Back to Journals » Cancer Management and Research » Volume 13
Current Progress in Investigating Mature T- and NK-Cell Lymphoma Gene Aberrations by Next-Generation Sequencing (NGS)
Authors Zhu L, Xie S ![]() , Yang C, Hua N, Wu Y, Wang L, Ni W, Tong X, Fei M, Wang S
, Yang C, Hua N, Wu Y, Wang L, Ni W, Tong X, Fei M, Wang S ![]()
Received 4 January 2021
Accepted for publication 14 June 2021
Published 2 July 2021 Volume 2021:13 Pages 5275—5286
DOI https://doi.org/10.2147/CMAR.S299505
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Xueqiong Zhu
Lifen Zhu,1,* Shufang Xie,1,2,* Chen Yang,1,3,* Nanni Hua,1,2 Yi Wu,4 Lei Wang,1 Wanmao Ni,1 Xiangmin Tong,1 Min Fei,5 Shibing Wang1
1Molecular diagnosis laboratory, Zhejiang Provincial People’s Hospital, Affiliated People’s Hospital, Hangzhou Medical College, Hangzhou, Zhejiang, People’s Republic of China; 2The Second Clinical Medical College, Zhejiang Chinese Medical University, Hangzhou, Zhejiang, People’s Republic of China; 3Department of Clinical Medicine, Qingdao University, Qingdao, Shandong, People’s Republic of China; 4Phase I clinical research center, Zhejiang Provincial People’s Hospital, Affiliated People’s Hospital, Hangzhou Medical College, Hangzhou, Zhejiang, People’s Republic of China; 5Center of Health Management, Zhejiang Provincial People’s Hospital, Affiliated People’s Hospital, Hangzhou Medical College, Hangzhou, Zhejiang, People’s Republic of China
*These authors contributed equally to this work
Correspondence: Min Fei
Center of Health Management, Zhejiang Provincial People’s Hospital, Affiliated People’s Hospital, Hangzhou Medical College, Hangzhou, Zhejiang, People’s Republic of China
Email [email protected]
Shibing Wang
Molecular diagnosis laboratory, Zhejiang Provincial People’s Hospital, Affiliated People’s Hospital, Hangzhou Medical College, Hangzhou, Zhejiang, People’s Republic of China
Email [email protected]
Abstract: Despite efforts to abrogate the severe threat to life posed by the profound malignancy of mature natural killer/T-cell lymphoma (NKTCL), therapeutic advances still require further investigation of its inherent regulatory biochemical processes. Next-generation sequencing (NGS) is an increasingly developing gene detection technique, which has been widely used in lymphoma genetic research in recent years. Targeted therapy based on the above studies has also generated a series of advances, making genetic mutation a new research hotspot in lymphoma. Advances in NKTCL-related gene mutations are reviewed in this paper.
Keywords: mature T-and NK-cell lymphoma, gene mutation, next generation sequencing
Introduction
Since 1977, Sanger’s invention of end termination sequencing,1 also known as DNA sequencing technology, has been used. As one of the most important analytical methods of molecular biology, so far, it has not only provided important data for basic biological research such as genetic information disclosure and gene expression regulation, but also played an important role in applied research such as gene-based diagnosis and therapy, for example, several studies have used NGS to analyze the genetic profile of lung cancer.2–4 Large clone classification by mapping of low resolution is known as first-generation sequencing technology, a term used to define the above sequencing approach which uses a dideoxy chain termination technique. Sanger sequencing cannot entirely satisfy research requirements due to the progressive advancement of medical technology. Therefore, next-generation sequencing (NGS) came into being in 2005.5 NGS relies on standard sequencing approaches from Sanger techniques, but there have been innovative improvements in other ways allowing millions of parallel sequencing reactions to be conducted6 (Figure 1). NGS can sequence smaller subclones at a high resolution and can identify genetic differences as the sequence of each base can be identified and numerous target loci (several genes) are detectable for simultaneous analysis.7 In addition, NGS is a high-throughput sequencing technology with higher flux and sensitivity,8 faster speed and lower cost9,10 (Figure 2). So far, NGS has three major sequencing platforms, including Solexa Genome Analyzer’s Illumina system, Applied Biosystems’ SOLID system and the 454 FLX system from Roche.11 Clinical research has evolved drastically due to progress in NGS technology.12
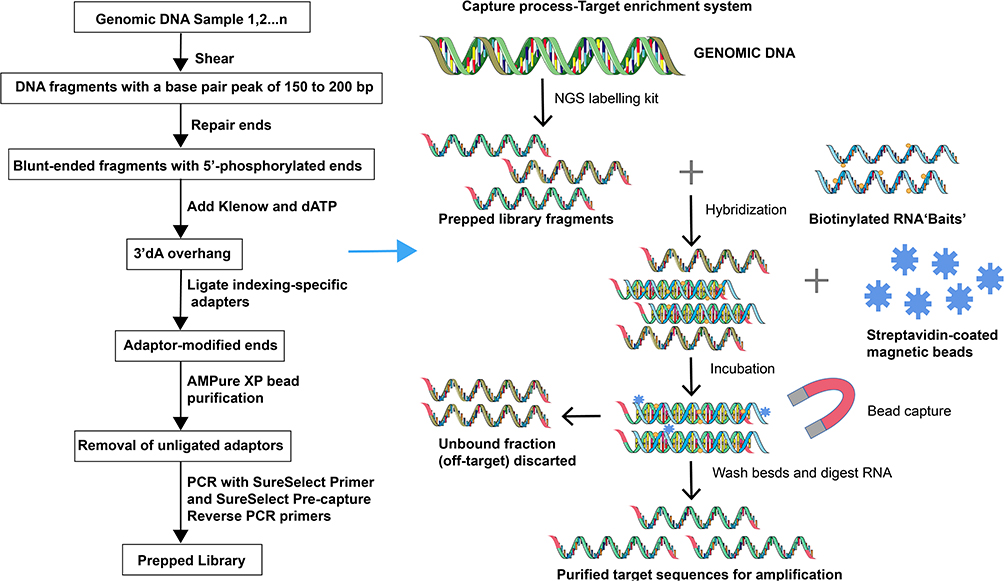 |
Figure 1 The construction of second-generation sequencing libraries and sequencing process. NGS library is prepared by fragmenting a gDNA sample and ligating specialized adapters to both fragment ends. Sequencing reagents, including fluorescently labeled nucleotides, are added and the first based is incorporated, The flow cell is imaged and the emission from each cluster is recorded. The emission wavelength and intensity are used to identify the base. This cycle is repeated “n” times to create a read length of “n” base. |
 |
Figure 2 Information analysis process. The library was sequenced and a large number of short fragments were obtained reads, Through bioinformatics analysis software, the reads was compared to the reference genome to obtain the location information of each read on the reference genome. Comprehensive reads comparison information to analyze the variation. Clinical NGS data analysis aims to detect, annotate, and provide a clear, professional and reliable report interpretation and ultimately as an important reference for clinical diagnosis and treatment. |
Lymphoma is the most common malignant tumor of the blood system in the world, which occurs in lymph nodes and other organs.13 Lymphoma occurs as a result of genetic changes within cells that predispose them to further genetic changes.14 Over time, other acquired abnormalities promote cloning, which delivers growth and/or survival advantages over other cells,15 and eventually develops into clinical lymphoma. Lymphomas were classified into non-Hodgkin’s lymphoma and Hodgkin’s lymphoma according to morphology and immunohistochemistry.16 Non-Hodgkin’s lymphoma is classified into T or B-cell lymphoma with respect to cell origin. Hodgkin’s lymphoma is divided into nodular lymphocytes and classic Hodgkin’s lymphoma, the former being the dominant type of the disease.17 NK/T extranodal cell lymphoma is a major NK cell lymphoma with a ratio of 10.4% in mature T cell lymphoma and NK cell lymphoma.18 Similar to other malignant proliferative diseases, lymphoma has genetic instability and chromosomal abnormalities, which together directly cause the development of malignancy.
NGS has been applied to accurate diagnosis of gene mutation, analysis of pathogenesis, prognosis monitoring and so on.19 Recent developments in lymphoma genome sequencing have led to a preliminary appreciation of somatic mutation complexity in these tumors as well as a number of findings of significant functional and clinical significance, including studies on the prognostic impact of some genes on lymphoma or potential as drug targets.20 Compared with cases of B cell lymphoma, there has been slower use of NGS for molecular studies in mature NK/T-cell lymphoma.21 This article reviews the application of NGS in scientific analysis and clinical detection of mature T- and NK-cell lymphoma (Table 1).
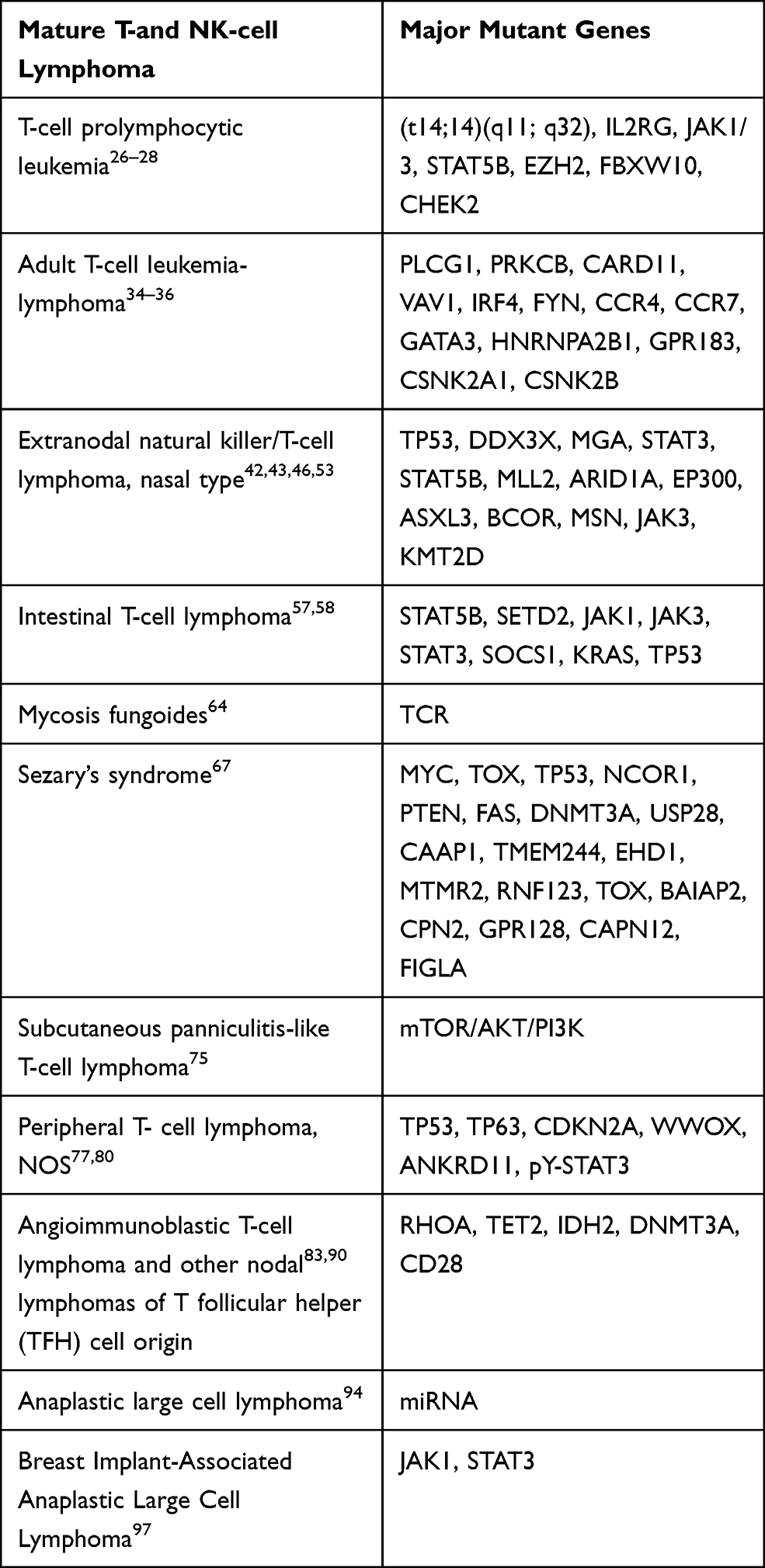 |
Table 1 Common Related Gene Mutations in Mature T- and NK-Cell Lymphoma |
T-Cell Prolymphocytic Leukemia
Prolymphocytic leukemia (PLL) is a highly malignant mature lymphoma,22 and available treatment options in this aggressive disease are largely inefficient and patient outcomes are highly dissatisfactory.23 Catovsky et al24 first reported T-cell prolymphocytic leukemia (T-PLL), a subtype of PLL in 1977. T-PLL accounted for about 80% of PLL and the average diagnosis age was between sixty-five and seventy years, mostly in men. Almost all T-PLL cases have cloning-specific T-cell receptor (TCR) gene rearrangement.25 Recombination of genes coding for receptors of T-cells suggests clonal T-cell proliferation. Cytogenetic analysis confirmed that T-PLL patients had multiple genetic changes. The common chromosomal abnormalities involved chromosome 14 (90%), including (tX;14)(q28; q11), inv (14) and (t14;14)(q11; q32).26 TCL1B and TCL1A involving MTCP1 and chromosome 14 in chromosome 14 transgenic animal models have confirmed that TCL1 and MTCP1 are carcinogenic, co-activating molecules of Akt kinases that promote cell proliferation and survival. Then chromosome 8 was reported to have idic (8p11), (t8;8) and trisomy 8q aberrations. Other reproducible cytogenetic abnormalities were −11q23,27 with −22q, −13q, 6q, −9p, 12p, 17p with 22q and 6p with −12p; visible in approximately 50% of T-PLL, and considered relevant to their occurrence and development. The application of second-generation sequencing technology further confirms that T-PLL carries of a variety of genetic molecular abnormalities such as IL2RG, JAK1/3, STAT5B, EZH2, FBXW10 and CHEK2 mutations28 which contribute to T-PLL pathology through several different pathways29 including affecting the repair of DNA, epigenetic transcriptional regulation and proteasome degradation.30 As more mutant genes are discovered, we can diagnose and treat T juvenile lymphoblastic leukemia more accurately. With the continuous development of sequencing technology, NGS technology provides a detailed overview of various genomic damages/lesions related to the pathogenesis of T-PLL.
Adult T Cell Lymphoma/Leukemia
While it is known to correlate with T cell-associated type 1 leukemia virus (HTLV-1) infection, the peripheral T cell tumor known as Adult T cell lymphoma/leukemia (ATL) still has no identified genetic causality.31–33 A comprehensive molecular analysis of the transcriptome and guided re-sequencing, exome, genome, methylation and copy number based on arrays was described by Keisuke Kataoka et al. The modifications found substantially correlate with the groups interacting with HTLV-1Tax and are enriched in signaling downstream of TCR-NF-kB, T cell mechanisms including transport and immune surveillance. Other notable roles include fusing ICOS-CD28 and CTLA4 -CD28genes as well as triggering aberrations in, IRF4, PRKCB, CKR7, FYN, VAV1, CCR4, PLCG1 and CARD11. It was additionally discovered that aberrations were present in CSnK2B, GPR183, CSNK1A1, HNRNPA2B1 and GATA3CSNK2A1 genes with repeated mutations and deletions inside CARD11 and IKZF2 genes. The discoveries give a novel understanding into the fundamental issues in ATL detection and therapy, yet additionally direct improvement of new strategies for the determination and treatment of this obstinate tumour.34–36
Extranodal NK/T Cell Lymphoma, Nasal Type
With over 90% of patients testing positive for the Epstein-Barr virus (EB) in its tumors.37 The virus has a clear association with nasal extranodal natural lymphatic tumor killer/T-cell (NKTCL), which is an invasive, but uncommon type of non-Hodgkin lymphoma (NHL) with a poor prognosis and is typified by cytotoxic cells.38 Often the upper digestive tract (eg paranasal sinuses, nasal cavity and nasopharynx) is affected by external tissue including the gastrointestinal tract, soft tissue and skin.39 It is the most common mature T-cell/NK type in Asia.40 But it is rare in the West. Nine percent of the total number of NHL patients in China was extranodal NK/T lymphomas.41
The conventional Sanger sequencing has identified numerous somatic gene alterations in NKTCL.42 Some epigenetic disorders and DDX3X mutations rarely found by traditional sequencing techniques have been revealed by NGS recently.43 Using full exome sequencing and validation in a large confirmatory cohort of 80 individuals via selective sequencing, Lu Jiang et al44 established somatic gene aberrations in twenty-five NKTCL patients. Recurring mutations occur primarily in JAK-STAT signaling molecules (STAT5B and STAT3), tumor suppressing genes (MGA and TP53), DDX3X RNA helicase genes (2/5 volunteers) and in epigenetic influencers (ARID1A, ASXL3 EP300 and MLL2). Poor clinical prognosis is associated with such DDX3X mutations. These discoveries help to understand NKTCL disease mechanisms.45
BCOR are typically the most common (16.9%) mutating genes, superior to DDX3X and TP53 (13.6% and 14.70%, respectively) as revealed in the report by Zhang et al.42 The study of such genes offers fresh perspectives on disease development and contributes to therapeutic goals or specific biomarker development. Montes-Mojarro et al investigated mutation profiles and various strains of EBV in seventy-one cases of ENKTL from South America and compared them with Asian populations. NGS mutation analysis covered common gene mutations previously identified in ENKTL. The results showed that STAT3 was the most common mutant gene (23%), followed by MSN (14%), BCOR (13%), DDX3X (8%), JAK3 (3%), TP53 (8%), STAT5B (1%) and MGA (4%). The DDX3X, BCOR and STAT3 mutations rarely appeared together, indicating divergent molecular signaling cascades were involved in the development of ENKTL. TP53, MGA and MSN mutations occur in conjunction with other mutations. Seventy-five percent of cases had type A EBV, with thirty base pairs. Overall, this ENKTL study of mutations in South Americans has shown that common gene mutations (twenty-five percent) induce activation of JAK-STAT signaling, most of which are STAT3 mutations. TP53, DDX3X and BCOR mutations were also identified, albeit at varying rates relative to similar populations in Asia. The findings also show that such mutations are not prognostic.46,47
Using clear sampling and immunohistochemistry, Sim et al examined JAK3 mutations and STAT3 genetic modifications. JAK3 mutations in the pseudokinase domain of 71 NTCL patients were present in five cases (7.0%): one JAK-3, one JAK-3[G589D], two JAK-3 [H583Y] and two JAK-3[A573V]. Tofatinib (tofactinib), the inhibitor of JAK3, blocks (JAK3 [G589D]) or JAK3 [H583Y] mutations which transduce Ba/F3 cell proliferation independently of IL-3.48,49 Full exome sequence and somatic activity mutations of JAK3 (A573V and A572V) were identified by Ghee Chong Koo et al in two of four patients receiving NKTCL. Further confirmation by Sanger sequence and high-resolution melting (HRM) examination detected the occurrence of JAK3 mutations in 61 more patients. Out of 65 patients, 23 (35.4%) had mutations of JAK3. Examination of the roles played by JAK3 mutations adds evidence that constant JAK/STAT stimulation in the absence of cytokines enhances cell growth. Furthermore, treatment of both wild-type and mutant NKTCL cell lines with novel pan-JAK inhibitor CP-690550 resulted in more apoptosis, less cell viability and less dose-dependent phosphorylation. Thus, an effective treatment for NKTCL patients could potentially target the dysregulated JAK/STAT pathways.50,51
Five NKTCL somatic mutations in tissue samples were documented by Dobachi et al Analysis based on sequencing found 21 genes with 25 mutations. The most common (4 out of 5) were histone-related modifications of KMT2D genes. These findings align with recent NGS studies showing that KMT2D genes are new NKTCL drivers.52 Chromatin reshaping genes TP53 and ARID1A, which have also been reported in recent studies using NGS, also showed mutations. Further mutations were detected in 18 new genetic candidates whose molecular activity may be linked to the progression of cancer and suggests that these genes may lead to numerous oncogenic instances. These could be used as possible NKTCL biomarkers in the coming years.53 Luyuan et al detected the mutation of 9 target genes in 29 ENKTL pathological specimens using a NGS technique. They analyzed the prognosis of patients with the mutated genes and found that 4 genes were closely related to the progression and prognosis of the disease, including KMT2D (31%), STST3 (24.1%), ARIDIA (34%) and TP53 (24%). The study suggests that high-frequency mutations of KMT2D genes in ENKTL, are associated with patient outcomes and may contribute significantly to the development of ENKTL as genes that normally suppress tumors. STAT3 mutations contribute to ENKTL tumor cell invasion and proliferation. TP53 and ARID1A mutations may be associated with ENKTL pathogenesis.
More and more studies use NGS technology to detect the target gene mutation of Extranodal NK/T-cell Lymphoma, and analyze its relationship with disease prognosis and clinical characteristics, so as to provide the basis for the pathogenesis, clinical diagnosis and targeted therapy of Extranodal NK/T-cell Lymphoma.
T-Cell Lymphoma in the Intestines
Lymphoma is a systemic disease in which a specific type of malignancy termed extranodal lymphoma (ENL) occurs in lymphoid organs or non-lymphatic aggregated organs outside lymph nodes.54 ENL has a high incidence and about 40%-50% of NHL occur outside the node. The most frequent site is the digestive tract.55 Among cancers of the digestive tract, intestinal T-cell lymphoma (ITCL) is quite uncommon.56 The initial study of the disease based on full-exome sequencing (WES) was conducted by Nairismagi et al, which demonstrated that STAT5B activation mutations were present in TCRβ and TCRαβ-derived EITL tumors using the most comprehensive set of the literature published to date. This study provides multipronged proof of highly activated G-protein coupling (GPCR) and JAK-STAT signaling pathways in EITL, most of which points to the ability to effectively reduce primary EITL cell viability using selective treatment.57 EATL genetic profiles have been identified by Moffitt et al through analyzing the whole-exome sequence of 69 EATL tumors. In EATL (32% of cases), SETD2 is the most frequent silencing gene. STAT5B, SOCS1, STAT3, JAK3 and JAK 1 are commonly mutated genes, all playing a role in the JAKSTAT pathway. The study also found TERT, TP53 and KRAS mutations. Type I and Type II (In the latest classification criteria, EATL type 2 has been classified as monomorphic epitheliotropic intestinal T-cell lymphoma) of EATL have significant genetic overlaps, suggesting a common pathogenesis mechanism. Final data provide the broadest genetic details concerning this uncommon yet deadly disease, and may guide future categorization initiatives.58
Lymphoma of Cutaneous T-Cells
Cutaneous T-cell lymphoma (CTCL), which develops primarily on the skin and is not normally subcutaneous, is a category of ectopic lymphoproliferative diseases.59 The most common form, comprising 55±5% of all CTCL is mycosis fungoides (MF). The leukemia type termed Sezary syndrome (SS) is commonly associated with erythrodermic lesions as well as peripheral blood and lymph node involvement. Most CTCL is therefore SS or MF.60 Whole genome sequencing for both SS and MF has shown that genes for epigenetic regulators, cell survival and fate, Th2 differentiation, cell cycle regulations, homologous recombination and TCR/NFκB had somatic mutations. A newly discovered primary skin lymphoma subtype, which is a primary T-cell lymphoma is called subcutaneous lipomatous T-cell lymphoma. Current profiles of SS and MF gene expression have been used to generate diagnostic approaches, which can consistently classify their genes.61
Mycosis Fungoides
MF is mostly skin-limited and rarely progresses into leukemia.62 It is one of the main types of nodular T-cell lymphoma and the most common skin T-cell lymphoma, which is believed to be caused by mature skin T-cell carcinogenesis.63 It commences with red plaques that look like scales containing tumorous T-cells, which may expand into tumors and eventually spread. Advanced MF is a prototype disease with the main features of peripheral T-cell lymphoma: chronic, recurrent growth, low proliferation, chemoresistance, and approximately 50% mortality over 5 years.
A detailed analysis using next-generation sequencing (NGS) was conducted by Hamrouni et al and found a large amount of clonal heterogeneity associated with TCR in MF samples. It also demonstrated that lymphoma cells with the same TCR gene sequence may have different TCR and DNA sequencing. The deletion of absolute TCR-α, -β, -γ monoclones was further confirmed by TCR amplification and sequencing from microdissected lymphoma cells. The study also found that, despite a lack of leukemic blood involvement, TCR rearrangements of lymphoma patients’ cells were characterized in the peripheral blood, but circulating TCRγclones did not always represent major skin clones.64 Sufficool et al determined clonality by a NGS-based method in which polymerase chain reaction was used to multiply the TCR-g variable regions and the identity of the rearranged variable and junction regions were determined by sequencing PCR products. Of the 35 MF cases tested, 29 (85%) patients showed clonal T-cell rearrangements through NGS, while the standard CE test gave a result of 15 (44%). Three patients with MF were followed up and the results showed the same clone TCR sequence in subsequent skin biopsy samples.65
Sezary’s Syndrome
SS is a type of T-cell lymphoma of the skin with grievous leukemia. It is distinguished by the presence of abnormal cells (CD45RO+CD4+) throughout the skin, blood and lymph nodes, bearing a phenotype of central memory T-cells (TCM), and serious lymphedema, pruritus, and erythroderma.66 SS is an extremely uncommon condition (occurrence: 0.1/100,000) and causes around 3% of CTCL, but its prevalence is rising. NGS and whole-genome analysis enabled KatarzynaI ż ykowska et al, to research the variation in number of copies of nine plugs and the impact of SS rearrangements on gene expression. Repetitive changes to the number of copies were found in 10q (PTEN, FAS), 8q (MIC, TOX), 9p (CAAP1), 2p (DNMT3A), 11q (USP28) and 17p (TP53, NCOR1), but recurrent modifications were not established. However, the expression of the five genes (TOX, RNF123, MTMR2, TMEM244 and EHD1) was modified in all patients. Fifteen modifications observed in SeAx and SS patients resulted in a new expression of fusion transcripts, of which nine were in the box (MAP4K3-FIGLA, DCP1A-CCL27, MBNL1-KIAA2018, TMEM66-BAIAP2, PTPRC-CPN2, EHD1-CAPN12, MYB-MBNL1, TFG-GPR128 and MBD4-PTPRC). However, 5 triggers of normal T cell ectopic gene expression were not raised (FIGLA, GPR128, CPN2, BAIAP2 and CAPN12). In addition to the results of this study revealing how complicated the cell genome of Sezary cancer is, they also display an unprecedented amount of new genetic modifications leading to ectopic genes expression and transcript fusion.67
A collection of pairs of normal tumor samples was compiled by Ana Carolina da Silva Almeida et al. Seventeen patients with CTCL and twenty-five patients with SS received full exon sequencing. The tests revealed a unique pattern of variation in the number of somatic copies of SS, including high-prevailing chromosomal deletions that included the following tumor inhibitors: CDKN1B, RB1, DNMT3A, TP53, and PTEN. T-cell receptor stimulation activity is regulated by the transmission of signals of somatic mutations like PRKG1, BRAF, mutated, CARD11, and MAPK1 mutations that propel the development of NFAT, NF kappa B, and MAPK. These were analyzed by a study determining mutations in core genes involved in epigenetic regulation (BRD9, MLL3, CHD3, SMARCA4, MLL2, CREBBP and TET2). Overall, new knowledge about mutational genetic elements of Sezary syndrome is provided by this study.68
The most commonly observed genetic abnormality in 58% of the cases using exome and RNA sequencing assays was found by Prasad et al to be mutation and/or elimination of the TP53 gene in the 17p chromosomal arm. Nevertheless, mutations affecting CARD11, GLI3, STAT5B and PLCG1 were found in a single individual. However, the study reported changes in the number of copies of several fusion and novel genes or single point mutations and forecasted biological associations. This work highlights the essence of genetic variation and the origins of human genetic variance. From cell to cell, Sezary’s cells will incorporate changes in the genes involved in T-cell, JAK and NF-κB69–71 signaling as well as the reaction to DNA disruption, chromatin restructuring, apoptosis regulation, and transcriptional route activation. The clinical importance of these potential targets should be tested by functional analyses. A recent study observed a 54/participant median cellular point mutation quantity, but few mutations (TP53, ITPR1, DSC1 and PRH) were identified in more than one individual. The most common genetic defects affected the gene TP53, with a missing 17p chromosomal arm with or without it being defective in 58% of cases, aligning with other NGS results from large studies. Many research teams have confirmed that the removal of TP53 in the 17p chromosomal arm is the most common SS deletion.72 TP53 clones with double knockouts of the gene were analyzed in two early identified patients, indicating that the reduction of TP53 can constitute a clinical SS subtype.73
Subcutaneous Panniculitis-Like T Cell Lymphoma
The disorder is sporadic and has a low overall prevalence.74 Li et al performed both selective and exome sequencing (WES) of several cases of Subcutaneous Panniculitis-like T-cell lymphoma (SPTCL). In fact, epigenetic SPTCL mutagens have been reported in 72% of cases. These mutagens are active epigenetically on almost all levels including functions in modifying histones (DOT1L, 2/18; KMT2D, 2/18 and CREBBP, 2/18), methylation of DNA (MBD1, 1/18) and chromatin assembly (ARID1B, 3/18; SMARCA4, 3/18; CHD4, 3/18). Recurring mutations in the gene for the mTOR/AKT/PI3K pathway have been identified in 44% of SPTCL cases. TSC1 and mTOR genes (3/18 cases) are the most frequent mutations that have been observed in the mTOR/AKT/PI3K signaling cascade, while AKT2, TSC2 and PI3KCB, (1/18 each), PI3KCA and PI3KCD (2/18 each) had a lower incidence. In this study, only one SPTCL patient had mutated TP53. Relative to other T-cell lymphomas, this showed a reduced incidence. In the mTOR/AKT/PI3K pathway and in SPTCL epigenetic mutagens, the team found multiple mutations. STPCL can potentially be treated by successful clinical strategies directed against these two pathways.75
Peripheral T- Cell Lymphoma, NOS
With violent properties, high mortality and poor prognosis, among other identifying characteristics, peripheral T-cell lymphoma (PTCL) is a malignant form of lymphoma with strong specificity, most of which develops after NK and T-cells mature in the thymus.76 Vasmatzis et al conducted a study that adapted previous preliminary bioinformatic algorithms to detect de novo PTCL chromosomal rearrangement. Thirteen of 21 PTCL patients were classified with recurrent rearrangements. Among these were five modifications affecting p53-related genes, including ANKRD11, WWOX, CDKN2A, TP63, and TP53. The current rearrangement of TP63 is very important,77 as it codes for a fusion protein which contains p63, that is n-truncated (Np63), which is almost the same as the normal Np63 subtype. And it is recognized for its carcinogenic traits and inhibits the p53 pathway through a strongly negative pathway.78 About 5.8% of patients had these rearrangements which correlated with poor survival outcomes in the ALK mutexed rearrangement. TP63 rearrangement correlates with poor PTCL survival outcomes. Compared to other malignant neoplasms, PTC53 is seldom caused by TP53 mutations and the results indicate that in PTCL, the suppressive function of p53 against tumors may be eliminated by other genetic aberrations.79 Emma I. Andersson et al sequencing patient samples of AITL (=30), ALCL (AITL(=21) and PTCL-NOS (AITL(=12) cases in one study, The pSTAT3, pMAPK and pAKT were amplified and sequenced and immunohistochemical staining. The team discovered mutations in 13% of AITL, 13% of ALKALCL, 38% of ALKALCL and 17% of PTCL-NOS cases. They found pY-STAT3 expression was highest in PTCL containing or mutated.80
Angioimmunoblastic T-Cell Lymphoma and Other Nodal Lymphomas of T Follicular Helper (TFH) Cell Origin
Angioimmunoblastic T-cell lymphoma (AITL) is an uncommon subtype of peripheral T-cell lymphoma, which arises primarily in older people and causes only 1 to 2% of NHL.81 In Europe, the incidence rate is greater (28.7%) than in Asia (17.9%). The recurring genetic mutations comonly found in AITL comprise CD28 (9.4–11.3%), DNMT3A (20–30%), IDH2 (20–45%), TET2 (47–83%) and RHOA mutations (50–70%).
G17V RHOA mutation has been shown to be extremely rare in other cancers of the blood system, and it is observed in about 60% of PTCL with the TFH phenotype. Nevertheless, G17V RHOA mutations are common and specific in AITL and lymph nodes.82 Somatic RHOA mutations that encode p.gly17val were reported to be expressed in 68% of AITL samples by Yanagimoto et al. It’s important to keep in mind that all patients with p.gelly17val coding mutations also had mutations in TET2. The p.gelly17val RHOA mutation was found exclusively in tumor cells,83 while TET2 mutation was discovered both in non-tumoral blood cells and tumor cells.84 RHOA controls several different biological mechanisms through encoding for a small GTPase.85 This study indicated that Gly17Val RHOA mutants fail to bind to GTP and block wild-type RHOA function. This finding suggests that the impaired RHOA function, along with the prior eradication of the TET2 activity, selectively contributes AITL pathogenesis.86 The consistency and sensitivities of G17V RHOA mutations identified by the methods of peptide nucleic acid locking of nucleic acid (pna-lna), NGS and droplet digital PCR (ddPCR) were compared by Nuhat et al Mutation of G17V RHOA was found in 40.3% of the NGS samples. Additionally, the samples observed by the pna-lna clamping system, ddPCR and NGS also identified the G17V mutation in 4 samples (46.3%). Moreover, the occurrence of mutated alleles using ddPCR showed a strong consistency to that of mutated alleles using NGS. RHOA mutations of pgly17val were present in 50.7% of the samples. The results of this study show that the combination of NGS with pna-lna clamping/ddPCR is the best method for aiding diagnosis of AITL by identification of p.gly17val RHOA mutations.87
Vallois et al based their findings on using deep sequencing to research mutations in a series of co-stimulation/TCR cascades in TFH-like AITL and PTCL. In addition to the prominent RHOA mutations, they also found that 49% of cases had mutations in the co-stimulatory/TCR pathway that were triggered frequently and almost mutually exclusively. Specifically, they found various CARD11 and PLCG1 mutations that they examined as in vitro functional activation. A thorough assessment of the gene expression profile demonstrated that samples with TCR mutations were abundant in biochemical markers, indicating higher T-cell proliferation and activation.88 Medically, patients who received anthracycline-based TCR mutations showed a greater risk of early development than patients lacking these aberrations. These findings show the possibility for the management of such lymphoma with medicines that abrogate signaling downstream of TCR.89
J Rohr et al studied twenty AITL cases and detected two repeated locations for mutation in D124 and CD124 T195 after carrying out full transcriptome sequencing (WTS). Later, 90 PTCL cases (including 5 AITL cases with WTS) were assigned to CD28 sequencing and functional modifications correlated with the two mutation sites of highest frequency were examined. It was eventually seen that they accentuated signaling cascades or receptor/ligand associations, which might lead to PTCL T-cell stimulation. This was backed up by an increase in secondary signal transduction by the CD28 mutant after binding ligands.90
Anaplastic Large Cell Lymphoma
Anaplastic large cell lymphoma (ALCL) is another NHL type of T-cell lymphoma.91 Two systemic diseases are known to be based on anaplastic kinase (ALK) lymphoma expression.92 In most instances ALK+ALCL is defined by the ALK gene and the chromosomal translocation (NPM) of t(2;5) (p23; Q35), culminating in the chimeric form of the fusion protein ALK and constitutive activation. Carcinogenic NPMALK can transform and activate many secondary signaling cascades, primarily JAK/STAT and PI3 K pathways, PLC control, MAPK/RAS, which are involved in cellular survival, specialization and propagation.93 NGS was used by Steinhilber et al, to study the differential expression of miRNA within normal T-cells, ALK−ALCL and ALK+ALCL. It established 106 miRNAs that were uniquely expressed between ALK+ and ALK−ALCL, and 228 miRNAs that were distinct between regular T-cells and ALK+ALCL cells. The research detected 56 miRNA signatures that differentiate between T cells, ALK+ALCL and ALK− ALCL. Seven down-regulated genes were among the most important candidate genes with significantly different expression between ALK−ALCL and ALK+: mir-155, mir-146a, mir-542-3p, mir-424*mir-503, mir-424, mir-196b. In comparison, 5 miRNAs were upregulated: mir-183, mi-182, mi-203 mir-340 and mi-135b. The ALK+ cells were upregulated in mir-17-92 clusters. Furthermore, the analysis identified three miRNAs characteristics greatly regulated by the C/EBP transcription factor, which is especially overexpressed by ALK+ALCL (such as the mir-181 family). Ironically, mir-181a, which controls TCR signal intensity and T-cell specialization, was significantly reduced in ALK+ALCL patients. To summarize, these findings revealed miRNA signals connecting ALK+ALCL to improper immune reactions, thereby partly arising from irregular ALK+ ALCL TCR expression of antigens.94
Large Cell Lymphoma That is Associated with Breast Implants and is Anaplastic
The connection between the fluid surrounding ALK−ALCL and the breast prosthesis developed in the capsule leads to the recognition of a unique clinicopathological entity known as ALCL related to breast implants (BIA−ALCL).95 BIA−ALCL is a sporadic form of T cell lymphoma that typically emerges after a relatively long incubation period following breast prosthesis.96 Blombery et al performed WES in two low-effusion BIA-ALCL patients in 2016, and observed acquired activation defects of JAK1 and STAT3 in both instances. This provides information on inherited lesions in this rare disease. In 2018, the team performed a comprehensive genome exploration of 11 BIA-ALCL cases based on NGS, including, whole-genome analysis of the number of copies, identifying changes in TCR structure by sequencing or measuring depth of TRB and the detecting of aberrations in 180 commonly mutated genes in hematologic malignant tumors. Defects were found JAK/STAT activation sequences in 10 out of 11 cases. Two mutations of the TP53 germ line were found in this research. Recent studies have shown that genetic changes in epigenetic modifiers and JAK-STAT signals are common in ALCL associated with breast implants.97 Moreover, there was frequent loss of RPL5 copy numbers and increase of PDGFRA, TMEM119, P2RX7, MYC and TNFRSF11A [RANK] copy numbers shown by two cases. Essentially, this study provides information on the fundamental disease pathways (TP53 and JAK/STAT and MYC) and establishes targets for potential therapeutic interventions in this rare disease, namely PDGRA and TNFRSF11A.98 The genomic data from Blombery et al showed that irregular BIA-ALCL signal transduction pathways were the same as sALCL’s, including abnormal BIA-ALCL signals. Nevertheless, the recorded gene aberrations causing this deregulation of signaling seem to be specific to BIA-ALCL, having 1p22 repeat deletions and greatly repetitive mutations of STAT3 influencing RPL5, which are not yet present in SALCL. Additionally, genomic data included anomalies in the transduced inflammatory body signals including Wnt/β-catenin, PKC and TGF-cell pathways that could be a future study area, to improve understanding of this rare lymphoma.99
Conclusion
Mature NK/T-cell lymphoma is aggressively malignant and as a result of its many classifications and low incidence, it has not been adequately studied. With the rapid development of NGS technology and more collaboration among medical research centers, more and more mutated genes of various classifications in mature T- and NK-cell lymphoma have been discovered. In this paper, the application of NGS technology in 9 types of mature T- and NK-cell lymphomas with a lot of related studies and the discovery of relevant gene mutations are summarized. The discovery of these mutated genes is conducive to the accurate diagnosis and treatment of diseases, so that these relatively rare lymphomas can get personalized treatment.
Acknowledgments
This article was supported by the Zhejiang provincial Medical Technology Plan Project (No. 2020KY052, 2017ZA007), Zhejiang Provincial Natural Science Foundation of China (No. LY19H160037, LY17H160062), Zhejiang provincial science and technology project (No. 2018C37078).
Disclosure
All authors have disclosed that they have no conflicts of interest in this work.
References
1. Sanger F, Nicklen S, Coulson AR. DNA sequencing with chain-terminating inhibitors. Proc Natl Acad Sci U S A. 1977;74(12):5463–5467. doi:10.1073/pnas.74.12.5463
2. Jin S, Zhou C, Hou X, et al. A multicenter real-world study of tumor-derived DNA from pleural effusion supernatant in genomic profiling of advanced lung cancer. Trans Lung Cancer Res. 2020;9(4):1507–1515. doi:10.21037/tlcr-20-882
3. Fang W, Huang Y, Gu W, et al. PI3K-AKT-mTOR pathway alterations in advanced NSCLC patients after progression on EGFR-TKI and clinical response to EGFR-TKI plus everolimus combination therapy. Trans Lung Cancer Res. 2020;9(4):1258–1267. doi:10.21037/tlcr-20-141
4. Chen H, Liu M, Dai Z, et al. EGFRConcomitant genetic alterations are associated with response to targeted therapy in patients with lung adenocarcinoma. Trans Lung Cancer Res. 2020;9(4):1225–1234. doi:10.21037/tlcr-20-679
5. Pareek CS, Smoczynski R, Tretyn A. Sequencing technologies and genome sequencing. J Appl Genet. 2011;52(4):413–435. doi:10.1007/s13353-011-0057-x
6. Muzzey D, Evans E, Lieber C. Understanding the basics of NGS: from mechanism to variant calling. Curr Genet Med Rep. 2015;3(4):158–165. doi:10.1007/s40142-015-0076-8
7. Yohe S, Thyagarajan B. Review of clinical next-generation sequencing. Arch Pathol Lab Med. 2017;141(11):1544–1557. doi:10.5858/arpa.2016-0501-RA
8. Choi S, Go JH, Kim EK, Lee H, Han K. Mutational analysis of extranodal NK/T-cell lymphoma using targeted sequencing with a comprehensive cancer. Genomics Inform. 2016;143(3):78–84.
9. Ross JS, Cronin M. Whole cancer genome sequencing by next-generation methods. Am J Clin Pathol. 2011;136(4):527–539. doi:10.1309/AJCPR1SVT1VHUGXW
10. Bewicke-Copley F, Kumar EA, Palladino G, et al. Applications and analysis of targeted genomic sequencing in cancer studies. Comput Struct Biotechnol J. 2019;17:1348–1359. doi:10.1016/j.csbj.2019.10.004
11. Mardis RE. Next-generation sequencing platforms. Ann Rev Anal Chem. 2013;6(1):287–303. doi:10.1146/annurev-anchem-062012-092628
12. Cronin M, Ross JS. Comprehensive next-generation cancer genome sequencing in the era of targeted therapy and personalized oncology. Biomark Med. 2011;5(3):293–305. doi:10.2217/bmm.11.37
13. Wang L, Qin W, Huo YJ, et al. Advances in targeted therapy for malignant lymphoma. Signal Transduct Target Ther. 2020;5:15.
14. Pearson PL, Luijt VD. The genetic analysis of cancer. J Intern Med. 1998;243(6):413–417. doi:10.1046/j.1365-2796.1998.00343.x
15. Feunteun J. [Hereditary predisposition to cancer]. Bull Acad Natl Med. 2005;189(5):797. [Portugese]
16. Lewis WD, Lilly S, Jones KL. Lymphoma: diagnosis and treatment. Am Fam Physician. 2020;101(1):34–41.
17. Wang H, Balakrishna J, Pittaluga S, Jaffe E. Diagnosis of Hodgkin lymphoma in the modern era. Br J Haematol. 2019;184(1):45–59. doi:10.1111/bjh.15614
18. Xiong J, Zhao WL. Advances in multiple omics of natural-killer/T cell lymphoma. J Hematol Oncol. 2018;11(1):134. doi:10.1186/s13045-018-0678-1
19. Araf S, Korfi K, Rahim T, et al. Advances in the molecular diagnosis of diffuse large B-cell lymphoma in the era of precision medicine. Expert Rev Mol Diagn. 2016;16(10):1093–1102. doi:10.1080/14737159.2016.1235974
20. McCabe MT, Ott HM, Ganji G. EZH2 inhibition as a therapeutic strategy for lymphoma with EZH2-activating mutations. Nature. 2012;492(7427):108–112. doi:10.1038/nature11606
21. Dietrich S, Glimm H, Andrulis M, et al. BRAF inhibition in refractory hairy-cell leukemia. N Engl J Med. 2012;366(21):2038–2040. doi:10.1056/NEJMc1202124
22. Staber P, Herling M, Bellido M, et al. Consensus criteria for diagnosis, staging, and treatment response assessment of T-cell prolymphocytic leukemia. Blood. 2019;134(14):1132–1143. doi:10.1182/blood.2019000402
23. Braun T, von Jan J, Wahnschaffe L, Herling M. Advances and perspectives in the treatment of T-PLL. Curr Hematol Malig Rep. 2020;15(2):113–124. doi:10.1007/s11899-020-00566-5
24. Robak T, Robak P. Current treatment options in prolymphocytic leukemia. Med Sci Monit. 2007;13(4):RA69–80.
25. Catovsky D, Okos A, Wiltshaw E, Galetto J, Galton DAG, Stathopoulos G. PRolymphocytic Leukemia of B and T cell type. Lancet. 1973;302(7823):232–234. doi:10.1016/S0140-6736(73)93135-8
26. Hu Z, Medeiros L, Fang L, et al. Prognostic significance of cytogenetic abnormalities in T-cell prolymphocytic leukemia. Am J Hematol. 2017;92(5):441–447. doi:10.1002/ajh.24679
27. Yuille M, Coignet L, Abraham S, et al. ATM is usually rearranged in T-cell prolymphocytic leukaemia. Oncogene. 1998;16(6):789–796.28.
28. Laribi K, Lemaire P, Sandrini J, Baugier de Materre A. Advances in the understanding and management of T-cell prolymphocytic leukemia. Oncotarget. 2017;8(61):104664–104686. doi:10.18632/oncotarget.22272
29. Sellner L, Brüggemann M, Schlitt M, et al. GvL effects in T-prolymphocytic leukemia: evidence from MRD kinetics and TCR repertoire analyses. Bone Marrow Transplant. 2017;52(4):656. doi:10.1038/bmt.2017.12
30. Kiel M, Velusamy T, Rolland D, et al. Integrated genomic sequencing reveals mutational landscape of T-cell prolymphocytic leukemia. Blood. 2014;124(9):1460–1472. doi:10.1182/blood-2014-03-559542
31. Kawamura K, Tanaka Y, Nakasone H, et al. Development of a unique T-cell receptor gene-transferred Tax-redirected T-cell immunotherapy for adult T-cell leukemia. Biol Blood Marrow Transplant. 2020;268(8):1377–1385. doi:10.1016/j.bbmt.2020.04.006
32. Tarokhian H, Rahimi H, Mosavat A, et al. HTLV-1-host interactions on the development of adult T cell leukemia/lymphoma: virus and host gene expressions. BMC Cancer. 2018;18(1):1287. doi:10.1186/s12885-018-5209-5
33. Harhaj E, Giam C. NF-κB signaling mechanisms in HTLV-1-induced adult T-cell leukemia/lymphoma. FEBS J. 2018;285(18):3324–3336. doi:10.1111/febs.14492
34. Bonn BR, Huge A, Rohde M, Oschlies I, Burkhardt B. Whole exome sequencing hints at a unique mutational profile of paediatric T-cell lymphoblastic lymphoma. Br J Haematol. 2014;168(2):308–313. doi:10.1111/bjh.13105
35. Kataoka K, Iwanaga M, Yasunaga J, et al. Prognostic relevance of integrated genetic profiling in adult T-cell leukemia/lymphoma. Blood. 2018;131(2):215–225. doi:10.1182/blood-2017-01-761874
36. Laurent C, Nicolae A, Laurent C, et al. Gene alterations in epigenetic modifiers and JAK-STAT signaling are frequent in breast implant-associated ALCL. Blood. 2020;135(5):360–370. doi:10.1182/blood.2019001904
37. Li Z, Xia Y, Feng LN, et al. Genetic risk of extranodal natural killer T-cell lymphoma: a genome-wide association study. Lancet Oncol. 2016;17(9):1240–1247. doi:10.1016/S1470-2045(16)30148-6
38. Kataoka K, Nagata Y, Kitanaka A. Integrated molecular analysis of adult T cell leukemia/lymphoma. Nat Genet. 2015;47(11):1304–1315. doi:10.1038/ng.3415
39. Xiong J, Zhao W. What we should know about natural killer/T-cell lymphomas. Hematol Oncol. 2019;37(S1):75–81. doi:10.1002/hon.2588
40. Avilès AA. Nasal NK/T-cell lymphoma. a comparative analysis of a Mexican population with the other populations of latin-america. Mediterr J Hematol Infect Dis. 2015;7(1):e2015052. doi:10.4084/mjhid.2015.052
41. Asano N, Kato S, Nakamura S. Epstein–Barr virus-associated natural killer/T-cell lymphomas. Best Pract Res Clin Haematol. 2013;26(1):15–21. doi:10.1016/j.beha.2013.04.002
42. Zhang Y, Li C, Xue W, Zhang M, Li Z. Frequent mutations in natural killer/T cell lymphoma. Cell Physiol Biochem. 2018;49(1):1–16. doi:10.1159/000492835
43. Jiang L, Gu Z-H, Yan Z-X, et al. Exome sequencing identifies somatic mutations of DDX3X in natural killer/T-cell lymphoma. Nat Genet. 2015;47(9):1061–1066. doi:10.1038/ng.3358
44. Chen B, Jiang L, Zhong ML, et al. Identification of fusion genes and characterization of transcriptome features in T-cell acute lymphoblastic leukemia. Proc Natl Acad Sci U S A. 2018;115(2):373–378.
45. Jiang L, Gu Z-H, Yan Z-X, et al. Exome sequencing identifies somatic mutations of DDX3X in natural killer/T-cell lymphoma. Nat Genet. 124(21).
46. Montes-Mojarro IA, Chen BJ, Ramirez-Ibarguen AF, et al. Mutational profile and EBV strains of extranodal NK/T-cell lymphoma, nasal type in Latin America. Mod Pathol. 2020;335(5):781–791.
47. Kim W, Montes-Mojarro I, Fend F, Quintanilla-Martinez L. Epstein-Barr virus-associated T and NK-cell lymphoproliferative diseases. Front Pediatr. 2019;7:71. doi:10.3389/fped.2019.00071
48. Sim SH, Kim S, Kim TM, Jeon YK, Heo DS. Novel JAK3 -activating mutations in extranodal nk/t-cell lymphoma, nasal type. Am J Pathol. 2017;187(5):5.
49. Sim S, Kim S, Kim T, et al. Novel JAK3-activating mutations in extranodal NK/T-cell lymphoma, nasal type. Am J Pathol. 2017;187(5):980–986. doi:10.1016/j.ajpath.2017.01.004
50. Koo GC, Tan SY, Tang T, et al. Janus kinase 3-activating mutations identified in natural killer/t-cell lymphoma. Cancer Discov. (7):591–597.
51. Koo G, Tan S, Tang T, et al. Janus kinase 3-activating mutations identified in natural killer/T-cell lymphoma. Cancer Discov. 2012;2(7):591–597. doi:10.1158/2159-8290.CD-12-0028
52. Küçük C, Wang J, Xiang Y, You H. Epigenetic aberrations in natural killer/T-cell lymphoma: diagnostic, prognostic and therapeutic implications. Ther Adv Med Oncol. 2020;12:1758835919900856. doi:10.1177/1758835919900856
53. Kataoka K, Miyoshi H, Sakata S, et al. Frequent structural variations involving programmed death ligands in Epstein-Barr virus-associated lymphomas. Leukemia. 2019;33(7):1687–1699. doi:10.1038/s41375-019-0380-5
54. Ferry J. Extranodal lymphoma. Arch Pathol Lab Med. 2008;132(4):565–578. doi:10.5858/2008-132-565-EL
55. Olszewska-Szopa M, Wróbel T. Gastrointestinal non-Hodgkin lymphomas. Adv Clin Exper Med. 2019;28(8):1119–1124. doi:10.17219/acem/94068
56. Hasnaoui H, El Bouhaddouti H, Mouaqit O, Benjelloun E, Ousadden A, Taleb K. [Acute intestinal intussusception revealing intestinal T-cell lymphoma in adults]. Pan Afr Med J. 2019;33:153. doi:10.11604/pamj.2019.33.153.18758 [Danish]
57. NairismGi ML, Tan J, Lim JQ, et al. JAK-STAT and G protein-coupled receptor signaling pathways are frequently altered in epitheliotropic intestinal T-cell lymphoma. Leukemia. 2016;30(6):1311–1319. doi:10.1038/leu.2016.13
58. Moffitt AB, Ondrejka SL, McKinney M, et al. Enteropathy-associated T cell lymphoma subtypes are characterized by loss of function of SETD2. J Exp Med. 214;5:1371–1386.
59. Pulitzer M. Cutaneous T-cell Lymphoma. Clin Lab Med. 2017;37(3):527–546. doi:10.1016/j.cll.2017.06.006
60. Mehta-Shah N, Horwitz SM, Ansell S. NCCN guidelines insights: primary cutaneous lymphomas, version 2.2020. J Natl Compr Canc Netw. 2020;18(5):522–536. doi:10.6004/jnccn.2020.0022
61. Choi J, Goh G, Walradt T, et al. Genomic landscape of cutaneous T cell lymphoma. Nat Genet. 2015;47(9):1011–1019. doi:10.1038/ng.3356
62. Larocca C, Kupper T. Mycosis fungoides and Sézary syndrome: an update. Hematol Oncol Clin North Am. 2019;33(1):103–120. doi:10.1016/j.hoc.2018.09.001
63. Yumeen S, Girardi M. Insights into the molecular and cellular underpinnings of cutaneous T cell lymphoma. Yale J Biol Med. 2020;93(1):111–121.
64. Hamrouni A, Fogh H, Zak Z, et al. Clonotypic diversity of the T-cell receptor corroborates the immature precursor origin of cutaneous T-cell lymphoma. Clin Cancer Re. 2019;2510(10):3104–3114.
65. Sud A, Dearden C. T-cell prolymphocytic leukemia[J]. Med Oncol. 2009;23(1):365–371.
66. Kiel M, Sahasrabuddhe A, Rolland D, et al. Genomic analyses reveal recurrent mutations in epigenetic modifiers and the JAK-STAT pathway in Sézary syndrome. Nat Commun. 2015;6(1):8470. doi:10.1038/ncomms9470
67. Iżykowska K, Przybylski GK, Gand C, Braun FC, Schmidt CA. Genetic rearrangements result in altered gene expression and novel fusion transcripts in Sézary syndrome. Oncotarget. 2017;8(24):39627. doi:10.18632/oncotarget.17383
68. Almeida ACDS, Abate F, Khiabanian H, Martinez-Escala E, Palomero T. The mutational landscape of cutaneous T cell lymphoma and Sézary syndrome. Nat Genet. 2015;47:12.
69. Wang L, Ni X, Covington K, et al. Genomic profiling of Sézary syndrome identifies alterations of key T cell signaling and differentiation genes. Nat Genet. 2015;47(12):1426–1434. doi:10.1038/ng.3444
70. Brouwer I, Out-Luiting J, Vermeer M, Tensen C. Cucurbitacin E and I target the JAK/STAT pathway and induce apoptosis in Sézary cells. Biochem Biophy Rep. 2020;24:100832. doi:10.1016/j.bbrep.2020.100832
71. Alexander-Savino C, Hayden M, Richardson C, Zhao J, Poligone B. Doxycycline is an NF-κB inhibitor that induces apoptotic cell death in malignant T-cells. Oncotarget. 2016;7(46):75954–75967. doi:10.18632/oncotarget.12488
72. Gros A, Laharanne E, Vergier M, et al. TP53 alterations in primary and secondary Sézary syndrome: a diagnostic tool for the assessment of malignancy in patients with erythroderma. PLoS One. 2017;12(3):e0173171. doi:10.1371/journal.pone.0173171
73. Chevret E, Merlio JP. Sézary syndrome: translating genetic diversity into personalized medicine. J Invest Dermatol. 2016;136(7):1319–1324. doi:10.1016/j.jid.2016.04.027
74. Ramachandran V, Park KE, Torres‐Cabala CA. Second primary malignancies in subcutaneous panniculitis-like T-cell lymphoma: a national database study. Clin Exp Dermatol. 2020;456(6).
75. Li Z, Lu L, Zhou Z, et al. Recurrent mutations in epigenetic modifiers and the PI3K/AKT/mTOR pathway in subcutaneous panniculitis-like T-cell lymphoma. Br J Haematol. 2018;181(3):406–410. doi:10.1111/bjh.14611
76. Zhang Y, Xu W, Liu H, Li J. Therapeutic options in peripheral T cell lymphoma. J Hematol Oncol. 2016;9(1):37. doi:10.1186/s13045-016-0267-0
77. Somerville T, Xu Y, Miyabayashi K, et al. TP63-mediated enhancer reprogramming drives the squamous subtype of pancreatic ductal adenocarcinoma. Cell Rep. 2018;25(7):1741–1755.e1747. doi:10.1016/j.celrep.2018.10.051
78. Ratovitski E, Patturajan M, Hibi K, Trink B, Yamaguchi K, Sidransky D. p53 associates with and targets Delta Np63 into a protein degradation pathway. Proc Natl Acad Sci U S A. 2001;98(4):1817–1822. doi:10.1073/pnas.98.4.1817
79. Vasmatzis G, Johnson SH, Knudson RA. Genome-wide analysis reveals recurrent structural abnormalities of TP63 and other p53-related genes in peripheral T-cell lymphomas. Blood. 2012;120(11):2280–2289. doi:10.1182/blood-2012-03-419937
80. Andersson EI, Brück O, Braun T, et al. STAT3 mutation is associated with STAT3 activation in CD30+ ALK ALCL[J]. Cancers. 2020;12(3):702. doi:10.3390/cancers12030702
81. Fujisawa M, Chiba S, Sakata-Yanagimoto M. Recent progress in the understanding of angioimmunoblastic T-cell lymphoma. J Clin Exper Hematopathol. 2017;57(3):109–119. doi:10.3960/jslrt.17019
82. Fukumoto K, Nguyen T, Chiba S, Sakata-Yanagimoto M. Review of the biologic and clinical significance of genetic mutations in angioimmunoblastic T-cell lymphoma. Cancer Sci. 2018;109(3):490–496. doi:10.1111/cas.13393
83. Fujisawa M, Sakata-Yanagimoto M, Nishizawa S, et al. Activation of RHOA-VAV1 signaling in angioimmunoblastic T-cell lymphoma. Leukemia. 2018;32(3):694–702. doi:10.1038/leu.2017.273
84. Feng Y, Li X, Cassady K, Zou Z, Zhang X. TET2 function in hematopoietic malignancies, immune regulation, and DNA repair. Front Oncol. 2019;9:210. doi:10.3389/fonc.2019.00210
85. Chiba S, Enami T, Ogawa S, Sakata-Yanagimoto M. G17V RHOA: genetic evidence of GTP-unbound RHOA playing a role in tumorigenesis in T cells. Small GTPases. 2015;6(2):100–103. doi:10.4161/21541248.2014.988088
86. Sakata-Yanagimoto M, Enami T, Yoshida K, et al. Somatic RHOA mutation in angioimmunoblastic T cell lymphoma. Nat Genet. 2014;46(2):171–175. doi:10.1038/ng.2872
87. Tanzima Nuhat S, Sakata‐Yanagimoto M, Komori D, et al. Droplet digital polymerase chain reaction assay and peptide nucleic acid-locked nucleic acid clamp method for RHOA mutation detection in angioimmunoblastic T-cell lymphoma. Cancer Sci. 2018;109(5):1682–1689. doi:10.1111/cas.13557
88. Vallois D, Dobay M, Morin R, et al. Activating mutations in genes related to TCR signaling in angioimmunoblastic and other follicular helper T-cell-derived lymphomas. Blood. 2016;128(11):1490–1502.
89. Vallois D, Dobay MPD, Morin RD. Activating mutations in genes related to TCR signaling in angioimmunoblastic and other follicular helper T-cell-derived lymphomas. Blood. 2016;128(11):1490–1502. doi:10.1182/blood-2016-02-698977
90. Rohr J, Guo S, Huo J, et al. Recurrent activating mutations of CD28 in peripheral T-cell lymphomas. Leukemia. 2016;30(5):1062–1070. doi:10.1038/leu.2015.357
91. Tsuyama N, Sakamoto K, Sakata S, Dobashi A, Takeuchi K. Anaplastic large cell lymphoma: pathology, genetics, and clinical aspects. J Clin Exper Hematopathol. 2017;57(3):120–142. doi:10.3960/jslrt.17023
92. Luchtel R, Zimmermann M, Hu G, et al. MSCRecurrent mutations in ALK-negative anaplastic large cell lymphoma. Blood. 2019;133(26):2776–2789. doi:10.1182/blood.2019000626
93. Merkel O, Hamacher F, Laimer D, et al. Identification of differential and functionally active miRNAs in both anaplastic lymphoma kinase (ALK)+ and ALK- anaplastic large-cell lymphoma. Proc Natl Acad Sci U S A. 2010;107(37):16228–16233. doi:10.1073/pnas.1009719107
94. Julia S, Michael B, Michael W, Falko F, Irina B, Leticia QM. Next-generation sequencing identifies deregulation of microRNAs involved in both innate and adaptive immune response in ALK+ ALCL. PLoS One. 2015;10(2):e0117780.
95. Sharma B, Jurgensen-Rauch A, Pace E, et al. Breast implant-associated anaplastic large cell lymphoma: review and multiparametric imaging paradigms. Radiographics. 2020;40(3):609–628. doi:10.1148/rg.2020190198
96. Jaffe ES, Ashar BS, Clemens MW, et al. Best practices guideline for the pathologic diagnosis of breast implant-associated anaplastic large-cell lymphoma. J Clin Oncol. 2020;38(10):1102–1111. doi:10.1200/JCO.19.02778
97. Kogure Y, Kataoka K. Genetic alterations in adult T-cell leukemia/lymphoma. Cancer Sci. 2017;108(9):1719–1725. doi:10.1111/cas.13303
98. Blombery P, Thompson ER, Jones K, et al. Whole exome sequencing reveals activating JAK1 and STAT3 mutations in breast implant-associated anaplastic large cell lymphoma anaplastic large cell lymphoma. Haematologica. 2016;101(9):e387–e390. doi:10.3324/haematol.2016.146118
99. Blombery P, Thompson E, Ryland GL, et al. Frequent activating STAT3 mutations and novel recurrent genomic abnormalities detected in breast implant-associated anaplastic large cell lymphoma. Oncotarget. 2018;9(90):36126–36136. doi:10.18632/oncotarget.26308
 © 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.