Back to Journals » Degenerative Neurological and Neuromuscular Disease » Volume 7
Current concepts in multiple sclerosis therapy
Authors Sedal L, Winkel A ![]() , Laing J, Law LY, McDonald E
, Laing J, Law LY, McDonald E
Received 27 March 2017
Accepted for publication 16 May 2017
Published 28 September 2017 Volume 2017:7 Pages 109—125
DOI https://doi.org/10.2147/DNND.S109251
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Thomas Müller
Leslie Sedal, Antony Winkel, Joshua Laing, Lai Yin Law, Elizabeth McDonald
Department of Neurology, St Vincent’s Hospital Melbourne, Melbourne, VIC, Australia
Abstract: Over the past 20 years, the available therapies for multiple sclerosis have expanded exponentially. With several more agents likely to be approved for public funding in Australia in the next 12 months on top of the existing multitude of Australian Pharmaceutical Benefits Scheme-subsidized therapies, the choice is becoming even more complex. This review summarizes the current state of available therapies and anticipates likely future directions, including an important focus on contemporary symptom management. For each agent, the major trials, side effects, and clinical utility are summarized, with a particular focus on the Australian experience of these therapies. It is hoped this review provides an up-to-date reference of the exciting current state of multiple sclerosis therapy.
Keywords: demyelination, disease modifying drugs, DMDs, no evidence of disease activity, NEDA, progressive multifocal leukoencephalopathy, PML
Introduction
2017 is a good year to review current concepts in the therapy of multiple sclerosis, as 2017 marks 20 years since the Australian Pharmaceutical Benefits Scheme first accepted IFNβ1b (Betaferon®; Bayer, Berlin, Germany) for support. At the start of 2017 there were 10 disease-modifying drugs supported by the scheme. For the purposes of this discussion, we have grouped them as “the injectables”, “the orals”, and “the infusions”.
To keep the article current, we have also chosen three drugs we think are very likely to go onto the Australian Pharmaceutical Benefits Scheme in the next year. By coincidence, they fall into the same three categories. However, as well as drugs that may alter the course of the disease, there is important research happening on the symptoms of the disease and what can be done to relieve them, and we have tried to give a brief survey of this aspect of multiple sclerosis care as well. Although other parts of the world will have had differing experience over the past two decades, we hope framing this discussion from an Australian point of view provides some international context for an ever-changing disease.
Approach to treatment
The goals of treatment have shifted drastically over time, with the availability of increasingly effective therapies reducing our tolerance for disease activity and progressive disability. Particularly in relapsing disease, the concept of “no evidence of disease activity” (NEDA) has come to the forefront, ie, a stabilization of the condition on therapy such that there is no clinical (relapse or progression of disability, as measured by a validated scale, such as the Expanded Disability Status Scale [EDSS]) or radiological evidence (new T2 or contrast-enhancing lesions) of activity over a period of observation. Although seemingly ambitious, many of the newer agents now have evidence of at least moderate rates of NEDA, which could be anticipated to substantially alter the long-term disease course. A difficulty with NEDA as a trial–outcome measure is that subtly different ways may be used to define it, and so direct intertrial comparisons must be made carefully. More advanced radiological analytic techniques, including brain volumes, atrophy, and gray-matter involvement, have also been examined as targets to monitor effectiveness of treatment.
In broad terms, once a diagnosis of multiple sclerosis is made, an assessment must be made about disease activity, ie, for a given patient, what is the expected risk and frequency of relapse, clinical worsening, or radiological progression? A number of prognostic factors can be looked at to help determine the long-term risk of progression, including African ancestry, incomplete recovery from first clinical relapse, more than one relapse in the first year of diagnosis, multifocal presentation and higher EDSS score prior to treatment, as well as imaging features at diagnosis.1,2 This prognostication then leads to a simpler therapeutic choice from a small handful of appropriate treatments based on severity of disease in an individual patient, rather than considering all agents, as many of the more efficacious therapies for aggressive disease are balanced against more onerous side-effect profiles and may not be appropriate for relatively inactive disease. In practice, the two most common strategies include a “step-up” approach (whereby therapy is increasingly escalated as breakthrough disease occurs) and “step down” (therapy is begun aggressively at diagnosis, and thereafter weaned, similar to an “induction”-type approach), and both have their merits and disadvantages.3,4 The Australian approach to this is molded somewhat by how the therapies were made available on government subsidy (and therefore mostly tending to favor a step-up method), but regional differences occur, and no one approach to treatment will be appropriate for every patient.
Finally, it is not uncommon for patients who have been stable on effective therapy for some time to request cessation. It should be noted that severe and even fatal flares of disease activity have been reported with several of the following agents, and this has been reported most prominently with fingolimod and natalizumab.5,6 Any interruptions to therapy must thus be considered and discussed carefully with the patient in the context of their clinical state.
The injectables
The first of the interferons was licensed in Australia in 1995, and injectable therapies have been a mainstay of the management of multiple sclerosis ever since. Although they are very safe in long-term use, their utility is offset by the newer availability of more efficacious therapies and relatively more burdensome side-effect profile. For the cohort of patients with relatively mild disease activity who are treated with these, however, they are a useful and cost-effective therapy without some of the unique drawbacks of newer therapies.
Interferons
IFNβ1b became available in Australia in 1995 after the publication of a pivotal trial in 1993, demonstrating a significant reduction in number and severity of relapses in relapsing–remitting multiple sclerosis.7 Thereafter, three further preparations of IFNβ1a were licensed in 1998, 2000, and 2014, with more convenient dosing regimens (Table 1).8–10 The biological and immunological mechanisms of action of the interferons in multiple sclerosis are incompletely understood, but enhanced T-helper (TH)-2-cell activity and inhibition of proinflammatory cytokines and blood–brain barrier permeability are the major postulated effects.11
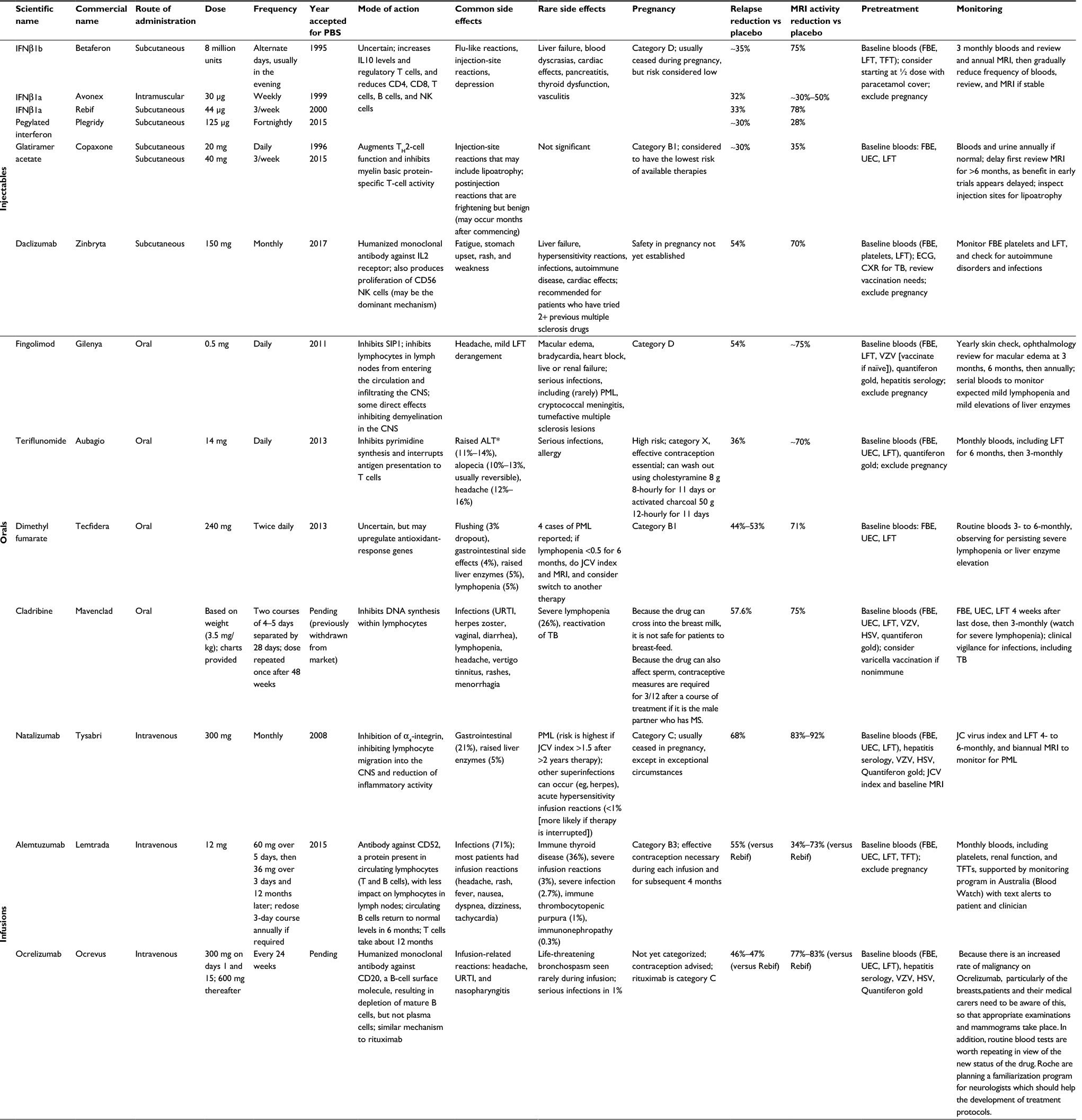 | Table 1 Disease-modifying therapies Notes: *If ALT is 2× normal, repeat the level weekly. If it reaches 3× normal, then the drug must be ceased. Betaferon®; Bayer, Berlin, Germany. Avonex®, Plegridy®, Zinbryta®, Tecfidera®, and Tysabri®; Biogen, Weston, MA, USA. Rebif® and Mavenclad®; Merck-Serono International, Darmstadt, Germany. Copaxone®; Teva Pharmaceutical Industries, Petach Tikva, Israel. Gilenya®; Novartis Pharma AG, Basel, Switzerland. Aubagio®; Sanofi-Aventis Paris, Paris, France. Lemtrada®; Sanofi-Genzyme, Cambridge, MA, USA. Ocrevus™; Roche-Genentech, South San Francisco, CA, USA. Abbreviations: PBS, Australian Pharmaceutical Benefits Scheme; NK, natural killer; FBE, full blood exam; LFT, liver-function test; TFT, thyroid-function test; MRI, magnetic resonance imaging; UEC, urea, electrolytes, creatinine; ECG, electrocardiography; CXR, chest X-ray; CNS, central nervous system; PML, progressive multifocal leukoencephalopathy; VZV, varicella zoster virus; JCV, John Cunningham virus; ALT, alanine aminotransferase; URTI, upper respiratory tract infection; TB, tuberculosis; HSV, herpes simplex virus. |
All of the interferons share the common side effects of flu-like symptoms and local injection-site reactions, as well as rarer effects on mood, transaminase elevations, cytopenias, and possibly seizures. Of these, flu-like symptoms affect almost half of all patients, and although the severity tends to decrease with duration of treatment, this remains a substantial limitation of these agents for many patients. The interferons are Australian pregnancy category D (US Food and Drug Administration [FDA] pregnancy category C), with animal data to suggest they act as abortifacients without significant teratogenicity. Human experience suggests the real-world risk is low, but contraception is firmly advised.
Although a lack of head-to-head trials hampers direct comparisons of efficacy, the availability of newer therapies means the interferons tend to be used in patients with mild disease, with rapid “uptitration” to more modern therapies if patients have evidence of active disease. The high prevalence of injection-related side effects limits their first-line use, though they remain an effective option for milder disease or for those patients who consider the newer agents’ risks to be too great.
Glatiramer acetate
Glatiramer acetate was added to the list of effective therapies after a pivotal trial was published in 1995.12 Initially developed as a drug to induce experimental autoimmune encephalitis, glatiramer acetate’s utility in multiple sclerosis emerged after it was found to act contrarily to its design.13 Its mechanism of beneficial effect in multiple sclerosis remains incompletely understood, though it is thought to augment TH2-cell function and inhibit myelin basic protein-specific T-cell activity.14 Regulatory approval in Australia followed only a few years following publication, and it has been a mainstay of therapy ever since. A combination of moderate efficacy and excellent tolerability helps to maintain its place in multiple sclerosis therapies. Injection-site reactions (including lipoatrophy) are the main side effect of note, although a minority will also experience unusual postinjection reactions of flushing, nausea, palpitations, anxiety, dyspnea, or chest pain. Although frightening for the patient, these reactions are benign and can occur even after many months of well-tolerated therapy. Importantly, glatiramer acetate is pregnancy category B1 (FDA category B), and is widely considered the safest multiple sclerosis drug in pregnancy, making it the therapy of choice for patients intending to conceive. A recent (February 2015) regulatory approval for a 40 mg thrice-weekly dose has improved convenience without sacrificing efficacy, though the drug’s moderate effect size and the need to inject subcutaneously limit its uptake as a first-line therapy. Glatiramer acetate retains an important place in pregnancy and among females of childbearing age, as well as in first-line therapy for patients eager to avoid the side effects of other therapies.
Daclizumab
Already widely used in transplant medicine as an anti-rejection drug but only recently approved for use in Australia for multiple sclerosis, daclizumab is a humanized monoclonal antibody against CD25, a subunit of the IL2 receptor. A resultant increase in CD56 killer T cells is thought likely to be the dominant mechanism of benefit in multiple sclerosis.15,16 Early studies comparing daclizumab to IFNβ1a demonstrated a favorable efficacy and side-effect profile.17 The subsequent placebo-controlled Phase III trial SELECT confirmed its effectiveness in relapsing–remitting multiple sclerosis and tested both 150 and 300 mg doses, given as subcutaneous injection monthly. These demonstrated a 54% (P<0.0001) and 50% (P=0.00015) reduction in annualized relapse rate compared to placebo, respectively.18 Importantly, given the emerging desire to see no progression on radiology, new or newly enlarged T2 lesions on magnetic resonance imaging (MRI) were reduced by 70% and 79%, respectively, at 52 weeks, and a separate trial to analyze the rate of NEDA compared to IFNβ1a found that almost twice as many patients met their criteria for NEDA over 96 weeks (24.6% vs 14.2%).19
The most common non-serious adverse effects have included upper respiratory tract infections (12%) and nasopharyngitis (12%). Rarer risks include serious infection (<1%), abnormal liver-function tests (1%), gastrointestinal side effects (2%), and malignancies (1%). At least 28% of patients will develop skin reactions, including eczema and urticaria, but 2% will suffer a severe reaction.20 It is recommended to avoid use if the liver-function tests are greater than twice the upper limit of normal at baseline. Daclizumab is FDA pregnancy category C (not yet classified in Australia). It is yet to be seen how daclizumab will fit into the therapeutic armamentarium, in view of the adverse effects and moderate efficacy.
The orals
The oral therapies have revolutionized treatment for multiple sclerosis, giving patients freedom from injections with the convenience of tablet medication and favorable side-effect profiles.
Fingolimod
In 2010 (Australian Pharmaceutical Benefits Scheme-listed in 2011), fingolimod became the first available oral treatment for relapsing–remitting multiple sclerosis. Fingolimod is a sphingosine-1-phosphate receptor modulator, a derivative from myriocin found in the fungus Isaria sinclairii, which is thought to activate sphingosine-1-phosphate receptor 1 and prevent lymphocytes egressing from lymph nodes. The FREEDOMS double-blind randomized trial (n=1,272) showed fingolimod had significantly reduced annualized relapse rates of 0.18 (0.5 mg dose) and 0.16 (1.25 mg dose) compared to placebo (0.4, P<0.001 for either dose vs placebo), a relative reduction of 54% for the smaller dose. Secondary end points included a significant reduction in disability progression at 24 months, with a hazard ratio of 0.7 and 0.68 for the 0.5 and 1.25 mg doses, respectively (P=0.02 vs placebo), in cumulative probability of disability progression (17.7% with 0.5 mg, 16.6% with 1.25 mg, and 24.1% with placebo) and in new MRI lesions and brain-volume loss.21 The TRANSFORMS study (n=1,292) comparing fingolimod to IFNβ1a showed similar results, with a significant reduction in annualized relapse rate of 0.16 for the 0.5 mg dose and 0.2 for the 1.125 mg dose compared with IFNβ1a, with a relapse rate of 0.33. No significant reduction was seen in disability progression in this study, although it only ran for 12 months.22 Long-term data over 4 years from the FREEDOMS extension study showed similar findings, with the annualized relapse rates being 0.19 for the 0.5 mg dose and 0.16 for the 1.25 mg dose compared with placebo (0.36, P<0.0001). Disability progression and new MRI lesions were also significantly reduced, as expected, at the end of the study.21
While there is little difference in efficacy between the two studied doses, side effects have been shown to be dose-dependent, and thus the recommended dose is 0.5 mg. In the TRANSFORMS study, 5.6% in the 0.5 mg group and 10% in the 1.25 mg group of patients withdrew due to adverse events. Bradycardia and atrioventricular block were seen within the TRANSFORMS study (3.6% in the 1.25 mg dose and 0.9% in the 0.5 mg dose), which are generally first-dose effects managed in practice by close cardiac monitoring for around 6 hours following the first dose, usually in a hospital setting. Viral infections also increased, with serious infections (2.5%) involving herpes and varicella, including two deaths from disseminated varicella and herpes encephalitis in the 1.25 mg-dose group in TRANSFORMS. Viral screening prior to treatment commencement has since been recommended, as well as consideration of varicella vaccination if nonimmune. Other known adverse effects include macular edema (<1%), raised alanine aminotransferase, reduction in lymphocyte count, and a modest increased risk of skin cancers. Pretreatment ophthalmology review and serial reviews thereafter are recommended to monitor for macular edema (which is reversible if detected early), though most of this risk seems to be within the first few months of therapy. Annual skin checks for malignancy are also advised. Fingolimod is a possible teratogen (Australian pregnancy category D, FDA category C) and should be stopped prior to conception (at least 2 months prior).23 The sudden occurrence of tumefactive lesions in nontumefactive multiple sclerosis has been reported with initiation of fingolimod, but this seems rare and has not been reported in the literature to any great extent in the past few years.24 A small number of cases of progressive multifocal leukoencephalopathy (PML) have also been reported.
Teriflunomide
In 2012, teriflunomide became the second oral therapy for multiple sclerosis. Teriflunomide is a derivative of leflunomide that inhibits pyrimidine synthesis and interrupts T-cell and B-cell function.25 The Phase III TEMSO study (n=1,088) showed a significant reduction in annualized relapse rate of 0.37 (7 or 14 mg dose) compared to placebo (0.54, P<0.001), amounting to a 37% relapse-rate reduction. There was also a modest reduction in disability progression with the higher dose of 14 mg.26 A follow-up trial, TOWER (n=1,169), showed a similar annualized relapse rate of 0.39 (7 mg dose) and 0.32 (14 mg dose) compared to placebo (0.5). A benefit was also seen with the higher dose in sustained reduction of disability,27 and so 14 mg is the currently marketed dose. A recently completed 9-year follow-up to the TEMSO study showed similar efficacy and improvements in those switching from placebo to teriflunomide.28 A Cochrane review supports the beneficial use of teriflunomide compared with placebo.29
High dropout rates in both the TEMSO and TOWER studies suggested poor tolerance to teriflunomide; however, the extended TEMSO study showed improved compliance and better tolerability.28 The most common adverse effects of teriflunomide are raised alanine transferase levels (11%–14%), hair thinning (10%–13%), and headache (12%–16%).27 It is suggested that teriflunomide be stopped if liver impairment develops, and it should not be used in those with liver disease. There was no significant risk in serious infections. Teriflunomide is pregnancy category X (FDA category X) and thus contraindicated in pregnancy. It is strongly advised in those at risk of pregnancy to avoid conception for at least 2 years following cessation of teriflunomide, due to residual circulation of the drug. The 9-year follow-up in the TEMSO study observed 14 pregnancies: 7 in women with multiple sclerosis on Teriflunomide, with 4 live births, and 7 in the female partners of men with multiple sclerosis on Teriflunomide, with 5 live births. 3 spontaneous abortions and 2 induced abortions accounted for the other pregnancies, with no birth defects being observed in the 9 live births.28 No cases of progressive multifocal leukoencephalopathy have been reported, to our knowledge.
Dimethyl fumarate
The third oral treatment is dimethyl fumarate, which has been shown to have immunomodulatory and neuroprotective effects. Phase III trials in 2012 included the DEFINE trial (n=1,234), which showed an annualized relapse rate at 2 years of 0.17 in the twice-daily group and 0.19 in the thrice-daily group compared to 0.36 in the placebo group (P<0.001), resulting in regulatory approval for the twice-daily dose. There was also a significant reduction in progression of disability and new MRI lesions in both treatment groups.30 The second study, CONFIRM (n=1,417), showed similar results for annualized relapse reduction: 0.22 for the twice-daily and 0.2 for the thrice-daily groups compared to the injectable glatiramer acetate (0.29) and placebo groups (0.4). Reductions in disability progression were not significant; however, there were reductions in new MRI lesions compared with placebo.31
While there have been no major safety complaints with dimethyl fumarate, there are some common complaints, including flushing (24%–31%) and gastrointestinal symptoms. Nausea and diarrhea occur in 10%–15% of patients, as well as upper abdominal pain in 10%. There have been no reported neoplasms. Lymphocyte counts drop slightly with treatment, although there is no significantly increased risk of infection.31 A few case reports of PML exist. Data are limited, but no adverse effects have been seen in fetal development or pregnancy outcomes, and the drug has been allocated pregnancy category B1 in Australia (FDA category C).32
Cladribine
Previously used as a chemotherapeutic in lymphoma, cladribine is a potent immunosuppressant, inhibiting DNA synthesis specifically in lymphocytes. The CLARITY study (n=1,326) showed a significant reduction in annualized relapse rate at 96 weeks: 0.14 for the low-dose (3.5 mg/kg) and 0.15 for the high-dose groups (5.25 mg/kg) compared with placebo (0.3, P<0.001). There was also reduction in disability progression and new MRI lesions.33 A prior study in progressive disease was not clinically effective but showed significant reductions in enhancing MRI lesions and was well tolerated.34
The safety of cladribine has been the limiting feature of its clinical approval, despite its known effectiveness. Despite initial approval in Australia in 2011, the drug was subsequently withdrawn from the market after both the FDA and the European Medicines Agency declined to provide approval for the drug, due to concerns over malignancy risk. This issue has subsequently been revisited, with a salient review and meta-analysis available suggesting the original trials overestimated malignancy risk, due to statistical handling of the unexpected zero rate of malignancy in the placebo group.35 The manufacturer is looking to make cladribine available again worldwide, with regulatory approval in Australia pending. The commercial name for oral Cladribine tablets has been changed from Movectro® to Mavenclad®.
There is expected dose-dependent lymphopenia (21.6% in the low-dose and 31.5% in the high-dose groups), resulting in an increased rate of viral infections, predominantly herpes zoster. The FDA has allocated a pregnancy category of D. Serious adverse events in CLARITY also included four deaths, with only one attributable to cladribine (tuberculosis reactivation), and ten neoplasms. These included three cases of malignant neoplasms, including a melanoma, a pancreatic carcinoma, and an ovarian carcinoma in the low-dose group and one cervical cancer in situ in the high-dose group, who had a known diagnosis of HPV16 prior to the study.33 As mentioned previously, this rate of malignancies is consistent with the general background population risk. A previous study using lower doses of subcutaneous cladribine showed a similar profile of lymphocyte suppression and only a small increase in respiratory infections.34
The infusions
The modern era has ushered in a raft of new therapies with which to manage multiple sclerosis. Chief among these, and fitting the niche of treating aggressive disease, are the infusional therapies. Administered via intravenous access at a varying schedule (Table 1), they have become an indispensable tool but do come at the cost of a variety of more severe possible side effects.
Natalizumab
A humanized monoclonal antibody against α4 integrin, natalizumab was approved by the Australian Therapeutic Goods Administration the same year as a pivotal trial demonstrating a dramatic risk reduction for clinical relapses, radiological activity, and progression of disability, heralding a new era for multiple sclerosis therapy.36 A dose of 300 mg is given 4-weekly via infusion, with the drug acting on α4 integrin to inhibit lymphocyte migration within the central nervous system, minimizing inflammation.37
The most effective of the available therapies at the time, natalizumab is remarkably well-tolerated. Acute infusion reactions, which occur in only a few percent, are the most common adverse effects. However, development of the severe and sometimes fatal PML is the most important adverse effect that has emerged, limiting more widespread use. Fortunately, serum measures of the John Cunningham virus (JCV) antibody titer, prior immunosuppression, and duration of therapy longer than 2 years are important predictors of this risk, allowing the use of this very effective therapy in most patients, even if only for a short time, and algorithms to predict risk are available.38,39 Treatment of PML consists predominantly of early detection and removing the drug from the circulation as soon as possible, but persisting neurological deficits can occur and immunoreconstitution inflammatory syndrome complicating drug withdrawal can also occur.40 Patients who are seronegative for JCV (and thus at very low risk of PML) are usually retested two to three times per year while on treatment to monitor their risk profile. It is also usually recommended that 6-monthly MRI scans are performed to monitor for this complication and assess disease activity.
Natalizumab is pregnancy category C (FDA category C), and is excreted in breast milk, requiring a careful risk–benefit discussion with the patient, including the problem of possible rebound relapse of disease on cessation.41,42 Around 6% of patients on therapy will develop neutralizing antibodies that reduce the effectiveness of therapy.43 Emerging data suggest less frequent infusions may retain efficacy while reducing side effects, which may increase the cost-effectiveness of this therapy.44 Only long-term follow up will tell whether this is sufficient and whether this less frequent dosing can reduce the frequency of progressive multifocal leukoencephalopathy. Rebound disease has also been reported on cessation, though it should be emphasized that the patient demographic for natalizumab tends to be active disease, and cessation (eg, for pregnancy) should thus generally be planned with substitution of another disease-modifying therapy as “cover” against rebound relapse. Natalizumab has found its niche as a very effective treatment for active disease, provided the threat of PML can be appropriately monitored and mitigated.
Alemtuzumab
The most recent infusional therapy and considered possibly the most potent of the subsidized therapies in Australia, alemtuzumab has a wide range of significant (and common) adverse effects generally restricting its use to a therapy of last resort in the face of very active disease. Three trials have established efficacy, and in striking contrast to other therapies’ placebo-controlled trials, the investigators of alemtuzumab compared it to high-dose beta interferon 1α.45–47 The drug is a monoclonal antibody against CD52, which depletes circulating lymphocytes, and is given as a 5-day course (12 mg/day) followed by a 3-day course 12 months later. Exactly how CD52 alters the disease course is not certain, but subsequent adverse effects consist mostly of autoimmune complications, particularly thyroid (>30% risk), but also immune thrombocytopenic purpura and glomerulonephritis (mediated by antiglomerular basement-membrane antibodies in most). These side effects require patient commitment to several years of close monitoring and monthly blood tests, and thus this treatment excludes the proportion of the population who cannot manage these stringent requirements. Infusion reactions to some degree are almost universal and necessitate covering infusions with methylprednisolone and other symptomatic treatments, though these only uncommonly require cessation of treatment. Alemtuzumab is pregnancy category B3 (FDA category C), but contraception is advised during infusions and for at least 4 months following.
Breakthrough disease following the full eight-dose regimen can be managed by retreatment with a 3-day course of infusions, as long as more than 12 months have elapsed since the last treatment.48 It is not yet clear what proportion of patients will require retreatment, although emerging data suggest only 40% of patients require retreatment over periods as long as 6–8 years of follow-up.49 For those patients who can bear the significant autoimmune risk and monthly blood-test inconvenience, however, alemtuzumab provides a sustained and powerful therapy for very aggressive disease.
B-cell therapies
Historically, multiple sclerosis has been considered a disease of T cells. There is now mounting interest in B cells and their relation to multiple sclerosis pathogenesis. B cells may contribute to inflammation and neurodegeneration through antigen presentation, autoantibody production, cytokine regulation, and formation of lymphoid-like structures in the meninges.50 The latter has been associated with meningeal inflammation, cortical demyelination, and microglial activation in progressive multiple sclerosis.51 Initial data using rituximab in Phase II trials were groundbreaking and paved the way for more aggressive investigation of this arm of therapy.52 Recent results from landmark Phase III randomized, double-blind, double-dummy-controlled trials of Ocrelizumab have demonstrated the efficacy of targeting B cells in both relapsing–remitting multiple sclerosis and also, for the first time, primary progressive multiple sclerosis.53,54
Ocrelizumab in relapsing–remitting multiple sclerosis
Trials have focused on targeting the CD20 antigen on pre-B, naïve, and memory B cells. Inherent to this specificity, such action is thought not to adversely affect repopulation of B cells or preexisting humoral immunity. Phase II trials have safely demonstrated the efficacy of rituximab and ocrelizumab.52,55 The rates of infusion reaction were higher for the former, and antichimeric antibodies were frequently detected, perhaps suggesting immunogenicity. The more humanized ocrelizumab is less immunogenic and considered a better modulator of the pathogenic response in multiple sclerosis.56 OPERA I and II were two international, multicenter, independently conducted trials using identical protocols.54 There were 1,656 trial participants. The comparator was high-dose IFNβ1a (44 mg thrice weekly). Participants had a clinicoradiological diagnosis of relapsing–remitting multiple sclerosis (2005 revised McDonald criteria) and “aggressive” disease (average 1.3 relapses in the past 12 months). Patients were excluded if they had neurologic worsening for 30 days before screening or previous exposure to other B-cell therapies (including rituximab), immunosuppression, or certain multiple sclerosis drugs. The mean age of the patients was 37 years, with an average EDSS score of 2.8 and immunotherapy-naïve.
The primary end point of annualized relapse rate by 96 weeks was significantly reduced (0.16 ocrelizumab vs 0.29 IFNβ1a). Prespecified secondary end points that reached significance were less disability progression (9.1% vs 13.6%) and more disability improvement (20.7% vs 15.6%). More patients achieved NEDA, defined as no relapse, no disability progression, no new or enlarging T2 lesions, and no contrast-enhancing lesions (OPERA I 47.9% vs 29.2%, OPERA II 47.5% vs 25.1%), but this was not considered significant following hierarchical analysis. MRI end points, including new or enlarging T2 lesions and gadolinium-enhancing lesions, favored ocrelizumab over interferon. The most common side effect was infusion-related reaction (34% vs 9.7%). There was one life-threatening nonfatal infusion reaction in the ocrelizumab group. Despite this, the authors suggested a low immunogenic potential, which was supported by only 0.4% detection of binding antibodies. Upper respiratory tract infections, nasopharyngitis, and herpesvirus infections were more common. Serious infections were reported in 1.3% vs 2.9% of patients treated with ocrelizumab compared to interferon. Importantly, fewer patients discontinued ocrelizumab because of adverse events.
Ocrelizumab in primary progressive multiple sclerosis
Before ocrelizumab, no immunotherapy had demonstrated efficacy in a Phase III trial for primary progressive multiple sclerosis. Despite not reaching the primary end point, subgroup analysis of primary progressive patients treated with rituximab (another CD20 monoclonal antibody) who were either aged less than 51 years or had gadolinium-enhancing lesions revealed significant delay in confirmed disease progression, forming the basis of the ocrelizumab trial ORATORIO.53,57 ORATORIO patients had primary progressive multiple sclerosis (2005 revised McDonald criteria), were older (median age 46 years), had an EDSS score of 4.7, and an elevated IgG index or unmatched oligoclonal bands. The comparator was placebo. The vast majority had not received prior immunotherapy. Secondary progressive multiple sclerosis patients were excluded. Enhancing lesions were observed in 27.5% of patients in the ocrelizumab group. Achieving the primary end point, there were significantly fewer ocrelizumab patients with 12-week confirmed disability progression (32.9% vs 39.3%) over at least 120 weeks. Secondary end points that reached significance in favor of ocrelizumab were 24-week confirmed disability progression and 25-foot (7.62 m) walk at week 120. The total volume of T2 lesions decreased (increased in placebo), and there was less decrease in mean brain-volume change in the ocrelizumab group compared to placebo. Overall, adverse events were slightly more common compared to placebo (95% vs 90%), but more patients taking ocrelizumab discontinued therapy (4.1% vs 3.3%). Infusion reactions with ocrelizumab were common (40%), but became less frequent with subsequent infusions. The number of serious infections was similar. Herpesvirus infections occurred in 4.7% of patients.
Ocrelizumab safety
There has been concern regarding a slightly higher rate of malignancy. Neoplasms occurred in 0.5% and 2.3% of ocrelizumab-treated patients compared to 0.2% and 0.8% of interferon and placebo patients in the OPERA and ORATORIO trials, respectively. Breast cancer appeared to be the most frequent malignancy. There was a slightly higher risk of herpesvirus-associated infections. An open-label extension phase is ongoing, which will provide long-term safety data and give insight into the durability of ocrelizumab’s benefit. To our knowledge, there are no reported cases of PML in patients treated solely with rituximab or ocrelizumab for MS.
Autologous hematopoietic stem-cell transplantation
Autologous hematopoietic stem-cell transplantation (AHSCT) uses the patient’s own cells to reconstitute the immune system, providing a “reset” function in cases of immunodysregulation, such as multiple sclerosis. Interest in the procedure for multiple sclerosis evolved rapidly through the 1990s and early 2000s as our understanding of the immunopathogenesis evolved. Although now over 800 cases treated with AHSCT exist in the literature, there have only been a few small Phase I and II randomized trials. Recently, several centers have published their extended experience with AHSCT, demonstrating that up to 90% of their cohorts remain relapse-free up to 5 years following transplant, a truly remarkable rate of success.58–61 The Australian experience of AHSCT is more limited, with only a small number of patients transplanted in large centers (registry data being collected, but not yet published), and unfortunately a small number of patients have also traveled to other countries to self-fund AHSCT, at times inappropriately.
Despite reasonable optimism, a limitation remains that the accrued experience thus far has been mostly observational data. Further, AHSCT is not a benign procedure, with early reports of transplant-related mortality of 5.3% in multiple sclerosis patients.62 The procedure has become safer in recent years, with less intensive conditioning regimens and improved care in experienced centers, but the most recent observational data still report mortality of 0–2.8%.58–60,63 Side effects of varying severity are experienced by almost all patients.
Another issue relates to patient selection. Particularly now that there exist highly effective infusional therapies, picking the patient with very active disease who will benefit more from AHSCT than, say, alemtuzumab, seems to be a challenging proposition, and more work is needed here. The increasing availability of the effective modern therapies outlined in the previous sections also warrants caution, because the only (small, Phase II) randomized study to compare AHSCT to another therapy found it did not alter disability progression relative to mitoxantrone (despite promising radiological results).64 Given this, most centers consider AHSCT after the failure of most or all other potent available therapies.
Overall, while portions of the multiple sclerosis community have welcomed AHSCT with open arms, the Australian approach is one of tempered enthusiasm, and we eagerly await randomized evidence to guide the treatment of highly aggressive or treatment-resistant disease. A consensus statement exists regarding an adequate trial to answer this critical question.65
Symptom management
Along with the increasing research and treatments available for multiple sclerosis, both with relapsing and progressive disease, there is increased awareness of the breadth and impact of the associated symptoms and diversity of research focusing on the management of these. A review conducted by MS Research Australia in 2016 sought to establish research priorities to inform future research strategy. Areas covered included varying types of research: basic, clinical, social, and applied and translational. Respondents included people with multiple sclerosis, those with a close personal connection, and health professionals and researchers working in the field of multiple sclerosis.
The top three priorities of the review were finding a cure, treatment of the disease process and prevention, and research to improve multiple sclerosis management and care: symptoms, rehabilitation, and support, particularly for those with increased disease severity. Specific symptoms highlighted in the review were walking and mobility, cognition, pain, fatigue, vision, speech, and swallowing. With milder disease, higher priority was given to stress management, lifestyle intervention, and diet, whereas more severe disease prioritized exercise, fatigue, depression, and physiotherapy to prevent disability. The following is an update of the management of multiple sclerosis symptoms.
Mobility
Mobility is a major issue, with natural history studies showing 50% of those with multiple sclerosis requiring a gait aid within 15 years. The Australian MS Longitudinal Study showed 73% of people with multiple sclerosis experience impaired balance and lower-limb weakness. In the absence of clinical disability, early gait and balance impairment can be detected.66
Changes in mobility can lead to reduced activity level, fall risk, fracture, reduced bone strength, secondary pain, fatigue, access issues, reduced self-esteem, and altered body image, together with economic problems related to cost of aids, transport, and loss of employment. Unemployment of those with multiple sclerosis is largely due to ineffective management of symptoms in the workplace rather than workplace-related factors.67
There is a need to consider many factors in addressing the management of impaired mobility. Improving general fitness and flexibility can improve endurance, increase bone strength, and reduce fatigue, whereas specific physical strategies can address spasticity, balance, and strength and focus on preventing secondary musculoskeletal problems.
Fampridine, a potassium-channel blocker, has been shown in clinical trials to improve speed of ambulation in 30% of subjects, with sustained response over years.68 A further study, ENABLE, assessed the effect of prolonged-release fampridine on patient-reported health impact. Improved patient-perceived physical and psychological health impact was seen over 48 weeks in those who initially responded with objective walking improvement at 4 weeks.69
Spasticity
While spasticity is commonly experienced by those with multiple sclerosis, it can vary in degree of severity in individuals and over time. Spasticity can be aggravated by changes in temperature, infections, bladder and bowel problems, and postural changes. Increase in tone can be a two-edged sword, as patients with considerable underlying weakness may benefit from the splinting effect that spasticity can provide, maintaining functional abilities, such as standing and walking. Individual assessment is required, and treatment usually involves physiotherapy and stretching routines in the first instance. If further treatment is indicated, baclofen (oral or intrathecal), benzodiazepines, and botulinum-toxin injections can be used, and a recent up-to-date review of these is available.70 A medicinal cannabis-based oral spray, Sativex® (GW Pharmaceuticals, Cambridge, UK), has been shown in clinical trials to reduce moderate–severe spasticity effectively in multiple sclerosis as an adjunct to current treatments.71
Pain
Pain in multiple sclerosis is either a direct result of the disease process or can be secondary to musculoskeletal problems. Accurate diagnosis is needed, and while treatment is usually successful with primary neurogenic pain using such medications as carbamazepine or amitriptyline, unfortunately some cases require multidisciplinary management and referral to pain clinics.
Fatigue
Fatigue, often referred to as the “hidden disability”, occurs in about 80% people with multiple sclerosis, and the exact cause is unknown. Overwhelming lassitude, commonly felt in the afternoon, is one type, the other being physical fatigue, with increased weakness seen with repetitive activity such as walking. It is important to rule out other causes, such as medical conditions, medications, poor general fitness, and sleep disorders, before putting fatigue down to multiple sclerosis. Medications have been shown to be of little benefit in treating multiple sclerosis fatigue.72
A multidisciplinary approach remains paramount, focusing on general fitness, good sleep routine, and energy-conservation techniques in activities of daily life. A review of clinical trials of the anti-Parkinsonian drug amantadine (Symmetrel®; Novartis Pharma AG, Basel, Switzerland) showed small and inconsistent improvement in 20%–40% of patients over the short term.73 The amphetamine-like drug modafinil (Provigil®; Teva Pharmaceutical Industries, Petach Tikva, Israel) has been used, but trial data are conflicting as to its efficacy and serious psychiatric side effects can occur.74
Cognition
The French neurologist Jean-Martin Charcot (1825–1893), in his clinical description of multiple sclerosis as a distinct disease, observed cognitive symptoms in his patients: “[…] there is marked enfeeblement of the memory; conceptions are formed slowly”. However, there was little acknowledgment of cognitive difficulties in multiple sclerosis until more recent times. Cognitive dysfunction occurs in up to 60% of people with multiple sclerosis and ranges from mild to moderate problems to (rarely) dementia. Cognitive problems can have a negative effect on employment, relationships, and overall quality of life of an individual. The most frequent symptoms are reduced speed of information processing, reduced memory, learning and concentration difficulties, reduced planning and organizational ability, and difficulty with problem solving.
Neuropsychological assessment can define the issues and assist with practical management ideas, such as use of diaries and alarms. Cognitive rehabilitation is an approach to cognitive retraining teaching compensatory strategies together with exercises to improve impaired cognitive function.75 Recent research is showing promising results using computer-aided programs promoting brain plasticity.76
Depression
Depression is more common in multiple sclerosis than in the general population, occurring in up to 50% of cases and at any time throughout the course of the disease. Structural changes on MRI and genetic, biochemical, immunological, and psychosocial factors have all been implicated in the etiology of depression in multiple sclerosis. There is often a complex association with coexisting fatigue, cognitive impairment, pain, and anxiety. Treatment with antidepressant medication is often combined with psychotherapy and cognitive behavioral therapy.77 A meta-analysis on the effect of exercise on depression in multiple sclerosis showed no conclusion that it prevented or reduced depressive symptoms, but suggested future well-designed studies are highly warranted.78
Tremor
Intention tremor, occurring in up to 20% of cases and the cause of marked functional disability, remains a difficult symptom to manage. Although there have been individual responses to a variety of drugs, including gabapentin, clonazepam, and isoniazid, there is no drug that is consistently effective. Recent trials of botulinum toxin have shown some benefit if the tremor is not too complex.79 Surgical treatment has never been as successful as in Parkinson’s disease; however, recently new targets have been identified for surgery and deep-brain stimulation that suggest a better outcome.80
Bladder and bowel dysfunction
Bladder symptoms are very common in multiple sclerosis, affecting 80% of patients. It is first important to determine the cause: failure to store urine (detrusor dysfunction), failure to empty urine (sphincter dysfunction), or a combination of the two. Reversible exacerbating factors, such as infections or constipation, should be identified and treated. A “failure to store” problem may warrant a trial of anticholinergics, and a failure to empty may warrant intermittent self-catheterization or permanent catheter placement if more severe. Occasional use of an antidiuretic hormone has been used in some centers to overcome urinary dysfunction for special occasions, but this is a challenging therapy to access for this indication in Australia. Botulinum-toxin intravesical injection may also be of assistance for detrusor overactivity if anticholinergics fail or are not tolerated.
Constipation is common in multiple sclerosis, resulting from the disease itself but often exacerbated by poor fluid intake, reduced physical activity, dietary changes, and medications. Less commonly, diarrhea and fecal incontinence can occur. Individual assessment is required, with attention to routine, diet, and fluid input, together with consideration of medications, stool softeners, aperients, or enemas.
Sexuality
Sexual difficulties are common in multiple sclerosis and can be due to neurological impairment, multiple sclerosis-related symptoms, such as fatigue, spasticity, or continence problems, medications, and psychosocial issues, or a combination of these. Management requires a broad biopsychosocial approach to assessment and management.81 The availability of several PDE5 inhibitors in recent years has added another treatment option for males with erectile dysfunction.82
Summary
As advances are seen in the treatment options available for people with relapsing–remitting multiple sclerosis and increasing focus on the progressive forms of multiple sclerosis, there is also further need to research and develop improved treatments for the diverse and complex symptoms associated with multiple sclerosis that impact on individuals’ quality of life.
Conclusion
The advances described herein have been remarkable and brought both relief and hope to many of our multiple sclerosis patients. However, many questions remain regarding the management of this complex disease.
Can the side effects of our two most powerful drugs (natalizumab and alemtuzumab) be controlled or ameliorated? With natalizumab, the main concern is regarding the complication of PML due to JCV. This has a 20% mortality and serious neurological sequelae in many of the survivors. Some progress has been made by the development of a test for JCV antibodies and the development of an antibody index that can be used to estimate the risk for the individual. In addition, the MRI studies of Wattjes et al have shown that that the disease behaves differently in multiple sclerosis to its occurrence in HIV, in that it appears to start in one lobe of the cerebral hemispheres and then spreads to others.83 If the drug is ceased while it is unilobar, the prognosis is much better. Unfortunately, these insights have not yet reduced the overall mortality of this complication, and work continues, with current hopes resting on refinement of risk stratification using L-selectin84 and the possible development of a vaccine against the virus. The total number of patients with multiple sclerosis affected by PML now numbers over 700 (unpublished data).
In the case of alemtuzumab, the main concern has been the development of autoimmune diseases, mainly thyroid (an incidence of a little over a third), but also immune thrombocytopenic purpura and immune glomerulonephritis. However, the 6-year follow-up presented at ECTRIMS in 2016 showed no increase in mortality or withdrawal from the study. Additionally, the autoimmune complications peaked in year 3 and then markedly reduced.
Obviously with the present number of drugs available and more in the pipeline, difficult questions arise as to which patients should receive which drugs. We will need better methods of prognosis verified by long-term follow-up. Freedman et al have developed a qualitative descriptive system that is now in its second iteration.85 Río et al have shown that clinical events or new MRI lesions in the first year of treatment are reliable indicators of prognosis, but more work in this area is needed.86 As a further complexity, the availability of multiple effective therapies for active disease means patients now recruited into trials of new agents tend to have less active disease than the initial trials of interferons, for example. This makes comparison of effect sizes difficult between the newer and older therapies, as it might be considered that it is less difficult to control less active disease, and thus demonstrate a more substantial relative risk reduction.
If we at last have in ocrelizumab the first drug shown to slow the progress of primary progressive multiple sclerosis in a Phase III trial, what about secondary progressive multiple sclerosis? We know that IFNβ1b showed a positive result in the European multicenter trial of secondary progressive multiple sclerosis, but an identical trial in the US showed no benefit.87
We now know that a lot of the secondary progressive multiple sclerosis patients in the European trial were still relapsing, though progressive, whereas the American trial had virtually all without relapses. Therefore, there is an urgent need to perform trials on secondary progressive multiple sclerosis patients without relapses using ocrelizumab and similar drugs, because these patients have a poorer prognosis.
We also need careful, very long-term follow-up of patients with apparently benign multiple sclerosis. We all see patients who, after a benign first decade with multiple sclerosis, have serious problems in the second decade, but what about patients first seen after more than 20 years of benign disease? Do they stay benign? What happens when aging in the central nervous system interacts with benign demyelination? We unfortunately have no medical details of their condition, but the intriguing fact is that in a 21-year follow-up of patients from the original IFNβ1b trial, those on the drug from the start lived on average 10 years longer than those in the placebo group, who went on the drug 6 years later.88 However, the reasons for this difference remain unclear.
What about AHSCT? This remains a controversial area and one where patients often disagree with their neurologist and if necessary travel overseas to obtain transplant. The problem is that there is only limited evidence so far. Freedman et al had published in 2016 a small Phase II trial with dramatic results, which received wide publicity and brought the question of hematopoietic transplantation back to the forefront of discussion about the treatment of severe multiple sclerosis.89 Their trial was not controlled, and the cytotoxic regimen used included cyclophosphamide, busulfan, and antithymocyte globulin. A total of 23 of the 24 patients did well, with some patients exhibiting actual improvement, but there was one death. All the patients were relatively early in their course, though obviously had very severe multiple sclerosis. This would accord with the impression given by the European Group for Blood and Marrow Transplantation registry of patients that if given early in the clinical course, hematopoietic transplantation can alter the course of the disease. Unfortunately, such methods have not shown to be of benefit for patients with established, advanced, and progressive disease. The European registry showed a striking difference between early severe relapsing–remitting patients and late secondary progressive patients, who did not benefit and had a high complication rate.62 Freedman et al feel that further controlled trials using more current immunosuppressants should proceed, but they warn about the dangers and advise against medical tourism to obtain the procedure. In talking to patients about their procedure, it is important to establish early on that the stem cells are blood stem cells and that the procedure is essentially the same as that used to treat acute leukemia, and not stem cells in the sense of cells that will travel to the central nervous system and effect repair. We hope that trials of this technique using more modern induction regimens will be explored in a controlled fashion.
With the advent of more powerful therapies for multiple sclerosis, the concept of NEDA has evolved. This refers to the proportion of patients with no evidence of clinical or radiological activity at the end of a clinical trial (ie, no relapses or increase in disability and no new MRI lesions). A NEDA grading scale has evolved, encompassing automated volume calculation based on MRI. However, as pointed out by the distinguished Dutch neuroradiologist Barkhof, these values are unreliable if applied to individuals as opposed to groups, because factors such as hydration can greatly influence them.90 Our main concern is that in evaluating a drug for a patient, there may be additional factors that are important and are not measured by NEDA. An example would be glatiramer for a multiple sclerosis patient thinking of pregnancy. Glatiramer predates the concept of NEDA, though would not be expected to have a high NEDA score. However, clearly for the patient contemplating pregnancy, glatiramer may be more appropriate than many other drugs with a theoretically higher NEDA score in trials.
Therefore, there are many questions that remain to be answered about this complex disease, both in understanding and in treating it. However, there is no doubt that significant progress has been made.
Acknowledgments
We are very grateful to our colleagues in the Department of Neurology at St Vincent’s Hospital Melbourne for their assistance and advice and to Professor Richard McDonnell of the Department of Neurology, Austin Hospital, Melbourne, for helpful discussion and provision of references.
Disclosure
Over the last 3-years, Dr Sedal has attended many meetings in Melbourne and Sydney to hear Australian and overseas speakers discuss various aspects of MS therapy. These meetings and speakers have been sponsored by all the companies with products in the field. Dr Sedal has been involved in many clinical trials, the most recent being the ocrelizumab trial in primary progressive MS. He received funding from Roche to attend a 1-day meeting of the Advisory board held in Sydney in early 2016. The other authors of this paper have nothing to declare and report no conflicts of interest in this work.
References
Vasconcelos CC, Aurenção JC, Thuler LC, Camargo S, Alvarenga MP, Alvarenga RM. Prognostic factors associated with long-term disability and secondary progression in patients with multiple sclerosis. Mult Scler Relat Disord. 2016;8:27–34. | ||
Leocani L, Rocca MA, Comi G. MRI and neurophysiological measures to predict course, disability and treatment response in multiple sclerosis. Cur Opin Neurol. 2016;29:243–253. | ||
Giovannoni G. Multiple sclerosis should be treated using a step-down strategy rather than a step-up strategy – yes. Mult Scler. 2016;22:1397–1400. | ||
Naismith RT. Multiple sclerosis should be treated using a step-down strategy rather than a step-up strategy – no. Mult Scler. 2016;22:1400–1402. | ||
Członkowska A, Smoliński Ł, Litwin T. Severe disease exacerbations in patients with multiple sclerosis after discontinuing fingolimod. Neurol Neurochir Pol. 2017;51:156–162. | ||
Fagius J, Feresiadou A, Larsson EM, Burman J. Discontinuation of disease modifying treatments in middle aged multiple sclerosis patients: first line drugs vs natalizumab. Mult Scler Relat Disord. 2017;12:82–87. | ||
IFNB Multiple Sclerosis Study Group. Interferon beta-1b is effective in relapsing-remitting multiple sclerosis I: clinical results of a multicenter, randomized, double-blind, placebo-controlled trial. Neurology. 1993;43:655–661. | ||
Jacobs LD, Cookfair DL, Rudick RA, et al. Intramuscular interferon beta-1a for disease progression in relapsing multiple sclerosis. Ann Neurol. 1996;39:285–294. | ||
Calabresi PA, Kieseier BC, Arnold DL, et al. Pegylated interferon β-1a for relapsing-remitting multiple sclerosis (ADVANCE): a randomised, phase 3, double-blind study. Lancet Neurol. 2014;13:657–665. | ||
Ebers GC. Randomised double-blind placebo-controlled study of interferon β-1a in relapsing/remitting multiple sclerosis. Lancet. 1998;352:1498–1504. | ||
Markowitz CE. Interferon-beta: mechanism of action and dosing issues. Neurology. 2007;68:S8–S11. | ||
Johnson KP, Brooks BR, Cohen JA, et al. Copolymer 1 reduces relapse rate and improves disability in relapsing-remitting multiple sclerosis: results of a phase III multicenter, double-blind, placebo-controlled trial. Neurology. 1995;45:1268–1276. | ||
Broadley SA, Barnett MH, Boggild M, et al. Therapeutic approaches to disease modifying therapy for multiple sclerosis in adults: an Australian and New Zealand perspective – part 1: historical and established therapies. J Clin Neurosci. 2014;21:1835–1846. | ||
Racke MK, Lovett-Racke AE. Glatiramer acetate treatment of multiple sclerosis: an immunological perspective. J Immunol. 2011;186:1887–1890. | ||
Sheridan JP, Zhang Y, Riester K, et al. Intermediate-affinity interleukin-2 receptor expression predicts CD56bright natural killer cell expansion after daclizumab treatment in the CHOICE study of patients with multiple sclerosis. Mult Scler. 2011;17:1441–1448. | ||
Martin JF, Perry JS, Jakhete NR, Wang X, Bielekova B. An IL-2 paradox: blocking CD25 on T cells induces IL-2-driven activation of CD56bright NK cells. J Immunol. 2010;185:1311–1320. | ||
Kappos L, Wiendl H, Selmaj K, et al. Daclizumab HYP versus interferon beta-1a in relapsing multiple sclerosis. N Engl J Med. 2015;373:1418–1428. | ||
Gold R, Giovannoni G, Selmaj K, et al. Daclizumab high-yield process in relapsing-remitting multiple sclerosis (SELECT): a randomised, double-blind, placebo-controlled trial. Lancet. 2013;381:2167–2175. | ||
Kappos L, Havrdova E, Giovannoni G, et al. No evidence of disease activity in patients receiving daclizumab versus intramuscular interferon beta-1a for relapsing-remitting multiple sclerosis in the DECIDE study. Mult Scler. Epub 2016 Dec 1. | ||
Gold R, Radue EW, Giovannoni G, et al. Safety and efficacy of daclizumab in relapsing-remitting multiple sclerosis: 3-year results from the SELECTED open-label extension study. BMC Neurol. 2016;16:117. | ||
Kappos L, O’Connor P, Radue EW, et al. Long-term effects of fingolimod in multiple sclerosis: the randomized FREEDOMS extension trial. Neurology. 2015;84:1582–1591. | ||
Cohen JA, Barkhof F, Comi G, et al. Oral fingolimod or intramuscular interferon for relapsing multiple sclerosis. N Engl J Med. 2010;362:402–415. | ||
Karlsson G, Francis G, Koren G, et al. Pregnancy outcomes in the clinical development program of fingolimod in multiple sclerosis. Neurology. 2014;82:674–680. | ||
Pilz G, Harrer A, Wipfler P, et al. Tumefactive MS lesions under fingolimod: a case report and literature review. Neurology. 2013;81:1654–1658. | ||
Bar-Or A, Pachner A, Menguy-Vacheron F, Kaplan J, Wiendl H. Teriflunomide and its mechanism of action in multiple sclerosis. Drugs 2014;74:659–674. | ||
O’Connor P, Wolinsky JS, Confavreux C, et al. Randomized trial of oral teriflunomide for relapsing multiple sclerosis. N Engl J Med. 2011;365:1293–1303. | ||
Confavreux C, O’Connor P, Comi G, et al. Oral teriflunomide for patients with relapsing multiple sclerosis (TOWER): a randomised, double-blind, placebo-controlled, phase 3 trial. Lancet Neurol. 2014;13:247–256. | ||
O’Connor P, Comi G, Freedman MS, et al. Long-term safety and efficacy of teriflunomide: nine-year follow-up of the randomized TEMSO study. Neurology. 2016;86:920–930. | ||
He D, Zhang C, Zhao X, et al. Teriflunomide for multiple sclerosis. Cochrane Database Syst Rev. 2016;3:CD009882. | ||
Gold R, Kappos L, Arnold DL, et al. Placebo-controlled phase 3 study of oral BG-12 for relapsing multiple sclerosis. N Engl J Med. 2012;367:1098–1107. | ||
Fox RJ, Miller DH, Phillips JT, et al. Placebo-controlled phase 3 study of oral BG-12 or glatiramer in multiple sclerosis. N Engl J Med. 2012;367:1087–1097. | ||
Gold R, Phillips JT, Havrdova E, et al. Delayed-release dimethyl fumarate and pregnancy: preclinical studies and pregnancy outcomes from clinical trials and postmarketing experience. Neurol Ther. 2015;4:93–104. | ||
Giovannoni G, Comi G, Cook S, et al. A placebo-controlled trial of oral cladribine for relapsing multiple sclerosis. N Engl J Med. 2010;362:416–426. | ||
Rice GP, Filippi M, Comi G. Cladribine and progressive MS: clinical and MRI outcomes of a multicenter controlled trial. Neurology. 2000;54:1145–1155. | ||
Pakpoor J, Disanto G, Altmann DR, et al. No evidence for higher risk of cancer in patients with multiple sclerosis taking cladribine. Neurol Neuroimmunol Neuroinflamm. 2015;2:e158. | ||
Polman CH, O’Connor PW, Havrdova E, et al. A randomized, placebo-controlled trial of natalizumab for relapsing multiple sclerosis. N Engl J Med. 2006;354:899–910. | ||
Rice GP, Hartung HP, Calabresi PA. Anti-α4 integrin therapy for multiple sclerosis: mechanisms and rationale. Neurology. 2005;64:1336–1342. | ||
Plavina T, Subramanyam M, Bloomgren G, et al. Anti-JC virus antibody levels in serum or plasma further define risk of natalizumab-associated progressive multifocal leukoencephalopathy. Ann Neurol. 2014;76:802–812. | ||
Bloomgren G, Richman S, Hotermans C, et al. Risk of natalizumab-associated progressive multifocal leukoencephalopathy. N Engl J Med. 2012;366:1870–1880. | ||
Tan IL, McArthur JC, Clifford DB, Major EO, Nath A. Immune reconstitution inflammatory syndrome in natalizumab-associated PML. Neurology. 2011;77:1061–1067. | ||
Fox RJ, Cree BA, de Seze J, et al. MS disease activity in RESTORE: a randomized 24-week natalizumab treatment interruption study. Neurology. 2014;82:1491–1498. | ||
Jokubaitis VG, Li V, Kalincik T, et al. Fingolimod after natalizumab and the risk of short-term relapse. Neurology. 2014;82:1204–1211. | ||
Calabresi PA, Giovannoni G, Confavreux C, et al. The incidence and significance of anti-natalizumab antibodies: results from AFFIRM and SENTINEL. Neurology. 2007;69:1391–1403. | ||
Ryerson LZ, Frohman TC, Foley J, et al. Extended interval dosing of natalizumab in multiple sclerosis. J Neurol Neurosurg Psychiatry. 2016;87:885–889. | ||
Coles AJ, Compston DA, Selmaj KW, et al. Alemtuzumab vs. interferon beta-1a in early multiple sclerosis. N Engl J Med. 2008;359:1786–1801. | ||
Coles AJ, Twyman CL, Arnold DL, et al. Alemtuzumab for patients with relapsing multiple sclerosis after disease-modifying therapy: a randomised controlled phase 3 trial. Lancet. 2012;380:1829–1839. | ||
Cohen JA, Coles AJ, Arnold DL, et al. Alemtuzumab versus interferon beta 1a as first-line treatment for patients with relapsing-remitting multiple sclerosis: a randomised controlled phase 3 trial. Lancet. 2012;380:1819–1828. | ||
Freedman MS, Rush CA. Severe, highly active, or aggressive multiple sclerosis. Continuum (Minneap Minn). 2016;22:761–784. | ||
Willis MD, Harding KE, Pickersgill TP, et al. Alemtuzumab for multiple sclerosis: long term follow-up in a multi-centre cohort. Mult Scler. 2016;22:1215–1223. | ||
Hauser SL. The Charcot Lecture –beating MS: a story of B cells, with twists and turns. Mult Scler. 2015;21:8–21. | ||
Howell OW, Reeves CA, Nicholas R, et al. Meningeal inflammation is widespread and linked to cortical pathology in multiple sclerosis. Brain. 2011;134:2755–2771. | ||
Hauser SL, Waubant E, Arnold DL, et al. B-cell depletion with rituximab in relapsing-remitting multiple sclerosis. N Engl J Med. 2008;358:676–688. | ||
Montalban X, Hauser SL, Kappos L, et al. Ocrelizumab versus placebo in primary progressive multiple sclerosis. N Engl J Med. 2017;376:209–220. | ||
Hauser SL, Bar-Or A, Comi G, et al. Ocrelizumab versus interferon beta-1a in relapsing multiple sclerosis. N Engl J Med. 2017;376:221–234. | ||
Kappos L, Li D, Calabresi PA, et al. Ocrelizumab in relapsing-remitting multiple sclerosis: a phase 2, randomised, placebo-controlled, multicentre trial. Lancet. 2011;378:1779–1787. | ||
Sørensen PS, Blinkenberg M. The potential role for ocrelizumab in the treatment of multiple sclerosis: current evidence and future prospects. Ther Adv Neuro Disord. 2016;9:44–52. | ||
Hawker K, O’Connor P, Freedman MS, et al. Rituximab in patients with primary progressive multiple sclerosis: results of a randomized double-blind placebo-controlled multicenter trial. Ann Neurol. 2009;66:460–471. | ||
Burt RK, Balabanov R, Han X, et al. Association of nonmyeloablative hematopoietic stem cell transplantation with neurological disability in patients with relapsing-remitting multiple sclerosis. JAMA. 2015;313:275–284. | ||
Shevchenko JL, Kuznetsov AN, Ionova TI, et al. Long-term outcomes of autologous hematopoietic stem cell transplantation with reduced-intensity conditioning in multiple sclerosis: physician’s and patient’s perspectives. Ann Hematol. 2015;94:1149–1157. | ||
Burman J, Iacobaeus E, Svenningsson A, et al. Autologous haematopoietic stem cell transplantation for aggressive multiple sclerosis: the Swedish experience. J Neurol Neurosurg Psychiatry. 2014;85:1116–1121. | ||
Nash RA, Hutton GJ, Racke MK, et al. High-dose immunosuppressive therapy and autologous HCT for relapsing-remitting MS. Neurology. 2017;88:842–852. | ||
Saccardi R, Kozak T, Bocelli-Tyndall C, et al. Autologous stem cell transplantation for progressive multiple sclerosis: update of the European Group for Blood and Marrow Transplantation autoimmune diseases working party database. Mult Scler. 2006;12:814–823. | ||
Muraro PA, Pasquini M, Atkins HL, et al. Long-term outcomes after autologous hematopoietic stem cell transplantation for multiple sclerosis. JAMA Neurol. 2017;74:459–469. | ||
Mancardi GL, Sormani MP, Gualandi F, et al. Autologous hematopoietic stem cell transplantation in multiple sclerosis: a phase II trial. Neurology. 2015;84:981–988. | ||
Saccardi R, Freedman MS, Sormani MP, et al. A prospective, randomized, controlled trial of autologous haematopoietic stem cell transplantation for aggressive multiple sclerosis: a position paper. Mult Scler. 2012;18:825–834. | ||
Martin CL, Phillips BA, Kilpatrick TJ, et al. Gait and balance impairment in early multiple sclerosis in the absence of clinical disability. Mult Scler. 2006;12:620–628. | ||
Simmons RD, Tribe KL, McDonald EA. Living with multiple sclerosis: longitudinal changes in employment and the importance of symptom management. J Neurol. 2010;257:926–936. | ||
Goodman AD, Brown TR, Krupp LB, et al. Sustained-release oral fampridine in multiple sclerosis: a randomised, double-blind, controlled trial. Lancet. 2009;373:732–738. | ||
Macdonell R, Nagels G, Laplaud DA, et al. Improved patient-reported health impact of multiple sclerosis: the ENABLE study of PR-fampridine. Mult Scler. 2016;22:944–954. | ||
Otero-Romero S, Sastre-Garriga J, Comi G, et al. Pharmacological management of spasticity in multiple sclerosis: systematic review and consensus paper. Mult Scler. 2016;22:1386–1396. | ||
Flachenecker P, Henze T, Zettl UK. Nabiximols (THC/CBD oromucosal spray, Sativex®) in clinical practice: results of a multicenter, non-interventional study (MOVE 2) in patients with multiple sclerosis spasticity. Eur Neurol. 2014;71:271–279. | ||
Kesselring J, Beer S. Symptomatic therapy and neurorehabilitation in multiple sclerosis. Lancet Neurol. 2005;4:643–652. | ||
Pucci E, Branãs P, D’Amico R, Giuliani G, Solari A, Taus C. Amantadine for fatigue in multiple sclerosis. Cochrane Database Syst Rev. 2007;14:CD002818. | ||
National Institute for Health and Care Excellence. Fatigue in Multiple Sclerosis: Modafinil. London: NICE; 2013. | ||
Chiaravalloti ND, Moore NB, Nikelshpur OM, DeLuca J. An RCT to treat learning impairment in multiple sclerosis: the MEMREHAB trial. Neurology. 2013;81:2066–2072. | ||
Bonavita S, Sacco R, Corte Della M, et al. Computer-aided cognitive rehabilitation improves cognitive performances and induces brain functional connectivity changes in relapsing remitting multiple sclerosis patients: an exploratory study. J Neurol. 2015;262:91–100. | ||
Feinstein A, Magalhaes S, Richard JF, Audet B, Moore C. The link between multiple sclerosis and depression. Nat Rev Neurol. 2014;10:507–517. | ||
Dalgas U, Sloth M, Stenager E. The effect of exercise on depressive symptoms in multiple sclerosis based on a meta-analysis and critical review of the literature. Eur J Neurol. 2015;22:443–456. | ||
Van Der Walt A, Sung S, Spelman T, et al. A double-blind, randomized, controlled study of botulinum toxin type A in MS-related tremor. Neurology. 2012;79:92–99. | ||
Xie T, Bernard J, Warnke P. Post subthalamic area deep brain stimulation for tremors: a mini-review. Transl Neurodegener. 2012;1:20. | ||
Kalb RC. Intimacy and Sexuality in MS. New York: National Multiple Sclerosis Society; 2012. | ||
Fowler CJ, Miller JR, Sharief MK, Hussain IF, Stecher VJ, Sweeney M. A double blind, randomised study of sildenafil citrate for erectile dysfunction in men with multiple sclerosis. J Neurol Neurosurg Psychiatry. 2005;76:700–705. | ||
Wattjes MP, Vennegoor A, Steenwijk MD, et al. MRI pattern in asymptomatic natalizumab-associated PML. J Neurol Neurosurg Psychiatry. 2014;86:793–798. | ||
Schwab N, Schneider-Hohendorf T, Pignolet B, et al. PML risk stratification using anti-JCV antibody index and L-selectin. Mult Scler. 2016;22:1048–1060. | ||
Freedman MS, Selchen D, Arnold DL, et al. Treatment optimization in MS: Canadian MS Working Group updated recommendations. Can J Neurol Sci. 2013;40:307–323. | ||
Río J, Rovira A, Tintorè M, et al. Disability progression markers over 6-12 years in interferon-β-treated multiple sclerosis patients. Mult Scler. Epub 2017 Mar 1. | ||
Kappos L, Weinshenker B, Pozzilli C, et al. Interferon beta-1b in secondary progressive MS: a combined analysis of the two trials. Neurology. 2004;63:1779–1787. | ||
Goodin DS, Reder AT, Ebers GC, et al. Survival in MS: a randomized cohort study 21 years after the start of the pivotal IFNβ-1b trial. Neurology. 2012;78:1315–1322. | ||
Atkins HL, Bowman M, Allan D, et al. Immunoablation and autologous haemopoietic stem-cell transplantation for aggressive multiple sclerosis: a multicentre single-group phase 2 trial. Lancet. 2016;388:576–585. | ||
Barkhof F. Brain atrophy measurements should be used to guide therapy monitoring in MS – no. Mult Scler. 2016;22:1524–1526. |
 © 2017 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2017 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.