")
Back to Journals » International Journal of Nanomedicine » Volume 18
Current Anti-Amyloid-β Therapy for Alzheimer’s Disease Treatment: From Clinical Research to Nanomedicine
Authors Zhao Z , Liu Y, Ruan S, Hu Y
Received 11 October 2023
Accepted for publication 12 December 2023
Published 20 December 2023 Volume 2023:18 Pages 7825—7845
DOI https://doi.org/10.2147/IJN.S444115
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Professor R.D.K. Misra
Zixuan Zhao,1,2,* Yun Liu,1,2,* Shirong Ruan,1,2 Yixuan Hu1,2
1Department of Neurosurgery, The Translational Research Institute for Neurological Disorders, The First Affiliated Hospital of Wannan Medical College (Yijishan Hospital of Wannan Medical College), Wuhu, 241000, People’s Republic of China; 2The Institute of Brain Science, Wannan Medical College, Wuhu, 241000, People’s Republic of China
*These authors contributed equally to this work
Correspondence: Zixuan Zhao, Department of Neurosurgery, The Translational Research Institute for Neurological Disorders, The First Affiliated Hospital of Wannan Medical College (Yijishan Hospital of Wannan Medical College), 2 West Zheshan Road, Wuhu, 241000, People’s Republic of China, Tel +86 18895631652, Email [email protected]
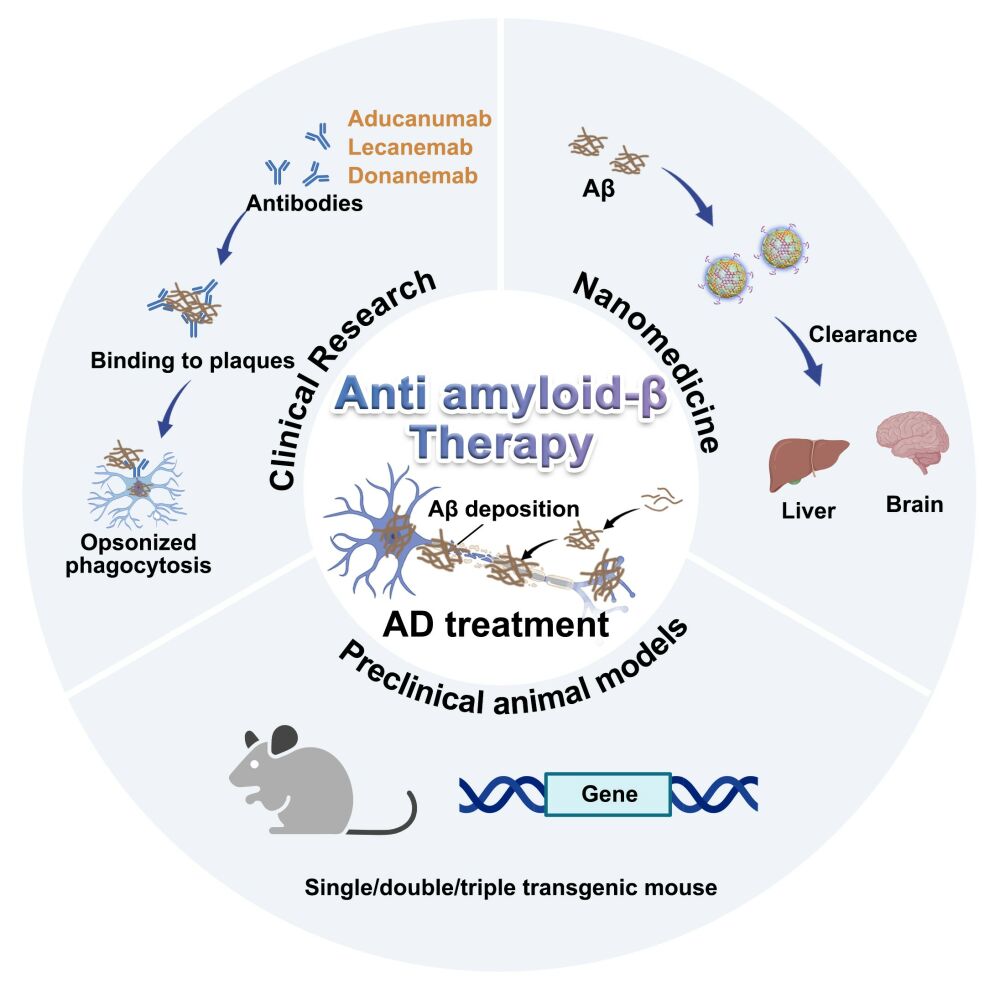
Abstract: Recent successive approval of anti-amyloid-β (Aβ) monoclonal antibodies as disease-modifying therapies against Alzheimer’s disease (AD) has raised great confidence in the development of anti-AD therapies; however, the current therapies still face the dilemma of significant adverse reactions and limited effects. In this review, we summarized the therapeutic characteristics of the approved anti-Aβ immunotherapies and dialectically analyzed the gains and losses from clinical trials. The review further proposed the reasonable selection of animal models in preclinical studies from the perspective of different animal models of Aβ deposition and deals in-depth with the recent advances of exploring preclinical nanomedical application in Aβ targeted therapy, aiming to provide a reliable systematic summary for the development of novel anti-Aβ therapies. Collectively, this review comprehensively dissects the pioneering work of Aβ-targeted therapies and proposed perspective insight into AD-modified therapies.
Keywords: Alzheimer’s disease, amyloid-β, anti-amyloid-β monoclonal antibody, disease-modifying therapeutics, nanomedicine
Graphical Abstract:
Introduction
Alzheimer’s disease (AD) is the most common type of dementia causing tens of millions of elderlies suffering from cognitive decline and behavioral impairment.1 Although it has been identified for more than 100 years, the cognitive deterioration of AD has remained frustratingly to develop disease-modifying therapeutics despite massive efforts. Most of the current research has been philosophically guided by the connection of the hallmark histopathology of AD, including the senile plaques (SP) formed by deposited amyloid-β (Aβ) and neurofibrillary tangles (NFTs) formed by hyperphosphorylated microtubule-associated protein tau. Before the first approval of Aβ antibody (aducanumab) in 2021 as the first disease-modifying therapy,2 the Food and Drug Administration (FDA) had only approved a handful of drugs to relieve the condition, including acetylcholine esterase (AChE) inhibitors and N-methyl-D-aspartic acid (NMDA) receptor modulators. This class of symptomatic remission drugs has been reported to be gradually discontinued by patients due to serious adverse reactions and weak disease remission,3 thus it is not hard to understand the encouraging success of the groundbreaking approval of aducanumab for confronting AD. Based on clarifying the shared role of Aβ deposition in AD as the clearest instance of cross-talk between neurodegenerative and cerebrovascular processes, lecanumab, the next generation of Aβ antibodies, was approved two years later with overwhelming support. These achievements have, to some extent, cleared the cloud in the development of AD therapies and represent the effectiveness of therapeutic strategies with Aβ as the mainstream intervention target.4 In this context, there is great potential to extend the entire life cycle of Aβ production and clearance metabolism and to develop advanced anti-AD therapies. Therefore, there is extremely necessary to systematically review the regulation of Aβ metabolism and clarify the key to the development of therapeutic strategies targeting Aβ.
The cerebral Aβ is the hydrolysate of amyloid precursor protein (APP) via proteolysis of β-/γ- secretase and is cleaved into 39–43 length peptides according to the different cutting sites (eg, Aβ1-40 or Aβ1-42).5 The extracellular Aβ monomers gradually aggregate to form soluble oligomers, fibrils and fibers until insoluble plaques and eventually lead to neuronal degeneration.6 The subsequent catabolism of Aβ was mediated by central or peripheral pathway to obtain the homeostasis of Aβ, including the glial immunophagocytosis in the central or direct draining by the circulatory system to the periphery.7–9 Abundant clues have suggested that the imbalance between the production and removal of anabolism and catabolism of Aβ is the main culprit of amyloid plaques.10 Therefore, therapies that block the production of Aβ or mobilize its clearance are the eternal theme of Aβ-targeting strategies, such as the current Aβ monoclonal antibodies which are specific IgG1s binding to Aβ with affinity and complete the degradation of Aβ by promoting cell recognition and phagocytosis. In addition, researchers have developed a series of small-molecule drugs, nucleic acid drugs, proteins/peptides, and others to regulate the level of enzyme activity of Aβ secretion or to mobilize the clearance and degradation of Aβ at the molecular biological level (Figure 1).11
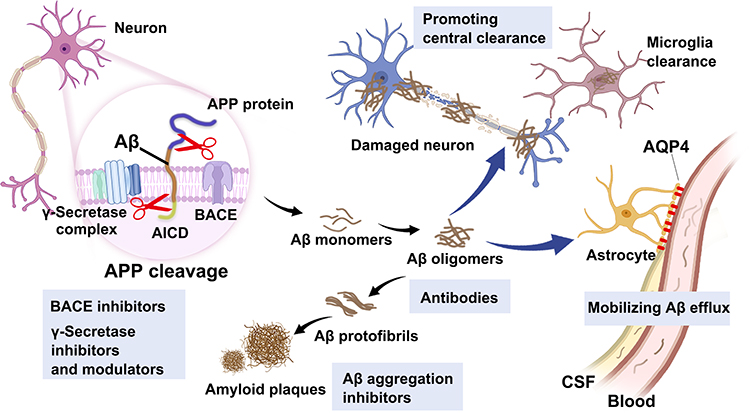 |
Figure 1 Illustration of the process of Aβ production, aggregation, and clearance in central and peripheral. APP is cleaved by β- and γ-secretase on neuron cells to release Aβ monomers, following self-aggregated into oligomers and fibrils to form plaques. Cerebral Aβ can be degraded by microglia phagocytosis or transported out of the brain for degradation. |
However, such treatment strategies are not without pitfalls. For example, amyloid-related imaging abnormalities (ARIA), the most controversial adverse effect of Aβ antibodies in clinical anti-Aβ immunotherapy, warn of the malignant events caused by excessive central burden in the process of Aβ clearance.12 The development of nanotechnology and targeted drug delivery systems in recent years has effectively facilitated the development of novel AD therapeutic strategies. In the past decade or so, nanomedicine for AD treatment has shown a continuous development process along with the progress of disease cognition. Previous nanomedicine focused on high-potency drug encapsulation to reduce peripheral adverse reactions of clinical medication or promoting central delivery by utilizing the advantageous size or functionalized surface to achieve the blood-brain barrier crossing, thus promoting the effective enrichment of potential anti-AD active drugs in the lesion.13,14 In recent years, as the elaboration of more detailed crosstalk relationship of AD pathogenic targets, the application of nanocarriers to efficiently support multiple therapeutic components has become a mainstream, especially the diverse drug carrying potential of the nanocarriers exhibits great advantages in the combinational delivery of biological macromolecules and active compounds. Meanwhile, with the successive approval of Aβ monoclonal antibodies, more studies have focused on the limitations of current antibody therapies, and a variety of synthetic nanoparticles have recently been demonstrated to act as highly efficient Aβ “chaperones” to regulate Aβ aggregation or facilitate clearance, attempting to provide alternatives to Aβ antibodies.15,16 Furthermore, the specific transport and enrichment rules of nanomedicine in vivo provide carrier tools for the systemic clearance of Aβ to realize the metabolism of Aβ outside the brain to achieve a safer and more efficient Aβ clearance pattern. Therefore, the rational application of nanomedicine strategies to reasonably design and construct AD-adapted Aβ targeted regulation of active components or delivery platforms to achieve safe and efficient Aβ clearance is the key challenge for the clinical transformation of Aβ nanomedicine. Finally, the success rate of drug clinical transformation is closely related to the selection of preclinical animal models. Therefore, it is essential to understand the complexity, advantages, and limitations of various animal models, as well as the irrational use, in order to enhance preclinical research on translational therapy for AD. In this review, we start from the gain and loss of clinical Aβ antibodies and elaborate on the key of targeted Aβ therapy. Following the introduction of applicable animal models in preclinical anti-Aβ studies, we have explored the endogenic clearance pathway of Aβ as well as the corresponding current AD modification strategies, aiming to provide a reliable approach for the development of Aβ-targeted therapies.
Literature Search Strategy
An electronic search was conducted using the PubMed, Clinical Trail, and Scopus databases, supplemented with a manual search using various combinations of Medical Subject Headings (MeSH) terms, including Alzheimer’s disease, amyloid-β, antibody, nanomedicine, and transgene mice model. The inclusion criteria for the search were as follows: articles published between 2010 and 2023, English as the language of publication, studies concerning Alzheimer’s disease, clinical studies, and nanomedicine. In order to include relevant studies on classic AD transgenic animal models, the search scope for this part was expanded to include articles published from January 1990 to September 2023. The exclusion criteria for the search included opinion papers, case reports, duplicate articles, and studies on biosensors or in vivo diagnostic reagents. For included articles, it should help answer at least one research question. Thus, the inclusion criteria developed relate to what the document covers:
- Analysis of the application of Aβ clinical therapy and the causes of adverse reactions;
- Construction method, characteristics and applicable scope of AD animal model;
- The application advantages of nanomedicine for AD compared with insufficient clinical treatment;
- Interaction between Aβ and other AD pathogenic targets, and the consideration of multi-drug combination.
Immunotherapies with Anti-Aβ Monoclonal Antibodies in AD Patients
The development of amyloid pathology in AD involves a series of steps, starting with the aggregation of Aβ monomers, followed by the formation of dimer/trimers, oligomers, protofibrils, fibrils, and ultimately the deposition of insoluble amyloid plaques.17 Aβ monoclonal antibodies (mAbs) can target different stages of this process and facilitate the clearance of Aβ.18 Recently, the FDA has approved two mAbs, aducanumab and lecanemab. Another promising mAb, donanemab, has submitted a marketing application to the FDA and received priority review by the Center for Drug Evaluation (CDE) in China in November 2023. The following sections provide a summary and discussion of the therapeutic mechanisms and clinical advancements of these antibodies, along with other mAbs that show promise for future approval.
Aducanumab
Aducanumab, the mAb designed to target residues 3–7 in the Aβ N-terminus, obtained accelerated approval from the FDA in June 2021, marking a significant milestone in AD treatment.19 Aducanumab exhibits low monovalent affinity but strong avidity for epitope-rich aggregates, allowing it to selectively bind to Aβ oligomeric or fibrillar aggregates.20 This binding activates microglia, leading to the clearance of Aβ from the brain.
The Phase I clinical trial (NCT01397539) revealed that patients generally tolerated doses up to 30 mg/kg well, with no occurrence of severe adverse events. A subsequent phase Ib clinical trial (PRIME, NCT01677572) further demonstrated that a dose range of 3 to 10 mg/kg of aducanumab reduced amyloid plaque and was safe enough to be used in the Phase III trials for efficacy.21 Based on the results of the PRIME trial, two clinical trials, EMERGE (NCT02484547) and ENGAGE (NCT02477800) trials, were initiated in 2015, both of which were phase III studies conducted over 18 months in individuals with prodromal and mild AD.22 Unfortunately, these two trials and an additional Phase II trial (EVOLVE, NCT03639987) that was designed to assess safety were prematurely ended following an interim futility analysis of phase III trial.23 In October 2019, Biogen announced that the interim futility analysis was incorrect. An analysis of a larger dataset showed that the high-dose aducanumab in the EMERGE trial met its primary endpoint, though the low-dose aducanumab did not exhibit significant benefits compared to the placebo group. Besides, while the ENGAGE trial did not meet its primary endpoint, it was observed that individuals who received 10 or more 10 mg/kg doses in the ENGAGE trail showed a slower decline similar to the participants in the EMERGE trial. The disparity in outcomes between the EMERGE and ENGAGE trials can be attributed to differences in the duration of exposure to high-dose aducanumab.
According to safety data from the EMERGE and ENGAGE trials, about a third of aducanumab-treated participants developed amyloid-related imaging abnormalities (ARIA), of which a quarter were symptomatic and 3% were severe.23 ARIA-E (vasogenic edema) and ARIA-H (microhemorrhages and hemosiderosis) were commonly observed in patients receiving a dosage of 10 mg/kg, with a 19.1% and 35% incidence rate, respectively. Besides, APOE ε4 carriers had a higher incidence compared to noncarriers.22 Notably, ARIA is a common phenomenon associated with anti-Aβ immunotherapies.24,25 This may be due to the antibody promoting the disaggregation of amyloid plaques into soluble forms when it binds with them, triggering glial cell-mediated neuroinflammation. At the same time, some soluble Aβ may enter blood vessels and deposit, leading to vascular pathology (Figure 2).
 |
Figure 2 The process of antibody-mediated Aβ clearance and production of vascular amyloid pathology. (A) Antibodies recognize and bind to amyloid plaques, prompting their transformation into a soluble form, which then promotes phagocytosis of microglia, during which microglia are activated and neuroinflammation occurs. (B) Soluble Aβ flows to the blood vessels and deposits, resulting in vascular pathology. |
Despite controversies, aducanumab received accelerated approval from the FDA based on the surrogate endpoint of Aβ reduction, representing a significant milestone in the field of AD therapeutics. It signifies the culmination of almost 25 years of research into the amyloid hypothesis and ushers in a new era for AD treatment, bringing hope to patients and researchers. However, the Centers for Medicare & Medicaid Services (CMS) currently limits coverage for aducanumab outside of approved clinical trials, reducing its accessibility in clinical settings. Moreover, it should be noted that further post-approval studies are still needed to confirm its clinical benefits.
Lecanumab
Lecanemab, a humanized IgG1 antibody derived from the mouse antibody mAb158, received accelerated approval by the FDA in January 2023 and full approval in July 2023 for the treatment of mild cognitive impairment (MCI) or mild dementia associated with AD.26 Lecanemab possesses an exceptional affinity for selectively binding to soluble Aβ aggregates (including oligomers and protofibrils), approximately 1000 times greater than Aβ monomers and 10–15 times than insoluble fibrils.27
The Phase I clinical trial (NCT01230853) of lecanemab conducted on 80 patients with mild-to-moderate AD, evaluated its safety, tolerability, and pharmacokinetics. Lecanemab demonstrated good tolerability at all tested doses, up to 10 mg/kg administered every two weeks for four months. The incidence of amyloid-related imaging abnormalities (ARIA-E, ARIA-H) was similar to placebo. Besides, pharmacokinetic analysis showed dose-proportional response when lecanemab dose was ≥10 mg/kg, with a serum elimination half-life of 7 days.28 Thereafter, a Phase IIb trial (NCT01767311) enrolled 856 subjects to determine the dosage and efficacy of the treatment. At the 12-month mark, the trial did not reach its primary endpoint or futility criteria. Hence, the trial continued until the 18-month mark. Results showed that patients receiving lecanemab at a dosage of 10 mg/kg twice a month experienced a statistically significant reduction in cognitive decline at 6, 12, and 18 months. This dosage led to a 93% reduction in brain amyloid plaques, a 47% slower decline in cognitive function as measured by ADAS-Cog, and a 30% slower decline in cognitive function as measured by ADCOMS.29
The phase III CLARITY AD trial (NCT03887455) enrolled 1795 participants with early AD, who were randomly assigned to receive either placebo or intravenous lecanemab at a dosage of 10 mg/kg every two weeks.30 The randomization process took into account clinical subgroups (MCI or mild AD) and concomitant use of approved AD symptomatic medication. Assessing clinical decline using CDR-SB as the primary endpoint, the lecanemab group showed a substantial reduction in clinical decline. Furthermore, lecanemab exhibited a highly statistically significant decrease in clinical decline on the global cognitive and functional scale at 18 months, with reduced amyloid plaque burden, a 24% deceleration in disease progression, and a 37% deceleration in decline in activities of daily living. Aside from ARIA-H and ARIA-E, infusion-related reactions are the most commonly reported adverse events, of which the majority reactions tended to be mild to moderate, with most occurring (75%) after the first dose. These results provide valuable insights into the efficacy and safety profile of lecanemab as a potential treatment for early AD.
Collectively, lecanemab stands out as a highly promising therapeutic option for AD due to its ability to reduce brain Aβ levels, mitigate cognitive decline, and exhibit a lower incidence of ARIA-E, thus ensuring improved safety. However, despite its moderate therapeutic effect and enhanced safety profile, lecanemab failed to demonstrate clinically significant benefits in patients with clinically manifest or prodromal dementia. Further research and investigation are essential to explore the efficacy and safety of lecanemab thoroughly.
Donanemab
Donanemab is a humanized IgG1 antibody designed to specifically target a pyroglutamated form of Aβ (N3pG-Aβ) that is aggregated in amyloid plaques.31 This truncated form of N-terminal Aβ is highly toxic and hydrophobic, leading to faster aggregation with β-sheet stabilization and resistance to degradation. The specificity of donanemab allows it to effectively bind these stable plaques, overcoming the issue of soluble Aβ forms that can impede recognition of deposited forms.32 Notably, donanemab has received priority review by CDE in November 2023 and is expected to become the third mAb approved by the FDA in the near future.
The Phase Ia clinical trial (NCT01837641) involved six different doses ranging from 0.3 to 10 mg/kg administered intravenously, and one group receiving donanemab through subcutaneous injection.33 The results showed that monthly intravenous administration of donanemab at a dose of 10 mg/kg reduced Aβ plaques by approximately 40–50%. The Phase Ib clinical trial (NCT02624778) enrolled 61 participants and used intravenous single or multiple dosing of donanemab at doses ranging from 10 to 40 mg/kg.34 All dosing cohorts showed a reduction in brain Aβ at 72 weeks. In a phase II trial (TRAILBLAZER-ALZ, NCT03367403) involving 272 patients with early symptomatic AD and confirmed tau and amyloid deposition, participants were randomly assigned to receive either donanemab or placebo intravenously every 4 weeks for up to 72 weeks. The primary outcome, measured by the change in the Integrated Alzheimer’s Disease Rating Scale (iADRS) score at 76 weeks, suggested that donanemab possessed a positive effect on cognitive and functional impairment. Donanemab also showed greater reductions in amyloid plaque levels and global tau load compared to placebo at 76 weeks. Notably, nearly complete clearance of Aβ plaques was observed within 6 months.35 In a Phase III trial, TRAILBLAZER-ALZ 2 (NCT04437511), 1736 patients were randomly assigned to either the donanemab treatment group (860 patients) or the placebo control group (876 patients). Over a 72-week period, patients received donanemab or placebo treatment every four weeks. At the 76-week mark, donanemab treatment showed a 35.1% reduction in disease progression risk compared to placebo in early AD patients with low/medium tau pathology, as measured by iADRS scores. Additionally, among early AD patients with low/medium tau pathology, 47% remained stable after one year of donanemab treatment, while only 29% of those receiving placebo treatment remained stable.
Donanemab was generally well tolerated but was reported to be associated with the occurrence of ARIA, with approximately 40% of participants in donanemab groups experiencing ARIA, and about 26.1% of them showing symptomatic ARIA. In contrast, symptomatic ARIA-E was observed in only 0.8% of participants receiving placebo.36 These data led to the FDA granting donanemab a breakthrough therapy designation and form the basis of donanemab’s application for traditional approval shortly thereafter.
Other Anti-Aβ mAb Candidates Under Clinical Trials
Remternetug, a humanized mAb developed by Eli Lilly, specifically targets N3pG-Aβ and represents a next-generation targeted therapy for AD. In July 2022, a global Phase III clinical trial (NCT05463731) was initiated to evaluate the safety, tolerability, and efficacy of Remternetug in early-stage AD patients. In 2023, Eli Lilly commenced an international multicenter Phase III trial (CTR20230785) employing a double-blind, randomized, placebo-controlled, parallel design to assess its safety and efficacy through subcutaneous injections in individuals with early AD symptoms. Additionally, the positive results of a Phase I clinical trial (NCT04451408) have been announced and indicated the efficacy of Remternetug in amyloid protein clearance.
Trontinemab is a humanized mAb as an improved vision of the gantenerumab developed by Genentech. Trontinemab combines the gantenerumab with a protein domain that can bind to transferrin receptors, aiming to enhance the antibody’s ability to cross the BBB in to the brain.37 Trontinemab possesses 50-fold higher BBB penetration and Aβ plaque binding efficiency compared to unmodified gantenerumab. Currently, Trontinemab is undergoing Phase II clinical trials (NCT04639050) for the treatment of mild-to-moderate AD. Preliminary results indicate that trontinemab can facilitate rapid amyloid plaque clearance at substantially lower doses than typical amyloid-targeting antibodies. After 28 weeks of treatment with the maximum dose of 1.8 mg/kg, 75% of patients exhibited a reduction of amyloid levels to below normal detectable limits.
ABBV-916 is a mAb developed by AbbVie, which recognizes the N3pG-Aβ aggregated in amyloid plaques. The preclinical data for this antibody has not been publicly disclosed yet. The clinical development of ABBV-916 began in August 2022, and the trial was initially listed as a phase I/II trial with an enrollment of 288 participants. It was later changed to a phase II trial (NCT05291234) with a target enrollment of 195 participants. This trial is randomized, placebo-controlled, double-blind, and conducted across multiple centers, which is being conducted in 88 centers across North America, Europe, and Japan, with an expected completion date of December 2024.
ACU193, a humanized mAb developed by Acumen, is the first Aβ oligomers-selective immunotherapeutic undergoing human clinical trials for the treatment of early-stage AD, aiming to investigate the Aβ oligomer hypothesis in AD.38 In clinical settings, the binding of anti-Aβ mAbs to amyloid plaques has been associated with a higher incidence of ARIA.39 ACU193 has a higher specificity for Aβ oligomers and shows minimal or no binding to amyloid plaques. Therefore, ACU193 is predicted to carry a lower risk of ARIA compared to other Aβ antibodies. In October 2022, ACU193 was granted Fast Track designation by the FDA for the treatment of MCI or early-stage AD. A randomized, placebo-controlled, double-blind Phase I clinical trial (NCT04931459) for ACU193 is complete.40 Data showed significant reductions in amyloid plaque levels as detectable by brain amyloid PET scans in patients receiving higher doses of ACU193 after 6–12 weeks. Besides, ACU193 demonstrated favorable tolerability and safety profiles, with 10.4% of patients experiencing ARIA-E, of which 2.1% were symptomatic. A Phase II clinical trial (NCT05291234) investigating the safety, efficacy, pharmacokinetics, and pharmacodynamics of ABBV-916 for the treatment of early-stage AD is currently recruiting participants.
In summary, Aβ monoclonal antibodies show promise in targeting Aβ pathology in AD (Table 1), despite challenges including disease stage limitations, target specificity, blood–brain barrier penetration,41 adverse effects,42 and the need for combination therapies.43 Ongoing research and advancements offer potential solutions to these challenges. The current immunotherapy approach for lowering amyloid levels in AD is inadequate in providing a comprehensive treatment. Consequently, there is a persistent quest for novel therapeutic options to address the complex nature of AD pathology.
 |
Table 1 The Therapeutic Mechanisms and Clinical Effect of Anti-Aβ mAbs |
Transgenic Mouse Models in Preclinical Studies
Despite concerted efforts from both academia and industry, a potent curative intervention for AD remains elusive.44 The high failure rate observed in clinical drug development is significantly related to the choice of animal models used in preclinical studies that demonstrate only limited aspects of AD pathology.45,46 Hence, understanding the intricacies, advantages, and limitations of each model while evaluating AD therapies across multiple models represents a crucial step in enhancing the translation of treatments from preclinical studies to patients. For that, we succinctly delineate several AD mouse models based on Aβ pathology, including mutation sites, pathological characteristics, advantages, disadvantages and applications, to provide reference and guiding significance for fundamental AD investigations (Table 2).
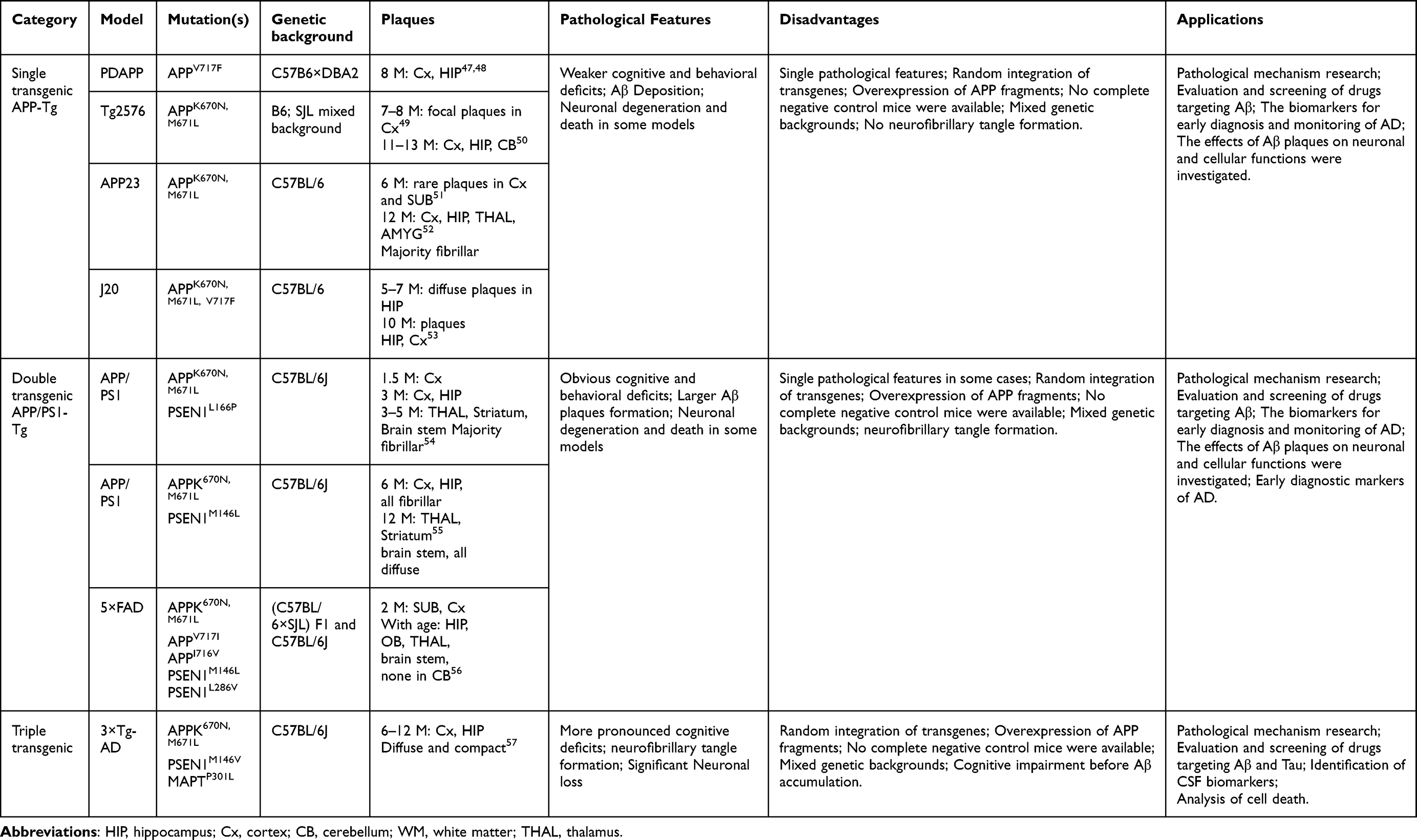 |
Table 2 Comparison of Current APP Mouse Models of AD |
Single Transgenic APP Mouse Model
The utilization of the APP single transgenic mouse emerges as a prominent animal model in AD research. This paradigm involves the integration of a mutated variant of the human APP gene within the murine genome, thus orchestrating an emulation of the aberrant Aβ deposition within the cerebral milieu.44 These mutations can be those associated with familial AD, such as the Swedish (K670N/M671L) mutation, which results in abnormal production and accumulation of Aβ.58 APP single transgenic murine models predominantly manifest an augmented accrual of Aβ, a cardinal pathological hallmark intricately linked to AD. In the cerebral milieu of these murine counterparts, Aβ progressively accumulates, orchestrating the emergence of amyloid plaques that potentially precipitate neuronal dysfunction and nerve impairment.59,60 These encompass deficits in learning and memory faculties, impediments in spatial orientation, and alterations in socio-behavioral tendencies. Concomitantly, within the milieu of APP single transgenic murine paradigms, the incipient onset of cellular detriment mechanisms becomes discernible, notably encompassing neuroinflammation and oxidative stress.47 These deleterious processes engender an intricate interplay that might further erode neuronal viability and functional efficacy.61 Analogous to the symptomatic manifestation of AD within human patients, these quintessential features proffer these murine models as a formidable armamentarium for delving into the realms of cognitive impairment etiology within the AD spectrum.
Although the APP single transgenic AD mouse model is important for the study of AD, it also has some shortcomings. AD, in its entirety, engenders a labyrinthine neurodegenerative pathos intrinsically underpinned by a myriad of pathological signatures, spanning neurofibrillary tangles, inflammatory cascades, and neuronal attrition. Thus, mimicking Aβ accumulation alone may not fully reflect multiple aspects of AD.62 In addition, APP single transgenic mouse models may be deficient in neurofibrillary tangles. Neurofibrillary tangles are strongly associated with abnormal Tau aggregation, which may not be fully simulated in a single transgenic model. However, Mesquita et al discovered that the single transgenic APP mouse (J20) has more pronounced lymphatic defects when investigating the connection between meningeal lymphatic vessels and cognitive dysfunction, making them a more suitable choice as a preclinical research model.63 Consequently, a judicious amalgamation of diverse models and methodological approaches is imperative to yield a panorama both comprehensive and veracious in its essence.
Double Transgenic APP/PS1 Mouse Model
APP/PS1 double transgenic mice, an animal model widely used to study AD, are unique in that they harbor mutations in both the human APP and Presenilin 1 (PS1) genes, thus mimicking multiple AD-related pathological features.64 Specifically, the mutations within the APP genes give rise to an abnormal Aβ production trajectory, while those in the PS1 genes, associated with familial AD, engender dysfunction within the γ-secretase complex, thereby engendering perturbations in Aβ metabolism and aggregation kinetics.57 Notably, APP/PS1 double transgenic mice typically manifest heightened cognitive and behavioral aberrations compared to their single transgenic counterparts, displaying potential anomalies in domains such as learning, memory, spatial cognition, and socio-behavioral tendencies. These deviations align more closely with the clinical symptomatic profile exhibited by human AD patients. APP/PS1 double transgenic mice are commonly used to evaluate various drug therapies. Because these mice mimic multiple AD pathological features, researchers can test drug effects on various aspects of Aβ accumulation, cognitive function, and more. Oakley et al invented 5×FAD mice with five FAD mutations in APP/PS1 double transgenic mice (APPK670N/M671L/V717I/I716V Tg and PSEN1M146L/L286V).44 The “5×FAD” mouse model was designed to introduce multiple genetic mutations into a single animal model to more fully mimic multiple pathological features of AD, especially Aβ plaque formation. Locci et al observed that the percentage of area occupied by plaques was significantly higher in 5xFAD and APP/PS1 mice compared to APPNLGF mice. Despite having the lowest Aβ plaque load, APPNLGF mice exhibit robust memory deficits, anxiety and depression-like behaviors, and impaired social behaviors, and are an effective preclinical model to study AD neuropathology, memory deficits, and affective behavioral changes.65 Therefore, it is essential to better understand the advantages and disadvantages of each model and to select the appropriate model for preclinical studies.
Triple Transgenic AD Mouse Model
This model configuration encompasses a triad of mutant genes, namely APP, PS1, and tau, thereby facilitating the manifestation of NFTs induced by Aβ deposition and perturbed tau phosphorylation, rendering it more consonant with the intricate spectrum of AD pathology.66 The 3×Tg-AD murine model has been meticulously devised to emulate a constellation of pathological features characteristic of AD, encompassing the genesis of Aβ plaques, the atypical aggregation of tau proteins, and the ensuing neurofibrillary tangle formations.57,67 This elaborate architecture permits a more comprehensive emulation of the multifaceted pathological panorama intrinsic to AD, thus furnishing an invaluable substrate for the explication of pathogenic mechanisms and the formulation of therapeutic modalities within the AD domain.
Some models may not fully mimic the complexity of the pathological development of AD in humans; Aβ deposition is not the only pathological feature of AD, and abnormal aggregation of tau protein is also closely related to the pathogenesis of AD.68,69 Therefore, rational selection of models, combining the advantages of multiple animal models, will contribute to a more comprehensive understanding of the pathogenesis of AD and the search for more effective treatment strategies. Taken together, when choosing the appropriate model, we should combine the research purpose and problem, and make full use of the advantages of different models to better promote the basic research of AD and provide a scientific basis and guidance for the treatment strategy of AD.
Exploration in Aβ-Targeted Preclinical Nanomedicine
The Intervention of Aβ Production
Though various interpretations including elevated tau phosphorylation, neuroinflammation, and so on have been proposed, the Aβ plaque is still a conspicuous pathological character and compelling cause for AD evolved.70 Especially, the imbalance between accumulation and removal of Aβ has been considered to contribute to accumulation a lot.71 Hence, intervention of Aβ production from the source would seem to be a promising way to decrease intracerebral Aβ. There are two main enzymes, beta-site amyloid precursor protein cleaving enzyme 1 (BACE1) following γ-secretase, cleave the transmembrane APP and achieve amyloidogenic cleavage (Figure 3A). Of them, the BACE1 plays a rate-limiting role in reducing Aβ production.72 The high enzymatic activity of BACE1 corresponding to the high levels of Aβ reconfirmed its vital position in the AD process. Given that numerous inhibitors have been extensively developed to lower the Aβ concentrations in patients. Due to the similar structure of BACE1 with other aspartyl proteases, and extensive location in the brain, high selectivity as well as high BBB penetration efficiency to implement minimal off-target side effects are crucial for inhibitor design.
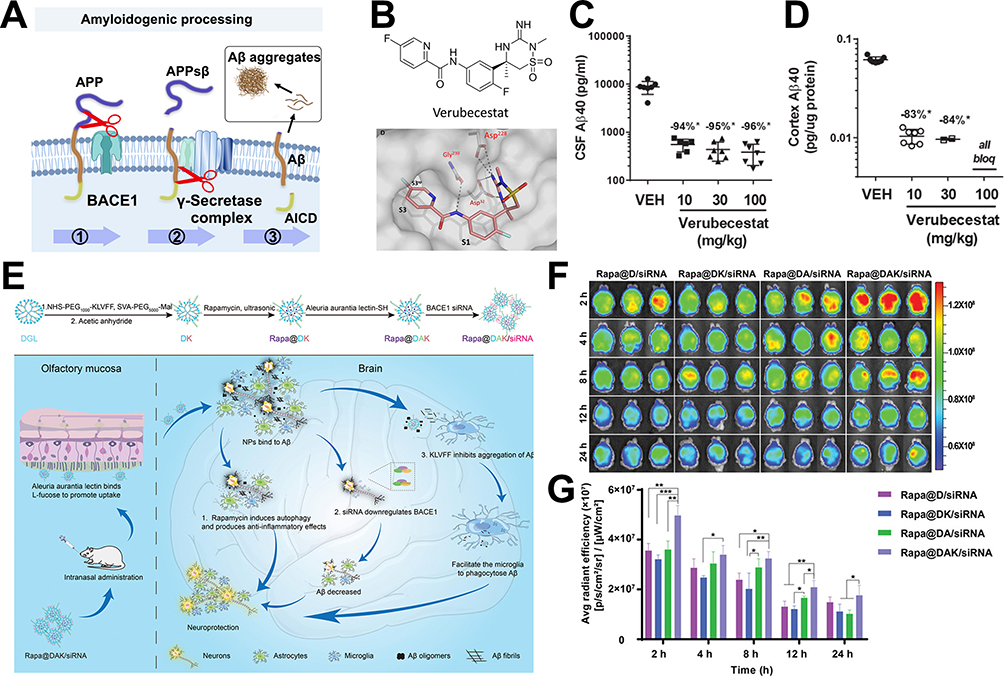 |
Figure 3 Inhibition of BACE1 is an effective AD treatment strategy to reduce Aβ production considering the rate-limiting role of BACE1. (A) Aβ is released from the APP after being cleaved by BACE1 and γ-secretases, subsequently self-aggregating into oligomers and fibrils to form plaques. (B) The chemical formula and structure-based drug design of verubecestat. (C) CSF levels of Aβ1-40 were reduced. (D) Cortex levels of Aβ1-40 were reduced. Reproduced with permission from Kennedy ME, Stamford AW, Chen X, et al. The BACE1 inhibitor verubecestat (MK-8931) reduces CNS β-amyloid in animal models and in Alzheimer’s disease patients. Sci Transl Med. 2016;8:363ra150.73 Copyright 2016, The American Association for the Advancement of Science. (E) Nanoparticles (Rapa@DAK/siRNA) for nasal administration and achieving BACE1 silence. (F) Ex vivo imaging of the brain tissue of AD model mice after intranasal administration with different preparations at series time points. (G) The semiquantitative analysis data of fluorescence intensity from (F). *p < 0.05 and **p < 0.01 and ***p < 0.001. Reproduced with permission from Yang X, Yang W, Xia X, et al. Intranasal Delivery of BACE1 siRNA and Rapamycin by Dual Targets Modified Nanoparticles for Alzheimer’s Disease Therapy. Small. 2022;18:e2203182.74 © 2022 Wiley‐VCH GmbH. |
Strategies to Inhibit BACE1
Over the past few decades, many pharmaceutical companies have invested time and effort in developing the BACE1 inhibitor pipeline, and 13 small-molecule inhibitors have been introduced into the clinic. Unfortunately, early clinical trial results did not support the BACE1 inhibitor, leading to its cessation of production. BACE1 inhibitors can be divided into two categories named peptidomimetic and non-peptidomimetic based on structure. The former class is characterized by an amide bond or its isosteres. Among them, OM99-2 is the earliest BACE1 inhibitor designed by mimicking the APP cleavage site sequence, firstly demonstrating a potent inhibitory activity of BACE1 and providing much insight into the development of subsequent inhibitors.75 However, due to the peptide-based analogs, the poor brain permeability, short half-life leading to rapid clearance, and low bioabsorption limit the in vivo therapeutic effect.76 Nonpeptidomimetic inhibitors, showing metabolic stability and sufficient lipophilicity to cross the both barriers of BBB and endosomal membranes, are usually found via high-throughput screening (HTS) and followed by lead optimization. For example, on the basis of isothiourea, a weak BACE1 ligand, the amidine moiety and diaryl amide substituent were modified to provide the high affinity for the continuous hydrophobic subsite of BACE1, as well as a high permeability for oral absorption, and MK-8931 (verubecestat) guanidine containing scaffold was designed (Figure 3B). In addition to its satisfying drug properties, it could significantly reduce Aβ1-40 content in cerebrospinal fluid and cortex by oral administration at the dose of 10 or 30 mg/kg (Figure 3C and D), thereby rescuing the memory loss of AD. Favorable data facilitated verubecestat to reach Phase III, but it was all overthrown by the unchanged cognitive ability along with adverse events.73 Other several of these BACE1 inhibitors have entered into clinical trials. The atabecestat and LY2886721 successfully alleviated the cognitive decline in the phase II/III trial but was discontinued owing to hepatic-related adverse events.77 A phase II trial of lanabecestat aiming at prodromal AD and mild AD participants was suspended because of futility.78 Clinical trials of BACE1 inhibitors can be described as a graveyard of new drug discovery for AD. Pharmaceutical companies seem to have given up on BACE1 inhibitors altogether as none are currently listed in any company’s experimental or clinical development pipeline.
In general, the safety of BACE1 inhibition therapy is a topic of concern. The over-inhibition of BACE1 may result in shortened lifespan, epilepsy and nerve desheathing according to transgene mice which are born to lack BACE1.79 However, recent research shows that this kind of BACE1-knockout therapy in mature mice or brain-target-knockout did not exhibit above adverse reaction, though the impairment of the formation of myelin was observed in the hippocampus, which may result in the Axon disorders in hippocampal mossy fiber pathway. Although the levels of BACE1 inhibition and Aβ reduction required for disease modification are unknown, they could be deduced from data collected in the clinical trials in progress. Data from clinical trials and preclinical works indicated that roughly 50% of BACE1 inhibition might be an appropriate therapeutic level.79–81 In sum, all the failure of latest drug candidates targeting BACE1 is mainly on account of its persistent side effects and the inefficacy in humans which might be in connection with genetic background discrepancy between species.82 Attention may be supposed to focus on appropriate dosage determination, avoiding negative results and pertinent animal models in further development.
Compared with small-molecule inhibitors, the small interfering RNA (siRNA) developed these years can directly block the disease-causing gene expression with appropriate dose and level to a certain extent to avoid the side effects and shows the advantages of good targeting ability with low effective doses. However, due to its poor stability and low permeability efficiency of BBB, nanomaterials for delivering siBACE1 are also being developed to propose solutions. A glycosylated modified nanocarrier was designed to utilize the glucose transporter-1 (GLUT1) to promote the BBB penetration efficiency of siRNA of BACE1.83 The stability was further enhanced by fluorinating the modified carrier. The obtained Gal-NP@siRNA nanodrugs had a 4.9-fold longer half-life in the blood than free siRNA, exhibiting excellent stability. Meanwhile, the brain accumulation of Gal-NP@Cy5-siRNA nanoparticles was 5.8-fold higher than the group without glycosylation modifications (NP@Cy5-siRNA), proving that improving BBB permeability through GLUT1 is a worthy method. In addition to vehicle modification, choosing the appropriate delivery method is also one of the ways to improve the efficiency of brain targeting and accumulation. Nasal administration is a method enabling bypass BBB and owns strong compliance, suitable for multiple administration of AD with a long course of disease. Gao et al designed an Aleuria aurantia lectin (AAL)-modified nanoparticle that could bind to fucose residue and locate at the nasal olfactory epithelium, and further enhance the ability to target Aβ lesion regions of the brain by modifying KLVFF (Figure 3E).74 The nanocarrier contains rapamycin capable of inducing microglial autophagy activation and siRNAs that silence the BACE1 gene. After nasal administration, AAL-modified nanoparticles accumulated in large quantities in the brain within 2 h (Figure 3F and G), indicating that AAL modification can improve nose-to-brain transport and promote brain-targeted delivery.
Strategies to Inhibit Aβ in Advance
Immunotherapy against Aβ includes active immunotherapy represented by vaccines, and passive immunotherapy which has been detailed in the “Immunotherapies with anti-Aβ monoclonal antibodies in AD patients” section. In contrast to passive immunotherapy, vaccination facilitates the endogenous production of therapeutically effective antibodies without monthly re-administration, which makes vaccination more affordable and compliant. These superiorities address many of these limitations and highlight the need for vaccines against pathologically specific targets in populations bearing the risk of AD.
In 1999, human full-length Aβ peptides mixing with adjuvants were first injected into transgenic mice and were found to produce antibodies, completely preventing plaque formation.84 As for the old mice, the Aβ deposition was observed significant reduction. As a result, Aβ active immunotherapy officially entered the stage of an AD treatment strategy. AN-1792 is the first vaccine reaching clinical trial, it showed Aβ plaque clearance in phase I trials.85 However, the meningitis that happened in phase II hindered the further trials, but it did not stop the researchers from exploring further. The CAD106 developed by Novartis consists of truncated Aβ fragments (Aβ1-6) to avoid T-cell activation.86 No signs of meningitis within the 52-week treatment of the Phase I trial demonstrated the success of this strategy in improving vaccine safety. The UB-311 vaccine designed by United Biomedical was optimized by linking the Aβ1-14 epitope to the helper T cell peptide epitope.87 This strategy was able to induce a B cell response to enhance immunogenicity while avoiding the inflammatory response of T cells. Although no Aβ-targeting vaccine has yet been approved for marketing, several candidates are undergoing clinical trials.
In addition to the most widely studied Aβ target, the N-terminally truncated Aβ peptide species harboring a pyroglutamate at position 3 (pE3-Aβ) was also focused by latest studies as a new promising target. It is derived from the subspecies pE3-Aβ which is a major component of intracerebral toxic plaques. Similar to Aβ1-42, pE3-Aβ begins to accumulate much prior to AD symptoms occurring, demonstrating its potential as a target for vaccine development.88 Recently, Ghochikyan et al decorated a C-terminal azide modifying pyroglutamated aa 3–11 peptide to the MultiTEP carrier protein utilizing copper-free click chemistry. This vaccine successfully reduced both pE3-Aβ and full-length Aβ in 5×FAD mice.89 FDA approval of the aducanumab has greatly promoted the research and development of other AD immunotherapies. Efforts still needed to be made to overcome the obstacles, including side effect events, target selection, and delivery platform, with the aim of developing more effective, even customized immunotherapies.
The Promotion of Aβ Clearance
Pathological results have shown that the impaired efficiency of Aβ clearance rate is the main inducement of a late-onset form of AD (LOAD) which accounts for a great proportion of total AD cases, indicating that improvement of Aβ elimination rate is another promising method to alleviate AD pathology.10 Recent evidence has revealed that intracerebral Aβ clearance is not only limited to the brain but consists of both central and peripheral pathways.90 The central way mainly includes glial cell-mediated catabolism and Aβ-degrading enzymes. Glial cell incorporates microglia and astrocyte. The former is a sort of innate immune cell in charge of cerebral microenvironment monitoring.91 Microglia are also a secreted source of some Aβ-degrading enzymes, such as insulin-degrading enzyme (IDE) and matrix metalloprotease-9 (MMP-9).92 Astrocyte is responsible for cell-to-cell communication and part of the nutrients that neurons require. The enzyme pathway requires many kinds of Aβ-degrading enzymes (ADEs), and a large number of ADEs have been discovered, including neprilysin (NEP), IDE, MMP-9, and so on. These enzymes can degrade full-length Aβ into smaller peptides with less toxicity, which are then cleared by glial cells or efflux out of the brain.93 As for the peripheral approach, the vasculature undertakes a critical role in ferrying cerebral-derived Aβ to peripheral organs (primarily the liver and kidney) to realize final degradation.94
As the disease progresses with age and progression of AD, both pathways of clearance are affected to varying degrees, resulting in a decrease in clearance. Therefore, accelerating the clearance of Aβ is also a major strategy for AD treatment.
Nanomedicine Strategies of Aβ Central Clearance
During brain development, microglia play the role of clearing excess synapses to maintain neuronal homeostasis, while toxic Aβ plaque would induce the state of microglia from quiescent condition to reactive condition.95 Proinflammatory cytokine released by hyperactivated microglia promotes astrocyte activation and contributes to Aβ-damaged neurons, also impairing synaptic function, and hindering cell communication and growth. Microglial regulation is, therefore, one of the attractive approaches for central Aβ clearance.
Previous studies have shown that a critical signal transformer, nuclear factor-kappa B (NF-κB) proteins, has a key role in microglia inflammation regulation.96 Once the NF-κB pathway is activated in microglia, it stimulates the secretion of ROS and pro-inflammatory cytokines, which in turn exert secondary neurotoxic effects. The team of Ding et al exploited an apoA-I-based HDL as an Aβ nano scavenger to achieve central clearance by microglia phagocytosis in the brain.97 Meanwhile, the natural compound curcumin is used to inhibit the NF-κB pathway of microglia and reduce the production of pro-inflammatory cytokines. These two complement each other to enable the continuous functioning of microglia (Figure 4A). After immunofluorescence labeling of microglia (Iba1 and CD86) and Aβ deposition, it can be seen in Figure 4B that pHDL/Cur-siBACE1 preparation could enhance microglial infiltration near plaques, in contrast to the lack of microglia next to plaques in AD mice. It was indicated that after treatment, the dysfunction of microglia phagocytosis Aβ was effectively reversed, and curcumin successfully regulated microglial function and improved Aβ clearance with silencing BACE1 (Figure 4C). In addition to inflammation, autophagy, and mitochondrial function regulation are also important strategies for microglia to target AD therapy. Among them, the Akt/mTOR/HIF-1α signaling pathway has been shown to be an important pathway to regulate microglial phenotypic transition and autophagy.Figure 4D Glutathione (GSH)-functionalized gold nanocage was designed and loaded with immunosuppressant fingolimod hydrochloride (FTY720) to form mTOR-mediated immunometabolic reprogramming nanoparticles (GAF NPs).98 Through CD86/CD206 double staining (Figure 4E), it was evident that GAF NPs were able to reduce the green fluorescence of the hippocampus and cortex representing CD86 positivity, indicating that the phenotype of microglia in the brain changed from M1 to M2, which is more conducive to Aβ clearance.
Emerging evidence has demonstrated that triggering receptors expressed on myeloid cell 2 (TREM2) deficient microglia tend to gather around Aβ deposition, and TREM2.99 The interrelationship between AD pathogenesis and several TREM2 variants has been suggested to be vital for microglial activation. It was previously reported that the rutin derivative NaR was able to enhance the infiltration of microglia around Aβ plaques.100 This is due to the enhancement of the number of phagocytosis receptors on the cell surface by NaR, specifically the expression and recycling of TREM2 (Figure 4F). To further investigate the potential regulatory pathway based on TREM2, Sheng et al proposed a hypothesis that TREM2 may have extracellular ligands and identified a group of lipoprotein particles with apoE as the main apolipoproteins.101 Results showed that, through lipid apoE interaction with TREM2, the extracellular Aβ clearance via microglial accelerated (Figure 4F). Due to the ambiguous relationships between inflammation and TREM2, it is still not clear how microglial lacking or overexpressing TREM2 is involved in phagocytosis in AD. Consequently, proper TREM2 function on AD needs to be clarified as a priority in future studies.
Astrocytes, a major member of intracerebral glial cell, contribute to synaptogenesis, maintenance of the neural circuit function, and formation of BBB.102 Astrocytes possess the ability to detect neurotransmitters and initiate a response by increasing intracellular calcium level. Additionally, they are capable of communicating with neurons through the release of gliotransmitters.103 Evidence showed that the reactivity of astrocytes has a close relationship with increased Aβ production, thereby inducing Aβ-triggered tau pathology.104 Hence, regulating the dysfunction of astrocytes has become a potential therapeutic avenue. Considering the participation of astrocytes in hippocampal neuronal abnormal activity, the earliest mark observed in both humans and mouse models, Aβ-dependent transient receptor potential ankyrin 1 (TRPA1) channels capable of activating astrocytes have been studied.105 After specifically blocking the pharmacology of TRPA1, the activities of both astrocytes and neurons were successfully normalized, thus preventing memory decline. In addition, AD therapeutic strategy was also developed based on astrocyte–microglia cross-talk.106 The microglia modulated by astrocytic interleukin-3 (IL-3) exhibited more effective Aβ clearance, limiting the progression of AD.
Nanomedicine Strategies of Aβ Peripheral Clearance
Recent studies have demonstrated the rapid flow of Aβ from the brain to the periphery, and the physiological breakdown of brain-derived Aβ in the peripheral system has been revealed in both humans and mice, providing a novel perspective for understanding the pathogenesis of AD and developing therapeutic approaches. The equilibrium between central and peripheral Aβ is maintained through continuous exchange,94,107 primarily involving drainage across the blood-brain barrier and cerebrospinal fluid (CSF)-interstitial fluid (ISF) convection via the lymphatic system.108 Specifically, cerebral Aβ is eliminated by low-density lipoprotein receptor-related protein-1 (LRP1), which is located in brain endothelium and mural cells, serving as a transport mechanism for Aβ across the blood-brain barrier. Additionally, soluble LRP1 (sLRP1) acts as a ligand in peripheral circulation to prevent free plasma Aβ from re-entering the brain.109 Building upon this pathway, Qu et al have developed a biomimetic nanozyme named CuxO@EM-K wrapped with erythrocyte membrane from 3×FAD mice and Aβ-specific sequence KLVFF, working synergistically with erythrocyte membrane to selectively recognize Aβ in blood.110 The CuxO core with superior multiple antioxidant enzyme-like activities plays a key role in stabilizing the outer erythrocyte membrane shell maintaining membrane integrity by relieving Aβ-induced membrane oxidative injury to enable prolonged blood circulation. The well-designed CuxO@EM-K successfully captures circulating Aβ followed by its elimination through liver metabolism. This rapid clearance of peripheral Aβ facilitates significant efflux of the central Aβ towards peripheral tissues due to the “sink in” phenomenon resulting in a reduced burden of Aβ accumulation within the brain. The genius of this design comes from the fully expanded application of inorganic materials such as copper nanoparticles in the treatment of AD, whereas this range of materials was previously thought to cause difficult metabolism and potential security issues in the treatment of central system diseases (Figure 5A and B). The highly safe peripheral circulation undoubtedly provides a more reliable therapeutic environment for such materials and expands the boundaries of anti-AD nanomedicine. In another study, Hyeon et al designed an extracorporeal Aβ cleansing system, where multifunctional magnetite/ceria nanoparticle assemblies are used to capture and remove Aβ from flowing blood in a dialysis-like manner. Aβ antibody-conjugated magnetite/ceria nanoparticles are introduced into the extracorporeal bloodstream circuit for specific capture of Aβ peptides and achieve facile magnetic separation by applying an external magnetic field. The treated blood then returns to the body after the magnetic separation of the nanoparticles and their bound Aβ peptides.111 5×FAD transgenic mice were used to demonstrate the blood Aβ cleansing treatment and showed that 71% of Aβ peptides in the blood on average were removed by the cleansing treatment resulting in a 77% decrease of Aβ plaques in cerebral cortices compared with the no-treatment group (Figure 5C–E). Both strategies are passive Aβ drainage using endogenous Aβ concentration gradient difference. This kind of treatment itself is affected by the course of AD disease. Specifically, Aβ in AD lesions is more in the form of aggregates that are difficult to flow, and the expression level of key affecting receptors is changed due to the lesions of the blood-brain barrier. Therefore, the application of this kind of passive effecting strategy in AD disease may be limited. In another recent study, Kim et al developed a chemical dissociator of brain plaques to drive the dissociation of Aβ aggregates in AD brains which release a substantial portion of soluble, smaller Aβ species to achieve outflow of dissociated amyloid burden from brain to blood (Figure 5F and G).112 This encouraging attempt provides an effective train of thought and tool for overcoming pathogenic Aβ effect impairment in AD progress, and further work is expected to expand this class of drugs from single dissociator to active drainage in further study.
 |
Figure 4 Strategies of Aβ central clearance via microglia. (A) The normalization of microglial dysfunction by blocking the NF-κB pathway to accelerate Aβ degradation. (B and C) pHDL/Cur-siBACE1 reduced microglia activation by inhibiting the NF-κB signal and decreased the Aβ deposition. Reproduced with permission from Zhang H, Jiang W, Zhao Y, et al. Lipoprotein-Inspired Nanoscavenger for the Three-Pronged Modulation of Microglia-Derived Neuroinflammation in Alzheimer’s Disease Therapy. Nano Lett. 2022;22:2450–2460. Copyright © 2022, American Chemical Society.97 (D) The illustration of how GAF NPs act on the Akt/mTOR/HIF-1α signaling pathway. (E) GAF NP treatment mobilized microglial polarization from the M1 to M2 in AD mouse brain. Reproduced with permission from Yang F, Zhao D, Cheng Met al mTOR-Mediated Immunometabolic Reprogramming Nanomodulators Enable Sensitive Switching of Energy Deprivation-Induced Microglial Polarization for Alzheimer’s Disease Management. ACS Nano. 2023. Copyright © 2023 American Chemical Society.98 (F) NaR treatment significantly increased the recycling of TREM2 in microglial cells, and NaR-mediated Aβ phagocytosis and clearance are not affected by microglial phagocytosis receptors. ***p < 0.001. Reproduced from Pan R-Y, Ma J, Kong X–X, et al. Sodium rutin ameliorates Alzheimer’s disease-like pathology by enhancing microglial amyloid-β clearance. Sci Adv. 2019;5:eaau6328. Copyright © 2019 The Authors, some rights reserved; exclusive licensee American Association for the Advancement of Science. No claim to original US Government Works. Distributed under a Creative Commons Attribution NonCommercial License 4.0 (CC BY-NC).100 |
 |
Figure 5 Strategies of Aβ peripheral clearance. (A) Schematic illustration of Aβ captures in the blood by CuxO@EM-K, followed by elimination of Aβ bound to CuxO@EM-K by the liver. Subsequently, clearance of peripheral Aβ facilitates a large efflux of Aβ from the brain into the blood through the sink effect, leading to the reduction of brain Aβ burden. (B) Blood Aβ levels were measured after treatment with CuxO@EM-K. *p < 0.05 and **p < 0.01. Reproduced with permission from Ma M, Liu Z, Gao N, et al. Self-Protecting Biomimetic Nanozyme for Selective and Synergistic Clearance of Peripheral Amyloid-β in an Alzheimer’s Disease Model. J Am Chem Soc. 2020;142:21702–21711.110 Copyright 2020 American Chemical Society. (C) Schematic illustration of Aβ capture and magnetic separation by magnetic nanoparticle. (D) MCNA concentration-dependent Aβ peptide capture efficiency. (E) Plasma Aβ concentrations were measured before and after the Aβ cleansing treatment with MCNAs. *p < 0.05. Reproduced with permission from Kim D, Kwon HJ & Hyeon T. Magnetite/Ceria Nanoparticle Assemblies for Extracorporeal Cleansing of Amyloid-β in Alzheimer’s Disease. Adv Mater. 2019;31:e1807965.111 © 2019 WILEY‐VCH Verlag GmbH & Co. KGaA, Weinheim (F) Individually normalized plasma Aβ tracking data of blood samples collected at weeks 5, 8, and 12 for each mouse in all groups using basal plasma Aβ and maximal plasma Aβ levels of individuals (Control, black; EPPS, red, Y. adult, Young-adult). (G) ThS stained Aβ plaques in the brain tissue of each mouse. Reproduced with permission from Lee D, Kim HV, Kim HY, et al. Chemical-Driven Outflow of Dissociated Amyloid Burden from Brain to Blood. Adv Sci (Weinh). 2022;9:e2104542.112 Copyright 2022 John Wiley & Sons, Inc. |
In addition to BBB-mediated Aβ efflux, the exchange between CSF and ISF plays a crucial role in removing soluble Aβ. Although the details of this pathway are not yet fully understood, this progress largely relies on the transportation of the glia-lymphatic (glymphatic) system and the draining of meningeal lymphatic fluid into deep cervical lymph nodes. The pia membrane penetrates the brain parenchyma with blood vessels (Virchow-Robin space),113 allowing cerebrospinal fluid to flow from the subarachnoid space into the perivascular space and exchange with interstitial fluid in the brain. The polarized aquaporin protein-4 (AQP4) on astrocyte foot processes drives the flow of solutes in the brain parenchyma toward the venous/perivascular Virchow-Robin space.114 The waste products then further flow back into the cerebrospinal fluid and are either recycled through the meningeal lymphatic canal or discharged into the systemic blood circulation through the arachnoid granules. This newly recognized pathway has been shown to be closely related to the metabolism of various pathogenic toxic proteins such as Aβ and tau. On the basis of these findings, there have been preliminary studies on the application of recombinant vascular endothelial growth factor-C (VEGF-C) and adeno-associated virus expression of VEGF-C in the treatment of AD by directly regulating lymphogenesis and lymphatic vessel function to promote lymphatic drainage and the excretion of toxic metabolites.115 In conclusion, the lymphatic-targeting system may be a viable therapeutic strategy for AD treatment; however, research on the lymphatic system in central nervous system diseases faces challenges, such as difficulty in capturing the anatomical structure and the lack of appropriate means to objectively evaluate the internal drainage function. Therefore, the use of novel imaging technology and nanomedicine platforms to investigate fluid homeostasis and substance drainage in the brain can provide a more intuitive understanding of the anatomy and function of the central lymphatic system, which would make a significant contribution to AD treatment.
Conclusion
Alzheimer’s disease (AD) has become one of the most significant health problems worldwide. With the recent emergence of two Aβ monoclonal antibodies approved by the FDA, the treatment of AD has made progress in overcoming the lack of available disease-modified therapies. Unquestionably, these encouraging developments have confirmed the correlation between Aβ clearance and cognitive improvement in clinical settings. However, adverse clinical responses also indicate that relying solely on Aβ clearance is flawed. On one hand, the rapid disintegration of Aβ plaques leads to difficulties in transporting and clearing a large amount of soluble Aβ. Therefore, any therapeutic strategy targeting Aβ must consider stimulating the internal metabolic system to clear Aβ plaques quickly and safely while also avoiding adverse reactions in the central nervous system.116 On the other hand, these antibodies can only improve mild cognitive impairment. This is because advanced stages of the disease have already caused significant neurological damage and other symptoms, making it necessary for many AD patients to undergo multidimensional drug combinations.117,118
In the research context, the highly controllable and intelligent advantages of nanomedicine provide ample means to address the complex symptoms of AD. However, it must be noted that the prerequisite for the clinical transformation of nanomedicine, that is, the appropriate selection of its research objects, is undoubtedly crucial. At present, most AD animal models can only simulate part of AD lesions, and animal models that fully conform to human AD lesions have not yet come out.119 However, with the development of molecular neurobiology, more effective targets and crosstalk relationships between targets have been discovered,120 which makes it difficult to propose a gold standard for AD disease models. Therefore, it is important to select the appropriate animal model according to the pathological process to be studied. In the recent study of the relationship between meningeal lymph and Aβ metabolism, for example, this lesion was difficult to detect in aggressive 5xFAD mice, although this animal model has been widely used in other Aβ pathological studies.63 This is precisely because of the inconsistency between the period of the onset of Aβ pathology and the pathological changes of physiological structure. In view of these considerations, we reviewed the animal models of AD characterized by Aβ deposition, and attempted to guide the selection of subsequent preclinical animal studies of nanomedical drugs through Aβ deposition level, deposition initiation time and cascade lesion characteristics. In subsequent drug development, more rational program design will inevitably be accompanied by the selection of reliable animal models, and even the pharmacodynamic research will be extended to the emerging human-derived induced pluripotent stem cell models, so as to more accurately reflect the development of disease in the human body.
Based on this premise, we summarized the design strategy of nanomedicine based on Aβ in the whole life cycle of Aβ, and interpreted the intervention strategy for the whole life cycle of Aβ from the perspective of inhibiting Aβ production (including inhibition of secretase and active immunoprophylaxis of Aβ production) and promoting Aβ clearance. Different from previous reviews that focused more on functional nanocarrier design, we describe the potential adverse reactions of excessive Aβ clearance and microglial inflammation, and propose a strategy of synergistic Aβ clearance and burden reduction through microglial polarization regulation. With the clarification of the metabolic pathways of Aβ inside and outside the brain, especially the breakthrough of central metabolic pathways such as meningeal lymphatic vessels, the metabolism of Aβ has become a systematic process, emphasizing the integrity of pathological intervention in AD.115 Therefore, the next generation of Aβ nanomedical drug development may pay more attention to the regulation of its transport process in multiple sites throughout the body by taking advantage of systematic elimination with greater potential.121 In summary, this paper aims to guide the development of nanomedicine from the perspective of AD pathology and promote the development of Aβ-based synergistic therapies.
Abbreviations
AD, Alzheimer’s disease; SP, senile plaques; Aβ, amyloid-beta; NFTs, neurofibrillary tangles; FDA, Food and Drug Administration; AChE, acetylcholine esterase; NMDA, N-methyl-D-aspartic acid; APP, amyloid precursor protein; ARIA, amyloid-related imaging abnormalities; BBB, blood–brain barrier; mAbs, Aβ monoclonal antibodies; CDR-SB, Clinical Dementia Rating Scale-Sum of Boxes; MCI, mild cognitive impairment; CTAD, Clinical Trials on Alzheimer’s Disease; iADRS, Integrated Alzheimer’s Disease Rating Scale; PS1, Presenilin 1; BACE1, beta-site amyloid precursor protein cleaving enzyme 1; HE, hydroxyethylene; HTS, high-throughput screening; siRNA, small interfering RNA; GLUT1, glucose transporter-1; AAL, aleuria aurantia lectin; pE3-Aβ, Aβ peptide species harboring a pyroglutamate at position 3; LOAD, late-onset form of AD; IDE, insulin-degrading enzyme; MMP-9, matrix metalloprotease-9; ADEs, Aβ-degrading enzymes; NEP, neprilysin; NF-ΚB, nuclear factor-kappa B; TREM2, triggering receptor expressed on myeloid cells 2; CSF, cerebrospinal fluid; ISF, interstitial fluid; LRP1, low-density lipoprotein receptor-related protein-1; AQP4, polarized aquaporin protein-4; VEGF-C, vascular endothelial growth factor-C.
Acknowledgments
This research did not receive any specific grant from funding agencies in the public, commercial, or not-for-profit sectors.
Disclosure
The authors declare that they have no conflicts of interest.
References
1. Gustavsson A, Norton N, Fast T, et al. Global estimates on the number of persons across the Alzheimer’s disease continuum. Alzheimers Dement. 2023;19:658–670. doi:10.1002/alz.12694
2. Dhillon S. Aducanumab: first Approval. Drugs. 2021;81:1437–1443. doi:10.1007/s40265-021-01569-z
3. Marucci G, Buccioni M, Ben DD, et al. Efficacy of acetylcholinesterase inhibitors in Alzheimer’s disease. Neuropharmacology. 2021;190:108352. doi:10.1016/j.neuropharm.2020.108352
4. Cummings J, Lee G, Nahed P, et al. Alzheimer’s disease drug development pipeline: 2022. Alzheimers Dement. 2022;8:e12295. doi:10.1002/trc2.12295
5. Hampel H, Vassar R, De Strooper B, et al. The β-Secretase BACE1 in Alzheimer’s Disease. Biol Psychiatry. 2021;89:745–756. doi:10.1016/j.biopsych.2020.02.001
6. Meisl G, Kirkegaard JB, Arosio P, et al. Molecular mechanisms of protein aggregation from global fitting of kinetic models. Nat Protoc. 2016;11:252–272. doi:10.1038/nprot.2016.010
7. Sahoo BR, Panda PK, Liang W, et al. Degradation of Alzheimer’s Amyloid-β by a catalytically inactive insulin-degrading enzyme. J Mol Biol. 2021;433:166993. doi:10.1016/j.jmb.2021.166993
8. Uddin MS, Lim LW. Glial cells in Alzheimer’s disease: from neuropathological changes to therapeutic implications. Ageing Res Rev. 2022;78:101622. doi:10.1016/j.arr.2022.101622
9. Grubman A, Choo XY, Chew G, et al. Transcriptional signature in microglia associated with Aβ plaque phagocytosis. Nat Commun. 2021;12:3015. doi:10.1038/s41467-021-23111-1
10. Mawuenyega KG, Sigurdson W, Ovod V, et al. Decreased clearance of CNS beta-amyloid in Alzheimer’s disease. Science. 2010;330:1774. doi:10.1126/science.1197623
11. Lu L, Zheng X, Wang S, et al. Anti-Aβ agents for mild to moderate Alzheimer’s disease: systematic review and meta-analysis. J Neurol Neurosurg Psychiatry. 2020;91:1316–1324. doi:10.1136/jnnp-2020-323497
12. Avgerinos KI, Ferrucci L, Kapogiannis D. Effects of monoclonal antibodies against amyloid-β on clinical and biomarker outcomes and adverse event risks: a systematic review and meta-analysis of phase III RCTs in Alzheimer’s disease. Ageing Res Rev. 2021;68:101339. doi:10.1016/j.arr.2021.101339
13. Qian K, Bao X, Li Y, et al. Cholinergic neuron targeting nanosystem delivering hybrid peptide for combinatorial mitochondrial therapy in Alzheimer’s disease. ACS Nano. 2022;16:11455–11472. doi:10.1021/acsnano.2c05795
14. Tang W, Fan W, Lau J, et al. Emerging blood-brain-barrier-crossing nanotechnology for brain cancer theranostics. Chem Soc Rev. 2019;48:2967–3014. doi:10.1039/C8CS00805A
15. Sun Y, Kakinen A, Wan X, et al. Spontaneous Formation of β-sheet nano-barrels during the early aggregation of Alzheimer’s amyloid beta. Nano Today. 2021;38:101125. doi:10.1016/j.nantod.2021.101125
16. Ye Z, Yan ZJ, Zhang C, et al. Charged tubular supramolecule boosting multivalent interactions for the drastic suppression of Aβ fibrillation. Nano Lett. 2021;21:10494–10500. doi:10.1021/acs.nanolett.1c04007
17. Mohammed AA, Barale SS, Kamble SA, et al. Molecular insights into the inhibition of early stages of Aβ peptide aggregation and destabilization of Alzheimer’s Aβ protofibril by dipeptide D-Trp-Aib: a molecular modelling approach. Int J Biol Macromol. 2023;242:124880. doi:10.1016/j.ijbiomac.2023.124880
18. Shi M, Chu F, Zhu F, et al. Impact of Anti-amyloid-β monoclonal antibodies on the pathology and clinical profile of Alzheimer’s disease: a focus on aducanumab and lecanemab. Front Aging Neurosci. 2022;14:870517. doi:10.3389/fnagi.2022.870517
19. Brockmann R, Nixon J, Love BL, et al. Impacts of FDA approval and Medicare restriction on antiamyloid therapies for Alzheimer’s disease: patient outcomes, healthcare costs, and drug development. Lancet Regional Health Am. 2023;20:100467. doi:10.1016/j.lana.2023.100467
20. Arndt JW, Qian F, Smith BA, et al. Structural and kinetic basis for the selectivity of aducanumab for aggregated forms of amyloid-β. Sci Rep. 2018;8:6412. doi:10.1038/s41598-018-24501-0
21. Sevigny J, Chiao P, Bussière T, et al. The antibody aducanumab reduces Aβ plaques in Alzheimer’s disease. Nature. 2016;537:50–56. doi:10.1038/nature19323
22. Budd Haeberlein S, Aisen PS, Barkhof F, et al. Two Randomized Phase 3 Studies of Aducanumab in Early Alzheimer’s Disease. J Prev Alzheimer’s Dis. 2022;9:197–210. doi:10.14283/jpad.2022.30
23. Salloway S, Chalkias S, Barkhof F, et al. Amyloid-related imaging abnormalities in 2 phase 3 studies evaluating aducanumab in patients with early Alzheimer disease. JAMA Neurol. 2022;79:13–21. doi:10.1001/jamaneurol.2021.4161
24. Barakos J, Purcell D, Suhy J, et al. Detection and management of amyloid-related imaging abnormalities in patients with Alzheimer’s disease treated with anti-amyloid beta therapy. J Prev Alzheimer’s Dis. 2022;9:211–220. doi:10.14283/jpad.2022.21
25. Sperling RA, Jack CR, Black SE, et al. Amyloid-related imaging abnormalities in amyloid-modifying therapeutic trials: recommendations from the Alzheimer’s association research roundtable workgroup. Alzheimer’s Demen. 2011;7:367–385. doi:10.1016/j.jalz.2011.05.2351
26. Jönsson L, Wimo A, Handels R, et al. The affordability of lecanemab, an amyloid-targeting therapy for Alzheimer’s disease: an EADC-EC viewpoint. Lancet Reg Health Europe. 2023;29:100657. doi:10.1016/j.lanepe.2023.100657
27. Tucker S, Möller C, Tegerstedt K, et al. The murine version of BAN2401 (mAb158) selectively reduces amyloid-β protofibrils in brain and cerebrospinal fluid of tg-ArcSwe mice. J Alzheimer’s Dis. 2015;43:575–588. doi:10.3233/JAD-140741
28. Logovinsky V, Satlin A, Lai R, et al. Safety and tolerability of BAN2401--a clinical study in Alzheimer’s disease with a protofibril selective Aβ antibody. Alzheimers Res Ther. 2016;8:14. doi:10.1186/s13195-016-0181-2
29. Swanson CJ, Zhang Y, Dhadda S, et al. A randomized, double-blind, phase 2b proof-of-concept clinical trial in early Alzheimer’s disease with lecanemab, an anti-Aβ protofibril antibody. Alzheimers Res Ther. 2021;13:80. doi:10.1186/s13195-021-00813-8
30. van Dyck CH, Swanson CJ, Aisen P, et al. Lecanemab in Early Alzheimer’s Disease. New Engl J Med. 2023;388:9–21. doi:10.1056/NEJMoa2212948
31. Jawhar S, Wirths O, Bayer TA. Pyroglutamate amyloid-β (Aβ): a hatchet man in Alzheimer disease. J Biol Chem. 2011;286:38825–38832. doi:10.1074/jbc.R111.288308
32. Bayer TA. Pyroglutamate Aβ cascade as drug target in Alzheimer’s disease. Mol Psychiatry. 2022;27:1880–1885. doi:10.1038/s41380-021-01409-2
33. Lowe SL, Willis BA, Hawdon A, et al. Donanemab (LY3002813) dose-escalation study in Alzheimer’s disease. Alzheimers Dement. 2021;7:e12112. doi:10.1002/trc2.12112
34. Lowe SL, Duggan Evans C, Shcherbinin S, et al. Donanemab (LY3002813) Phase 1b study in Alzheimer’s disease: rapid and sustained reduction of brain amyloid measured by florbetapir F18 imaging. J Prev Alzheimers Dis. 2021;8:414–424. doi:10.14283/jpad.2021.56
35. Mintun MA, Lo AC, Duggan Evans C, et al. Donanemab in early Alzheimer’s disease. New Engl J Med. 2021;384:1691–1704. doi:10.1056/NEJMoa2100708
36. Yadollahikhales G, Rojas JC. Anti-amyloid immunotherapies for Alzheimer’s disease: a 2023 clinical update. Neurotherapeutics. 2023;20:914–931. doi:10.1007/s13311-023-01405-0
37. Grimm HP, Schumacher V, Schäfer M, et al. Delivery of the Brainshuttle™ amyloid-beta antibody fusion trontinemab to non-human primate brain and projected efficacious dose regimens in humans. MAbs. 2023;15:2261509. doi:10.1080/19420862.2023.2261509
38. Krafft GA, Jerecic J, Siemers E, et al. ACU193: an immunotherapeutic poised to test the amyloid β oligomer hypothesis of Alzheimer’s disease. Front Neurosci. 2022;16:848215. doi:10.3389/fnins.2022.848215
39. Withington CG, Turner RS. Amyloid-related imaging abnormalities with anti-amyloid antibodies for the treatment of dementia due to Alzheimer’s disease. Front Neurol. 2022;13:862369. doi:10.3389/fneur.2022.862369
40. Siemers E, Hitchcock J, Sundell K, et al. ACU193, a monoclonal antibody that selectively binds soluble Aß oligomers: development rationale, phase 1 trial design, and clinical development plan. J Prev Alzheimers Dis. 2023;10:19–24. doi:10.14283/jpad.2022.93
41. Gustavsson T, Metzendorf NG, Wik E, et al. Long-term effects of immunotherapy with a brain penetrating Aβ antibody in a mouse model of Alzheimer’s disease. Alzheimer’s Res Ther. 2023;15:90. doi:10.1186/s13195-023-01236-3
42. Jeremic D, Navarro-López JD, Jiménez-Díaz L. Efficacy and safety of anti-amyloid-β monoclonal antibodies in current Alzheimer’s disease phase III clinical trials: a systematic review and interactive web app-based meta-analysis. Ageing Res Rev. 2023;90:102012. doi:10.1016/j.arr.2023.102012
43. Congdon EE, Sigurdsson EM. Tau-targeting therapies for Alzheimer disease. Nat Rev Neurol. 2018;14:399–415. doi:10.1038/s41582-018-0013-z
44. Alzheimer’s Association Report. 2023 Alzheimer’s disease facts and figures. Alzheimers Dement. 2023;19:1598–1695. doi:10.1002/alz.13016
45. Gregory S, Saunders S, Ritchie CW. Science disconnected: the translational gap between basic science, clinical trials, and patient care in Alzheimer’s disease. Lancet Healthy Longev. 2022;3:e797–e803. doi:10.1016/S2666-7568(22)00219-7
46. Cummings J, Zhou Y, Lee G, et al. Alzheimer’s disease drug development pipeline: 2023. Alzheimers Dement. 2023;9:e12385. doi:10.1002/trc2.12385
47. Cohen RM, Rezai-Zadeh K, Weitz TM, et al. A transgenic Alzheimer rat with plaques, tau pathology, behavioral impairment, oligomeric aβ, and frank neuronal loss. J Neurosci. 2013;33:6245–6256. doi:10.1523/JNEUROSCI.3672-12.2013
48. Frost JL, Le KX, Cynis H, et al. Pyroglutamate-3 amyloid-β deposition in the brains of humans, non-human primates, canines, and Alzheimer disease-like transgenic mouse models. Am J Pathol. 2013;183:369–381. doi:10.1016/j.ajpath.2013.05.005
49. Ghosal K, Vogt DL, Liang M, et al. Alzheimer’s disease-like pathological features in transgenic mice expressing the APP intracellular domain. Proc Natl Acad Sci U S A. 2009;106:18367–18372. doi:10.1073/pnas.0907652106
50. Elfenbein HA, Rosen RF, Stephens SL, et al. Cerebral beta-amyloid angiopathy in aged squirrel monkeys. Histol Histopathol. 2007;22:155–167. doi:10.14670/HH-22.155
51. Karch CM, Goate AM. Alzheimer’s disease risk genes and mechanisms of disease pathogenesis. Biol Psychiatry. 2015;77:43–51. doi:10.1016/j.biopsych.2014.05.006
52. Kim DH, Yeo SH, Park J-M, et al. Genetic markers for diagnosis and pathogenesis of Alzheimer’s disease. Gene. 2014;545:185–193. doi:10.1016/j.gene.2014.05.031
53. Hartley D, Blumenthal T, Carrillo M, et al. Down syndrome and Alzheimer’s disease: common pathways, common goals. Alzheimers Dement. 2015;11:700–709. doi:10.1016/j.jalz.2014.10.007
54. Holtzman DM, Fagan AM, Mackey B, et al. Apolipoprotein E facilitates neuritic and cerebrovascular plaque formation in an Alzheimer’s disease model. Ann Neurol. 2000;47:739–747. doi:10.1002/1531-8249(200006)47:6<739::AID-ANA6>3.0.CO;2-8
55. Cuyvers E, Sleegers K. Genetic variations underlying Alzheimer’s disease: evidence from genome-wide association studies and beyond. Lancet Neurol. 2016;15:857–868. doi:10.1016/S1474-4422(16)00127-7
56. Heuer E, Rosen RF, Cintron A, et al. Nonhuman primate models of Alzheimer-like cerebral proteopathy. Curr Pharm Des. 2012;18:1159–1169. doi:10.2174/138161212799315885
57. Holcomb L, Gordon MN, McGowan E, et al. Accelerated Alzheimer-type phenotype in transgenic mice carrying both mutant amyloid precursor protein and presenilin 1 transgenes. Nat Med. 1998;4:97–100. doi:10.1038/nm0198-097
58. Citron M, Oltersdorf T, Haass C, et al. Mutation of the beta-amyloid precursor protein in familial Alzheimer’s disease increases beta-protein production. Nature. 1992;360:672–674. doi:10.1038/360672a0
59. Wisniewski T, Sigurdsson EM. Murine models of Alzheimer’s disease and their use in developing immunotherapies. Biochim Biophys Acta. 2010;1802:847–859. doi:10.1016/j.bbadis.2010.05.004
60. Dujardin S, Colin M, Buée L. Invited review: animal models of tauopathies and their implications for research/translation into the clinic. Neuropathol Appl Neurobiol. 2015;41:59–80. doi:10.1111/nan.12200
61. Guerreiro R, Hardy J. Genetics of Alzheimer’s disease. Neurotherapeutics. 2014;11:732–737. doi:10.1007/s13311-014-0295-9
62. De Strooper B, Simons M, Multhaup G, et al. Production of intracellular amyloid-containing fragments in hippocampal neurons expressing human amyloid precursor protein and protection against amyloidogenesis by subtle amino acid substitutions in the rodent sequence. EMBO J. 1995;14:4932–4938. doi:10.1002/j.1460-2075.1995.tb00176.x
63. Da Mesquita S, Louveau A, Vaccari A, et al. Functional aspects of meningeal lymphatics in ageing and Alzheimer’s disease. Nature. 2018;560:185–191. doi:10.1038/s41586-018-0368-8
64. Mucke L, Masliah E, Yu GQ, et al. High-level neuronal expression of abeta 1-42 in wild-type human amyloid protein precursor transgenic mice: synaptotoxicity without plaque formation. J Neurosci. 2000;20:4050–4058. doi:10.1523/JNEUROSCI.20-11-04050.2000
65. Locci A, Orellana H, Rodriguez G, et al. Comparison of memory, affective behavior, and neuropathology in APPNLGF knock-in mice to 5xFAD and APP/PS1 mice. Behav Brain Res. 2021;404:113192. doi:10.1016/j.bbr.2021.113192
66. Andorfer C, Kress Y, Espinoza M, et al. Hyperphosphorylation and aggregation of tau in mice expressing normal human tau isoforms. J Neurochem. 2003;86:582–590. doi:10.1046/j.1471-4159.2003.01879.x
67. Davis J, Xu F, Deane R, et al. Early-onset and robust cerebral microvascular accumulation of amyloid beta-protein in transgenic mice expressing low levels of a vasculotropic Dutch/Iowa mutant form of amyloid beta-protein precursor. J Biol Chem. 2004;279:20296–20306. doi:10.1074/jbc.M312946200
68. Molnár Z, Clowry G. Cerebral cortical development in rodents and primates. Prog Brain Res. 2012;195:45–70.
69. Nithianantharajah J, Grant SGN. Cognitive components in mice and humans: combining genetics and touchscreens for medical translation. Neurobiol Learn Mem. 2013;105:13–19. doi:10.1016/j.nlm.2013.06.006
70. Xi Y, Chen Y, Jin Y, et al. Versatile nanomaterials for Alzheimer’s disease: pathogenesis inspired disease-modifying therapy. J Control Release. 2022;345:38–61. doi:10.1016/j.jconrel.2022.02.034
71. Wang J, Gu BJ, Masters CL, et al. A systemic view of Alzheimer disease - insights from amyloid-β metabolism beyond the brain. Nat Rev Neurol. 2017;13:612–623. doi:10.1038/nrneurol.2017.111
72. Moussa-Pacha NM, Abdin SM, Omar HA, et al. BACE1 inhibitors: current status and future directions in treating Alzheimer’s disease. Med Res Rev. 2020;40:339–384. doi:10.1002/med.21622
73. Kennedy ME, Stamford AW, Chen X, et al. The BACE1 inhibitor verubecestat (MK-8931) reduces CNS β-amyloid in animal models and in Alzheimer’s disease patients. Sci Transl Med. 2016;8:363ra150. doi:10.1126/scitranslmed.aad9704
74. Yang X, Yang W, Xia X, et al. Intranasal delivery of BACE1 siRNA and rapamycin by dual targets modified nanoparticles for Alzheimer’s disease therapy. Small. 2022;18:e2203182.
75. Hu B, Xiong B, B-y Q, et al. Construction of a small peptide library related to inhibitor OM99-2 and its structure-activity relationship to beta-secretase. Acta Pharmacol Sin. 2006;27:1586–1593. doi:10.1111/j.1745-7254.2006.00432.x
76. Zhu Y-P, Xiao K, H-P Y, et al. Discovery of potent beta-secretase (bace-1) inhibitors by the synthesis of isophthalamide-containing hybrids. Acta Pharmacol Sin. 2009;30:259–269. doi:10.1038/aps.2008.26
77. Henley D, Raghavan N, Sperling R, et al. Preliminary results of a trial of atabecestat in preclinical Alzheimer’s disease. N Engl J Med. 2019;380:1483–1485. doi:10.1056/NEJMc1813435
78. Wessels AM, Tariot PN, Zimmer JA, et al. Efficacy and safety of lanabecestat for treatment of early and mild Alzheimer disease: the AMARANTH and DAYBREAK-ALZ randomized clinical trials. JAMA Neurol. 2020;77:199–209. doi:10.1001/jamaneurol.2019.3988
79. Yan R, Vassar R. Targeting the β secretase BACE1 for Alzheimer’s disease therapy. Lancet Neurol. 2014;13:319–329. doi:10.1016/S1474-4422(13)70276-X
80. Ou-Yang MH, Kurz JE, Nomura T, et al. Axonal organization defects in the hippocampus of adult conditional BACE1 knockout mice. Sci Transl Med. 2018;10. doi:10.1126/scitranslmed.aao5620
81. Hu X, Hu J, Dai L, et al. Axonal and Schwann cell BACE1 is equally required for remyelination of peripheral nerves. J Neurosci. 2015;35:3806–3814. doi:10.1523/JNEUROSCI.5207-14.2015
82. Moussa CEH. Beta-secretase inhibitors in phase I and phase II clinical trials for Alzheimer’s disease. Expert Opin Investig Drugs. 2017;26:1131–1136. doi:10.1080/13543784.2017.1369527
83. Zhou Y, Zhu F, Liu Y, et al. Blood-brain barrier-penetrating siRNA nanomedicine for Alzheimer’s disease therapy. Sci Adv. 2020;6:1. doi:10.1126/sciadv.abc7031
84. Schenk D, Barbour R, Dunn W, et al. Immunization with amyloid-β attenuates Alzheimer-disease-like pathology in the PDAPP mouse. Nature. 1999;400(6740):173–177. doi:10.1038/22124
85. Ferrer I, Boada Rovira M, Sánchez Guerra ML, et al. Neuropathology and pathogenesis of encephalitis following amyloid-beta immunization in Alzheimer’s disease. Brain Pathol. 2004;14:11–20. doi:10.1111/j.1750-3639.2004.tb00493.x
86. Winblad B, Andreasen N, Minthon L, et al. Safety, tolerability, and antibody response of active Aβ immunotherapy with CAD106 in patients with Alzheimer’s disease: randomised, double-blind, placebo-controlled, first-in-human study. Lancet Neurol. 2012;11:597–604. doi:10.1016/S1474-4422(12)70140-0
87. Wang CY, Wang P-N, Chiu M-J, et al. UB-311, a novel UBITh® amyloid β peptide vaccine for mild Alzheimer’s disease. Alzheimers Dement. 2017;3:262–272. doi:10.1016/j.trci.2017.03.005
88. Vermunt L, Sikkes SAM, van den Hout A, et al. Duration of preclinical, prodromal, and dementia stages of Alzheimer’s disease in relation to age, sex, and APOE genotype. Alzheimers Dement. 2019;15:888–898. doi:10.1016/j.jalz.2019.04.001
89. Zagorski K, King O, Hovakimyan A, et al. Novel vaccine against pathological pyroglutamate-modified amyloid beta for prevention of Alzheimer’s disease. Int J Mol Sci. 2023;24. doi:10.3390/ijms24129797
90. Tarasoff-Conway JM, Carare RO, Osorio RS, et al. Clearance systems in the brain-implications for Alzheimer disease. Nat Rev Neurol. 2015;11:457–470. doi:10.1038/nrneurol.2015.119
91. Leng F, Edison P. Neuroinflammation and microglial activation in Alzheimer disease: where do we go from here? Nat Rev Neurol. 2021;17:157–172. doi:10.1038/s41582-020-00435-y
92. Hickman SE, Allison EK, El Khoury J. Microglial dysfunction and defective beta-amyloid clearance pathways in aging Alzheimer’s disease mice. J Neurosci. 2008;28:8354–8360. doi:10.1523/JNEUROSCI.0616-08.2008
93. Dinda B, Dinda M, Kulsi G, et al. Therapeutic potentials of plant iridoids in Alzheimer’s and Parkinson’s diseases: a review. Eur J Med Chem. 2019;169:185–199. doi:10.1016/j.ejmech.2019.03.009
94. Cheng Y, Tian D-Y, Wang Y-J. Peripheral clearance of brain-derived Aβ in Alzheimer’s disease: pathophysiology and therapeutic perspectives. Transl Neurodegener. 2020;9:16. doi:10.1186/s40035-020-00195-1
95. Hansen DV, Hanson JE, Sheng M. Microglia in Alzheimer’s disease. J Cell Biol. 2018;217:459–472. doi:10.1083/jcb.201709069
96. Rahimifard M, Maqbool F, Moeini-Nodeh S, et al. Targeting the TLR4 signaling pathway by polyphenols: a novel therapeutic strategy for neuroinflammation. Ageing Res Rev. 2017;36:11–19. doi:10.1016/j.arr.2017.02.004
97. Zhang H, Jiang W, Zhao Y, et al. Lipoprotein-inspired nanoscavenger for the three-pronged modulation of microglia-derived neuroinflammation in Alzheimer’s disease therapy. Nano Lett. 2022;22:2450–2460. doi:10.1021/acs.nanolett.2c00191
98. Yang F, Zhao D, Cheng M, et al. mTOR-mediated immunometabolic reprogramming nanomodulators enable sensitive switching of energy deprivation-induced microglial polarization for Alzheimer’s disease management. ACS Nano. 2023;2023:1.
99. Song W, Hooli B, Mullin K, et al. Alzheimer’s disease-associated TREM2 variants exhibit either decreased or increased ligand-dependent activation. Alzheimers Dement. 2017;13:381–387. doi:10.1016/j.jalz.2016.07.004
100. Pan R-Y, Ma J, Kong -X-X, et al. Sodium rutin ameliorates Alzheimer’s disease-like pathology by enhancing microglial amyloid-β clearance. Sci Adv. 2019;5:eaau6328. doi:10.1126/sciadv.aau6328
101. Yeh FL, Wang Y, Tom I, et al. TREM2 binds to apolipoproteins, including APOE and CLU/APOJ, and thereby facilitates uptake of amyloid-beta by microglia. Neuron. 2016;91:328–340. doi:10.1016/j.neuron.2016.06.015
102. Tuzer F, Torres C. Involvement of astrocyte senescence in Alzheimer’s disease. Curr Opin Neurobiol. 2022;76:102594. doi:10.1016/j.conb.2022.102594
103. Brandebura AN, Paumier A, Onur TS, et al. Astrocyte contribution to dysfunction, risk and progression in neurodegenerative disorders. Nat Rev Neurosci. 2023;24:23–39. doi:10.1038/s41583-022-00641-1
104. Bellaver B, Povala G, Ferreira PCL, et al. Astrocyte reactivity influences amyloid-β effects on tau pathology in preclinical Alzheimer’s disease. Nat Med. 2023;29:1775–1781. doi:10.1038/s41591-023-02380-x
105. Paumier A, Boisseau S, Jacquier-Sarlin M, et al. Astrocyte-neuron interplay is critical for Alzheimer’s disease pathogenesis and is rescued by TRPA1 channel blockade. Brain. 2022;145:388–405. doi:10.1093/brain/awab281
106. McAlpine CS, Park J, Griciuc A, et al. Astrocytic interleukin-3 programs microglia and limits Alzheimer’s disease. Nature. 2021;595:701–706. doi:10.1038/s41586-021-03734-6
107. Roberts KF, Elbert DL, Kasten TP, et al. Amyloid-β efflux from the central nervous system into the plasma. Ann Neurol. 2014;76:837–844. doi:10.1002/ana.24270
108. Rasmussen MK, Mestre H, Nedergaard M. The glymphatic pathway in neurological disorders. Lancet Neurol. 2018;17:1016–1024. doi:10.1016/S1474-4422(18)30318-1
109. Storck SE, Meister S, Nahrath J, et al. Endothelial LRP1 transports amyloid-β(1-42) across the blood-brain barrier. J Clin Invest. 2016;126:123–136. doi:10.1172/JCI81108
110. Ma M, Liu Z, Gao N, et al. Self-protecting biomimetic nanozyme for selective and synergistic clearance of peripheral amyloid-β in an Alzheimer’s disease model. J Am Chem Soc. 2020;142:21702–21711. doi:10.1021/jacs.0c08395
111. Kim D, Kwon HJ, Hyeon T. Magnetite/ceria nanoparticle assemblies for extracorporeal cleansing of amyloid-β in Alzheimer’s disease. Adv Mater. 2019;31:e1807965. doi:10.1002/adma.201807965
112. Lee D, Kim HV, Kim HY, et al. Chemical-driven outflow of dissociated amyloid burden from brain to blood. Adv Sci. 2022;9:e2104542. doi:10.1002/advs.202104542
113. Perosa V, Oltmer J, Munting LP, et al. Perivascular space dilation is associated with vascular amyloid-β accumulation in the overlying cortex. Acta Neuropathol. 2022;143:331–348. doi:10.1007/s00401-021-02393-1
114. Hablitz LM, Plá V, Giannetto M, et al. Circadian control of brain glymphatic and lymphatic fluid flow. Nat Commun. 2020;11:4411. doi:10.1038/s41467-020-18115-2
115. Da Mesquita S, Papadopoulos Z, Dykstra T, et al. Meningeal lymphatics affect microglia responses and anti-Aβ immunotherapy. Nature. 2021;593:255–260. doi:10.1038/s41586-021-03489-0
116. Stower H. Meningeal lymphatics in aging and Alzheimer’s disease. Nat Med. 2018;24:1781.
117. Mangialasche F, Solomon A, Winblad B, et al. Alzheimer’s disease: clinical trials and drug development. Lancet Neurol. 2010;9:702–716. doi:10.1016/S1474-4422(10)70119-8
118. Hunsberger JG, Rao M, Kurtzberg J, et al. Accelerating stem cell trials for Alzheimer’s disease. Lancet Neurol. 2016;15:219–230. doi:10.1016/S1474-4422(15)00332-4
119. Tcw J, Qian L, Pipalia NH, et al. Cholesterol and matrisome pathways dysregulated in astrocytes and microglia. Cell. 2022;185:2213–2233.e2225. doi:10.1016/j.cell.2022.05.017
120. Gao Y, Wang Y, Lei H, et al. A novel transgenic mouse line with hippocampus-dominant and inducible expression of truncated human tau. Transl Neurodegener. 2023;12:51. doi:10.1186/s40035-023-00379-5
121. Cheng Y, He CY, Tian DY, et al. Physiological β-amyloid clearance by the liver and its therapeutic potential for Alzheimer’s disease. Acta Neuropathol. 2023;145:717–731. doi:10.1007/s00401-023-02559-z
 © 2023 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2023 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.