Back to Journals » Drug Design, Development and Therapy » Volume 9
Current and emerging treatment options for ANCA-associated vasculitis: potential role of belimumab and other BAFF/APRIL targeting agents
Received 3 November 2014
Accepted for publication 24 November 2014
Published 7 January 2015 Volume 2015:9 Pages 333—347
DOI https://doi.org/10.2147/DDDT.S67264
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 4
Editor who approved publication: Professor Shu-Feng Zhou
Video abstract presented by Lenert
Views: 1384
Aleksander Lenert,1 Petar Lenert2
1Division of Rheumatology, University of Kentucky, Kentucky Clinic, Lexington, KY, USA; 2Division of Immunology, Department of Internal Medicine, The University of Iowa, Iowa City, IA, USA
Abstract: Antineutrophil cytoplasmic antibody (ANCA)-associated vasculitis (AAV) comprises several clinical entities with diverse clinical presentations, outcomes, and nonunifying pathogenesis. AAV has a clear potential for relapses, and shows unpredictable response to treatment. Cyclophosphamide-based therapies have remained the hallmark of induction therapy protocols for more than four decades. Recently, B-cell depleting therapy with the anti-CD20 antibody rituximab has proved beneficial in AAV, leading to Food and Drug Administration approval of rituximab in combination with corticosteroids for the treatment of AAV in adults. Rituximab for ANCA-associated vasculitis and other clinical trials provided clear evidence that rituximab was not inferior to cyclophosphamide for remission induction, and rituximab appeared even more beneficial in patients with relapsing disease. This raised hopes that other B-cell-targeted therapies directed either against CD19, CD20, CD22, or B-cell survival factors, B-cell activating factor of the tumor necrosis factor family (BAFF) and a proliferation-inducing ligand could also be beneficial for the management of AAV. BAFF neutralization with the fully humanized monoclonal antibody belimumab has already shown success in human systemic lupus erythematosus and, along with another anti-BAFF reagent blisibimod, is currently undergoing Phase II and III clinical trials in AAV. Local production of BAFF in granulomatous lesions and elevated levels of serum BAFF in AAV provide a rationale for BAFF-targeted therapies not only in AAV but also in other forms of vasculitis such as Behcet’s disease, large-vessel vasculitis, or cryoglobulinemic vasculitis secondary to chronic hepatitis C infection. BAFF-targeted therapies have a very solid safety profile, and may have an additional benefit of preferentially targeting newly arising autoreactive B cells over non-self-reactive B cells.
Keywords: B-cell-activating factor of the TNF family, a proliferation-inducing ligand, antineutrophil cytoplasmic antibody-associated vasculitis, granulomatosis with polyangiitis, microscopic polyangiitis, B cells
Insight into the classification, pathogenesis, and current management of AAV
Antineutrophil cytoplasmic antibody (ANCA)-associated vasculitis (AAV) includes several life-threatening forms of vasculitis: granulomatosis with polyangiitis (GPA), microscopic polyangiitis (MPA), eosinophilic granulomatosis with polyangiitis (EGPA), and renal-limited vasculitis. The connecting pathologic feature of this group of diseases is a necrotizing small-vessel vasculitis commonly affecting multiple organs, including lungs and kidneys (pulmonary–renal syndromes).1 Despite grouping them together under the umbrella of AAV, there are significant clinical and pathophysiologic differences between these diseases with implications for treatment. These diseases typically present with high titer ANCA. Two major ANCA targets are proteinase 3 (PR3-ANCA), giving rise to cytosplasmic (C)-ANCA pattern, and myeloperoxidase (MPO-ANCA), which gives rise to perinuclear (P)-ANCA pattern on ethanol-fixed neutrophils. These antigens are found within the cytoplasm of neutrophils, but can also be found on the cell surface of a subset of neutrophils.1,2 Occasionally, other autoantigens can be targeted by ANCA, such as cathepsin G, lactoferrin, lysozyme, bacterial permeability increasing factor, hLAMP-2, and elastase. Atypical P-ANCA staining can sometimes be found in other diseases, such as inflammatory bowel disease, rheumatoid arthritis (RA), cystic fibrosis, and primary sclerosing cholangitis. ANCA can even coexist with ANA, as reported in cases of drug-induced vasculitis associated with chronic hydralazine or minocycline use.3 The role of B cells in AAV extends way beyond their role in ANCA production. B cells are excellent antigen-presenting cells for antigens delivered via their B-cell receptor for antigen. When costimulated through their innate receptors (eg, Toll-like receptors 4, 7, and 9), B cells can upregulate costimulatory molecules of the B7 family, allowing them to provide a second signal necessary for the cognate T-cell activation. They can also secrete proinflammatory cytokines, such as interleukin (IL)-6 and tumor necrosis factor (TNF), that can downregulate the function of regulatory T cells and boost the differentiation of effector T cells. Indeed, the complex and delicate interplay between T cells – including circulating follicular helper T cells and regulatory T cells – and B cells has been observed in GPA patients treated with rituximab. Treatment with rituximab, but not conventional therapy, resulted in restored balance between follicular helper T cells and regulatory T cells, similar to the one seen in healthy controls.4 Increased frequencies of effector memory T cells, and particularly IL-21-producing follicular helper T cells, have been observed in patients with GPA and were restricted to ANCA-positive patients.5 Once released, IL-21 enhanced in vitro production of immunoglobulin G (IgG) and ANCA in GPA patients. Finally, B cells may also have an important regulatory function, which is diminished in AAV.6
GPA is a complex systemic disease characterized by granulomatous inflammation of the upper airways and lungs, together with a predominant small-vessel vasculitis. GPA is clinically associated with the presence of ANCA-targeting PR3-ANCA. A recent large-scale genome-wide association study has shown strong genetic predisposition for making PR3-ANCA versus MPO-ANCA antibodies.7 In addition to airway disease, pauci-immune necrotizing glomerulonephritis can be seen in up to three-fourths of the patients, leading to end-stage renal disease in 20%–25% of patients within 5 years. Over the same time period, clinical relapses are seen in up to 50% of patients.2 Unfortunately, there are currently no reliable disease biomarkers that can sensitively predict flares of GPA in an individual patient. Management of GPA varies greatly from one case to other based on the extent of systemic involvement (localized/limited vs multisystemic disease) and relapsing nature of the disease. Further difficulties in the management of GPA come from a need for prolonged maintenance therapy and drug- and disease-related comorbidities.
In contrast to GPA, MPA is characterized by systemic small vasculitis, including a very common pauci-immune glomerulonephritis, but without evidence of systemic granulomatous disease. Serologically, MPA presents with MPO-ANCA. Chronic lung damage in MPA, while common, tends to assume a more restrictive fibrosing pattern different from that seen in GPA and EGPA. Another distinctive feature is that MPA has a significantly lower relapse rate compared to GPA.2
While the ultimate goal of the treatment of AAV should aim at restoring the abnormal immunologic tolerance to ANCA-associated autoantigens, such a goal is currently unrealistic. We need to better understand what leads to defective immunologic checkpoints at different stages of lymphoid development, allowing escape and subsequent survival of autoreactive T and B cells.
Since its original description of GPA and up to the early 1970s, the prognosis of GPA was very poor with a very low survival rate in critically ill patients. Combined cyclophosphamide and corticosteroid treatment, along with hemodialysis and ventilator support, was the first promising therapy for this deadly disease, first introduced at the National Institutes of Health in the early 1970s.8 Thereafter, cyclophosphamide-based regimens became the standard of care for remission induction in GPA, MPA, and severe cases of EGPA. However, high cumulative dose of cyclophosphamide has been associated with serious side effects including infections, bone marrow toxicity, infertility, and cancer (particularly bladder cancer; acute myeloid leukemia, and non-melanoma skin cancer).9 In line with this, a recent study, surprisingly, showed that the early mortality in GPA was more commonly associated with secondary infections due to immunosuppression rather than to active vasculitis.10 Early mortality during the first year of treatment thus remains a significant clinical problem, and novel therapies are therefore desperately needed.
Treatment of AAV (both GPA and MPA) can be divided into two phases: induction of remission and maintenance. In the first phase, oral cyclophosphamide (dosed 2 mg/kg/day up to 150 mg/day and adjusted for renal insufficiency) and high-dose corticosteroids (pulse IV methylprednisolone followed by prednisone 1 mg/kg/day) are employed to rapidly reduce inflammation and prevent permanent organ damage. In the remission maintenance phase, use of less toxic immunosuppression is aimed at reducing the incidence of relapses. The toxicity is especially severe in elderly patients and those who present with severe renal involvement. Studies have shown that cyclophosphamide toxicity can be reduced by switching from oral cyclophosphamide to azathioprine once remission is achieved, usually within the 3–6 months period. Use of IV cyclophosphamide is associated with lower cumulative dose and reduced toxicity. However, while a similar remission induction rate was observed, the relapse rate was unfortunately higher in those treated with IV cyclophosphamide.2
Methotrexate has also been used in early induction phase, but it is less effective than cyclophosphamide and is reserved for those with localized/limited disease or those without major organ involvement.
Plasma exchange is frequently used in AAV patients, particularly in those presenting with severe renal involvement resulting in rapidly deteriorating renal function.11 The rationale for plasma exchange is to rapidly remove ANCA and other inflammatory mediators, before the effect of immunosuppressive/anti-inflammatory agents comes into play. PEXIVAS, an international, multicenter clinical trial, is currently evaluating the benefits from plasma exchange in renal recovery and in patients with pulmonary hemorrhage (Clinicaltrials.gov NCT00987389, study is recruiting participants, no study results provided).
A major breakthrough in the management of the induction phase of AAV, as an alternative to cyclophosphamide, came from Rituximab in ANCA-associated vasculitis (RAVE) and RITUXVAS (an international, randomized, open-label trial comparing a rituximab-based regimen with a standard cyclophosphamide/azathioprine regimen in the treatment of active, “generalized” AAV) studies employing a B-cell-depleting agent rituximab.12,13 Rituximab (chimeric human/mouse anti-CD20 antibody) in combination with corticosteroids was not inferior to cyclophosphamide and corticosteroids for remission induction in AAV (GPA and MPA). The RAVE study enrolled 197 patients with AAV (newly diagnosed or relapsing GPA or MPA) allocated to induction therapy with rituximab or to daily oral cyclophosphamide (2 mg/kg/day) in addition to corticosteroids. After remission, cyclophosphamide was replaced with azathioprine. Rituximab dose was 375 mg/m2 and was administered once weekly for a period of 4 weeks. The primary endpoint was complete disease remission and complete tapering of prednisone at 6 months. Sixty-four percent of patients in the rituximab group versus 53% in the cyclophosphamide group (P<0.0001 for non-inferiority) achieved complete remission and were steroid-free at 6 months. Furthermore, rituximab appeared more effective in inducing remission in a subgroup of patients with relapsing disease (67% vs 42%, P=0.01).12 However, it is unclear whether rituximab is as effective in cases of severe AAV, because patients with severe renal failure (creatinine >4 mg/dL) and severe respiratory involvement requiring mechanical ventilation were excluded from the trial. Thus, current vasculitis experts prefer the use of oral cyclophosphamide for cases with severe multisystem disease at presentation. Adverse rates were not different between treatment groups. In the RITUXVAS study, the non-inferiority of rituximab to cyclophosphamide was studied in 44 patients with newly diagnosed GPA or MPA complicated with glomerulonephritis. The primary endpoints were sustained remission at 12 months and severe adverse events. Both groups showed nonsignificant differences. These two studies have led to Food and Drug Administration (FDA) approval of rituximab in combination with glucocorticoids for adults with GPA and MPA. Rituximab should therefore be considered equivalent to cyclophosphamide as a first-line therapy in patients with GPA and MPA. In those who fail both agents, open-label studies suggested a potential benefit from alemtuzumab (anti-CD52), anti-TNF antagonists (with the caveat that etanercept failed to provide benefits in a larger study),14 mycophenolate mofetil, complement inhibitors, 15-deoxyspergualin, CTLA4-Ig, or IV immunoglobulins.2,15
The cumulative effect of rituximab in AAV is unknown, but few cases of John Cunningham (JC) virus infection leading to progressive multifocal leukoencephalopathy have been reported.16 Besides rituximab, other fully humanized anti-CD20 antibodies have been developed (eg, ofatumumab, ocrelizumab, and veltuzumab), but none has been approved for the treatment of AAV. Another antibody, epratuzumab, targets the human CD22 on B cells and works by inducing a negative regulation of B cells stimulated via their B-cell receptor for antigen and, while it is currently undergoing a trial in systemic lupus erythematosus (SLE), it is unclear at this time whether it can be beneficial in AAV.
Regarding maintenance therapy for GPA, studies have suggested that azathioprine and methotrexate are equivalent in maintaining remission,17 with the caveat that methotrexate cannot be used in patients with significant renal dysfunction. Leflunomide (high dose of 30 mg/day) could be considered as another alternative agent to methotrexate,18 while, surprisingly, mycophenolate mofetil was less effective in maintaining remission compared to azathioprine.19 The optimal duration of maintenance therapy for GPA is unknown and is currently undergoing clinical evaluation.
Management of the localized disease remains a very challenging task and requires a team-based approach between rheumatologists, ENT specialists, and ophthalmologists. There is a suggestion that rituximab maybe less effective in treating localized granulomatous disease compared to generalized disease, particularly in those with orbital masses and pachymeningitis.20 However, anecdotal clinical experience with rituximab for localized mass lesions has shown promise. Older literature suggests that treatment with trimethoprim–sulfamethoxazole for 24 months may reduce the incidence of relapses in upper respiratory GPA, likely through an effect on nasal carriage of Staphylococcus aureus.21 Obstructive tracheobronchial disease can lead to permanent scarring, and is another example of poor responsiveness to systemic treatment. Early tracheobronchial disease sometimes responds well to intralesional corticosteroids with or without intralesional mitomycin-C and endoluminal dilatation.22,23 Tracheal and bronchial stenosis can predispose patients to recurrent chest infections. Reconstructive surgery for saddle nose deformity is currently recommended only for patients in clinical remission. Local management with intranasal glucocorticoids and regular saline washes can help patients with chronic nasal crusting and sinusitis. Surgical intervention should be considered in patients who develop obstruction of the middle ear.24 However, these recommendations are based on limited evidence usually from small case series or individual reports.
Minor relapses can be managed with increasing dose of oral glucocorticoids or by optimizing the maintenance immunosuppressive therapy. Unfortunately, major relapses may require a repeat of the induction therapy. Lung and upper respiratory involvement in GPA is associated with higher relapse rates, and, interestingly, previous relapses are predictive of future flares.2,25 In relapsing patients, scheduled maintenance therapy with rituximab (MAINRITSAN study) appears to be very effective for remission maintenance and is superior to azathioprine (5% vs 29% at month 28).26 In that study, low-dose rituximab 500 mg was administered at days 0 and 14, at 6 months, 12 months, and 18 months for the total of five infusions. However, this observation by the French Vasculitis groups has yet to be verified in a prospective clinical trial (RITAZAREM study). This study will compare conventional DMARD treatment with fixed-interval courses of rituximab for prevention of disease flares.
Therefore, the success of B-cell-depleting therapy with rituximab in both induction phase and maintenance phase of AAV has opened the entryway for other B-cell-targeted therapies. The purpose of this review is to explore the rationale for targeting BAFF, a B-cell survival factor. Neutralization of BAFF with the anti-BAFF antibody belimumab has recently been approved by FDA for the treatment of SLE and is currently undergoing Phase II/III clinical trials in vasculitis.
Rationale for targeting BAFF in vasculitis
Role of BAFF in B-cell maturation
BAFF is a member of the TNF family, also known as BLyS. Other commonly used names for this molecule are TNFSF13b, TALL-1, THANK, and zTNF4. BAFF plays a crucial role in B-cell development by promoting B-cell survival and transition from the immature to mature B-cell stage. It also plays a role in Ig-class switching and subsequent antibody production in vivo. BAFF can costimulate B-cell proliferation and splenic B-cell survival in vitro.27–29
BAFF is a transmembrane protein from which, by action of furin protease, a biologically active protein is generated (soluble BAFF).30At this time, a role for membrane BAFF is unknown. Soluble BAFF binds to three different TNF receptors: B-cell maturation antigen (BCMA), transmembrane activator and calcium modulator and cyclophilin ligand interactor (TACI), and BAFF-R (BR3). BCMA and TACI, but not BAFF-R, are also receptors for another B-cell survival ligand – a proliferation-inducing ligand (APRIL) (Figure 1).27 Binding of BAFF to its high-affinity BAFF-R activates the NF-κB pathway (both classical and noncanonical pathways) and MAPK pathway, leading to the expression of genes essential for B-cell survival.31 Besides B cells, BAFF can also augment certain Th1 responses in vivo.32
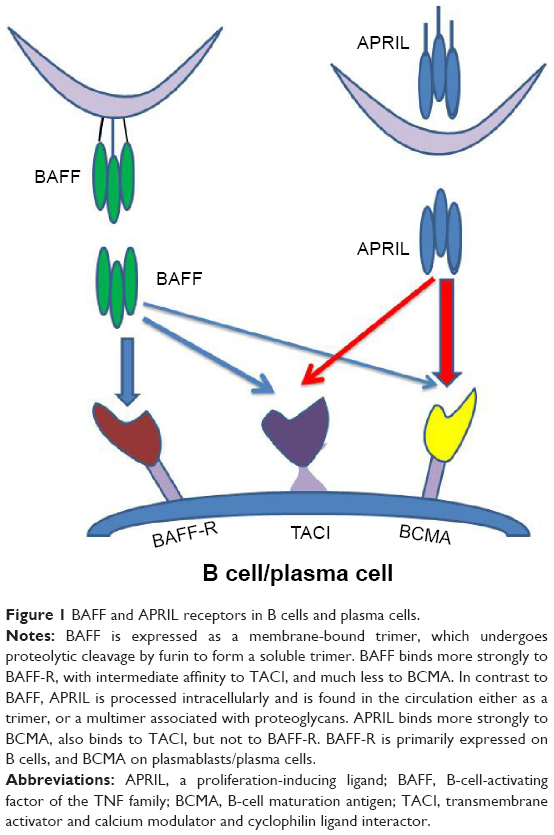 | Figure 1 BAFF and APRIL receptors in B cells and plasma cells. Notes: BAFF is expressed as a membrane-bound trimer, which undergoes proteolytic cleavage by furin to form a soluble trimer. BAFF binds more strongly to BAFF-R, with intermediate affinity to TACI, and much less to BCMA. In contrast to BAFF, APRIL is processed intracellularly and is found in the circulation either as a trimer, or a multimer associated with proteoglycans. APRIL binds more strongly to BCMA, also binds to TACI, but not to BAFF-R. BAFF-R is primarily expressed on B cells, and BCMA on plasmablasts/plasma cells. Abbreviations: APRIL, a proliferation-inducing ligand; BAFF, B-cell-activating factor of the TNF family; BCMA, B-cell maturation antigen; TACI, transmembrane activator and calcium modulator and cyclophilin ligand interactor. |
While BAFF appears to have a primary role in promoting survival of immature B cells, APRIL appears to act at later stages of B-cell development supporting the maintenance of plasma cells. Interestingly, switched human memory B cells (CD27 +IgD−) may not depend on either BAFF or APRIL.33
A variety of cell types have been shown to be capable of making BAFF. While cells of the monocyte/macrophage lineage appear to be a primary source of BAFF production in vitro, under certain stimulatory conditions neutrophils can also express and release BAFF.34
BAFF – possible pathogenic role in systemic autoimmune diseases: animal models
Elevated BAFF levels favor positive selection of autoreactive B cells and abnormal autoantibody production in animals.35 While BAFF overexpression cannot rescue highly autoreactive B cells, which are typically deleted during early stages of B-cell development, they can save those self-reactive B cells normally deleted at the late T2 stage of peripheral B-cell development. This has been nicely demonstrated in a model of anti-HEL self-reactive B cells.36 BAFF-transgenic mice overexpressing BAFF develop a systemic disease closely mimicking human SLE and Sjögren’s syndrome characterized by excessive autoantibody production, increased peripheral B-cell numbers, and hypergammaglobulinemia.37 These animals also develop lymphadenopathy and splenomegaly and may suffer from arthritis and immune-complex-mediated glomerulonephritis.
Several strains of mice that spontaneously develop a lupus-like disease (eg, MRL-Fas
On the contrary, mice deficient in BAFF lack transitional T2-B cells as well as mature marginal zone and follicular B cells, and have significantly reduced spleen weights. BAFF-deficient mice appear to have adequate number of T1-B cells and B1 cells, and their T-cell zones appear normal. BAFF−/− mice have a ten-fold reduction in total serum Ig level and mount diminished T-cell independent and T-cell dependent antibody responses.29
BAFF in human systemic and organ-specific autoimmune diseases
Like mice, humans with the BAFF-R gene deletion have severe B-cell lymphopenia. B cells are arrested at the transitional B-cell stage and this condition presents with adult onset antibody-deficiency syndrome.41 Humans with this condition have diminished numbers of mature B cells, eg, follicular, marginal zone, and memory B cells, and their T-independent immune responses are severely impaired.41
In relation to the possible role of BAFF in autoimmunity, several rheumatic diseases such as SLE, Sjögren’s syndrome, systemic sclerosis, and RA have all been shown to have elevated serum levels of BAFF.42–46 Particularly high levels of serum BAFF were observed in patients with Sjögren’s syndrome.43,44 High levels of BAFF have been associated with high double-stranded DNA (dsDNA) antibody titers in SLE, anti-SS-A in primary Sjögren’s syndrome, and rheumatoid factor (RF) levels in RA.
Several studies in humans have shown a clear association between elevated serum BAFF levels and SLE.47–49 Zhang was the first to observe elevated levels of soluble BAFF in SLE (and also in RA).42 Patients with higher BAFF levels tended to have higher anti-dsDNA antibody levels. Later studies confirmed this observation and found a good relationship between the elevated BAFF levels and subsequent increase in lupus disease activity scores, thus identifying BAFF as a valid target for SLE treatment.50
In addition to systemic autoimmune disease, BAFF is also elevated in organ-specific autoimmune diseases such as Graves’ disease, anti-GBM disease, autoimmune pancreatitis, myasthenia gravis, idiopathic thrombocytopenic purpura, and multiple sclerosis.32 Interestingly, independent of the autoimmune disease, increased soluble BAFF levels were also found in B-cell malignancies and certain primary antibody deficiencies (BTK, BAFF-R, or TACI deficiency).51
BAFF studies in AAV
ANCA directed against PR3 is the principal autoimmune target in patients with GPA (formerly Wegener’s granulomatosis). It is believed that ANCA binding to cytokine-primed neutrophils (eg, granulocyte-colony stimulating factor, IFN-γ or TNF-primed neutrophils) or fMLP-treated nonprimed neutrophils may initiate neutrophil adhesion, transmigration, and endothelial cell injury, as well as MPO and PR3 upregulation, in vitro.52 Binding of ANCA to these cells may lead to superoxide generation and release of lytic enzymes and proinflammatory cytokines/chemokines, such as IL-8. Furthermore, short pretreatment with anti-PR3 IgG antibodies results in significant cell-surface expression of BAFF followed by its shedding into the culture medium. Such culture supernatants could further promote BAFF-dependent survival of Burkitt’s lymphoma-derived centroblast cell line L3055, thus pointing toward possible pathogenic mechanism in AAV.53
Intravenous injection of mouse antibodies specific for MPO can induce pauci-immune crescentic glomerulonephritis that mimics human disease.54 Another layer of evidence for potential pathogenic role of anti-MPO antibodies comes from maternal–fetal transfer studies.55 Unfortunately, an animal model for PR3-AAV is still missing and has yet to be developed. While the persistence of positive ANCA in GPA patients after induction of remission with cyclophosphamide correlates with increased risk of relapse, there is no clear-cut linear correlation between ANCA positivity and disease activity, thus raising a need for more sensitive disease biomarkers.56
It has been well established that patients with active AAV have increased numbers of activated B cells. Autoreactive B cells within granulomas may act as antigen-presenting cells and may promote T-cell activation through direct interaction and/or secretion of proinflammatory cytokines (eg, IL-6, TNF-α). Germinal centers, typical of formation of tertiary lymphoid organs, have been detected in granulomatous lesions from patients with GPA.57 More importantly, autoreactive B cells, when compared to those without self-reactivity, appear to have heightened dependency on BAFF. Excessive BAFF production can rescue at least some autoreactive B cells from peripheral deletion, allowing them to enter forbidden niches within lymphoid organs.36 While the above data support a possible role for B cells in GPA, they do not rule out a role for antigen-specific T cells, particularly Th17 cells, in line with recent observation of elevated IL-17 and IL-23 levels in GPA.58
BAFF in GPA (Wegener’s granulomatosis)
Edberg, in 2004, was the first to observe elevated levels of BAFF in patients with GPA.59 It was later confirmed by Krumbholz, who found evidence of elevated serum levels of BAFF in a cohort of 46 GPA patients from the Mayo Clinic.60 Difference in BAFF levels was even more pronounced when patients with active untreated disease were compared with those treated with glucocorticoids or controls. Interestingly, a small subset of healthy donors had a very high serum BAFF level, which was stable over time, probably reflecting a genetic polymorphism.61 A small study showed no difference in BAFF levels between patients with limited GPA and those with generalized GPA.62 Based on these studies, Krumbholz first suggested that neutralization of BAFF may provide potential benefits to a subset of chronically relapsing GPA patients.
Evidence of T-cell activation, as reflected by elevation of soluble IL-2R and soluble CD30, was found in patients with active GPA. These markers were also associated with elevated BAFF levels in a longitudinal study where GPA patients were followed for 24 months. However, while soluble IL-2R, soluble CD30, and IL-10 levels were higher at the time of diagnosis and during a relapse, this was not the case with BAFF.63 BAFF levels did not differ significantly between patients who had a relapse and those without a relapse. It also failed to correlate with ANCA positivity.62
Important evidence for the role of BAFF/APRIL in the pathogenesis of GPA came from studying mucosal biopsy samples, which showed evidence of activated B cells alongside the PR3-expressing cells and B-cell survival factors BAFF and APRIL.64
BAFF in MPO-associated vasculitis and eosinophilic GPA (formerly Churg–Strauss syndrome)
Schneeweis compared BAFF levels in patients with GPA, MPA, and EGPA.65 Elevated levels were observed only in those with GPA. In contrast, VCAM-1, a marker for endothelial cell activation, was elevated in all patients with AAV. Opposing results were found in a study directed by Xin in People’s Republic of China, which showed significant elevation of BAFF in patients with MPO-AAV. Out of 121 patients with MPO-ANCA that were studied, 100 of them had MPA, 18 were diagnosed with GPA, and three had renal-limited vasculitis. When compared to healthy controls, BAFF levels were significantly elevated in both active disease and in remission, although patients with active disease still had significantly higher levels than those in remission.66 Serum BAFF correlated well with the Birmingham Vasculitis Activity Score (BVAS) and erythrocyte sedimentation rate (ESR), but failed to correlate with the MPO-ANCA titer. Another B-cell survival factor APRIL, along with BAFF, was studied in 37 Japanese patients with MPO-ANCA-associated renal vasculitis. Serum BAFF (but not APRIL) appeared be a very useful marker for distinguishing patients with active vasculitis from those with inactive vasculitis and from those with infectious complications.67
BAFF in large-vessel vasculitis (Takayasu arteritis and giant-cell arteritis)
Takayasu arteritis (TAK) affects large elastic arteries with predilection for the aorta and its main branches. TAK may have a very silent clinical course and is typically diagnosed late, often at the time when irreversible damage has already occurred. Furthermore, the disease may even progress despite treatment with glucocorticoids and normal acute-phase reactants. Even in those patients deemed to be in clinical remission, new angiographic changes could be detected in up to 60% of patients. In a small study performed on nine patients with TAK, BAFF levels were higher in TAK patients compared to healthy controls. BAFF levels tended to diminish with treatment.68
Giant-cell arteritis (GCA) is the most common form of systemic vasculitis in the western world. While GCA is traditionally viewed as a T-cell-mediated disease with additional roles for dendritic cells and macrophages, a recent study evaluated the relationship between B-cell numbers, B-cell phenotype, and serum BAFF levels in patients with newly diagnosed GCA.69 Active disease was characterized by an inverse relationship between decreased B-cell numbers, especially low B effector (but not regulatory B) cells, and increased serum levels of BAFF. Following corticosteroid treatment, serum BAFF and the number of B effector cells normalized. One explanation for these findings is that circulating B cells were redistributed during the active phase, but quickly returned to peripheral blood during remission. These B effector cells showed enhanced ability to make IL-6. Few B cells could be found in temporal artery biopsy specimens. It is believed that B-cell-derived IL-6 may potentiate T-cell-mediated autoimmune responses (Th17 responses), linking these two arms of the immune system together. Indeed, this study found a positive correlation between serum BAFF and IL-6 levels. It would be of interest to see whether B cell depletion therapy, BAFF neutralization, IL-6 receptor blockade, or IL-17A blockade can ameliorate disease in patients with GCA.
BAFF in other forms of vasculitis
BAFF has also been studied in Behcet’s disease, Kawasaki disease, and hepatitis C-related mixed cryoglobulinemia.
Vasculitis in Behcet’s disease may affect arterial and venous blood vessels of any size. Serum BAFF levels were found to be elevated in patients with active Behcet’s disease compared to healthy subjects.70–72 There was also a positive correlation between serum BAFF levels, skin BAFF mRNA expression, and extent of skin involvement. Patients with vasculitic features tended to have higher BAFF-R expression on their B cells. In vitro treatment of B lymphocytes from active Behcet’s disease with BAFF resulted in enhanced IgG and IL-6 secretion. Increased BAFF levels were further found in cerebrospinal fluid (CSF) in patients with neuro-Behcet’s disease. BAFF levels correlated with progressive dementia and psychosis, but not with myelopathy, aseptic meningitis, or serum BAFF levels.73 Another study found a correlation between BAFF levels and uveitis.72
A case report found elevated BAFF in a 2-year-old girl with Kawasaki’s disease refractory to IVIG treatment.74 Another report recently confirmed this observation.75
Finally, host genetic background related to BAFF promoter polymorphism and elevated serum BAFF levels were found in a subset of patient with hepatitis C virus infection complicated with mixed cryoglobulinemia.76,77 This is of particular interest, as B-cell-depleting therapy with rituximab has shown benefits in this condition.
Is BAFF a potential biomarker for systemic autoimmune disease and vasculitis?
In SLE, relatively good correlation exists between serum BAFF, autoantibody levels, and disease activity.42,45
With regard to AAV, reports suggest a possible difference between patients with GPA and MPO-associated vasculitis when it comes to correlation between BAFF and disease activity. For example, BAFF, although elevated, did not correlate with C-reactive protein (CRP) levels, BVAS, disease extent index, or vasculitis damage index scores in GPA patients.62 Moreover, individual serum BAFF levels appeared stable over time in up to two-thirds of patients. Similar lack of correlation with disease activity was found in other studies performed in GPA patients.63 Fluctuations in BAFF levels did occur in a minority of patients, but were not associated with changes in BVAS scores or other disease activity measures.62 In contrast, in renal MPO-ANCA positive vasculitis patients, there was a significant correlation between BVAS and serum BAFF (r=0.737). However, the sensitivity of serum BAFF levels was inferior to serum CRP levels, questioning its potential usefulness as a disease biomarker.
While BAFF levels were elevated in a subgroup of untreated GPA patients, Krumbholz could not find any correlation between serum BAFF and C-ANCA titers, or BAFF and disease activity.60 Similar lack of correlation between BAFF and ANCA was found in a subsequent longitudinal study57 and in more recent studies from Norway and Germany.62,65 There was actually an inverse correlation between ANCA and BAFF in a study by Bader et al.62 Again, in contrast to GPA, a study in renal vasculitis patients found a significant correlation between serum BAFF and ANCA titers.67
Changes in BAFF in patients treated with rituximab and in those with decreased B-cell numbers
In humans, serum levels of BAFF are highly dependent on a number of circulating B cells and expression of BAFF receptors. Patients with severe primary antibody deficiencies have higher levels of BAFF compared to controls. In line with this, mice expressing human BAFF had high levels of BAFF in the absence of B cells.51 Similar observations were made in several clinical trials that utilized anti-CD20 antibody treatment (rituximab) to deplete B cells.53,78,79 For example, Holden found increased levels of serum BAFF in PR3-ANCA-positive vasculitis patients.53 He compared BAFF levels before and 1–3 months after rituximab treatment at the time when the patients were in clinical remission (BVAS <2) and had no detectable peripheral blood B cells. Interestingly, the already high BAFF levels were further increased after rituximab treatment. One can wonder whether increased levels of BAFF may favor selection of autoreactive B cells,35 potentially increasing the likelihood of GPA relapse. It would be of great interest to see whether anti-CD20-mediated B-cell depletion followed by treatment with BAFF antagonists may provide a longer lasting remission in AAV and decrease the number of relapses compared to current standard of care.
Clinical trials with belimumab and other anti-BAFF agents in SLE and other rheumatic diseases
Belimumab (Lympho-Stat-B, Benlysta®; GlaxoSmithKline Plc, London, UK), a monoclonal antibody against BAFF, has recently been approved for the treatment of human SLE. Furthermore, several other BAFF/APRIL blocking agents are currently undergoing clinical trials in SLE, RA, and other autoimmune diseases.
Belimumab was the first clinically tested BAFF-neutralizing humanized monoclonal antibody directed against soluble BAFF. Noticeably, it does not bind to the membrane form. It was obtained by initial screening of a human phage display library against human BAFF. It acts by preventing the binding of BAFF to its high-affinity receptors on B cells.80 Consequently, this leads to apoptotic B-cell death and reduction in circulating B-cell numbers. Administration of Lympho-Stat-B to cynomolgus monkeys resulted in decreased numbers of B cells in both spleens and mesenteric lymph nodes. BAFF-blocking antibody primarily depletes naïve IgM+IgD+CD27– B cells in humans and their counterparts in mice.31,81
In March 2011, belimumab was approved for the treatment of SLE, thus becoming the first FDA-approved medication for SLE in the past 50 years. Clinical trials of SLE patients with belimumab (plus standard of care) showed improved clinical disease activity scores and reduced peripheral B-cell numbers. Serologically positive patients with SLE responded to this treatment better than serologically negative patients. As traditional lupus activity indices such as Selena-SLEDAI (Safety of Estrogens in Lupus Erythematosus National Assessment – Systemic Lupus Erythematosus Disease Activity Index) and BILAG (British Isles Lupus Assessment Group) were not sensitive enough to capture partial changes in disease activity, a novel SLE Responder Index (SRI) was developed to better access the efficacy of belimumab in SLE patients.82 BLISS-52 (BLISS – A Study of Belimumab in Subjects With Systemic Lupus Erythematosus) and BLISS-76 were two randomized, multicentric, placebo-controlled Phase III clinical trials that assessed the safety and efficacy of belimumab (plus standard of care) in patients with active (seropositive) SLE.83,84 Patients were treated with 1 or 10 mg/kg of belimumab or placebo on days 0, 14, 28, and every 28 days thereafter. The primary endpoint was the change in SRI from baseline to 52 weeks. Both treatment groups reached the primary endpoint. Group receiving 10 mg/kg of belimumab started showing significant difference at week 16. BLISS-76 was conducted with patients enrolled in Europe and North America, who were followed for a total of 76 weeks. Only the 10 mg/kg group reached a statistically significant response at week 52 (43.2% vs 33.5%). Both treatment groups showed reduced relapse rate, increased time to first flare, no worsening in PGA scores, reduced prednisone dose, and decreased serologic outcomes. Importantly, no safety concerns were noticed between the groups. Long-term safety/efficacy study performed over the period of 7 years showed sustained improvement in lupus disease activity, decreased flares, and reduction in autoantibodies.81,85,86
It is estimated that approximately 5% of all SLE patients in the US have been treated with belimumab, at least once (~15,000 patients).87 Post-marketing experience with belimumab showed that arthritis, rash, and serositis were the most common clinical manifestations requiring treatment with belimumab with a favorable clinical outcome at 12 months and with the ability to taper of corticosteroids. Small number of patients received belimumab for stable lupus nephritis and CNS lupus with some encouraging results. Results from an ongoing clinical trial evaluating belimumab in severe lupus nephritis have not yet been released.
In addition to belimumab, other BAFF (and APRIL) neutralizing therapeutic agents have recently been developed (Tables 1–4): tabalumab (LY2127399), blisibimod (AMG623), and atacicept (TACI-Ig).
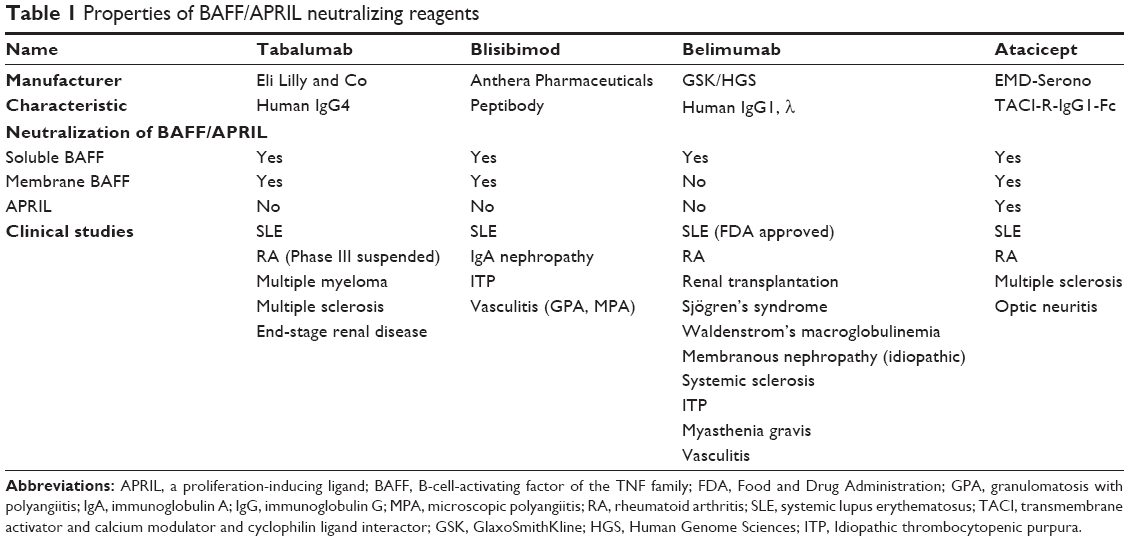 | Table 1 Properties of BAFF/APRIL neutralizing reagents Abbreviations: APRIL, a proliferation-inducing ligand; BAFF, B-cell-activating factor of the TNF family; FDA, Food and Drug Administration; GPA, granulomatosis with polyangiitis; IgA, immunoglobulin A; IgG, immunoglobulin G; MPA, microscopic polyangiitis; RA, rheumatoid arthritis; SLE, systemic lupus erythematosus; TACI, transmembrane activator and calcium modulator and cyclophilin ligand interactor; GSK, GlaxoSmithKline; HGS, Human Genome Sciences; ITP, Idiopathic thrombocytopenic purpura. |
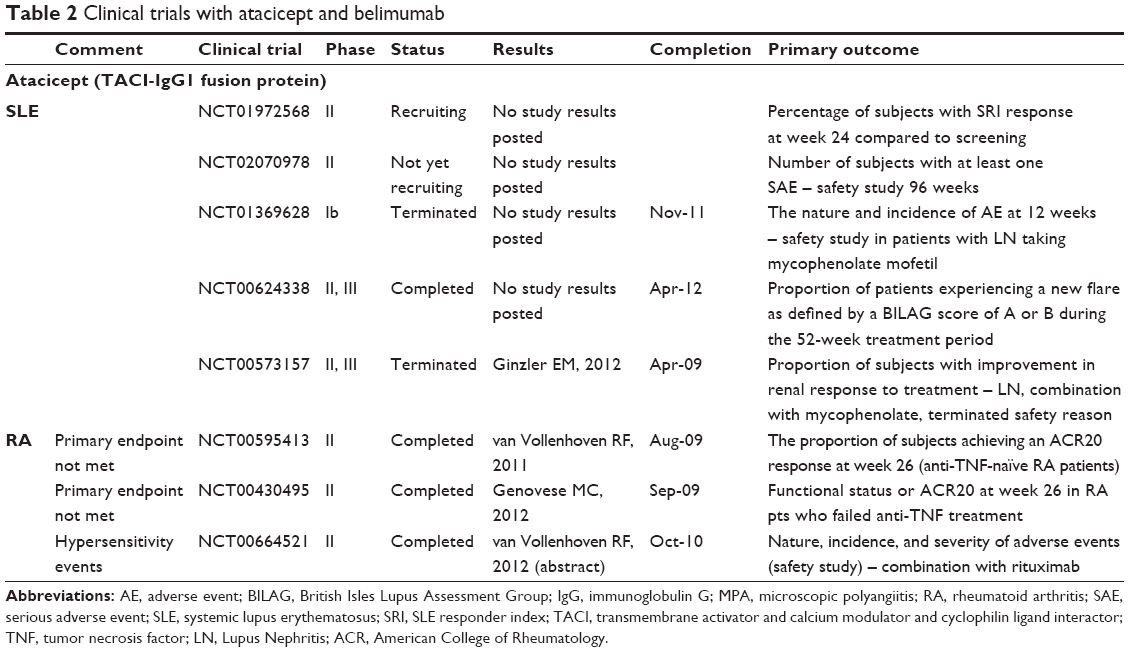 | Table 2 Clinical trials with atacicept and belimumab Abbreviations: AE, adverse event; BILAG, British Isles Lupus Assessment Group; IgG, immunoglobulin G; MPA, microscopic polyangiitis; RA, rheumatoid arthritis; SAE, serious adverse event; SLE, systemic lupus erythematosus; SRI, SLE responder index; TACI, transmembrane activator and calcium modulator and cyclophilin ligand interactor; TNF, tumor necrosis factor; LN, Lupus Nephritis; ACR, American College of Rheumatology. |
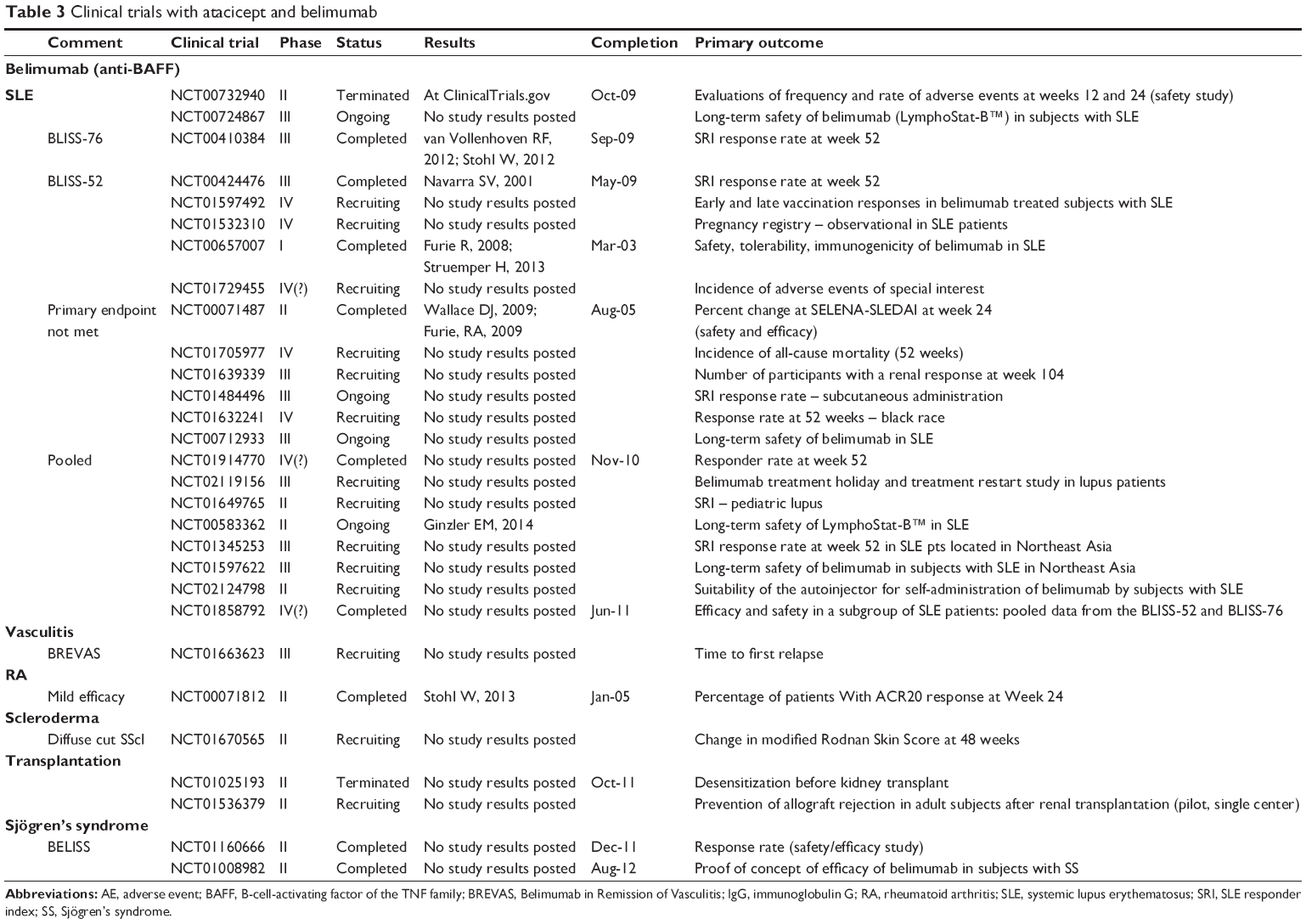 | Table 3 Clinical trials with atacicept and belimumab Abbreviations: AE, adverse event; BAFF, B-cell-activating factor of the TNF family; BREVAS, Belimumab in Remission of Vasculitis; IgG, immunoglobulin G; RA, rheumatoid arthritis; SLE, systemic lupus erythematosus; SRI, SLE responder index; SS, Sjögren’s syndrome. |
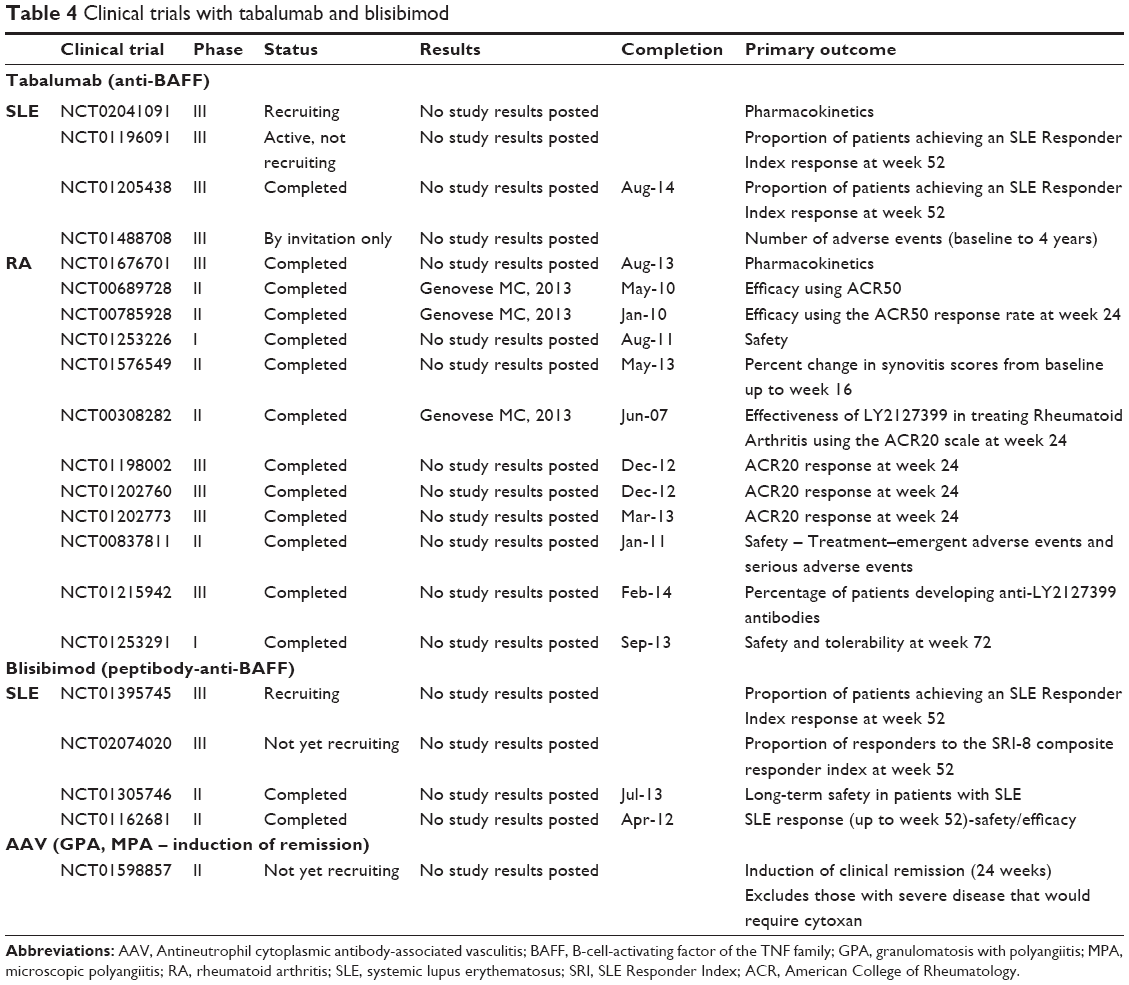 | Table 4 Clinical trials with tabalumab and blisibimod Abbreviations: AAV, Antineutrophil cytoplasmic antibody-associated vasculitis; BAFF, B-cell-activating factor of the TNF family; GPA, granulomatosis with polyangiitis; MPA, microscopic polyangiitis; RA, rheumatoid arthritis; SLE, systemic lupus erythematosus; SRI, SLE Responder Index; ACR, American College of Rheumatology. |
Tabalumab is a fully humanized monoclonal antibody of the IgG4 subclass that is capable of neutralizing both soluble and membrane-bound forms of BAFF, but not APRIL.88 It is unclear at this time which biological role a membrane BAFF may have. For example, mice expressing membrane BAFF, but lacking soluble BAFF, have similar phenotype to BAFF-deficient mice showing marked reduction in B cells and serum Ig levels.30 It remains to be determined whether combined neutralization of soluble and membrane bound BAFF has any advantages over solely blocking soluble BAFF. Tabalumab is currently undergoing clinical trials in SLE, RA, relapsing or refractory multiple myeloma, multiple sclerosis, and end-stage renal disease. A Phase III trial in RA has recently been suspended by the manufacturer, even though a previous trial, at week 16, showed efficacy of tabalumab in RA patients with inadequate response to methotrexate.88 This study showed a transient reduction in naïve B cells and simultaneous increase in CD27+ memory B cells along with decreased serum Immunoglobulin M levels. Its safety profile was acceptable. A Phase III study in SLE and a Phase II study in relapsing remitting multiple sclerosis were recently completed, but no clinical results have been posted yet.
Blisibimod is a peptibody produced in bacteria (E. coli) which, similar to tabalumab, targets both soluble and membrane BAFF. It is undergoing clinical trials in SLE, Immunoglobulin A nephropathy, and immune thrombocytopenic purpura. Two Phase II trials have been completed, but no published data are available. An important caveat, in case of Blisibimod, is that the BAFF-binding domain of peptibody is completely synthetic and likely immunogenic to the host. Neutralizing antibody response may potentially develop and decrease the potency of Blisibimod.
Atacicept is a chimeric fusion protein made of the extracellular domain of the TACI receptor attached to the human IgG1 Fc domain. It is different from the above-mentioned BAFF-specific reagents by its ability to neutralize not only BAFF but also APRIL and heterotrimers made of BAFF/APRIL combinations. Furthermore, it is the only agent that can significantly deplete plasma cells.32 SLE-prone mice responded favorably to TACI-Ig, as well as humans with SLE in phase Ib studies. However, because of increased rate of infection and significant decrease in serum IgG, a recent Phase II/III trial in active lupus nephritis was halted. Noticeably, in this trial, patients were first started on corticosteroids and mycophenolate mofetil, and reduction of serum IgG was observed even before atacicept was added.89 Another Phase II/III study, which evaluated the effect of atacicept in SLE patients without active CNS or renal disease, has been completed, but data have not been released yet. Atacicept was also studied in relapsing multiple sclerosis (terminated), optic neuritis (terminated), and in RA patients who had either inadequate response to methotrexate or in those who failed anti-TNF treatment. Disease activity actually worsened in multiple sclerosis patients, reminding us of the possible role of regulatory B cells in animal models of multiple sclerosis and lupus.90,91 Both Phase II RA studies failed to meet the primary endpoint despite significant reduction in rheumatoid factor levels (but not anti-CCP levels),92,93 while a trial of atacicept in combination with rituximab in RA patients resulted in significantly more allergic events. Thus, it appears that atacicept has the greatest potential of causing unacceptable toxicities. A monoclonal antibody solely targeting APRIL potentially may be more beneficial for lupus, at least based on its effect in animal models of lupus.94
As a next step in targeting BAFF, one can also envision development of small-molecule inhibitors of BAFF. For example, an exon-skipping approach was used to generate ΔBAFF, a minor alternative splicing variant of BAFF that works as a physiologic inhibitor of BAFF. This was beneficial in a mouse model of Sjögren’s syndrome, which is characterized by overexpression of BAFF and clinical sialoadenitis.95
Why to target BLys/BAFF over B-cell depletion in AAV
The question remains: what is the potential advantage of indirect targeting of B cells via withdrawal of an important survival factor (BAFF) over direct depletion of B cells?
One important reason (already elaborated above) is that (some) autoreactive B cells may have a greater dependency on BLyS/BAFF for their survival over B cells with non-autoreactive properties. A favorable safety signal observed over a period of 7 years in patients with SLE and diminished autoantibody levels are in line with this observation. In contrast, nonselective B-cell depletion (with rituximab) has been associated with rare but devastating cases of progressive multifocal encephalopathy.96
Furthermore, BAFF may also have a direct effect on T cells, and may be involved in generation of Th17 or Th1 T cells that are believed to have an important role in pathogenesis.
Finally, selective preservation of B cells with regulatory properties may have a potential role in fine-tuning B-cell responses in autoimmune systemic diseases. However, these postulates have yet to be verified clinically.
There are presently two ongoing clinical trials designed to address the role of BAFF in AAV. BIANCA-SC (A Study of the Efficacy, Safety, and Tolerability of Blisibimod in Addition to Methotrexate During Induction of Remission in Subjects With ANCA-Associated Small Vessel Vasculitis) is a Phase II trial to determine the efficacy and safety of blisibimod in addition to methotrexate for induction of remission in patients with AAV. It is designed to exclude patients with severe disease requiring cyclophosphamide treatment. This study is not yet recruiting participants. Belimumab in Remission of Vasculitis is a Phase III study focused on the efficacy and safety of belimumab (10 mg/kg) in combination with azathioprine for maintenance of remission in GPA and MPA. The primary outcome is time to first relapse. This study is currently open for enrollment.
Conclusion
Based on overwhelming evidence of increased serum and tissue levels of BAFF in AAV and favorable therapeutic and safety profile of belimumab in patients with SLE, BAFF and possibly APRIL appear to be promising targets for treatment of AAV. While AAV is obviously the first choice for treatment with BAFF antagonists, other vasculitides including TAK, GCA, and Behcet’s disease may be considered too. In contrast, BAFF as a potential biomarker in AAV appears to be less reliable compared to more traditional disease activity markers (eg, ESR and CRP). BAFF levels also failed to correlate with ANCA titers. We believe that induction therapy with a B-cell-depleting agent (eg, rituximab) followed by maintenance therapy with anti-BAFF reagents may result in diminished numbers of relapses and provide a safer control of AAV compared to currently available treatment protocols. Further clinical trials are needed to assess clinical efficacy of anti-BAFF agents in AAV.
Disclosure
The authors declare no conflicts of interest in this work.
References
Kallenberg CGM. Advances in pathogenesis and treatment of ANCA-associated vasculitis. Discov Med. 2014;18(99):195–201. | ||
Tarzi RM, Pusey CD. Current and future prospects in the management of granulomatosis with polyangiitis (Wegener’s granulomatosis). Ther Clin Risk Manag. 2014;10:279–293. | ||
Lenert P, Icardi M, Dahmoush L. ANA (+) ANCA (+) systemic vasculitis associated with the use of minocycline: case-based review. Clin Rheumatol. 2013;32(7):1099–1106. | ||
Zhao Y, Lutalo PM, Thomas JE, et al. Circulating T follicular helper cell and regulatory T cell frequencies are influenced by B-cell depletion in patients with granulomatosis with polyangiitis. Rheumatology (Oxford). 2014;53(4):621–630. | ||
Abdulahad WH, Lepse N, Stegeman CA, et al. Increased frequency of circulating IL-21 producing Th-cells in patients with granulomatosis with polyangiitis (GPA). Arthritis Res Ther. 2013;15(3):R70. | ||
Wilde B, Thewissen M, Damoiseaux J, et al. Regulatory B cells in ANCA-associated vasculitis. Ann Rheum Dis. 2013;72(8):1416–1419. | ||
Lyons PA, Rayner TF, Trivedi S, et al. Genetically distinct subsets within ANCA-associated vasculitis. N Engl J Med. 2012;367(3):214–223. | ||
Fauci AS, Wolff SM. Wegener’s granulomatosis: studies in eighteen patients and a review of the literature. Medicine (Baltimore). 1973;52(6):535–561. | ||
Hoffman GS, Kerr GS, Leavitt RY, et al. Wegener granulomatosis: an analysis of 158 patients. Ann Intern Med. 1992;116(6):488–498. | ||
Little MA, Nightingale P, Verburgh CA, et al; European Vasculitis Study (EUVAS) Group. Early mortality in systemic vasculitis: relative contribution of adverse events and active vasculitis. Ann Rheum Dis. 2010;69(6):1036–1043. | ||
Jayne DR, Gaskin G, Rasmussen N, et al; European Vasculitis Study Group. Randomized trial of plasma exchange or high-dosage methylprednisolone as adjunctive therapy for severe renal vasculitis. J Am Soc Nephrol. 2007;18(7):2180–2188. | ||
Stone JH, Merkel PA, Spiera R, et al; RAVE-ITN Research Group. Rituximab versus cyclophosphamide for ANCA-associated vasculitis. N Engl J Med. 2010;363(3):221–232. | ||
Jones RB, Tervaert JW, Hauser T, et al; European Vasculitis Study Group. Rituximab versus cyclophosphamide in ANCA-associated renal vasculitis. N Engl J Med. 2010;363(3):211–220. | ||
Wegener’s Granulomatosis Etanercept Trial (WGET) Research Group. Etanercept plus standard therapy for Wegener’s granulomatosis. N Engl J Med. 2005;352(4):351–361. | ||
Golbin JM, Specks U. Targeting B lymphocytes as therapy for ANCA-associated vasculitis. Rheum Dis Clin North Am. 2007;33(4):741–754. | ||
Molloy ES, Calabrese LH. Progressive multifocal leukoencephalopathy associated with immunosuppressive therapy in rheumatic diseases: evolving role of biologic therapies. Arthritis Rheum. 2012;64(9):3043–3051. | ||
Pagnoux C, Mahr A, Hamidou MA, et al; French Vasculitis Study Group. Azathioprine or methotrexate maintenance for ANCA-associated vasculitis. N Engl J Med. 2008;359(26):2790–2803. | ||
Metzler C, Miehle N, Manger K, et al; German Network of Rheumatic Diseases. Elevated relapse rate under oral methotrexate versus leflunomide for maintenance of remission in Wegener’s granulomatosis. Rheumatology (Oxford). 2007;46(7):1087–1091. | ||
Hiemstra TF, Walsh M, Mahr A, et al; European Vasculitis Study Group (EUVAS). Mycophenolate mofetil vs azathioprine for remission maintenance in antineutrophil cytoplasmic antibody-associated vasculitis: a randomized controlled trial. JAMA. 2010;304(21):2381–2388. | ||
Holle JU, Dubrau C, Herlyn K, et al. Rituximab for refractory granulomatosis with polyangiitis (Wegener’s granulomatosis): comparison of efficacy in granulomatous versus vasculitic manifestations. Ann Rheum Dis. 2012;71(3):327–333. | ||
Stegeman CA, Tervaert JW, de Jong PE, Kallenberg CG. Trimethoprim-sulfamethoxazole (co-trimoxazole) for the prevention of relapses of Wegener’s granulomatosis. Dutch Co-Trimoxazole Wegener Study Group. N Engl J Med. 1996;335(1):16–20. | ||
Langford CA, Sneller MC, Hallahan CW, et al. Clinical features and therapeutic management of subglottic stenosis in patients with Wegener’s granulomatosis. Arthritis Rheum. 1996;39(10):1754–1760. | ||
Hoffman GS, Thomas-Golbanov CK, Chan J, Akst LM, Eliachar I. Treatment of subglottic stenosis, due to Wegener’s granulomatosis, with intralesional corticosteroids and dilation. J Rheumatol. 2003;30(5):1017–1021. | ||
Hernández-Rodríguez J, Hoffman GS, Koening CL. Surgical interventions and local therapy for Wegener’s granulomatosis. Curr Opin Rheumatol. 2010;22(1):29–36. | ||
Walsh M, Flossmann O, Berden A, et al; European Vasculitis Study Group. Risk factors for relapse of antineutrophil cytoplasmic antibody-associated vasculitis. Arthritis Rheum. 2012;64(2):542–548. | ||
Guillevin L, Pagnoux C, Karras A, et al; French Vasculitis Study Group. Rituximab versus azathioprine for maintenance in ANCA-associated vasculitis. N Engl J Med. 2014;371(19):1771–1780. | ||
Mackay F, Schneider P, Rennert P, Browning J. BAFF and APRIL: a tutorial on B cell survival. Annu Rev Immunol. 2003;21:231–264. | ||
Schneider P, MacKay F, Steiner V, et al. BAFF, a novel ligand of the tumor necrosis factor family, stimulates B-cell growth. J Exp Med. 1999;189(11):1747–1756. | ||
Schiemann B, Gommerman JL, Vora K, et al. An essential role for BAFF in the normal development of B cells through a BCMA-independent pathway. Science. 2001;293(5537):2111–2114. | ||
Bossen C, Tardivel A, Willen L, et al. Mutation of the BAFF furin cleavage site impairs B-cell homeostasis and antibody responses. Eur J Immunol. 2011;41(3):787–797. | ||
Rauch M, Tussiwand R, Bosco N, Rolink AG. Crucial role for BAFF-BAFF-R signaling in the survival and maintenance of mature B cells. PLoS One. 2009;4(5):e5456. | ||
Vincent FB, Saulep-Easton D, Figgett WA, Fairfax KA, Mackay F. The BAFF/APRIL system: emerging functions beyond B-cell biology and autoimmunity. Cytokine Growth Factor Rev. 2013;24(3):203–215. | ||
Baker KP. BLys – an essential survival factor for B cells: basic biology, links to pathology and therapeutic target. Autoimmun Rev. 2004;3(5):368–375. | ||
Scapini P, Nardelli B, Nadali G, et al. G-CSF-stimulated neutrophils are a prominent source of functional BLyS. J Exp Med. 2003;197(3):297–302. | ||
Ota M, Duong BH, Torkamani A, et al. Regulation of the B-cell receptor repertoire and self-reactivity by BAFF. J Immunol. 2010;185(7):4128–4136. | ||
Thien M, Phan TG, Gardam S, et al. Excess BAFF rescues self-reactive B cells from peripheral deletion and allows them to enter forbidden follicular and marginal zone niches. Immunity. 2004;20(6):785–798. | ||
Mackay F, Woodcock SA, Lawton P, et al. Mice transgenic for BAFF develop lymphocytic disorders along with autoimmune manifestations. J Exp Med. 1999;190(11):1697–1710. | ||
Gross JA, Johnston J, Mudri S, et al. TACI and BCMA are receptors for a TNF homologue implicated in B-cell autoimmune disease. Nature. 2000;404(6781):995–999. | ||
Jacob CO, Pricop L, Putterman C, et al. Paucity of clinical disease despite serological autoimmunity and kidney pathology in lupus-prone New Zealand mixed 2328 mice deficient in BAFF. J Immunol. 2006;177(4):2671–2680. | ||
Jacob N, Guo S, Mathian A, et al. B-cell and BAFF dependence of IFN-α-exaggerated disease in systemic lupus erythematosus-prone NZM 2328 mice. J Immunol. 2011;186(8):4984–4993. | ||
Warnatz K, Salzer U, Rizzi M, et al. B-cell activating factor receptor deficiency is associated with an adult-onset antibody deficiency syndrome in humans. Proc Natl Acad Sci U S A. 2009;106(33):13945–13950. | ||
Zhang J, Roschke V, Baker KP, et al. Cutting edge: a role for B lymphocyte stimulator in systemic lupus erythematosus. J Immunol. 2001;166(1):6–10. | ||
Mariette X, Roux S, Zhang J, et al. The level of BLyS (BAFF) correlates with the titre of autoantibodies in human Sjögren’s syndrome. Ann Rheum Dis. 2003;62(2):168–171. | ||
Groom J, Kalled SL, Cutler AH, et al. Association of BAFF/BLyS overexpression and altered B-cell differentiation with Sjögren’s syndrome. J Clin Invest. 2002;109(1):59–68. | ||
Cheema GS, Roschke V, Hilbert DM, Stohl W. Elevated serum B lymphocyte stimulator levels in patients with systemic immune-based rheumatic diseases. Arthritis Rheum. 2001;44(6):1313–1319. | ||
Tan SM, Xu D, Roschke V, et al. Local production of B lymphocyte stimulator protein and APRIL in arthritic joints of patients with inflammatory arthritis. Arthritis Rheum. 2003;48(4):982–992. | ||
Vincent FB, Morand EF, Schneider P, Mackay F. The BAFF/APRIL system in SLE pathogenesis. Nat Rev Rheumatol. 2014;10(6):365–373. | ||
Liu Z, Davidson A. BAFF inhibition: a new class of drugs for the treatment of autoimmunity. Exp Cell Res. 2011;317(9):1270–1277. | ||
Md Yusof MY, Vital EM, Emery P. B-cell-targeted therapies in systemic lupus erythematosus and ANCA-associated vasculitis: current progress. Expert Rev Clin Immunol. 2013;9(8):761–772. | ||
Petri M, Stohl W, Chatham W, et al. Association of plasma B lymphocyte stimulator levels and disease activity in systemic lupus erythematosus. Arthritis Rheum. 2008;58(8):2453–2459. | ||
Kreuzaler M, Rauch M, Salzer U, et al. Soluble BAFF levels inversely correlate with peripheral B-cell numbers and the expression of BAFF receptors. J Immunol. 2012;188(1):497–503. | ||
Falk RJ, Jennette JC. ANCA are pathogenic – oh yes they are! J Am Soc Nephrol. 2002;13(7):1977–1979. | ||
Holden NJ, Williams JM, Morgan MD, et al. ANCA-stimulated neutrophils release BLyS and promote B-cell survival: a clinically relevant cellular process. Ann Rheum Dis. 2011;70(12):2229–2233. | ||
Xiao H, Heeringa P, Hu P, et al. Antineutrophil cytoplasmic autoantibodies specific for myeloperoxidase cause glomerulonephritis and vasculitis in mice. J Clin Invest. 2002;110(7):955–963. | ||
Bansal PJ, Tobin MC. Neonatal microscopic polyangiitis secondary to transfer of maternal myeloperoxidase-antineutrophil cytoplasmic antibody resulting in neonatal pulmonary hemorrhage and renal involvement. Ann Allergy Asthma Immunol. 2004;93(4):398–401. | ||
Slot MC, Tervaert JW, Boomsma MM, Stegeman CA. Positive classic antineutrophil cytoplasmic antibody (C-ANCA) titer at switch to azathioprine therapy associated with relapse in proteinase 3-related vasculitis. Arthritis Rheum. 2004;51(2):269–273. | ||
Voswinkel J, Müller A, Lamprecht P. Is PR3-ANCA formation initiated in Wegener’s granulomatosis lesions? Granulomas as potential lymphoid tissue maintaining autoantibody production. Ann N Y Acad Sci. 2005;1051:12–19. | ||
Nogueira E, Hamour S, Sawant D, et al. Serum IL-17 and IL-23 levels and autoantigen-specific Th17 cells are elevated in patients with ANCA-associated vasculitis. Nephrol Dial Transplant. 2010;25(7):2209–2217. | ||
Edberg JC, Zhou T, Aksi K, et al. Levels of circulating B lymphocyte stimulator (BLys) are elevated in patients with Wegener’s granulomatosis (abstract). Kidney Blood Press Res. 2004;26:256. | ||
Krumbholz M, Specks U, Wick M, Kalled SL, Jenne D, Meinl E. BAFF is elevated in serum of patients with Wegener’s granulomatosis. J Autoimmun. 2005;25(4):298–302. | ||
Kawasaki A, Tsuchiya N, Fukuzawa T, Hashimoto H, Tokunaga K. Analysis on the association of human BLys (BAFF, TNFSF113B) polymorphisms with systemic lupus erythematosus and rheumatoid arthritis. Genes Immun. 2002;3(7):424–429. | ||
Bader L, Koldingsnes W, Nossent J. B-lymphocyte activating factor levels are increased in patients with Wegener’s granulomatosis and inversely correlated with ANCA titer. Clin Rheumatol. 2010;29(9):1031–1035. | ||
Sanders JS, Huitma MG, Kallenberg CG, Stegeman CA. Plasma levels of soluble interleukin 2 receptor, soluble CD30, interleukin 10 and B-cell activator of the tumor necrosis factor family during follow-up in vasculitis associated with proteinase 3-antineutrophil cytoplasmic antibodies: associations with disease activity and relapse. Ann Rheum Dis. 2006;65(11):1484–1489. | ||
Zhao Y, Odell E, Choong LM, et al. Granulomatosis with polyangiitis involves sustained mucosal inflammation that is rich in B-cell survival factors and autoantigen. Rheumatology (Oxford). 2012;51(9):1580–1586. | ||
Schneeweis C, Rafalowicz M, Feist E, et al. Increased levels of BLyS and sVCAM-1 in anti-neutrophil cytoplasmatic antibody (ANCA)-associated vasculitides (AAV). Clin Exp Rheumatol. 2010;28(1 suppl 57):62–66. | ||
Xin G, Chen M, Su Y, Xu LX, Zhao MH, Li KS. Serum B-cell activating factor in myeloperoxidase-antineutrophil cytoplasmic antibodies-associated vasculitis. Am J Med Sci. 2014;348(1):25–29. | ||
Nagai M, Hirayama K, Ebihara I, Shimohata H, Kobayashi M, Koyama A. Serum levels of BAFF and APRIL in myeloperoxidase anti-neutrophil cytoplasmic autoantibody-associated renal vasculitis: association with disease activity. Nephron Clin Pract. 2011;118(4):c339–c345. | ||
Nishino Y, Tamai M, Kawakami A, et al. Serum levels of BAFF for assessing the disease activity of Takayasu arteritis. Clin Exp Rheumatol. 2010;28(1 suppl 57):14–17. | ||
van der Geest KS, Abdulahad WH, Chalan P, et al. Disturbed B-cell homeostasis in newly diagnosed giant cell arteritis and polymyalgia rheumatica. Arthritis Rheumatol. 2014;66(7):1927–1938. | ||
Hamzaoui K, Houman H, Hentati F, Hamzaoui A. BAFF is up-regulated in central nervous system of neuro-Behçet’s disease. J Neuroimmunol. 2008;200(1–2):111–114. | ||
Hamzaoui K, Houman H, Ben Dhifallah I, Kamoun M, Hamzaoui A. Serum BAFF levels and skin mRNA expression in patients with Behçet’s disease. Clin Exp Rheumatol. 2008;26(4 suppl 50):S64–S71. | ||
Gheita TA, Raafat H, Khalil H, Hussein H. Serum level of APRIL/BLyS in Behçet’s disease patients: clinical significance in uveitis and disease activity. Mod Rheumatol. 2013;23(3):542–546. | ||
Sumita Y, Murakawa Y, Sugiura T, Wada Y, Nagai A, Yamaguchi S. Elevated BAFF levels in the cerebrospinal fluid of patients with neuro-Behçet’s disease: BAFF is correlated with progressive dementia and psychosis. Scand J Immunol. 2012;75(6):633–640. | ||
Yoshida S, Sakurai Y, Takeda T, Fukuda K. Changes in BAFF/APRIL levels in a 2-year-old girl with Kawasaki disease refractory to intravenous immunoglobulin therapy. J Investig Allergol Clin Immunol. 2013;23(1):52–53. | ||
Park SJ, Shin JI. Interleukin-17 and B-cell-activating factor in Kawasaki disease and juvenile systemic lupus erythematosus. Lupus. 2012;21(11):1260. | ||
Gragnani L, Piluso A, Giannini C, et al. Genetic determinants in hepatitis C virus-associated mixed cryoglobulinemia: role of polymorphic variants of BAFF promoter and Fcγ receptors. Arthritis Rheum. 2011;63(5):1446–1451. | ||
Fabris M, Quartuccio L, Sacco S, et al. B-Lymphocyte stimulator (BLyS) up-regulation in mixed cryoglobulinaemia syndrome and hepatitis-C virus infection. Rheumatology (Oxford). 2007;46(1):37–43. | ||
Vallerskog T, Heimbürger M, Gunnarsson I, et al. Differential effects on BAFF and APRIL levels in rituximab-treated patients with systemic lupus erythematosus and rheumatoid arthritis. Arthritis Res Ther. 2006;8(6):R167. | ||
Pollard RP, Abdulahad WH, Vissink A, et al. Serum levels of BAFF, but not APRIL, are increased after rituximab treatment in patients with primary Sjogren’s syndrome: data from a placebo-controlled clinical trial. Ann Rheum Dis. 2013;72(1):146–148. | ||
Baker KP, Edwards BM, Main SH, et al. Generation and characterization of LymphoStat-B, a human monoclonal antibody that antagonizes the bioactivities of B lymphocyte stimulator. Arthritis Rheum. 2003;48(11):3253–3265. | ||
Jacobi AM, Huang W, Wang T, et al. Effect of long-term belimumab treatment on B cells in systemic lupus erythematosus: extension of a phase II, double-blind, placebo-controlled, dose-ranging study. Arthritis Rheum. 2010;62(1):201–210. | ||
Furie RA, Petri MA, Wallace DJ, et al. Novel evidence-based systemic lupus erythematosus responder index. Arthritis Rheum. 2009;61(9):1143–1151. | ||
Navarra SV, Guzmán RM, Gallacher AE, et al; BLISS-52 Study Group. Efficacy and safety of belimumab in patients with active systemic lupus erythematosus: a randomised, placebo-controlled, phase 3 trial. Lancet. 2011;377(9767):721–731. | ||
Furie R, Petri M, Zamani O, et al; BLISS-76 Study Group. A phase III, randomized, placebo-controlled study of belimumab, a monoclonal antibody that inhibits B lymphocyte stimulator, in patients with systemic lupus erythematosus. Arthritis Rheum. 2011;63(12):3918–3930. | ||
Wallace DJ, Navarra S, Petri MA, et al; BLISS-52 and -76, and LBSL02 Study Groups. Safety profile of belimumab: pooled data from placebo-controlled phase 2 and 3 studies in patients with systemic lupus erythematosus. Lupus. 2013;22(2):144–154. | ||
Stohl W, Hiepe F, Latinis KM, et al; BLISS-52 Study Group; BLISS-76 Study Group. Belimumab reduces autoantibodies, normalizes low complement levels, and reduces select B-cell populations in patients with systemic lupus erythematosus. Arthritis Rheum. 2012;64(7):2328–2337. | ||
Askanase AD, Yazdany J, Molta CT. Post-marketing experiences with belimumab in the treatment of SLE patients. Rheum Dis Clin North Am. 2014;40(3):507–517. | ||
Genovese MC, Bojin S, Biagini IM, et al. Tabalumab in rheumatoid arthritis patients with an inadequate response to methotrexate and naive to biologic therapy: a phase II, randomized, placebo-controlled trial. Arthritis Rheum. 2013;65(4):880–889. | ||
Ginzler EM, Wax S, Rajeswaran A, et al. Atacicept in combination with MMF and corticosteroids in lupus nephritis: results of a prematurely terminated trial. Arthritis Res Ther. 2012;14(1):R33. | ||
Fillatreau S, Sweenie CH, McGeachy MJ, Gray D, Anderton SM. B cells regulate autoimmunity by provision of IL-10. Nat Immunol. 2002;3(10):944–950. | ||
Lenert P, Brummel R, Field EH, Ashman RF. TLR-9 activation of marginal zone B cells in lupus mice regulates immunity through increased IL-10 production. J Clin Immunol. 2005;25(1):29–40. | ||
Genovese MC, Kinnman N, de La Bourdonnaye G, Pena Rossi C, Tak PP. Atacicept in patients with rheumatoid arthritis and an inadequate response to tumor necrosis factor antagonist therapy: results of a phase II, randomized, placebo-controlled, dose-finding trial. Arthritis Rheum. 2011;63(7):1793–1803. | ||
van Vollenhoven RF, Kinnman N, Vincent E, Wax S, Bathon J. Atacicept in patients with rheumatoid arthritis and an inadequate response to methotrexate: results of a phase II, randomized, placebo-controlled trial. Arthritis Rheum. 2011;63(7):1782–1792. | ||
Huard B, Tran NL, Benkhoucha M, Manzin-Lorenzi C, Santiago-Raber ML. Selective APRIL blockade delays systemic lupus erythematosus in mouse. PLoS One. 2012;7(2):e31837. | ||
Roescher N, Vosters JL, Alsaleh G, et al. Targeting the splicing of mRNA in autoimmune diseases: BAFF inhibition in Sjögren’s syndrome as a proof of concept. Mol Ther. 2014;22(4):821–827. | ||
Carson KR, Focosi D, Major EO, et al. Monoclonal antibody-associated progressive multifocal leucoencephalopathy in patients treated with rituximab, natalizumab, and efalizumab: a review from the research on adverse drug events and reports (RADAR) Project. Lancet Oncol. 2009;10(8):816–824. |
 © 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.