay that was comparable to that seen in CMV-driven expression of the wild-type.43 This sophisticated strategy of target (direct tumor transcriptional targeting) and transgene optimization (the S187del mutant had 5–6 times more cytotoxic potency than the wild-type transgene) highlights the options that are available to the enterprising gene therapist.
Transcriptional regulation of gene expression was achieved in an elegant approach that employed PEGylated PEI polyplexes functionalized with a MUC-1 nanobody. The complex was used to deliver the cytotoxic tBid transgene under the control of the MUC-1 promoter, commonly overexpressed in cancer. The nanobody/promoter-targeting successfully evoked cell killing in MCF-7, T47D, and SKBR3 breast cancer cells; replacement of the MUC-1 promoter with the synapsin 1 promoter (expression of which is confined to the central nervous system) abolished the cytotoxic effect of the complex, underlining the therapeutic potential of tumor promoter targeting.44
Tumor-specific promoters have also been used to improve target specificity of viral vectors. PRC1 and RRM2 were overexpressed in MCF-7 and T47D breast cancer cell lines, compared with nontransformed MCF-10As, and were also overexpressed in 48% and 58%, respectively, of tumor samples, but not adjacent normal tissue, from a cohort of breast cancer patients. Both promoters evoked impressive reporter gene expression in MCF-7 and MDA-MB-231 cells, and were as active as CMV-promoted expression when transduced using a recombinant adeno-associated virus vector.45 The human fatty acid synthase promoter, expression of which is again limited to transformed tissue, was used to drive expression of HSV thymidine kinase in an adenoviral vector. SKBR3 cells treated with Ad-FAS-TK therapy before implantation into mice failed to develop into tumors, while vector controls successfully developed into xenografts.46 T47D xenograft regression was seen in mice that received adenoviral cytosine deaminase gene therapy under transcriptional control of the alpha lactalbumin promoter with 5-FC; the regression of tumors was of the same magnitude as seen when promotion by CMV was performed. Again, transgene expression was limited to breast cancer cells, and absent from nontransformed MCF10A breast cells and U2OS osteosarcoma cells.47
While in principle there are numerous options for controlling expression through the use of tumor-specific promoters, the rate-limiting factor is that delivery of naked DNA correlates to extremely poor gene expression and delivery vehicles are required for all but the most superficial of lesions. Of course, this presents a new set of complex interactions that need to be fully characterized in terms of efficiency of overcoming the biological barriers, immunogenicity, cost-effectiveness, and the probability of gaining regulatory approval for human use.
RNA interference therapies
The aim of the majority of the gene therapy strategies above has been the delivery of nucleic acid to evoke expression of a therapeutic gene. Another viable genetic therapy strategy is interference with the expression of unappealing genes. siRNA and shRNA can be used for the transient or long-term abrogation of the expression of a target gene, and have themselves received a degree of attention for experimental treatment of breast cancer.48 The therapeutic potential of siRNA targeting of the survivin gene in 4T1 breast cancer xenografts using a nonviral TAT-g-CS was recently reported. Nanoparticulate formulations of the therapy delivered intratumorally to xenograft-bearing mice evoked significant slowing of tumor development.49 A reduction-sensitive linear cationic click polymer (RCP) used to deliver shRNA targeting the multidrug resistance (MDR) gene restored sensitivity to doxorubicin in resistant MCF-7 breast cancer cells in a mouse xenograft model. The complexes were delivered systemically, and xenograft volume was significantly lessened in mice receiving MDR interference plus doxorubicin compared to those that received doxorubicin alone.50
A competitive advantage that nonviral vehicles have over viral is the possibility to deliver more than just the nucleic acid therapeutic; a cationic core–shell nanoparticle used to deliver Bcl-2 siRNA along with paclitaxel resulted in improved cytotoxicity toward MDA-MB-231 breast cancer cells in vitro, while the same vehicle delivered IL-12 pDNA with paclitaxel, and slowed tumor development more efficiently than either cargo delivered singly.51 In a separate study, local IL-12 gene therapy delivered using a water-soluble lipoprotein (WSLP) improved response to systemically delivered paclitaxel in 4T1 breast cancer xenografts, boosting survival of mice and diminishing the number of metastases developed.52
Although the HER-2 receptor is the most popular among gene therapy targeting strategies, others are gaining in popularity. Targeting the heparin-binding epidermal growth factor-like growth factor (HB-EGF) receptor for the delivery of siRNA was an effective strategy in HB-EGF-expressing MDA-MB-231 in vitro.53 Indirect inhibition of tumor progression has also been achieved using RNA interference technology; when siRNA targeted against vascular endothelial growth factor (VEGF) was delivered using reconstituted high-density lipoprotein to mice bearing MCF-7 xenografts, significant slowing of tumor progression was noted. The systemically delivered therapy evoked downregulation of VEGF protein, and a dose-dependent lessening of the number and density of microvessels in the tumors.54 Similarly, impressive results were observed when a core shell nanoparticle was used to deliver VEGF-directed siRNA along with paclitaxel. MCF-7 xenograft tumor progression was slowed by nonfunctionalized nanoparticles carrying the dual cargo; functionalization with vapreotide, a ligand for somatostatin receptors, commonly upregulated in cancer, significantly improved the response seen in nonfunctionalized therapy. Again, tumor microvessel infiltration was repressed in xenografts that received the VEGF RNA-interfering therapy.55 Silica nanoparticles carrying p53 and functionalized with transferrin achieved impressive tumor growth retardation in MCF-7 xenografts,56,57 as did micellar nanoparticles carrying acid ceramidase siRNA in BT474 xenografts.57
Use of recombinant multifunctional nonviral delivery systems
The delivery strategies detailed above involve complex chemical and physical processes to develop vehicles capable of multifunctionality. The last few years have seen the development of recombinant vector technology; competent cells are transformed with bio-inspired plasmids that contain motifs arranged specifically to render the resultant recombinant biomimetic vehicles capable of binding, condensing, and delivering nucleic acid cargo to target cells. One such vector was generated to contain the adenoviral penton base, which is critical for viral internalization and endosome disruption. A recombinant gene was constructed to contain wild-type Ad5 penton flanked at the N-terminal by a peptide sequence for targeting to the HER-2 receptor, and at the C-terminal by a decalysine with responsibility for DNA binding and condensation. The resultant vehicle was capable of delivering reporter gene plasmids to breast cancer cells, and preferentially to those cells that overexpressed HER-2.58 Another recombinant vector was used to deliver TNF-related apoptosis-inducing ligand (TRAIL)59 to ZR-75-1 breast cancer cells. The vector contained a DNA-condensing motif, a breast cancer cell-targeting peptide, a pH-responsive endosome disruption peptide, and a nuclear localization signal. The recombinant vehicle condensed pTRAIL DNA into nanoscale particles that, when exposed to ZR-75-1 breast cancer cells, evoked impressive cell kill (up to 62%).59 Similar technology was used to condense and deliver iNOS pDNA to ZR-75-1 breast cancer cells in vitro.60 Clonogenic assay analysis revealed impressive potency of the recombinant vector complexed with iNOS pDNA, with a viable surviving fraction only of 0.4% of control after transfection of ZR-75-1.60
Another HER-2 motif-expressing recombinant vector was used to target breast cancer, and was capable of preferentially targeting a recombinant vector carrying the β-galactosidase gene toward MDA-MB-453 breast cancer cells,61 and yet another such recombinant protein, KALA-2H1-NLS-TP, that differs in the DNA-condensing motif was recently reported from the same group.62 While the developments made in the designer biomimetic vector are impressive, evaluation of the delivery systems in vivo is ultimately required to determine their therapeutic potential.
A HER-2 antibody/protamine fusion protein expressed in and purified from Escherichia coli was capable of binding and delivering siRNA-targeting DNA methyltransferases to BT474 breast cancer cells. The authors of the study were able to report impressive tumor growth delay evoked by intravenous delivery of their therapy, resultant of the silencing of DNA methyltransferases, with consequent restoration of tumor suppressor gene function.63 Similarly, intratumoral injection of a tissue factor-targeting shRNA, complexed using a recombinant system and targeted toward the HER-2 receptor, significantly diminished tumor progression in an MDA-MB-453 xenograft model in mice.64 Low cost and ease of production of these recombinant vectors make them highly attractive to the biopharmaceutical gene therapist.
DNA vaccination
Of course, many of the issues with traditional nonviral gene therapy are circumvented with DNA vaccination, as there is no need for long-term gene expression. Rather, the aim is to prime and boost to activate the patient’s own immune response to target a particular set of antigens. Issues with safety and side effects have rendered conventional pathogenic vaccines obsolete. Current developments are focused on vaccines developed from purified subunits, recombinant proteins, or synthetic peptides. Although these vaccines have an excellent safety profile, their immunogenic potency can be significantly compromised with the induction of only an antibody response. For many pathogens, especially intracellular, the effects of these vaccines are minimal. Therefore, there is a “need” to activate the cell-mediated immune response primarily through cytotoxic T lymphocytes (CTLs). It has been well-documented that DNA vaccination is a highly potent strategy whereby pDNA encodes a particular antigen for a disease.4,65,66 In order for the desired antigen to be expressed, the pDNA should be delivered to the nucleus of cells to ensure transcription and translation of the protein antigen in the cytoplasm of the host cell. The DNA should also ideally be delivered to an antigen-presenting cell (APC). This is so that the antigen expressed in the cytoplasm can be presented to the immune system via the MHC I pathway. The consequences of this are that there will be a cell-mediated immune response. This renders DNA vaccines much more effective than current vaccines in evoking a long-term immune response. In addition to a prophylactic response, DNA vaccines can also be therapeutic, are inexpensive to manufacture, and do not require cold chain storage. Many cancers are characterized by tumor-associated antigens (TAAs) that provide unique targets for vaccination. Oncept® is the first licensed product for cancer in either humans or animals for canine melanoma based upon delivery of cDNA for human tyrosinase (huTyr), which evokes a greater immune response than canine tyrosinase.67
There are a plethora of TAAs that could be potential targets for breast cancer. Such targets include HER-2, MUC-1, CEA, mammaglobin A, and hTERT.68–72 As a transmembrane receptor, HER-2 can be easily exposed to humoral and cell-mediated response. Furthermore, the differential expression of HER-2 between breast tumors and normal tissue negates the risk of an autoimmune response. The HER-2 DNA vaccine was delivered with considerable success to FVB-huHER-2 transgenic mice that form spontaneous mammary tumors. The vaccination consisted of two intramuscular injections of 50 μg of chimeric human/rat HER-2 plasmid followed by electroporation at 2-week intervals; 65% of mice were tumor-free after 90 weeks compared to 45 weeks for mock vaccinated controls.73 Frequently though adjuvants are also required for a successful immune response. For example, patients with metastatic breast cancer were given full-length HER-2-pDNA with GM-CSF and IL-2 cytokines as adjuvants. The dosing schedule involved three cycles, comprising 270 μg HER-2-pDNA delivered intramuscularly and 30 μg intracutaneously. Two days prior to receiving the vaccine, patients were administered 3×40 μg daily intracutaneous injections of GM-CSF molgramostim as an adjuvant. In addition, 24 hours after vaccination, IL-2 aldesleukin (1 μg/kg) was given to the patients subcutaneously for 4 consecutive days. There were 4 weeks between each HER-2-pDNA vaccination. Of the six patients that completed the three vaccination cycles, two survived in excess of 4 years, and overall, the vaccination was well-tolerated with cell-mediated and humoral immune responses observed.74
Encouraging results have also been reported with mammaglobin-A DNA vaccination. Mammaglobin-A is present in up to 80% of primary and metastatic breast cancers and expression is not stage dependent, rendering it an ideal target for vaccination.71 Narayanan et al demonstrated for the first time in vivo that vaccination with mammaglobin-A cDNA (four doses of intramuscular injections of 100 μg of cDNA at 2-week intervals) resulted in a CD8+ response against mammaglobin-A positive tumors, and that passive transfer of CD8+ CTLs were able to regress such tumors in vivo.75 In this instance, there was no electroporation to enhance cellular uptake, and without some sort of delivery assistance, cellular uptake can be greatly reduced. Nevertheless, the preclinical data demonstrated the efficacy of this vaccine, and the mammaglobin cDNA was evaluated in a Phase I clinical trial.76 Seven patients with stage IV metastatic breast cancer and HLA-A2+ status were given three doses of the mammaglobin-A DNA vaccine 28 days apart. Although most reports detail the CD8+ and antibody response, this clinical trial focused on the CD4+ population, identified by the expression of the inducible costimulator molecule (ICOS) on recently activated T-cells. Results indicated a significant expansion in CD4+ ICOS T-cells coupled with a reduction in regulatory T-cells, which is indicative of antitumor immunity, although further studies are required to evaluate the clinical outcome.76
Challenges
It is clear from existing preclinical and early-stage trials that DNA vaccination holds promise for breast cancer patients but that there are a number of challenges and developments required before this becomes an approved product. One of the first challenges lies in which TAAs should be selected for a DNA vaccine for breast cancer. With an increased emphasis on personalized medicine and the screening of biopsies, it is possible to create a patient- or tumor-specific DNA vaccine where multiple antigens are delivered. In addition, the delivery of fusion sequences of DNA coding for more than one antigen and adjuvants such as GM-CSF also increases the chances of incurring a therapeutic immune response.
Another challenge lies in improving the efficacy of the DNA vaccine. Although the “gold standard” mode of delivery is currently injection followed by electroporation, it is questionable whether this will gain regulatory approval, and perhaps more importantly, achieve an acceptable level of patient compliance. Given the number of exciting nonviral technologies available, it is only a matter of time before a relatively straightforward delivery system becomes a frontrunner to replace needle injection and electroporation. Such a delivery system should work with the body to overcome the extra- and intracellular barriers to deliver the DNA payload to the nucleus of the immune cells. Of noteworthy importance, however, is the fact that optimal formulations in vitro rarely correlate to the optimum immune response in vivo. A study by Endmann et al examined the ratios of MIDGE-Th1 (minimalistic immunogenically defined gene expression) DNA vaccine with the cationic lipid SAINT-18 in vitro and in vivo. The MIDGE vector resulted in optimal transfection in vitro at a DNA:lipid weight per weight ratio of 1:4.8, yet in vivo, there was no immune response using the same ratio. The highest immune response was found to be at a weight per weight ratio of 1:0.5 (MIDGE: SAINT-18).77 Although time consuming, once the correct formulation of delivery system is determined, the immune response can be greater than naked DNA.77 It is likely that such a system could take longer to gain regulatory approval but the resultant highly potent DNA vaccine should reduce the frequency of administrations and have a greater chance of being successful.
DNA vaccines are typically plasmids that retain nonessential information such as an origin of replication and an antibiotic resistance gene. In addition, they are grown in genetically modified bacteria, thereby increasing the number of quality control issues and manufacturing steps.78 In a DNA vaccine, the critical components are a promoter, antigen, and polyA sequence, and a study by Walters et al has demonstrated that a minimal closed double-stranded linear DNA construct can produce comparable humoral and cell-mediated immune response to conventional pDNA.79 Touchlight Genetics synthesize these “doggybone” DNAs in an enzymatic process which should render them highly desirable from a regulatory and clinical perspective. Such minimal DNA systems could become the future of approved human DNA vaccines.
Conclusion
Generation of an appropriate delivery vehicle is a critical obstacle that needs to be surmounted for a genetic-based therapy to be accepted and gain regulatory approval. Despite the progress that has been made in more than 40 years of research into using nucleic acids as drugs, there are still concerns over the vectors used to deliver these therapies. Immunological concerns that surround viral delivery, less potent delivery by nonviral methodologies, and concerns over the manufacture of recombinant vectors must all be addressed to placate regulatory bodies. The recent push for personalized medicine offers opportunities for gene therapy researchers, with targeting strategies that were highlighted here ripe for exploitation. Ever evolving bioinformatics tools are increasing our knowledge and understanding of the genomics of breast cancer, opening new avenues for exploration. Despite this though, the translation of impressive bench-generated data has not been as rapid as desired. The clinical gene therapies for breast cancer patients documented here (both viral and nonviral) were generally well-tolerated, with few serious side effects reported. It is hoped that continued generation of meaningful preclinical data and delivery vector improvement will lead to advancements of clinical testing of gene therapies for breast cancer patients.
Disclosure
The authors report no conflicts of interest in this work.
References
Official Journal of the European Union. L 159: Commission Directive 2003/63/EC of 25 June 2003 amending Directive 2001/83/EC of the European Parliament and of the Council on the Community code relating to medicinal products for human use. Available from: http://eur-lex.europa.eu/LexUriServ/LexUriServ.do?uri=OJ:L:2003:159:0046:0094:en:PDF. Accessed October 15, 2014. | |
Kan O, Griffiths L, Baban D, et al. Direct retroviral delivery of human cytochrome P450 2B6 for gene-directed enzyme prodrug therapy of cancer. Cancer Gene Ther. 2001;8(7):473–482. | |
Braybrooke JP, Slade A, Deplanque G, et al. Phase I study of MetXia-P450 gene therapy and oral cyclophosphamide for patients with advanced breast cancer or melanoma. Clin Cancer Res. 2005;11(4):1512–1520. | |
Gordon EM, Levy JP, Reed RA, et al. Targeting metastatic cancer from the inside: a new generation of targeted gene delivery vectors enables personalized cancer vaccination in situ. Int J Oncol. 2008;33(4):665–675. | |
Gordon EM, Hall FL. Noteworthy clinical case studies in cancer gene therapy: tumor-targeted Rexin-G advances as an efficacious anti-cancer agent. Int J Oncol. 2010;36(6):1341–1353. | |
Hartkopf AD, Fehm T, Wallwiener D, Lauer UM. Oncolytic virotherapy of breast cancer. Gynecol Oncol. 2011;123(1):164–171. | |
Takakuwa H, Goshima F, Nozawa N, et al. Oncolytic viral therapy using a spontaneously generated herpes simplex virus type 1 variant for disseminated peritoneal tumor in immunocompetent mice. Arch Virol. 2003;148(4):813–825. | |
Teshigahara O, Goshima F, Takao K, et al. Oncolytic viral therapy for breast cancer with herpes simplex virus type 1 mutant HF 10. J Surg Oncol. 2004;85(1):42–47. | |
Kimata H, Imai T, Kikumori T, et al. Pilot study of oncolytic viral therapy using mutant herpes simplex virus (HF10) against recurrent metastatic breast cancer. Ann Surg Oncol. 2006;13(8):1078–1084. | |
Sahin TT, Kasuya H, Nomura N, et al. Impact of novel oncolytic virus HF10 on cellular components of the tumor microenviroment in patients with recurrent breast cancer. Cancer Gene Ther. 2012;19(4):229–237. | |
Russell SJ, Federspiel MJ, Peng KW, et al. Remission of disseminated cancer after systemic oncolytic virotherapy. Mayo Clin Proc. 2014;89(7):926–933. | |
Hu JC, Coffin RS, Davis CJ, et al. A phase I study of OncoVEXGM-CSF, a second-generation oncolytic herpes simplex virus expressing granulocyte macrophage colony-stimulating factor. Clin Cancer Res. 2006;12(22):6737–6747. | |
Liu BL, Robinson M, Han ZQ, et al. ICP34.5 deleted herpes simplex virus with enhanced oncolytic, immune stimulating, and anti-tumour properties. Gene Ther. 2003;10(4):292–303. | |
Guffey MB, Parker JN, Luckett WS Jr, et al. Engineered herpes simplex virus expressing bacterial cytosine deaminase for experimental therapy of brain tumors. Cancer Gene Ther. 2007;14(1):45–56. | |
Papanastassiou V, Rampling R, Fraser M, et al. The potential for efficacy of the modified (ICP 34.5(-)) herpes simplex virus HSV1716 following intratumoural injection into human malignant glioma: a proof of principle study. Gene Ther. 2002;9(6):398–406. | |
MacKie RM, Stewart B, Brown SM. Intralesional injection of herpes simplex virus 1716 in metastatic melanoma. Lancet. 2001;357(9255):525–526. | |
Cody JJ, Scaturro P, Cantor AB, Yancey Gillespie G, Parker JN, Markert JM. Preclinical evaluation of oncolytic deltagamma(1)34.5 herpes simplex virus expressing interleukin-12 for therapy of breast cancer brain metastases. Int J Breast Cancer. 2012;2012:628697. | |
Colombo MP, Trinchieri G. Interleukin-12 in anti-tumor immunity and immunotherapy. Cytokine Growth Factor Rev. 2002;13(2):155–168. | |
Siegl G. Biology and pathogenicity of autonomous parvoviruses. In: Berns KI, editor. The Parvoviruses. 1st ed. New York: Springer US; 1984:264–297. | |
Cornelis JJ, Chen YQ, Spruyt N, et al. Susceptibility of human cells to killing by the parvoviruses H-1 and minute virus of mice correlates with viral transcription. J Virol. 1990;64(6):2537–2544. | |
Sieben M, Schafer P, Dinsart C, Galle PR, Moehler M. Activation of the human immune system via toll-like receptors by the oncolytic parvovirus H-1. Int J Cancer. 2013;132(11):2548–2556. | |
Muharram G, Le Rhun E, Loison I, et al. Parvovirus H-1 induces cytopathic effects in breast carcinoma-derived cultures. Breast Cancer Res Treat. 2010;121(1):23–33. | |
Angelova AL, Aprahamian M, Grekova SP, et al. Improvement of gemcitabine-based therapy of pancreatic carcinoma by means of oncolytic parvovirus H-1PV. Clin Cancer Res. 2009;15(2):511–519. | |
Enderlin M, Kleinmann EV, Struyf S, et al. TNF-alpha and the IFN-gamma-inducible protein 10 (IP-10/CXCL-10) delivered by parvoviral vectors act in synergy to induce antitumor effects in mouse glioblastoma. Cancer Gene Ther. 2009;16(2):149–160. | |
Norman KL, Coffey MC, Hirasawa K, et al. Reovirus oncolysis of human breast cancer. Hum Gene Ther. 2002;13(5):641–652. | |
Gollamudi R, Ghalib MH, Desai KK, et al. Intravenous administration of Reolysin, a live replication competent RNA virus is safe in patients with advanced solid tumors. Invest New Drugs. 2010;28(5):641–649. | |
Vidal L, Pandha HS, Yap TA, et al. A phase I study of intravenous oncolytic reovirus type 3 Dearing in patients with advanced cancer. Clin Cancer Res. 2008;14(21):7127–7137. | |
Harrington KJ, Karapanagiotou EM, Roulstone V, et al. Two-stage phase I dose-escalation study of intratumoral reovirus type 3 dearing and palliative radiotherapy in patients with advanced cancers. Clin Cancer Res. 2010;16(11):3067–3077. | |
Nanni P, Gatta V, Menotti L, et al. Preclinical therapy of disseminated HER-2(+) ovarian and breast carcinomas with a HER-2-retargeted oncolytic herpesvirus. PLoS Pathog. 2013;9(1):e1003155. | |
Cody JJ, Markert JM, Hurst DR. Histone deacetylase inhibitors improve the replication of oncolytic herpes simplex virus in breast cancer cells. PLoS One. 2014;9(3):e92919. | |
Walther W, Siegel R, Kobelt D, et al. Novel jet-injection technology for nonviral intratumoral gene transfer in patients with melanoma and breast cancer. Clin Cancer Res. 2008;14(22):7545–7553. | |
Shibata MA, Ito Y, Morimoto J, Kusakabe K, Yoshinaka R, Otsuki Y. In vivo electrogene transfer of interleukin-12 inhibits tumor growth and lymph node and lung metastases in mouse mammary carcinomas. J Gene Med. 2006;8(3):335–352. | |
Miyata K, Nishiyama N, Kataoka K. Rational design of smart supramolecular assemblies for gene delivery: chemical challenges in the creation of artificial viruses. Chem Soc Rev. 2012;41(7):2562–2574. | |
Yoo GH, Hung MC, Lopez-Berestein G, et al. Phase I trial of intratumoral liposome E1A gene therapy in patients with recurrent breast and head and neck cancer. Clin Cancer Res. 2001;7(5):1237–1245. | |
Hortobagyi GN, Ueno NT, Xia W, et al. Cationic liposome-mediated E1A gene transfer to human breast and ovarian cancer cells and its biologic effects: a phase I clinical trial. J Clin Oncol. 2001;19(14):3422–3433. | |
Gribben JG, Ryan DP, Boyajian R, et al. Unexpected association between induction of immunity to the universal tumor antigen CYP1B1 and response to next therapy. Clin Cancer Res. 2005;11(12):4430–4436. | |
McCrudden C, McCarthy H. Cancer gene therapy – key biological concepts in the design of multifunctional non-viral delivery systems. In: Molina FM, editor. Cancer Gene Therapy. 1st ed. Rijeka, Croatia: InTech; 2013:213–235. | |
Robson T, Hirst DG. Transcriptional targeting in cancer gene therapy. J Biomed Biotechnol. 2003;2003(2):110–137. | |
Bianchini G, Gianni L. The immune system and response to HER2-targeted treatment in breast cancer. Lancet Oncol. 2014;15(2):e58–e68. | |
McCarthy HO, Coulter JA, Worthington J, Robson T, Hirst DG. Human osteocalcin: a strong promoter for nitric oxide synthase gene therapy, with specificity for hormone refractory prostate cancer. J Gene Med. 2007;9(6):511–520. | |
Coulter JA, Page NL, Worthington J, Robson T, Hirst DG, McCarthy HO. Transcriptional regulation of inducible nitric oxide synthase gene therapy: targeting early stage and advanced prostate cancer. J Gene Med. 2010;12(9):755–765. | |
Pandha HS, Martin LA, Rigg A, et al. Genetic prodrug activation therapy for breast cancer: a phase I clinical trial of erbB-2-directed suicide gene expression. J Clin Oncol. 1999;17(7):2180–2189. | |
Garg H, Salcedo R, Trinchieri G, Blumenthal R. Improved nonviral cancer suicide gene therapy using survivin promoter-driven mutant Bax. Cancer Gene Ther. 2010;17(3):155–163. | |
Sadeqzadeh E, Rahbarizadeh F, Ahmadvand D, Rasaee MJ, Parhamifar L, Moghimi SM. Combined MUC1-specific nanobody-tagged PEG-polyethylenimine polyplex targeting and transcriptional targeting of tBid transgene for directed killing of MUC1 over-expressing tumour cells. J Control Release. 2011;156(1):85–91. | |
Yun HJ, Cho YH, Moon Y, et al. Transcriptional targeting of gene expression in breast cancer by the promoters of protein regulator of cytokinesis 1 and ribonuclease reductase 2. Exp Mol Med. 2008;40(3):345–353. | |
Yan C, Wen-Chao L, Hong-Yan Q, Rui Z, Wei-Lin J, Hua H. A new targeting approach for breast cancer gene therapy using the human fatty acid synthase promoter. Acta Oncol. 2007;46(6):773–781. | |
Anderson LM, Krotz S, Weitzman SA, Thimmapaya B. Breast cancer-specific expression of the Candida albicans cytosine deaminase gene using a transcriptional targeting approach. Cancer Gene Ther. 2000;7(6):845–852. | |
Rao DD, Vorhies JS, Senzer N, Nemunaitis J. siRNA vs shRNA: similarities and differences. Adv Drug Deliv Rev. 2009;61(9):746–759. | |
Yang F, Huang W, Li Y, et al. Anti-tumor effects in mice induced by survivin-targeted siRNA delivered through polysaccharide nanoparticles. Biomaterials. 2013;34(22):5689–5699. | |
Gao Y, Chen L, Zhang Z, Chen Y, Li Y. Reversal of multidrug resistance by reduction-sensitive linear cationic click polymer/iMDR1-pDNA complex nanoparticles. Biomaterials. 2011;32(6):1738–1747. | |
Wang Y, Gao S, Ye WH, Yoon HS, Yang YY. Co-delivery of drugs and DNA from cationic core-shell nanoparticles self-assembled from a biodegradable copolymer. Nat Mater. 2006;5(10):791–796. | |
Janat-Amsbury MM, Yockman JW, Lee M, et al. Combination of local, nonviral IL12 gene therapy and systemic paclitaxel treatment in a metastatic breast cancer model. Mol Ther. 2004;9(6):829–836. | |
Okamoto A, Asai T, Kato H, et al. Antibody-modified lipid nanoparticles for selective delivery of siRNA to tumors expressing membrane-anchored form of HB-EGF. Biochem Biophys Res Commun. 2014;449(4):460–465. | |
Ding Y, Wang Y, Zhou J, et al. Direct cytosolic siRNA delivery by reconstituted high density lipoprotein for target-specific therapy of tumor angiogenesis. Biomaterials. 2014;35(25):7214–7227. | |
Feng Q, Yu MZ, Wang JC, et al. Synergistic inhibition of breast cancer by co-delivery of VEGF siRNA and paclitaxel via vapreotide-modified core-shell nanoparticles. Biomaterials. 2014;35(18):5028–5038. | |
Rejeeth C, Kannan S. P53 gene therapy of human breast carcinoma: using a transferrin-modified silica nanoparticles. Breast Cancer. Epub May 4, 2014. | |
Mao CQ, Du JZ, Sun TM, et al. A biodegradable amphiphilic and cationic triblock copolymer for the delivery of siRNA targeting the acid ceramidase gene for cancer therapy. Biomaterials. 2011;32(11):3124–3133. | |
Medina-Kauwe LK, Maguire M, Kasahara N, Kedes L. Nonviral gene delivery to human breast cancer cells by targeted Ad5 penton proteins. Gene Ther. 2001;8(23):1753–1761. | |
Mangipudi SS, Canine BF, Wang Y, Hatefi A. Development of a genetically engineered biomimetic vector for targeted gene transfer to breast cancer cells. Mol Pharm. 2009;6(4):1100–1109. | |
McCarthy HO, Zholobenko AV, Wang Y, et al. Evaluation of a multi-functional nanocarrier for targeted breast cancer iNOS gene therapy. Int J Pharm. 2011;405(1–2):196–202. | |
Jeyarajan S, Xavier J, Rao NM, Gopal V. Plasmid DNA delivery into MDA-MB-453 cells mediated by recombinant Her-NLS fusion protein. Int J Nanomedicine. 2010;5:725–733. | |
Soltani F, Sankian M, Hatefi A, Ramezani M. Development of a novel histone H1-based recombinant fusion peptide for targeted non-viral gene delivery. Int J Pharm. 2013;441(1–2):307–315. | |
Dou S, Yao YD, Yang XZ, et al. Anti-Her2 single-chain antibody mediated DNMTs-siRNA delivery for targeted breast cancer therapy. J Control Release. 2012;161(3):875–883. | |
Govindarajan S, Sivakumar J, Garimidi P, et al. Targeting human epidermal growth factor receptor 2 by a cell-penetrating peptide-affibody bioconjugate. Biomaterials. 2012;33(8):2570–2582. | |
Tang DC, DeVit M, Johnston SA. Genetic immunization is a simple method for eliciting an immune response. Nature. 1992;356(6365):152–154. | |
Ulmer JB, Fu TM, Deck RR, et al. Protective CD4+ and CD8+ T cells against influenza virus induced by vaccination with nucleoprotein DNA. J Virol. 1998;72(7):5648–5653. | |
Grosenbaugh DA, Leard AT, Bergman PJ, et al. Safety and efficacy of a xenogeneic DNA vaccine encoding for human tyrosinase as adjunctive treatment for oral malignant melanoma in dogs following surgical excision of the primary tumor. Am J Vet Res. 2011;72(12):1631–1638. | |
Slamon D, Eiermann W, Robert N, et al. Adjuvant trastuzumab in HER2-positive breast cancer. N Engl J Med. 2011;365(14):1273–1283. | |
Chung MA, Luo Y, O’Donnell M, et al. Development and preclinical evaluation of a Bacillus Calmette-Guérin-MUC1-based novel breast cancer vaccine. Cancer Res. 2003;63(6):1280–1287. | |
Hodge JW. Carcinoembryonic antigen as a target for cancer vaccines. Cancer Immunol Immunother. 1996;43(3):127–134. | |
Watson MA, Dintzis S, Darrow CM, et al. Mammaglobin expression in primary, metastatic, and occult breast cancer. Cancer Res. 1999;59(13):3028–3031. | |
Bednarek AK, Sahin A, Brenner AJ, Johnston DA, Aldaz CM. Analysis of telomerase activity levels in breast cancer: positive detection at the in situ breast carcinoma stage. Clin Cancer Res. 1997;3(1):11–16. | |
De Giovanni C, Nicoletti G, Quaglino E, et al. Vaccines against human HER2 prevent mammary carcinoma in mice transgenic for human HER2. Breast Cancer Res. 2014;16(1):R10. | |
Norell H, Poschke I, Charo J, et al. Vaccination with a plasmid DNA encoding HER-2/neu together with low doses of GM-CSF and IL-2 in patients with metastatic breast carcinoma: a pilot clinical trial. J Transl Med. 2010;8:53. | |
Narayanan K, Jaramillo A, Benshoff ND, et al. Response of established human breast tumors to vaccination with mammaglobin-A cDNA. J Natl Cancer Inst. 2004;96(18):1388–1396. | |
Tiriveedhi V, Fleming TP, Goedegebuure PS, et al. Mammaglobin-A cDNA vaccination of breast cancer patients induces antigen-specific cytotoxic CD4+ICOShi T cells. Breast Cancer Res Treat. 2013;138(1):109–118. | |
Endmann A, Oswald D, Riede O, et al. Combination of MIDGE-Th1 DNA vaccines with the cationic lipid SAINT-18: studies on formulation, biodistribution and vector clearance. Vaccine. 2014;32(27):3460–3467. | |
Mairhofer J, Cserjan-Puschmann M, Striedner G, Nobauer K, Razzazi-Fazeli E, Grabherr R. Marker-free plasmids for gene therapeutic applications – lack of antibiotic resistance gene substantially improves the manufacturing process. J Biotechnol. 2010;146(3):130–137. | |
Walters AA, Kinnear E, Shattock RJ, et al. Comparative analysis of enzymatically produced novel linear DNA constructs with plasmids for use as DNA vaccines. Gene Ther. 2014;21(7):645–652. | |
Canine BF, Wang Y, Hatefi A. Biosynthesis and characterization of a novel genetically engineered polymer for targeted gene transfer to cancer cells. J Control Release. 2009;138(3):188–196. |
 © 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.