Back to Journals » International Journal of Chronic Obstructive Pulmonary Disease » Volume 11 » Issue 1
Curcumin modulates the effect of histone modification on the expression of chemokines by type II alveolar epithelial cells in a rat COPD model
Authors Gan L, Li C, Wang J, Guo X
Received 31 May 2016
Accepted for publication 1 August 2016
Published 7 November 2016 Volume 2016:11(1) Pages 2765—2773
DOI https://doi.org/10.2147/COPD.S113978
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 4
Editor who approved publication: Prof. Dr. Richard Russell
Lixing Gan,1 Chengye Li,2 Jian Wang,1 Xuejun Guo3
1Department of Respiratory Medicine, Shanghai Ninth People’s Hospital, Shanghai Jiao Tong University School of Medicine, Shanghai, 2Department of Respiratory Medicine, The First Affiliated Hospital of Wenzhou Medical University, Wenzhou, 3Department of Respiratory Medicine, Xin Hua Hospital Affiliated to Shanghai Jiao Tong University School of Medicine, Shanghai, People’s Republic of China
Background: Studies have suggested that histone modification has a positive impact on various aspects associated with the progression of COPD. Histone deacetylase 2 (HDAC2) suppresses proinflammatory gene expression through deacetylation of core histones.
Objective: To investigate the effect of histone modification on the expression of chemokines in type II alveolar epithelial cells (AEC II) in a rat COPD model and regulation of HDAC2 expression by curcumin in comparison with corticosteroid.
Materials and methods: The rat COPD model was established by cigarette smoke exposure and confirmed by histology and pathophysioloy. AEC II were isolated and cultured in vitro from the COPD models and control animals. The cells were treated with curcumin, corticosteroid, or trichostatin A, and messenger RNA (mRNA) expression of interleukin-8 (IL-8), monocyte chemoattractant protein-1 (MCP-1), and macrophage inflammatory protein-2α (MIP-2α) was assessed by quantitative real-time polymerase chain reaction (RT-PCR). The expression of HDAC2 was measured by Western blot. Chromatin immunoprecipitation was used to detect H3/H4 acetylation and H3K9 methylation in the promoter region of three kinds of chemokine genes (IL-8, MCP-1, and MIP-2α).
Results: Compared to the control group, the mRNAs of MCP-1, IL-8, and MIP-2α were upregulated 4.48-fold, 3.14-fold, and 2.83-fold, respectively, in the AEC II from COPD model. The protein expression of HDAC2 in the AEC II from COPD model was significantly lower than from the control group (P<0.05). The decreased expression of HDAC2 was negatively correlated with the increased expression of IL-8, MCP-1, and MIP-2α mRNAs (all P<0.05). The level of H3/H4 acetylation was higher but H3K9 methylation in the promoter region of chemokine genes was lower in the cells from COPD model than from the control group (all P<0.05). Curcumin downregulated the expression of MCP-1, IL-8, and MIP-2α, and the expression was further enhanced in the presence of corticosteroid. Moreover, curcumin restored HDAC2 expression, decreased the levels of H3/H4 acetylation, and increased H3K9 methylation in the promoter region of chemokine in the presence or absence of dexamethasone (all P<0.05).
Conclusion: Curcumin may suppress chemokines and restore corticosteroid resistance in COPD through modulating HDAC2 expression and its effect on histone modification.
Keywords: chronic obstructive pulmonary disease, type II alveolar epithelial cell, histone deacetylase, curcumin, corticosteroid
Introduction
Cigarette smoke is the major cause of COPD and stimulates the generation of inflammatory chemokines in the lung resident cells such as alveolar epithelial cells as well as inflammatory cells such as alveolar macrophages. The expression of proinflammatory and anti-inflammatory cytokines and chemokines is determined by a balance between histone acetylation, which activates transcription, and deacetylation, which switches off transcription.1
While COPD is characterized as a chronic airway inflammation, existing anti-inflammatory treatments such as corticosteroids have no proven disease modifying effect.2 The mechanisms underlying this resistance are largely unknown, especially in the smokers with chronic airway disease. Histone acetyltransferase (HAT) and histone deacetylases (HDACs) are enzymes that upregulate or downregulate proinflammatory gene transcription in various kinds of cells through regulating histone acetylation and deacetylation.3 HDAC2 is required by corticosteroids to switch off activated inflammatory genes, and HDAC2 was reduced in lung biopsies and alveolar macrophages of COPD patients.3–5 However, there have not been any studies identifying HDAC2 expression in the type II alveolar epithelial cells (AEC II).
AEC II have been considered as tissue stem cells that have potency to self-proliferate and differentiate into type I alveolar epithelial cells (AEC I), which are essential for lung development and remodeling. AEC II could secrete inflammatory chemokines such as monocyte chemoattractant protein-1 (MCP-1), interleukin-8 (IL-8), and macrophage inflammatory protein-2α (MIP-2α), which are important in immune defense and inflammatory responses in the lung.
Curcumin (1,7-bis(4-hydroxy-3-methoxyphenyl)-1,6-heptadiene-3,5-dione), a yellow pigment obtained from Curcuma longa (turmeric plant), has a wide spectrum of therapeutic properties and a remarkable range of protective effects in various diseases. It has been reported to have both anticancer and anti-inflammatory properties and to inhibit a wide range of inflammatory and signaling molecules.2,6,7 However, the effect of curcumin on HDAC2 and gene expression of chemokines in AEC II isolated from a rat COPD model has not been defined. Since HDAC2 plays an important role in modulating synthesis of proinflammatory cytokines and curcumin has anti-inflammatory effect, the present study was designed to determine the effect of curcumin on the gene expression of chemokines as well as the expression of HDAC2 in AEC II isolated from the cigarette smoke-induced rat COPD models.
Materials and methods
Reagents
Fetal bovine serum (FBS) and Dulbecco’s Modified Eagle’s Medium were purchased from (Thermo Fisher Scientific, Waltham, MA, USA); chromatin immunoprecipitation (ChIP) kit from Millipore (EMD Millipore, Billerica, MA, USA); anti-HDAC2 from Abcam (Cambridge, UK); anti-GAPDH from KangChen Bio-tech Inc. (Shanghai, People’s Republic of China); elastase (pancreatic from porcine pancreas) and curcumin (≥94% curcuminoid content, ≥80% curcumin) from Sigma-Aldrich Co. (St Louis, MO, USA); trichostatin A (TSA) and Cell Counting Kit-8 (CCK-8) from Beyotime Institute of Biotechnology (Haimen, People’s Republic of China); bicinchoninic acid (BCA) protein assay kit from Pierce (Rockford, IL, USA); real-time polymerase chain reaction (RT-PCR), SYBR Premix Ex Taq II (Perfect Real Time) from Takara (Tokyo, Japan); and Immobilon Western Chemiluminescent HRP Substrate from Millipore (EMD Millipore). All other chemicals were of reagent grade.
Animals
Thirty-two 6-week-old, male, specific-pathogen free (SPF)-grade Sprague Dawley rats were purchased from Shanghai Laboratory Animal Research Center and bred in the animal center of the Xinhua Hospital. The study was approved by the medical ethics committee of Xinhua Hospital. All animal experimental procedures were approved according to the Guide for Central Animal Care and Use of the Committee of Shanghai Jiao Tong University School of Medicine.
Cigarette smoke exposure
The Sprague Dawley rats were randomly divided into two groups: control group (16 rats) and COPD model group (16 rats). Animals of control group inhaled clean air, while the animals of COPD model group inhaled cigarette smoke. Briefly, animals were passively exposed to cigarette smoke in a chamber at a rate of ~15 minutes per cigarette and ten cigarettes each day for 90 successive days. The cigarette was purchased from Shanghai Tobacco Corporation (trade name: Da Qianmen, Shanghai, People’s Republic of China) and contains 12 mg tar (equivalent to 1.3-fold of tar quantity in the Kentucky Research Cigarette 2R4F) and 0.8 mg nicotine (equivalent to 1.1-fold of nicotine quantity in the Kentucky Research Cigarette 2R4F) per cigarette.
Histological examination
After 90 days of cigarette smoke exposure, the animals were sacrificed. The lung tissues were fixed with 10% neutral formalin, embedded in paraffin, and sectioned into 5 μm tissue slides. After deparaffinization and rehydration, tissues were stained with hematoxylin and eosin (H&E) for evaluation of inflammatory cell infiltration under the light microscopy.
Measurement of lung function
Airway resistance and dynamic compliance were determined. Briefly, animals were anesthetized, tracheostomized, and placed at supine position inside a plexiglass whole-body plethysmograph. The flow rate was monitored with a fisher tube connected to the airways in a pressure transducer, and the changes in lung volume were measured by detecting pressure changes in the plethysmographic chamber through a port in the connecting tube with a pressure transducer. To measure pleural pressure, a needle with a multiholed tip was directly inserted into pleural cavity through a port in the connecting tube with a differential pressure transducer. Transpulmonary pressure was calculated as the difference between mouth and pleural pressure. The signals from all pressure transducers were continuously processed (MedLab, Nanjing Biotech Instruments, Nanjing, People’s Republic of China) by fitting flow, volume, and pressure to an equation of motion.
Isolation and culture of AEC II
AEC II were isolated from the lungs of Sprague Dawley male rats as previously published8 with miner modification. Rats were anesthetized and then subjected to anticoagulation and tracheal intubation. The left lobe of the lung was used to isolate AEC II. The culture was maintained in complete growth medium containing 10% FBS, 100 U/mL penicillin, and 100 mg/mL streptomycin at 37°C in a humidified atmosphere with 5% CO2.
Alkaline phosphatase (AKP) assay and electron microscopy were performed to characterize the AEC II. Briefly, AKP was determined by the modified Kaplow method following the manufacture’s instruction. For electron microscope examination, the collected cells were fixed with 3% glutaric dialdehyde and then examined under transmission electron microscope.
Cell treatment
After 24-hour culture, cells were treated with or without curcumin (100 μM), TSA (100 nM), or dexamethasone (100 nM) and incubated for additional 24 hours. Cell toxicity was monitored by CCK-8 following the manufacture’s instruction.
RNA extraction and RT-PCR
Following the treatment, the AEC II were harvested, and the total RNA was extracted using the RNeasy mini kit (Qiagen, Hilden, German). RNA integrity and yield were analyzed and quantified using the BioPhotometer Plus (Eppendorf AG, Hamburg, Germany). Total RNA (1 μg) was reverse-transcribed to cDNA, and RT-PCR was performed using IL-8-, MCP-1-, MIP-2α-, and GAPDH-specific primers and SYBR Premix Ex Taq II PCR kit (Takara) on a 7500 Real-Time PCR System (Thermo Fisher Scientific). Data were expressed by comparative CT method (also known as the 2−ΔΔCT method).
Western blotting
Total protein of cell lysate was extracted with modified radioimmunoprecipitation assay (RIPA) buffer (Merck KGaA, Darmstadt, Germany). The protein amount was determined by the BCA protein assay kit (Pierce) and subjected to electrophoresis in sodium dodecyl sulfate-polyacrylamide gel. After transfer to a protran nitrocellulose hybridization transfer membrane (EMD Millipore), it was blocked with 10% nonfat dry milk in Tris-buffered saline with 0.1% Tween 20 followed by incubation with mouse monoclonal anti-rat HDAC2 at 4°C overnight. After washing, the levels of HDAC2 and GAPDH protein were detected with horseradish peroxidase-conjugated rabbit anti-mouse antibody (Jackson Lab, West Grove, PA, USA). Blots were visualized using enhanced chemiluminiscence fluid (EMD Millipore), and image density was analyzed using Image-pro plus 6.0 (Media Cybernetics, Inc, Bethesda, MD, USA).
ChIP assay
ChIP assay was performed following the manufacture’s (EMD Millipore) instruction. Briefly, cells were fixed with 0.4% formaldehyde at 22°C for 5 minutes. Cell lysates were sonicated to give a DNA size of ~600 bp, and supernatants were diluted with dilution buffer containing protease inhibitors. The solutions were precleared with salmon sperm DNA/protein G agarose slurry and then treated with antibody and incubated overnight at 4°C. The anti-acetyl-histone H3, anti-acetyl-histone H4, and anti-dimethyl-histone H3 (Lys9) antibodies were applied, and the immune complexes were then collected by adding a salmon sperm DNA/protein G agarose slurry. The beads were then washed sequentially in the following buffers: low-salt wash buffer, high-salt wash buffer, LiCl wash buffer, and Tris–EDTA buffer. Immune complexes were separated from the beads, and then digested and reversed by cross-linking. DNA was extracted with a PCR purification kit. The −222 to −134 bp region of the rat IL-8 proximal promoter (translation start site designated as +1) was amplified by PCR, so did the −189 to −31 bp region of MCP-1 and −240 to −123 bp region of MIP-2α proximal promoter. Equal amounts of input DNA were controlled by gel electrophoresis. PCR products were separated by agarose gel electrophoresis and detected by ethidium bromide staining.
Statistical analysis
Statistical analysis was performed by using SPSS l2.0 software package (SPSS Inc., Chicago, IL, USA). Data are expressed as mean ± standard deviation. Statistical analysis was performed with one-way analysis of variance (ANOVA), and Student’s t-test was used for calculation of the difference between the groups. Significance was considered at the level of P<0.05. Experiments were performed independently at least twice; results were qualitatively identical, and representative results were presented.
Results
Characterization of COPD model
Histologic evaluation of the H&E-stained rat lung tissue revealed the presence of pigmented macrophages in the lung parenchyma of the rats exposed to smoke. Infiltration of inflammatory cells, predominantly lymphocytes, was found in both the lung parenchyma and around terminal bronchioles (Figure 1). Semiquantitative scoring on tissue sections of rats exposed to smoke showed that both the number and the size of these inflammatory infiltrates increased during prolonged exposure to cigarette smoke (data not shown).
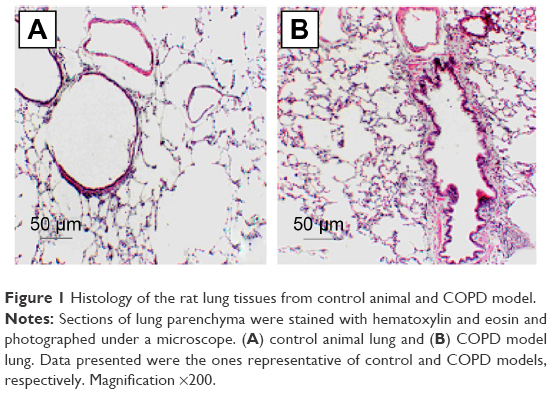 | Figure 1 Histology of the rat lung tissues from control animal and COPD model. |
Effect of curcumin on the expression of IL-8, MCP-1, and MIP-2α in AEC II isolated from COPD model
The messenger RNA (mRNA) expression of IL-8 (4.48-fold increase), MCP-1 (3.14-fold increase), and MIP-2α (2.83-fold increase) was significantly upregulated in the AEC II isolated from COPD model compared to that of the cells isolated from control animals (P<0.05, Figure 2A).
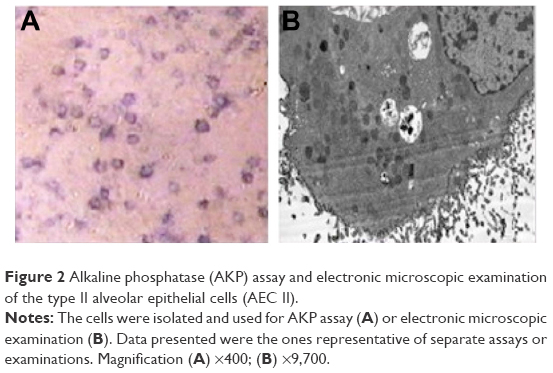 | Figure 2 Alkaline phosphatase (AKP) assay and electronic microscopic examination of the type II alveolar epithelial cells (AEC II). |
In vitro treatment of the AEC II from COPD animals with dexamethasone failed to suppress the upregulation of IL-8, MCP-1, and MIP-2α mRNA expression in the AEC II from COPD animal models (P>0.05, Figure 2B). TSA, a specific HDAC inhibitor, significantly upregulated mRNA expression of the IL-8, MCP-1, and MIP-2α in the AEC II isolated from the COPD model (Figure 2B, P<0.01). In contrast, curcumin significantly downregulated the expression of IL-8, MCP-1, and MIP-2α mRNA in the AEC II isolated from the COPD model, and this inhibitory effect was further augmented in the presence of dexamethasone (Figure 2B, P<0.05). However, curcumin could not suppress the upregulation of the chemokine mRNAs in the presence of TSA (Figure 2B). In vitro treatment with 100 μM curcumin, 100 nM dexamethasone, or 100 nM TSA did not show significant cytotoxicity to the cells as measured by CCK assay (data not shown).
Effect of curcumin on HDAC2 protein expression in AEC II isolated from COPD model
The protein level of HDAC2 in AEC II from COPD group was significantly lower than from control group (P<0.05, Figure 3A). The average reduction of HDAC2 protein (−4.04±0.11) was negatively correlated with the increase of IL-8 (4.52±0.43), MCP-1 (4.54±0.54), and MIP-2α (3.94±0.95) mRNA expression (all P<0.05, Table 1).
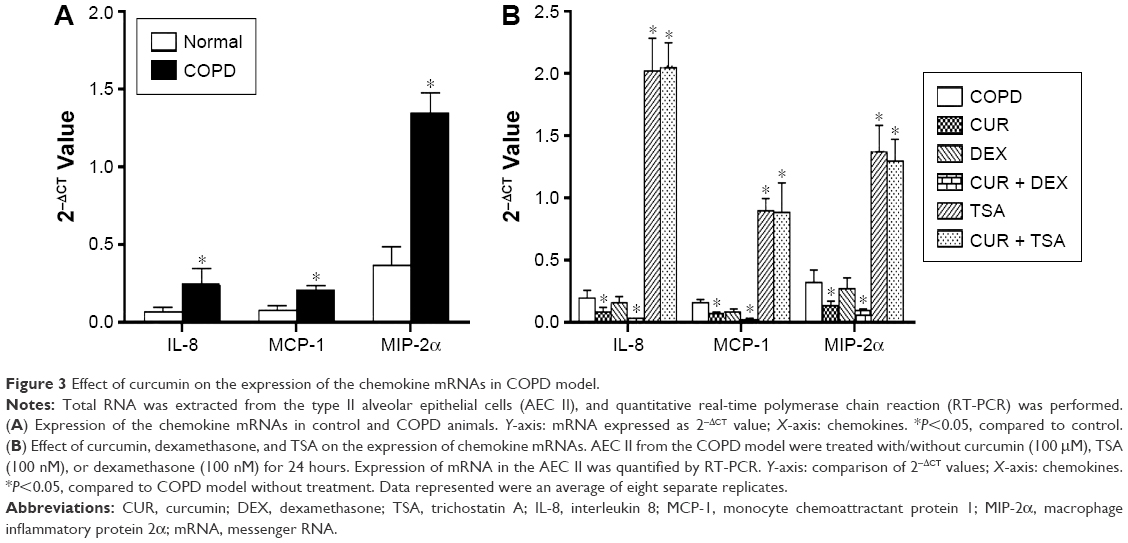 | Figure 3 Effect of curcumin on the expression of the chemokine mRNAs in COPD model. |
 | Table 1 Correlation between expression of HDAC2 and the chemokine mRNAs |
While dexamethasone could not alter HDAC2 protein level in the AEC II from COPD animals, curcumin could restore the downregulated HDAC2 protein in the AEC II from COPD animals (P<0.05). Curcumin, however, could not block TSA inhibition on HDAC2 protein in AEC II from COPD model animals (P<0.05, Figure 3B).
Histone modification by cigarette smoke and its modulation by curcumin in the AEC II from COPD model
The levels of H3 or H4 acetylation were higher in the rat COPD model than in the normal group, while H3K9 dimethylation level was lower in the COPD model than in the normal group in the promoter region of IL-8, MCP-1, and MIP-2α genes in AEC II (P<0.05, Figure 4; Table 2).
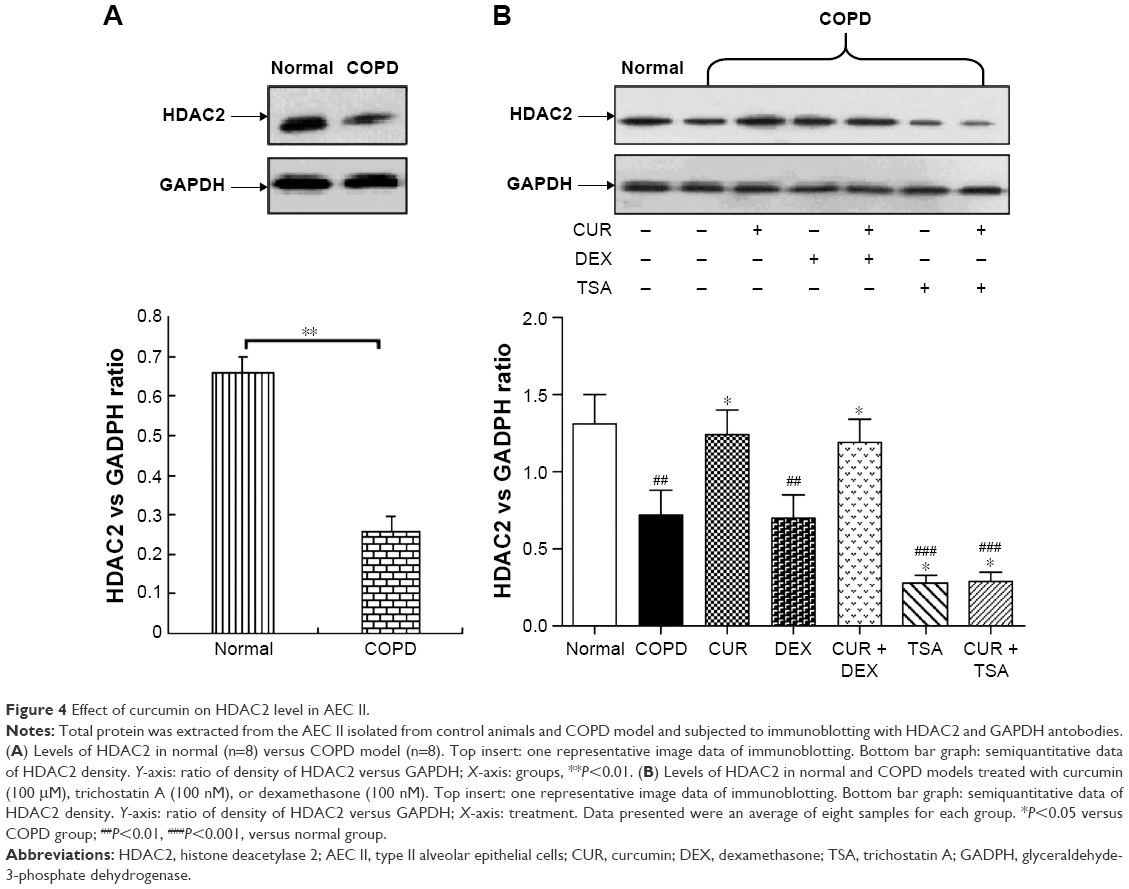 | Figure 4 Effect of curcumin on HDAC2 level in AEC II. |
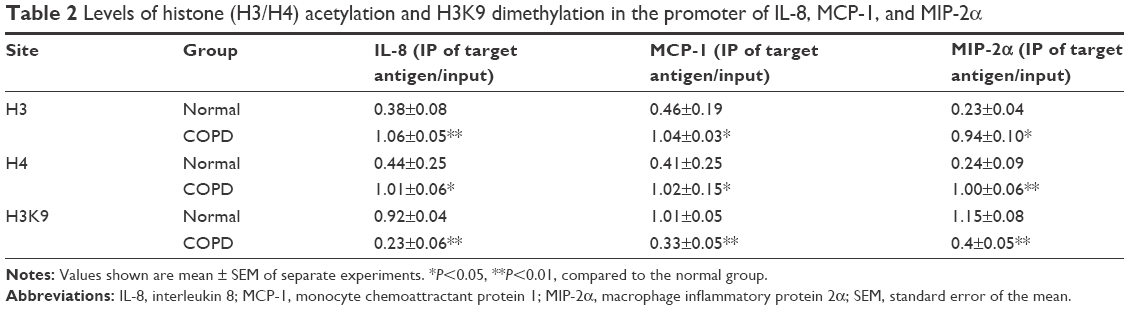 | Table 2 Levels of histone (H3/H4) acetylation and H3K9 dimethylation in the promoter of IL-8, MCP-1, and MIP-2α |
While neither the status of H3/H4 acetylation nor the status of H3K9 dimethylation was altered by dexamethasone in the promoter region of IL-8, MCP-1, and MIP-2α genes of the AEC II from COPD model, While neither the status were significantly decreased (H3/H4 acetylation) or increased (H3K9 dimethylation) when the cells were treated with curcumin for 24 hours (Figure 5; Table 3). TSA had similar effect as curcumin, but there was no synergetic or additive effect when TSA and curcumin were combined (Figure 6; Table 3).
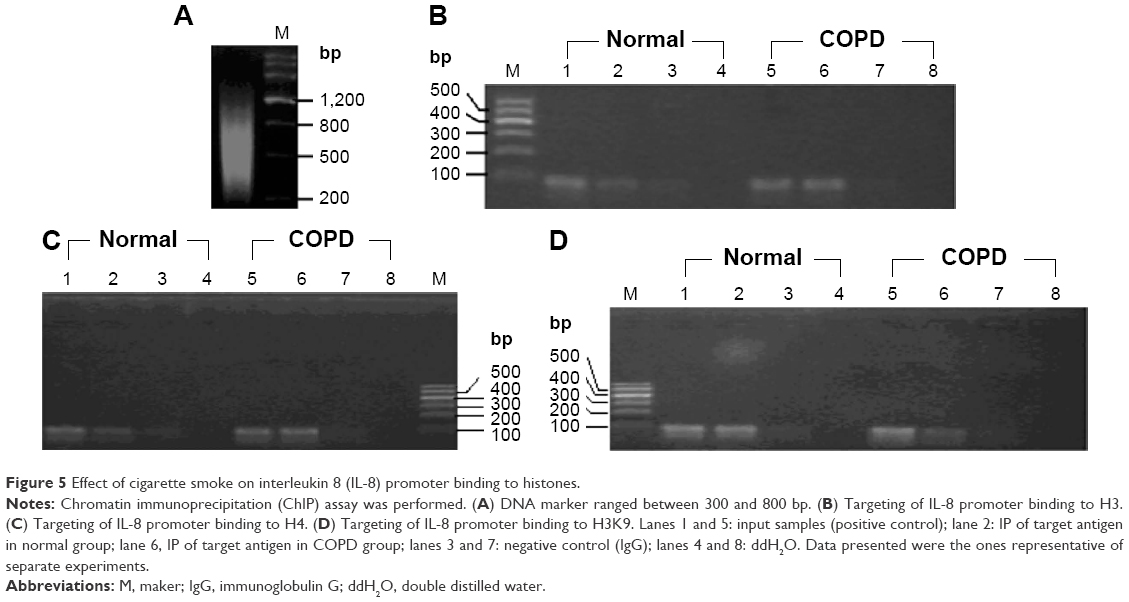 | Figure 5 Effect of cigarette smoke on interleukin 8 (IL-8) promoter binding to histones. |
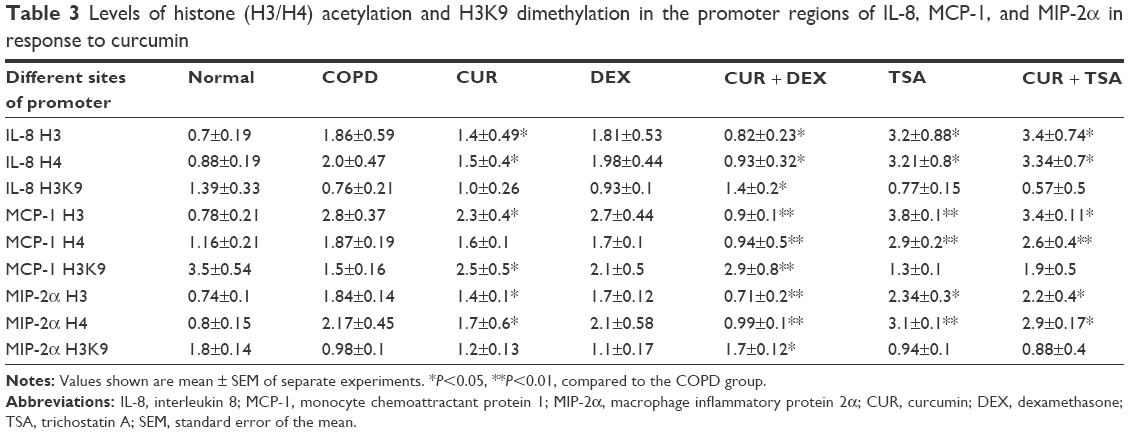 | Table 3 Levels of histone (H3/H4) acetylation and H3K9 dimethylation in the promoter regions of IL-8, MCP-1, and MIP-2α in response to curcumin |
 | Figure 6 Effect of curcumin on interleukin 8 (IL-8) promoter binding to histones. |
Discussion
Corticosteroids are often used in the treatment of chronic airway inflammation including COPD and asthma. However, unlike their effect in asthma, corticosteroids are much less effective in controlling the chronic inflammation of COPD. Reduction of HDAC2 is believed to play an important role in mediating corticosteroid resistance in chronic airway inflammation. The present study investigated the effect of curcumin in modulating HDAC2 level in the AEC II isolated from rat model of cigarette smoke-induced COPD animals. It was found that mRNA of proinflammatory chemokines, including IL-8, MCP-1, and MIP-2α, was significantly upregulated in the AEC II isolated from COPD model compared to that from the control animals. Curcumin significantly blocked the upregulation of these proinflammatory chemokines. Moreover, HDAC2 level was significantly reduced in the AEC II isolated from COPD model compared to that from the control animals. Curcumin significantly blocked HDAC2 reduction in the COPD models while dexamethasone did not. The levels of H3/H4 acetylation were higher in the IL-8 promoter region in the AEC II from COPD model than from control animals, while H3K9 dimethylation level was lower in the COPD model than in the control group. Curcumin significantly decreased H3/H4 acetylation but increased H3K9 dimethylation in the promoter region of the cytokines from COPD model. These findings suggested that curcumin inhibits proinflammatory chemokine expression in the AEC II of COPD model through modulating HDAC2 expression and histone acetylation of the promoters of the proinflammatory chemokines.
Cigarette smoke is the major environmental risk factor for COPD development. Inhaled cigarette smoke and other noxious particles lead to airway inflammation through activation of proinflammatory transcription factors, such as NF-κB and AP-1,6,9 which turn on gene expression of various kinds of proinflammatory cytokines and chemokines in lung parenchyma resident cells, including airway epithelial cells as well as inflammatory cells such as alveolar macrophages and neutrophils.10–12 Consistent with previous reports, this study demonstrated that cigarette smoke exposure in rat resulted in airway inflammation and upregulation of mRNAs of the proinflammatory chemokines including IL-8, MCP-1, and MIP-2α. IL-8 is believed to mediate neutrophil migration and infiltration into the airway tissues,13 while MCP-1 mediates monocytes and macrophage migration and accumulation in the alveoli and14 MIP-2α, also called chemokine (C-X-C motif) ligand 2 (CXCL2), is chemotactic for polymophonuclear leukocytes.15 These proinflammatory chemokines were significantly upregulated in the AEC II isolated from rat COPD model, and more importantly, the upregulation of these proinflammatory chemokines was significantly blocked by curcumin, suggesting curcumin is a potent inhibitor of chronic airway inflammation in COPD.
Therapeutic effect of corticosteroids in COPD is limited due to steroid resistance. One of the several mechanisms of corticosteroid resistance is reduction in HDAC2 activity and expression. Reversal of corticosteroid resistance in COPD patients by restoring HDAC2 levels has proved to be effective. In this regard, it has been reported that theophylline and phosphoinositide-3 kinase inhibitors could reverse steroid resistance by increasing HDAC2 expression.16,17 Consistent with previous reports,18,19 the present study demonstrated that cigarette smoke inhibited HDAC2 expression. While dexamethasone did not alter cigarette smoke inhibition on HDAC2 expression, curcumin significantly blocked cigarette smoke-induced HDAC2 suppression, suggesting curcumin could counteract cigarette smoke effect on HDAC2 expression. However, the mechanisms of curcumin blockade on cigarette smoke inhibition of HDAC2 expression remain to be defined, which was one of the limitations of the present study.
HDACs are critically involved in the biological activities of many transcription factors, receptors, and histones. The expression levels and activities of HDACs play important role in regulating pro- and anti-inflammatory effects. Currently, HDACs are classified as class I (1,2,3,8), class II (4, 5, 6, 7, 9, 10), and class IV (11).20 Different classes of HDACs have distinct activities on histones H2A, H2B, H3, and H4.21 HDAC7 is proinflammatory and stimulates IL-6 expression,22 whereas HDAC6 and HDAC11 suppress anti-inflammatory cytokine, IL-10, expression in antigen-presenting cells.23 In contrast, HDAC2 and HDAC3 are anti-inflammatory and suppress proinflammatory cytokine gene expression. In this context, HAT leads to histone acetylation and proinflammatory gene transcription, such as tumor necrosis factor-alpha (TNF-α), IL-8, and IL-1β. HDAC2 and HDAC3 counteract with HAT activity through deacetylation of histone on target gene promoter region, and decrease transcriptional activity through tightening of chromatin.19 In this study, we further demonstrated that cigarette smoke-induced H3/H4 acetylation in the promoter region of IL-8, but inhibited H3K9 dimethylation, and by this mechanism, at least partially, cigarette smoke stimulated mRNA expression of IL-8. Moreover, curcumin significantly blocked cigarette smoke effect on H3/H4 acetylation and H3K9 dimethylation in the promoter region of IL-8, suggesting curcumin modulates proinflammatory cytokine gene expression through modulating histone acetylation and deacetylation process.
Curcumin is a compound derived from the turmeric plant that has been used as a potential drug for the treatment of chronic diseases such as cancer, asthma, diabetes, and neurodegenerative disorders.24 In vitro and in vivo studies have demonstrated that curcumin significantly suppresses the expression of proinflammatory cytokines such as IL-1β, IL-6, IL-8, and TNF-α25,26 and that curcumin possesses antioxidant properties. Consistent with the previous reports, this study demonstrated that curcumin significantly blocks mRNA expression of IL-8, MCP-1, or MIP-2α in the AEC II isolated from cigarette smoke-induced rat COPD model, and that curcumin exerts its anti-inflammatory effect through modulating HDAC2 expression in the cells.
Conclusion
Taken together, the present study reports that proinflammatory cytokines were upregulated, but HDAC2 expression was significantly downregulated in the AEC II isolated from cigarette smoke-induced COPD animal models compared to that from control animals. Curcumin significantly blocked suppression of HDAC2 expression by the cigarette smoke in the AEC II. Furthermore, curcumin blocked cigarette smoke-induced alteration of H3/H4 acetylation and H3K9 dimethylation status in the AEC II. These findings suggest that curcumin exerts anti-inflammatory effect in the animal models of COPD through modulating HDAC2 expression and activity.
Acknowledgment
This work was supported by the key program of Shanghai Nature Science Foundation (10jc1411300). The abstract of this paper was presented at the 16th Congress of the Asian Pacific Society of Respirology, November 3–6, 2011, Shanghai International Convention Centre, Shanghai, People’s Republic of China, as an abstract with interim findings. The abstract was published in Respirology, Special Issue, Volume 16, Issue Supplement s2, pages 1–326, November 2011.
Disclosure
The authors report no conflicts of interest in this work.
References
Reddy R, Buckley S, Doerken M, et al. Isolation of a putative progenitor subpopulation of alveolar epithelial type 2 cells. Am J Physiol Lung Cell Mol Physiol. 2004;286(4):L658–L667. | ||
Barnes PJ, Adcock IM. Glucocorticoid resistance in inflammatory diseases. Lancet. 2009;373(9678):1905–1917. | ||
Barnes PJ, Adcock IM, Ito K. Histone acetylation and deacetylation: importance in inflammatory lung diseases. Eur Respir J. 2005;25(3):552–563. | ||
Adenuga D, Yao H, March TH, Seagrave J, Rahman I. Histone deacetylase 2 is phosphorylated, ubiquitinated, and degraded by cigarette smoke. Am J Respir Cell Mol Biol. 2009;40(4):464–473. | ||
Ito K, Ito M, Elliott WM, et al. Decreased histone deacetylase activity in chronic obstructive pulmonary disease. N Engl J Med. 2005;352(19):1967–1976. | ||
Cheng SE, Luo SF, Jou MJ, et al. Cigarette smoke extract induces cytosolic phospholipase A2 expression via NADPH oxidase, MAPKs, AP-1, and NF-kappaB in human tracheal smooth muscle cells. Free Radic Biol Med. 2009;46(7):948–960. | ||
Adcock IM, Tsaprouni L, Bhavsar P, Ito K. Epigenetic regulation of airway inflammation. Curr Opin Immunol. 2007;19(6):694–700. | ||
Dobbs LG, Gonzalez R, Williams MC. An improved method for isolating type II cells in high yield and purity. Am Rev Respir Dis. 1986;134(1):141–145. | ||
Manna SK, Rangasamy T, Wise K, et al. Long term environmental tobacco smoke activates nuclear transcription factor-kappa B, activator protein-1, and stress responsive kinases in mouse brain. Biochem Pharmacol. 2006;71(11):1602–1609. | ||
Liu X, Togo S, Al-Mugotir M, et al. NF-kappaB mediates the survival of human bronchial epithelial cells exposed to cigarette smoke extract. Respir Res. 2008;9:66. | ||
Moretto N, Bertolini S, Iadicicco C, et al. Cigarette smoke and its component acrolein augment IL-8/CXCL8 mRNA stability via p38 MAPK/MK2 signaling in human pulmonary cells. Am J Physiol Lung Cell Mol Physiol. 2012;303(10):L929–L938. | ||
Gaschler GJ, Zavitz CC, Bauer CM, et al. Cigarette smoke exposure attenuates cytokine production by mouse alveolar macrophages. Am J Respir Cell Mol Biol. 2008;38(2):218–226. | ||
Govindaraju V, Michoud MC, Al-Chalabi M, Ferraro P, Powell WS, Martin JG. Interleukin-8: novel roles in human airway smooth muscle cell contraction and migration. Am J Physiol Cell Physiol. 2006;291(5):C957–C965. | ||
Yoshimura T. Discovery of IL-8/CXCL8 (The Story from Frederick). Front Immunol. 2015;6:278. | ||
Kobayashi Y. The role of chemokines in neutrophil biology. Front Biosci. 2008;13:2400–2407. | ||
Barnes PJ. Corticosteroid resistance in patients with asthma and chronic obstructive pulmonary disease. J Allergy Clin Immunol. 2013;131(3):636–645. | ||
Sun X, Li Q, Gong Y, Ren L, Wan H, Deng W. Low-dose theophylline restores corticosteroid responsiveness in rats with smoke-induced airway inflammation. Can J Physiol Pharmacol. 2012;90(7):895–902. | ||
Tamimi A, Serdarevic D, Hanania NA. The effects of cigarette smoke on airway inflammation in asthma and COPD: therapeutic implications. Respir Med. 2012;106(3):319–328. | ||
Malhotra D, Thimmulappa RK, Mercado N, et al. Denitrosylation of HDAC2 by targeting Nrf2 restores glucocorticosteroid sensitivity in macrophages from COPD patients. J Clin Invest. 2011;121(11):4289–4302. | ||
Bradley EW, Carpio LR, van Wijnen AJ, McGee-Lawrence ME, Westendorf JJ. Histone deacetylases in bone development and skeletal disorders. Physiol Rev. 2015;95(4):1359–1381. | ||
Winkler AR, Nocka KN, Williams CM. Smoke exposure of human macrophages reduces HDAC3 activity, resulting in enhanced inflammatory cytokine production. Pulm Pharmacol Ther. 2012;25(4):286–292. | ||
Shakespear MR, Hohenhaus DM, Kelly GM, et al. Histone deacetylase 7 promotes Toll-like receptor 4-dependent proinflammatory gene expression in macrophages. J Biol Chem. 2013;288(35):25362–25374. | ||
Cheng F, Lienlaf M, Perez-Villarroel P, et al. Divergent roles of histone deacetylase 6 (HDAC6) and histone deacetylase 11 (HDAC11) on the transcriptional regulation of IL10 in antigen presenting cells. Mol Immunol. 2014;60(1):44–53. | ||
Gupta SC, Patchva S, Koh W, Aggarwal BB. Discovery of curcumin, a component of golden spice, and its miraculous biological activities. Clin Exp Pharmacol Physiol. 2012;39(3):283–299. | ||
Jung KK, Lee HS, Cho JY, et al. Inhibitory effect of curcumin on nitric oxide production from lipopolysaccharide-activated primary microglia. Life Sci. 2006;79(21):2022–2031. | ||
Zhang L, Wu C, Zhao S, et al. Demethoxycurcumin, a natural derivative of curcumin attenuates LPS-induced pro-inflammatory responses through down-regulation of intracellular ROS-related MAPK/NF-kappaB signaling pathways in N9 microglia induced by lipopolysaccharide. Int Immunopharmacol. 2010;10(3):331–338. |
 © 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.