Back to Journals » Neuropsychiatric Disease and Treatment » Volume 17
CSF TNF-α Levels Were Associated with Longitudinal Change in Brain Glucose Metabolism Among Non-Demented Older People
Received 8 November 2020
Accepted for publication 19 April 2021
Published 26 May 2021 Volume 2021:17 Pages 1659—1666
DOI https://doi.org/10.2147/NDT.S291020
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Yu-Ping Ning
Pan Fu, Bihong Zhu, Yangping Huang On behalf of Alzheimer’s Disease Neuroimaging Initiative
Department of Neurology, Taizhou First People’s Hospital, Taizhou, Zhejiang, People’s Republic of China
Correspondence: Yangping Huang
Department of Neurology, Taizhou First People’s Hospital, 218 Hengjie Road, Huangyan District, Taizhou City, Zhejiang Province, People’s Republic of China
Email [email protected]
Purpose: Emerging studies have suggested that tumor necrosis factor-alpha (TNF-α) is implicated in the pathogenesis of Alzheimer’s disease (AD), and that cerebral glucose hypometabolism is a key feature of AD. However, the association of CSF TNF-α levels with changes in cerebral glucose metabolism has not been studied among non-demented older people.
Patients and Methods: At baseline, there were a total of 214 non-demented older people from Alzheimer’s Disease Neuroimaging Initiative (ADNI) study. We examined the cross-sectional and longitudinal associations of CSF TNF-α with global cognition (as assessed by mini-mental state examination), verbal memory (as assessed by Rey Auditory Verbal Learning Test-total learning score), and cerebral glucose metabolism (as measured by FDF-PET). Linear mixed-effects models were used to examine the longitudinal association of CSF TNF- α with change in each outcome over time with adjustment of age, educational level, gender, and APOE4 status.
Results: In the cross-sectional study, CSF TNF-α was negatively associated with MMSE scores, but not verbal memory or FDG-PET. In the longitudinal study, higher CSF TNF- α at baseline was associated with a faster decline in cerebral glucose metabolism, but not MMSE scores or RAVLT total learning scores.
Conclusion: Higher CSF TNF-α levels were associated with a steeper decline in cerebral glucose metabolism among non-demented older people.
Keywords: mild cognitive impairment, Alzheimer’s disease, TNF-α, cerebral glucose metabolism, FDG-PET
Introduction
Alzheimer’s disease (AD) is a neurodegenerative disease characterized by cognitive impairment and the formation of two core pathological markers (β-amyloid plaques and neurofibrillary tangles).1 Emerging evidence has suggested that neuroinflammation plays an important role in the pathogenesis of AD.2 Observational studies have shown that the use of anti-inflammatory drugs is associated with a decreased risk of developing AD dementia.3 Tumor necrosis factor-alpha (TNF- α) is one of the most important inflammatory cytokines expressed by activated microglia and astrocytes, and is elevated in several brain regions of patients with AD dementia.4 In addition, a previous study found that higher levels of TNF- α were associated with incident AD among cognitively normal community-dwelling older people.5
Reduced cerebral glucose metabolism, assessed by FDG-PET, is a core feature of AD dementia.6,7 However, to the best of our knowledge, the association of CSF TNF-α levels with longitudinal changes in brain glucose metabolism (reflecting neuronal and synaptic function) over time has not been investigated among non-demented older people. If this association does exist, it may provide some insights into the effect of TNF- α on neuronal and synaptic function. Therefore, we aimed to examine the cross-sectional and longitudinal associations of CSF TNF- α with global cognition, verbal memory, and brain glucose metabolism (as assessed by FDG-PET) among non-demented older people from Alzheimer’s Disease Neuroimaging Initiative (ADNI) study.
Methods
Alzheimer’s Disease Neuroimaging Initiative (ADNI) Study
Cross-sectional and longitudinal data used in the preparation of this work were extracted from the ADNI database (adni.loni.usc.edu). The ADNI study was initiated in 2003 with the primary goal of examining whether positron emission tomography (PET), serial magnetic resonance imaging (MRI), biological markers, and neuropsychological assessments could be integrated to measure the progression of mild cognitive impairment (MCI) and early AD.
Participants
At baseline, there were a total of 214 non-demented older people, including 85 subjects with normal cognition (NC) and 129 subjects with MCI. These participants had baseline CSF TNF-α samples and follow-up examinations of global cognition, verbal memory, and brain glucose metabolism. At ADNI centers, participants provided written informed consents, and local institutional review board approved the ADNI study. The official or affiliated names of the approving local ethics committee could be found at the website: http://adni.loni.usc.edu/wp-content/themes/freshnews-dev-v2/documents/policy/ADNI_Acknowledgement_List%205-29-18.pdf.
Diagnostic Criteria
Individuals with NC had a Clinical Dementia Rating (CDR)8 score of 0 and a Mini-mental State Examination (MMSE)9 score of 24 or higher. Individuals with MCI had a CDR score of 0.5, an MMSE score of 24 or higher, essentially preserved activities of daily living, verbal memory impairment as evidenced by delayed recall scores of the Wechsler Memory Scale Logical Memory II, and an absence of dementia.
Neuropsychological Assessments
The ADNI participants underwent a comprehensive neuropsychological assessment. In this study, we selected two primary cognitive outcomes: MMSE scores and Rey auditory verbal learning test (RAVLT)10 total learning scores. MMSE scores and RAVLT total learning scores were utilized to examine global cognition and verbal memory, respectively.
FDG Standardized Uptake Value Ratio (SUVR) Measurement
Levels of brain glucose metabolism were examined by FDG PET, details of which have been described previously11 and can also be found at the ADNI website (http://adni.loni.usc.edu/methods/pet-analysis/). The mean of FDG uptake was determined within middle/inferior temporal gyrus, bilateral angular gyri, and bilateral posterior cingulate gyrus due to the fact that low levels of glucose metabolism within these regions have been found to be associated with cognitive deficits in patients with MCI and AD dementia.12,13 SUVRs were determined by averaging uptakes of these regions and dividing by cerebellum.
Measurement of CSF TNF-α Levels
Levels of CSF TNF-α were determined using commercially available multiplex immunoassays (Millipore Sigma, Burlington, MA) by the Hu lab (Dr. William Hu), Department of Neurology, Emory University. Initially, a total of 15 analytes were examined, whereas only levels of TNF-α in CSF were reported because it is our variable of interest in the present study. Samples were randomized across twelve 96-well plates for each of the five assays (60 plates total) encompassing 15 analytes. All CSF samples were run in duplicate along with six CSF standards on each plate. Samples were normalized across plates using CSF standard values. The inter-plate CV (%) of the measurement of TNF-α was 9.38. The median sample concentration of TNF-α was 1.7 pg/mL. The LLOD (MFI) was 49.2. The median MFI was 53.53. Values were given as pg/mL.
Statistical Analysis
Descriptive statistics were utilized to summarize the variables of our study sample, including demographics and clinical variables. The distribution of MMSE was negatively skewed, whereas the distribution of other variables (TNF- α, RAVLT total learning score and FDG) was largely normal based on the histograms of these variables. Therefore, Pearson’s correlation tests were used to examine the cross-sectional associations of baseline CSF TNF- α levels with RAVLT total learning score and FDG SUVR. However, Spearman correlation test was used to examine the relationship between TNF-α and MMSE. In the longitudinal analysis, several linear mixed models were utilized to examine the associations of baseline CSF TNF- α levels (categorized into the High and Low groups based on the median) with changes in each outcome (MMSE sores, RAVLT total learning scores, and FDG SUVR) over time among non-demented older people. Each model included the main effects of baseline CSF TNF-α, age, gender, educational level, APOE4 genotype and their interactions with time, along with a random intercept and a random slope for each subject. All statistical work was performed with R software (version 3.6.0).14 The “lme4” package15 was used to conduct linear mixed models.
Results
Demographics and Clinical Variables
In the present study, a total of 214 non-demented older people were included. Socio-demographic and clinical variables are summarized in Table 1. The mean age (mean ± SD) for the whole study sample was 74.9 ± 7 years. Eighty-seven (40.7%) participants were females. Ninety-one (42.5%) participants were APOE4 carriers and 129 (60.3%) were MCI patients.
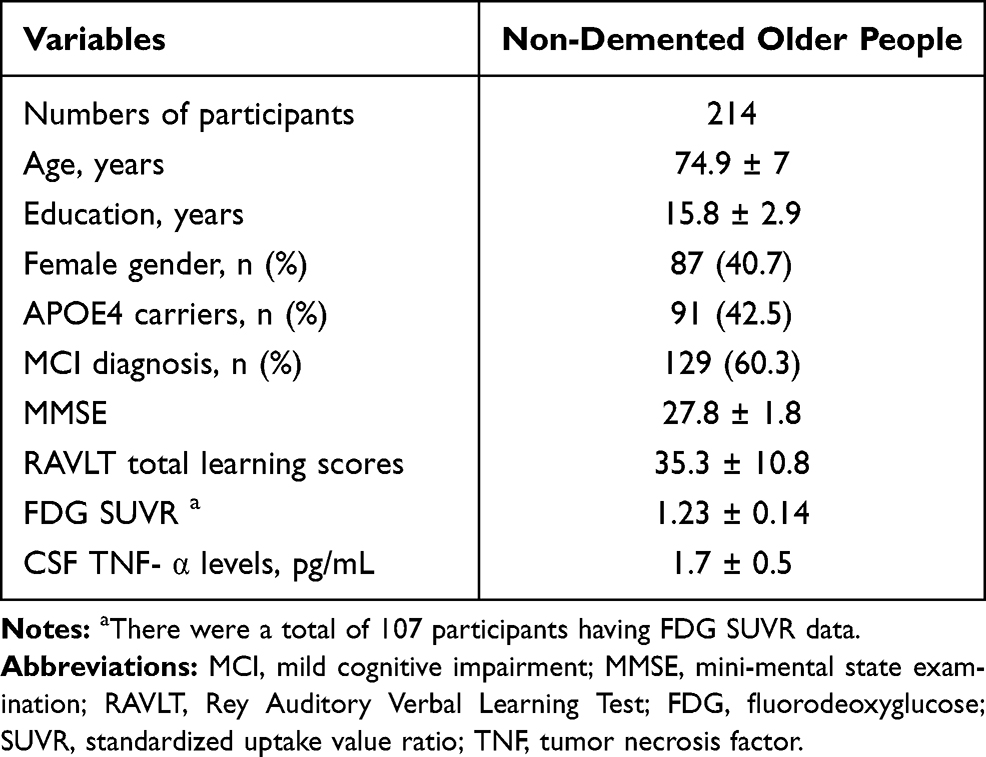 |
Table 1 Demographic and Clinical Characteristics of 214 Non-Demented Older People |
Cross-Sectional Associations of CSF TNF-α with Global Cognition, Memory, and FDG SUVR Among Non-Demented Older People
To examine the cross-sectional associations of baseline CSF TNF-α with MMSE scores, RAVLT total learning scores, and FDG SUVR among non-demented older people, Pearson’s correlation tests were used. As shown in Table 2, CSF TNF-α levels were negatively associated with MMSE scores (rho = −0.17, p = 0.011), but not RAVLT total learning scores (r = −0.13, p = 0.053) or FDG SUVR (r = −0.12, p = 0.22). Further, CSF TNF-α was associated with age (r = 0.39, p < 0.001).
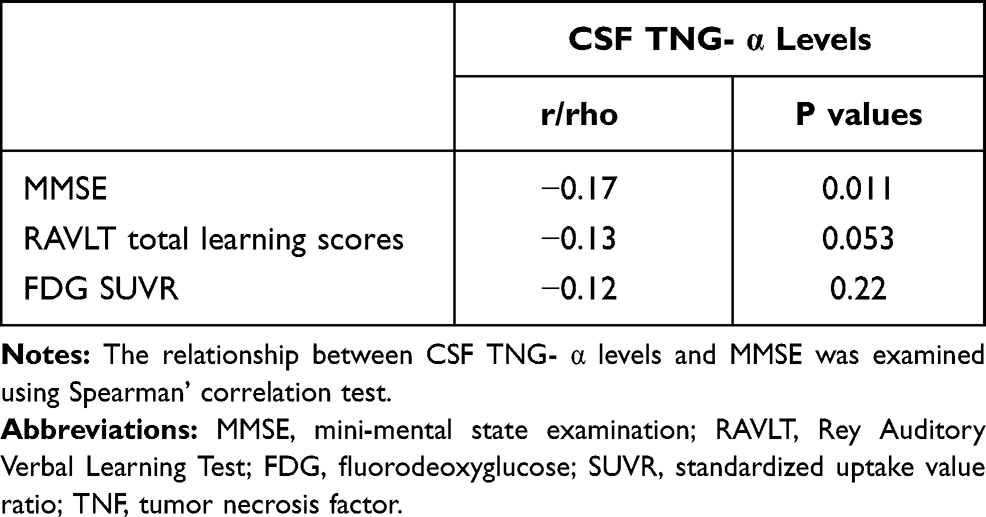 |
Table 2 Cross-Sectional Relationships Between CSF TNF- α and AD-Related Makers |
Longitudinal Associations of Baseline CSF TNF-α with Global Cognition, Memory, and FDG SUVR Among Non-Demented Older People
To examine the associations of baseline CSF TNF-α levels with changes in MMSE scores (Table 3), RAVLT total learning scores (Table 4), and FDG SUVR (Table 5) over time among non-demented older people, several linear mixed models were fitted for each outcome. As shown in Tables 3–5 and Figure 1, we found that among non-demented older people, higher CSF TNF-α levels were associated with a faster decline in FDG SUVR (estimate = −0.0089, se= 0.0043, p value = 0.0385), but not MMSE scores (estimate = −0.1246, se= 0.1592, p value = 0.4341) or RAVLT total learning scores (estimate = 0.0105, se= 0.2333, p value = 0.9641).
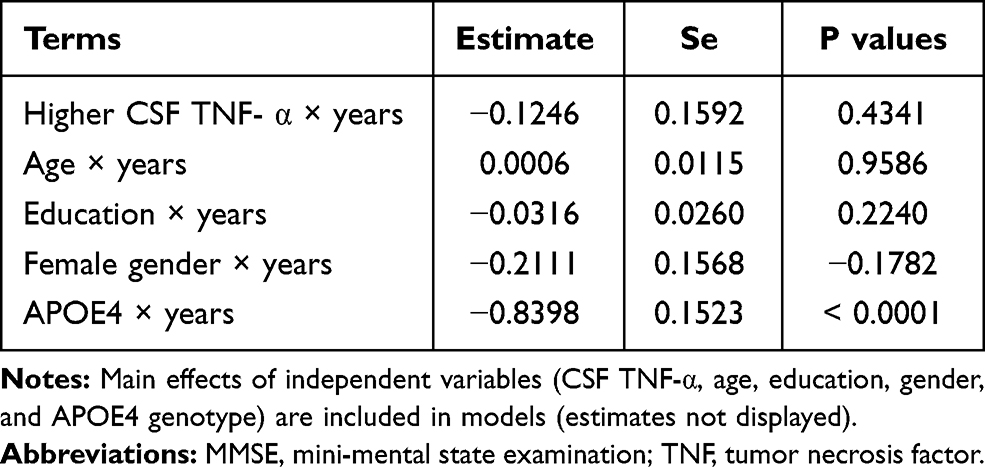 |
Table 3 Longitudinal Association of Baseline CSF TNF-α with MMSE Scores |
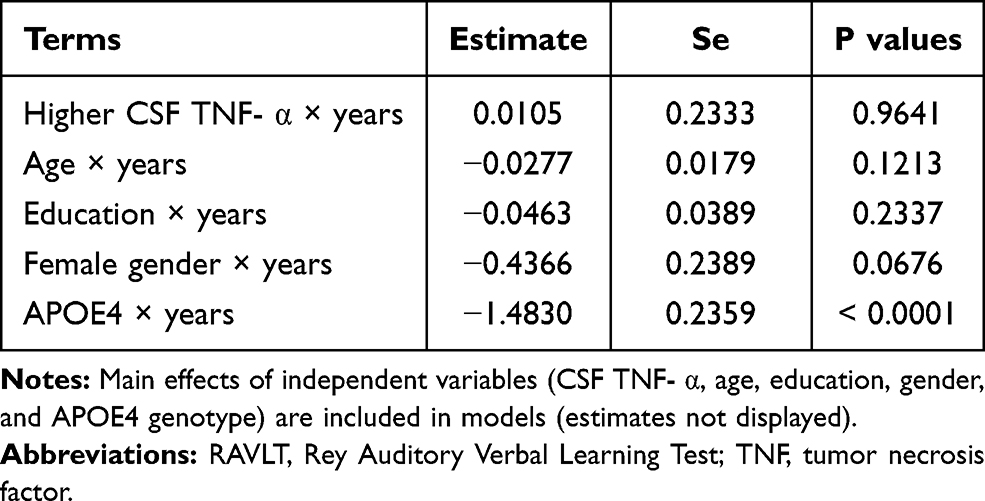 |
Table 4 Longitudinal Association of Baseline CSF TNF-α with RAVLT Total Learning Scores |
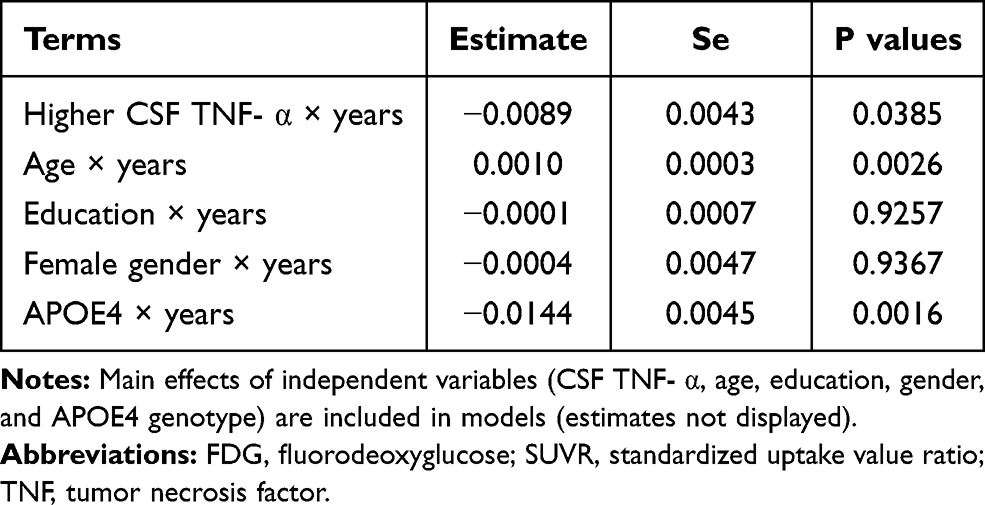 |
Table 5 Longitudinal Association of Baseline CSF TNF-α with FDG SUVR |
 |
Figure 1 Associations of CSF TNF- α with change in AD-related markers over time. Higher CSF TNF-α levels were associated with a faster decline in FDG SUVR (estimate = −0.0089, se= 0.0043, p value = 0.0385, (C) but not MMSE scores (estimate = −0.1246, se= 0.1592, p value = 0.4341, (A) or RAVLT total learning scores (estimate = 0.0105, se= 0.2333, p value = 0.9641, (B). Abbreviations: MMSE, mini-mental state examination; RAVLT, Rey Auditory Verbal Learning Test; FDG, fluorodeoxyglucose; SUVR, standardized uptake value ratio; TNF, tumor necrosis factor. |
Supplementary Analyses
We further examined whether amyloid status (positive vs negative) can modify the association of CSF TNF- α with change in MMSE score, RAVLT total learning score and FDG SUVR over time among MCI subjects. At baseline, patients with MCI were further classified into two groups according to their amyloid status (amyloid-positive: CSF Aβ42 < 192 pg/mL; amyloid-negative: CSF Aβ42 ≥ 192 pg/mL): 40 amyloid-negative MCI subjects and 89 amyloid-positive MCI subjects. Several linear mixed models were utilized to examine the associations of this interaction term (amyloid status*CSF TNF-α) with change in each outcome (MMSE sores, RAVLT total learning scores, and FDG SUVR) over time among older people with MCI. Each model included the amyloid status*CSF TNF- α interaction term, age, gender, educational level, APOE4 genotype and their interactions with time, along with a random intercept and a random slope for each subject. We found that this interaction term was significant for FDG, but not for MMSE or RAVLT total learning score (Data not shown), indicating that amyloid status may modify the association of CSF TNF- α with change in FDG over time among MCI subjects. Therefore, an amyloid-stratified analysis was further performed. In the amyloid-positive group, CSF TNF-α was not associated with change in FDG over time in MCI (p > 0.05, Table 6, Figure 2A). However, higher CSF TNF-α levels were associated with a steeper decline in FDG SUVR in the amyloid-negative group (p < 0.05, Table 6, Figure 2B).
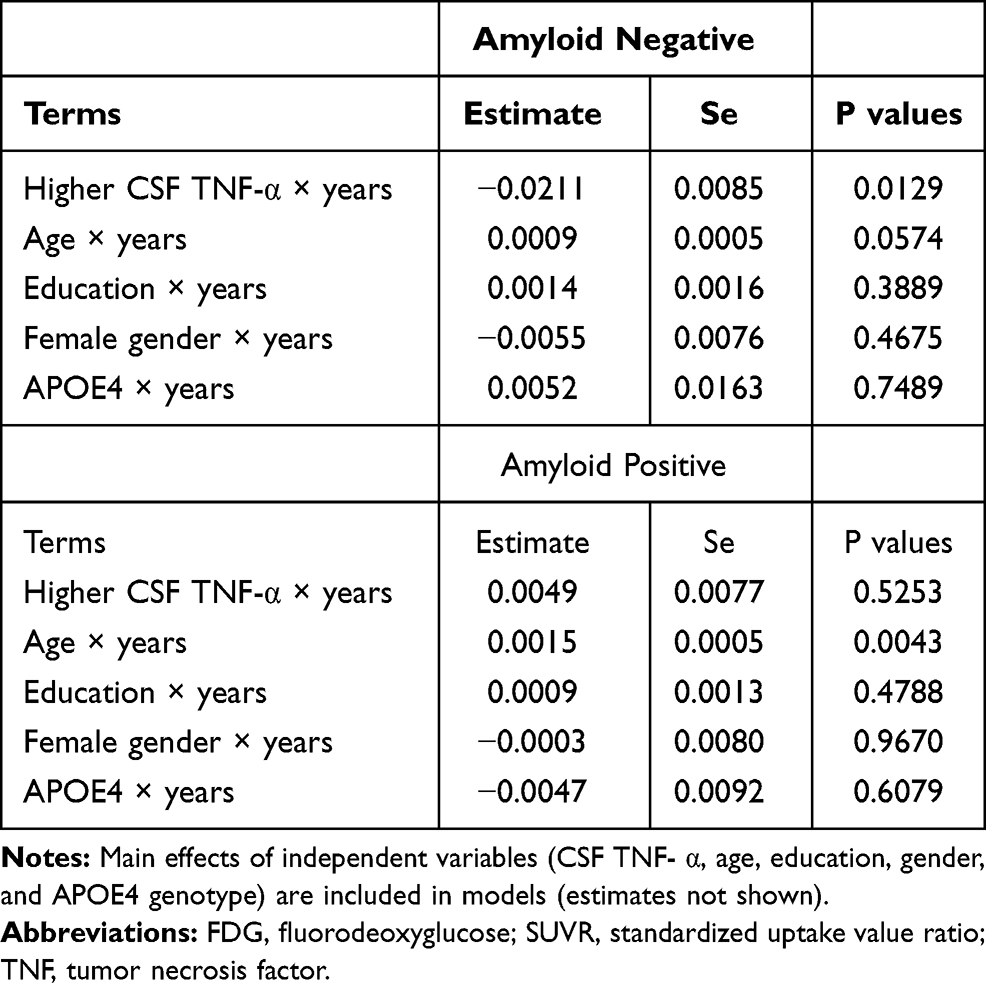 |
Table 6 Longitudinal Association of CSF TNF- α with Change in FDG SUVR in the Amyloid Negative MCI Subjects and Amyloid Positive MCI Subjects |
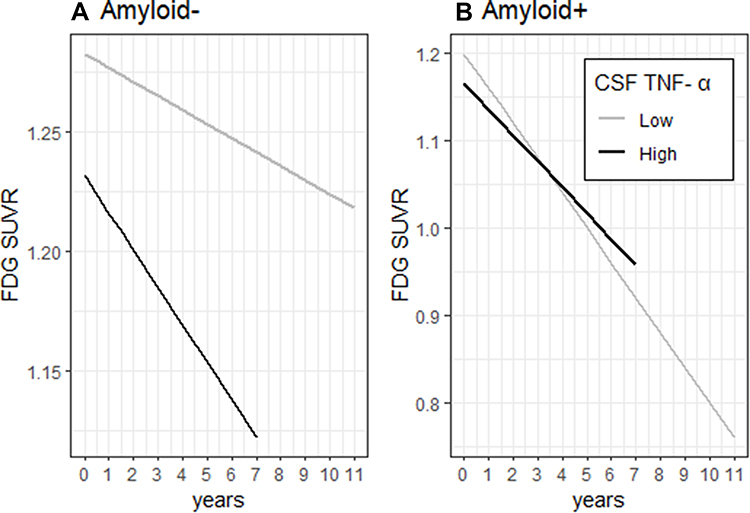 |
Figure 2 Associations of CSF TNF- α with change in FDG SUVR over time in the amyloid negative MCI subjects and amyloid positive MCI subjects. In the amyloid-positive group, CSF TNF-α was not associated with change in FDG over time in MCI (p > 0.05, A). However, higher CSF TNF-α levels were associated with a steeper decline in FDG SUVR in the amyloid-negative group (p < 0.05, B). |
In addition, we examined whether the clinical diagnosis (MCI vs normal cognition) can modify the association of CSF TNF- α with change in MMSE score, RAVLT total learning score and FDG SUVR over time among non-demented older people. Several linear mixed models were utilized to investigate the association of the clinical diagnosis*CSF TNF-α interaction term with change in each outcome (MMSE sores, RAVLT total learning scores, and FDG SUVR) over time among non-demented older people. Each model included the clinical diagnosis*CSF TNF- α interaction term, age, gender, educational level, APOE4 genotype and their interactions with time, along with a random intercept and a random slope for each subject. However, this interaction term was not significant for any outcomes (all p > 0.05, data not shown), suggesting that there was no significant difference in the association of CSF TNF-α with change in MMSE score, RAVLT total learning score or FDG SUVR over time between normal cognition and MCI.
Discussion
To the best of our knowledge, this is the first study to examine the association of baseline CSF TNF-α levels with longitudinal changes in MMSE scores, RAVLT total learning scores, and brain glucose metabolism (as assessed by FDG-SUVR) among non-demented older people. We found that higher CSF TNF-α levels were linked to a steeper decline in FDG-SUVR, but not MMSE or RAVLT total learning scores among non-demented older people.
In terms of the biology of TNF-α, it and its receptors participate in a variety of biological functions in the body, such as immune responses to fight microbial infections, immune surveillance, and apoptosis.16–18 One previous study showed that levels of CSF TNF-α in AD patients are higher than those of normal controls.19 This was further supported by elevated levels of TNF-α observed in the blood of patients with AD compared to normal controls.20 Furthermore, in previous cross-sectional studies, higher levels of blood TNF-α were found to be associated with worse cognitive performance among AD patients.21,22 In addition, preclinical studies have also been suggested that TNF-α could activate the astroglial response, contributing to the neuroinflammatory process.23,24 TNF-α could stimulate the NF-kB pathway that leads to the elevation of Aβ accumulation.25 Taken together, both preclinical and clinical studies support the notion that TNF-α is an important cytokine that plays a critical role in the pathogenesis of AD.
Reduced cerebral glucose metabolism is a core feature of AD dementia.6,7 In the present study, we found that higher CSF TNF-α levels were associated with a steeper decline in FDG-SUVR among non-demented older people, indicating that TNF-α may play an important role in neuronal and synaptic dysfunction in AD. There are several possible explanations for this finding. First, the adverse effect of TNF-α on FDG-PET may be Aβ-mediated. For example, it has been reported that TNF-α increases apoptosis of neurons administrated with Aβ and impacts Aβ production in cell culture studies.26–28 Further, microglia are unable to efficiently phagocytose Aβ in the presence of increased TNF levels.29 Given that the close relationship between Aβ accumulation in the brain and cerebral glucose hypometabolism,30 it is possible that the detrimental effect of TNF-α on cerebral glucose metabolism may be Aβ-mediated.
Second, TNF-α may directly affect synaptic and neuronal functions and contribute to neurodegeneration. For instance, numerous studies have suggested that TNF-α could mediate activation of oxidative stress cascades, neuronal and synaptic dysfunction, and exacerbation of neurodegeneration.31,32
Third, it is also possible that TNF-α-related neuroinflammation may contribute to hyperphosphorylation and accumulation of tau, thus leading to axonal malfunction and neurodegeneration.33
Interestingly, we found that among non-demented older people, higher CSF TNF-α levels were associated with a steeper decline in FDG SUVR, but not cognitive assessments. Based on the amyloid cascade hypothesis, amyloid accumulation precedes cerebral glucose hypometabolism, which is followed by cognitive impairment.11,34 It is possible that among non-demented older people, dysregulation of neuroinflammation may be a contributing factor for cerebral glucose hypometabolism (synaptic and neuronal dysfunction), but not for cognitive impairment. However, evidence on the effect of neuroinflammation on cognitive impairment and clinical progression has been inconsistent.2 For example, a recent study examining the association of the levels of 27 cytokines in CSF with cognitive impairment and clinical progression in AD patients found that some of cytokines may be “protective”.35 It has been reported that neuroinflammation is not exclusively beneficial or detrimental in the pathogenesis of AD.36 Further studies should be conducted to further clarify the relationship between CSF TNF-α and AD-related markers at different stages of the disease.
There are several limitations in this study. First, the observational nature of this study does not allow us to address the causal relationship between CSF TNF-α and cerebral glucose metabolism among non-demented older people. Second, because most of the ADNI participants were white and well educated, sample selection bias may affect the interpretation of our findings. Third, in the present study, only one cytokine (CSF TNF-α) was used to examine its relation with AD-related markers. Further studies should utilize a group of cytokines to investigate the association of the pattern of various cytokines with cognitive impairment and clinical progression. Additionally, this approach should also be applied at different stages of the disease.
In conclusion, higher CSF TNF- α levels were associated with a steeper decline in cerebral glucose metabolism among non-demented older people.
Acknowledgments
Data collection and sharing for this project were funded by the Alzheimer’s Disease Neuroimaging Initiative (ADNI) (National Institutes of Health Grant U01 AG024904) and DOD ADNI (Department of Defense award number W81XWH-12-2-0012). ADNI is funded by the National Institute on Aging, the National Institute of Biomedical Imaging and Bioengineering, and through generous contributions from the following: AbbVie, Alzheimer’s Association; Alzheimer’s Drug Discovery Foundation; Araclon Biotech; BioClinica, Inc.; Biogen; Bristol-Myers Squibb Company; CereSpir, Inc.; Cogstate; Eisai Inc.; Elan Pharmaceuticals, Inc.; Eli Lilly and Company; EuroImmun; F. Hoffmann-La Roche Ltd and its affiliated company Genentech, Inc.; Fujirebio; GE Healthcare; IXICO Ltd.; Janssen Alzheimer Immunotherapy Research & Development, LLC.; Johnson & Johnson Pharmaceutical Research & Development LLC.; Lumosity; Lundbeck; Merck & Co., Inc.; Meso Scale Diagnostics, LLC.; NeuroRx Research; Neurotrack Technologies; Novartis Pharmaceuticals Corporation; Pfizer Inc.; Piramal Imaging; Servier; Takeda Pharmaceutical Company; and Transition Therapeutics. The Canadian Institutes of Health Research is providing funds to support ADNI clinical sites in Canada. Private sector contributions are facilitated by the Foundation for the National Institutes of Health (www.fnih.org). The grantee organization is the Northern California Institute for Research and Education, and the study is coordinated by the Alzheimer’s Therapeutic Research Institute at the University of Southern California. ADNI data are disseminated by the Laboratory for Neuro Imaging at the University of Southern California. Data used in preparation of this article were obtained from the Alzheimer’s Disease Neuroimaging Initiative (ADNI) database (adni.loni.usc.edu). As such, the investigators within the ADNI contributed to the design and implementation of ADNI and/or provided data but did not participate in analysis or writing of this report. A complete listing of ADNI investigators can be found at: http://adni.loni.usc.edu/wp-content/uploads/how_to_apply/ADNI_Acknowledgement_List.pdf.
Disclosure
The authors declare that they have no conflicts of interest.
References
1. Edwards FA. A unifying hypothesis for alzheimer’s disease: from plaques to neurodegeneration. Trends Neurosci. 2019;42(5):310–322. doi:10.1016/j.tins.2019.03.003
2. Kinney JW, Bemiller SM, Murtishaw AS, Leisgang AM, Salazar AM, Lamb BT. Inflammation as a central mechanism in alzheimer’s disease. Alzheimers Dement. 2018;4:575–590.
3. McGeer PL, Schulzer M, McGeer EG. Arthritis and anti-inflammatory agents as possible protective factors for alzheimer’s disease: a review of 17 epidemiologic studies. Neurology. 1996;47(2):425–432. doi:10.1212/WNL.47.2.425
4. Zhao M, Cribbs DH, Anderson AJ, et al. The induction of the TNFα death domain signaling pathway in alzheimer’s disease brain. Neurochem Res. 2003;28(2):307–318. doi:10.1023/A:1022337519035
5. Tan Z, Beiser A, Vasan R, et al. Inflammatory markers and the risk of Alzheimer disease: the Framingham Study. Neurology. 2007;68(22):1902–1908. doi:10.1212/01.wnl.0000263217.36439.da
6. Minoshima S, Giordani B, Berent S, Frey KA, Foster NL, Kuhl DE. Metabolic reduction in the posterior cingulate cortex in very early alzheimer’s disease. Ann Neurol. 1997;42(1):85–94. doi:10.1002/ana.410420114
7. Herholz K. Cerebral glucose metabolism in preclinical and prodromal alzheimer’s disease. Expert Rev Neurother. 2010;10(11):1667–1673. doi:10.1586/ern.10.136
8. Morris JC. The Clinical Dementia Rating (CDR): current version and scoring rules. Neurology. 1993;43(11):2412–2414. doi:10.1212/WNL.43.11.2412-a
9. Folstein MF, Folstein SE, McHugh PR. “Mini-mental state”. A practical method for grading the cognitive state of patients for the clinician. J Psychiatr Res. 1975;12(3):189–198. doi:10.1016/0022-3956(75)90026-6
10. Schmidt M. Rey Auditory Verbal Learning Test: A Handbook. Los Angeles, CA: Western Psychological Services; 1996.
11. Landau SM, Mintun MA, Joshi AD, et al. Amyloid deposition, hypometabolism, and longitudinal cognitive decline. Ann Neurol. 2012;72(4):578–586. doi:10.1002/ana.23650
12. Jagust WJ, Bandy D, Chen K, et al. The alzheimer’s disease neuroimaging initiative positron emission tomography core. Alzheimers Dement. 2010;6(3):221–229. doi:10.1016/j.jalz.2010.03.003
13. Landau SM, Harvey D, Madison CM, et al. Associations between cognitive, functional, and FDG-PET measures of decline in AD and MCI. Neurobiol Aging. 2011;32(7):1207–1218. doi:10.1016/j.neurobiolaging.2009.07.002
14. Team RC. R: A Language and Environment for Statistical Computing; 2013.
15. Bates D, Mächler M, Bolker B, Walker S. Fitting linear mixed-effects models using lme4. arXiv Preprint arXiv. 2014.
16. Fischer R, Maier O. Interrelation of oxidative stress and inflammation in neurodegenerative disease: role of TNF. Oxid Med Cell Longev. 2015;2015:1–18. doi:10.1155/2015/610813
17. Clark IA, Alleva LM, Vissel B. The roles of TNF in brain dysfunction and disease. Pharmacol Ther. 2010;128(3):519–548.
18. Locksley RM, Killeen N, Lenardo MJ. The TNF and TNF receptor superfamilies: integrating mammalian biology. Cell. 2001;104(4):487–501. doi:10.1016/S0092-8674(01)00237-9
19. Tarkowski E, Liljeroth A-M, Minthon L, Tarkowski A, Wallin A, Blennow K. Cerebral pattern of pro-and anti-inflammatory cytokines in dementias. Brain Res Bull. 2003;61(3):255–260. doi:10.1016/S0361-9230(03)00088-1
20. Fillit H, Ding W, Buee L, et al. Elevated circulating tumor necrosis factor levels in alzheimer’s disease. Neurosci Lett. 1991;129(2):318–320. doi:10.1016/0304-3940(91)90490-K
21. Alvarez XA, Franco A, Fernández-Novoa L, Cacabelos R. Blood levels of histamine, IL-1β, and TNF-α in patients with mild to moderate Alzheimer disease. Mol Chem Neuropathol. 1996;29(2–3):237–252. doi:10.1007/BF02815005
22. Holmes C, Cunningham C, Zotova E, et al. Systemic inflammation and disease progression in Alzheimer disease. Neurology. 2009;73(10):768–774. doi:10.1212/WNL.0b013e3181b6bb95
23. Hensley K. Neuroinflammation in alzheimer’s disease: mechanisms, pathologic consequences, and potential for therapeutic manipulation. J Alzheimers Dis. 2010;21(1):1–14. doi:10.3233/JAD-2010-1414
24. Heneka MT, Carson MJ, El Khoury J, et al. Neuroinflammation in alzheimer’s disease. Lancet Neurol. 2015;14(4):388–405. doi:10.1016/S1474-4422(15)70016-5
25. Chen C-H, Zhou W, Liu S, et al. Increased NF-κB signalling up-regulates BACE1 expression and its therapeutic potential in alzheimer’s disease. Int J Neuropsychopharmacol. 2012;15(1):77–90. doi:10.1017/S1461145711000149
26. Blasko I, Marx F, Steiner E, Hartmann T, Grubeck-Loebenstein B. TNFalpha plus IFNgamma induce the production of Alzheimer beta-amyloid peptides and decrease the secretion of APPs. FASEB J. 1999;13(1):63–68. doi:10.1096/fasebj.13.1.63
27. Blasko I, Schmitt TL, Steiner E, Trieb K, Grubeck-Loebenstein B. Tumor necrosis factor alpha augments amyloid beta protein (25–35) induced apoptosis in human cells. Neurosci Lett. 1997;238(1–2):17–20. doi:10.1016/S0304-3940(97)00845-8
28. Blasko I, Veerhuis R, Stampfer-Kountchev M, Saurwein-Teissl M, Eikelenboom P, Grubeck-Loebenstein B. Costimulatory effects of interferon-gamma and interleukin-1beta or tumor necrosis factor alpha on the synthesis of Abeta1-40 and Abeta1-42 by human astrocytes. Neurobiol Dis. 2000;7(6Pt B):682–689. doi:10.1006/nbdi.2000.0321
29. Koenigsknecht-Talboo J, Landreth GE. Microglial phagocytosis induced by fibrillar beta-amyloid and IgGs are differentially regulated by proinflammatory cytokines. J Neurosci. 2005;25(36):8240–8249. doi:10.1523/JNEUROSCI.1808-05.2005
30. Pascoal TA, Mathotaarachchi S, Shin M, et al. Amyloid and tau signatures of brain metabolic decline in preclinical alzheimer’s disease. Eur J Nucl Med Mol Imaging. 2018;45(6):1021–1030. doi:10.1007/s00259-018-3933-3
31. Cunningham C, Campion S, Lunnon K, et al. Systemic inflammation induces acute behavioral and cognitive changes and accelerates neurodegenerative disease. Biol Psychiatry. 2009;65(4):304–312. doi:10.1016/j.biopsych.2008.07.024
32. Medeiros R, Prediger RD, Passos GF, et al. Connecting TNF-alpha signaling pathways to iNOS expression in a mouse model of alzheimer’s disease: relevance for the behavioral and synaptic deficits induced by amyloid beta protein. J Neurosci. 2007;27(20):5394–5404. doi:10.1523/JNEUROSCI.5047-06.2007
33. Kitazawa M, Oddo S, Yamasaki TR, Green KN, LaFerla FM. Lipopolysaccharide-induced inflammation exacerbates tau pathology by a cyclin-dependent kinase 5-mediated pathway in a transgenic model of alzheimer’s disease. J Neurosci. 2005;25(39):8843–8853. doi:10.1523/JNEUROSCI.2868-05.2005
34. Bateman RJ, Xiong C, Benzinger TL, et al. Clinical and biomarker changes in dominantly inherited alzheimer’s disease. N Engl J Med. 2012;367(9):795–804. doi:10.1056/NEJMoa1202753
35. Taipa R, Das Neves SP, Sousa AL, et al. Proinflammatory and anti-inflammatory cytokines in the CSF of patients with alzheimer’s disease and their correlation with cognitive decline. Neurobiol Aging. 2019;76:125–132. doi:10.1016/j.neurobiolaging.2018.12.019
36. Michaud JP, Rivest S. Anti-inflammatory signaling in microglia exacerbates Alzheimer’s disease-related pathology. Neuron. 2015;85(3):450–452. doi:10.1016/j.neuron.2015.01.021
 © 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.