Back to Journals » OncoTargets and Therapy » Volume 12
Cryptotanshinone inhibits esophageal squamous-cell carcinoma in vitro and in vivo through the suppression of STAT3 activation
Authors Ji Y ![]() , Liu Y
, Liu Y ![]() , Xue N, Du T, Wang L, Huang R, Li L, Yan C, Chen X
, Xue N, Du T, Wang L, Huang R, Li L, Yan C, Chen X
Received 16 September 2018
Accepted for publication 3 January 2019
Published 29 January 2019 Volume 2019:12 Pages 883—896
DOI https://doi.org/10.2147/OTT.S187777
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Jianmin Xu
Yubin Ji,1 Yichen Liu,1,2 Nina Xue,2 Tingting Du,2 Liyuan Wang,2 Rui Huang,2 Ling Li,2 Chunhong Yan,3 Xiaoguang Chen2
1Research Center on Life Sciences and Environmental Sciences, Harbin University of Commerce, Harbin 150076, People’s Republic of China; 2State Key Laboratory of Bioactive Substances and Functions of Natural Medicines, Institute of Materia Medica, Chinese Academy of Medical Sciences and Peking Union Medical College, Beijing 100050, People’s Republic of China; 3Georgia Cancer Center, Augusta University, Augusta, GA, USA
Purpose: Esophageal squamous-cell carcinoma (ESCC) is the most common subtype of esophageal cancer, with a poor clinical outcome. Cryptotanshinone (CTS) is the main bioactive compound from the root of Salvia miltiorrhiza Bunge. Our study aimed to investigate the anti-cancer effects and molecular mechanisms of CTS on ESCC.
Materials and methods: We investigated the anti-tumor activity of CTS on ESCC in vitro and in vivo. Activation of the STAT3 signaling pathway was evaluated in ESCC and HEK-Blue™ IL-6 cells. Cell viability was assessed by the MTT assay. Apoptosis and cell cycle arrest were assessed using flow cytometry. Cell migration was detected by a scratch wound assay.
Results: CTS inhibited STAT3 expression and IL-6-mediated STAT3 activation in esophageal cancer cells. Subsequently, CTS dose-dependently inhibited the proliferation of esophageal cancer cells via induction of cell apoptosis. Furthermore, CTS suppressed the migration of esophageal cancer cells. In vivo, CTS inhibited tumor growth of EC109 cell in xenograft mice without any obvious effect on body weight.
Conclusion: Our results indicated that STAT3 inhibition may be a therapeutic target for esophageal cancer. CTS could provide a potential approach for esophageal cancer therapy by influencing the janus kinase-2/STAT3 signaling pathway.
Keywords: xenograft, CTS, ESCC, proliferation, apoptosis, migration
Introduction
There are two major subtypes of esophageal cancer, esophageal squamous-cell carcinoma (ESCC) and esophageal adenocarcinoma.1 Esophageal adenocarcinoma has become the predominant type of esophageal cancer in North America and Europe, but ESCC still accounts for approximately 90% of cases of global esophageal cancer.2 Most cases of ESCC occur in Asia, especially in northern Iran through the Central Asian republics to north-central China.2–4 As ESCC is a high-grade invasive tumor, although ESCC patients have received comprehensive treatment based on surgery, the therapeutic effect is not satisfactory: the 5-year overall survival rate ranges from 30% to 50%, without significant improvement during the past few years, and more than half of patients suffer recurrence within 2–3 years after surgery.5 Therefore, improvement and exploration of more effective therapeutic approaches are desired for ESCC therapy.
Janus kinase (JAK)-mediated STAT signaling is one of the most promising new targets for cancer therapy.6 In this pathway, STAT3 protein, as an important signal switch, generally regulates many pathological patterns of cancer cells, including proliferation, survival, differentiation and metastasis. It has been reported that abnormal expression of STAT3 is correlated with the occurrence and development of many cancer types, such as breast cancer, ovarian cancer and colorectal cancer.7–9 For example, Alvarez et al reported that constitutive activation of STAT3 facilitates cell proliferation in non-small-cell lung cancer.10 Zamo et al described that overexpressed STAT3 was related to apoptosis inhibition in anaplastic large-cell lymphomas.11 However, the role of STAT3 in esophageal cancer has rarely been discussed. Bhat et al12 and Zhang et al13 found STAT3 overexpression in esophageal cancer tissues, resulting in the proliferation of esophageal cancer cells. These findings indicated that the dysfunction of STAT3 signaling also participated in the modulation of aggravation of esophageal cancer.
Cryptotanshinone (CTS) is a quinoid diterpene purified from the root of medicinal plant Salvia miltiorrhiza Bunge (DanShen),14 which has been widely used in the clinic for treatment of multiple diseases, including inflammatory conditions,15 cardiac fibrosis16 and Alzheimer’s disease,17 without obvious adverse effects. CTS is a potent anti-cancer agent, reducing cell proliferation by suppressing STAT3 signals.18 However, the anti-tumor activity of CTS on esophageal cancer and whether it relates to the blockade of STAT3 signaling have not been elucidated.
In this study, we evaluated the anti-tumor efficacy and molecular mechanism of CTS on esophageal cancer in vitro and in vivo. CTS inhibits cell proliferation, induces cell apoptosis and suppresses cell migration in ESCC. These results suggest that STAT3 signals may be a target and CTS shows potential in esophageal cancer treatment.
Materials and methods
Reagents
CTS was purchased from Aladdin (Shanghai, People’s Republic of China). To prepare working solutions, CTS was dissolved in 100% dimethyl sulfoxide (DMSO) to create a stock solution (20 mmol/L) and stored at −20°C. CTS was further diluted in culture media for all in vitro experiments. MTT was obtained from Sigma (St Louis, MO, USA). Annexin V–fluorescein isothiocyanate (FITC) and propidium iodide (PI) were purchased from GenStar (Beijing, People’s Republic of China). Antibodies to phosphorylated STAT3 (p-STAT3; Tyr705), STAT3, p-JAK2, JAK2, cleaved caspase3 and KI-67 were purchased from Cell Signaling Technologies (Shanghai, People’s Republic of China); β-actin antibody, mouse and rabbit IgG were purchased from ZSGB-Bio (Beijing, People’s Republic of China).
Cell lines and cell culture
Two human esophageal cancer cell lines, EC109 and CAES17, were purchased from the Cell Resource Center at the Institute of Medical Sciences, Peking Union Medical College. EC109 and CAES17 cells were cultured in RPMI medium 1640 or DMEM, respectively. HEK-Blue™ IL-6 cells were purchased from InvivoGen (San Diego, CA, USA) and cultured in DMEM according to the manufacturer’s instructions. The media for the cell lines were supplemented with 10% FBS (Gibco, Grand Island, NY, USA), 100 μg/mL streptomycin and 100 U/mL penicillin (Hyclone, Logan, UT, USA) and maintained at 37°C in a humidified atmosphere with 5% CO2.
Cell viability assay
Cell viability was assessed by the MTT assay.19 Cells were seeded at a density of 2,000 cells per well in a flat-bottomed 96-well plate and cultured overnight. Then, the cells were treated with CTS at various concentrations (0, 1.25, 5, 10, 20 and 40 μmol/L) for 24, 48 and 72 hours. Subsequently, 20 μL MTT (5 mg/mL) reagent was added into each well and incubated for another 4 hours until the purple dye was visible. The culture medium was then replaced with 150 μL of DMSO, and the absorbance at 450 and 570 nm was measured by a Synergy microplate reader (BioTek, Winooski, VT, USA). Results represent the average of three parallel samples. The cell viability ratio was calculated by the following formula: Cell viability (%) = (Average absorbance of treated group/Average absorbance of control group) ×100%.
Western blot assay
EC109 and CAES17 cells were seeded into six-well plates and treated with CTS (0, 1, 2.5, 5, 10 and 20 μmol/L). Then, the cells were harvested and lysed in RIPA buffer (50 mM Tris, pH 7.4, 150 mM NaCl, 1 mM PMSF, 1 mM EDTA, 1% Triton X-100, 0.1% SDS, 1 mM Na3VO4, 1 mM NaF and protease inhibitor cocktail; ZSGB-Bio, Beijing, People’s Republic of China). Protein concentration was determined by the Enhanced BCA Protein Assay Kit (Beyotime, Beijing, People’s Republic of China). For Western blot analysis,20 samples were separated on an 8%–10% SDS-PAGE gel, transferred to a polyvinylidene fluoride membrane (Immobilon; Millipore, USA) by semi-wet electrophoresis, then probed with primary antibodies: p-STAT3 (Tyr705), STAT3, p-JAK2 and JAK2. Signals were detected using a Luminescent Image Analyzer (GE Healthcare, Little Chalfont, UK). The experiments were independently performed at least twice, each in triplicate.
CTS-induced STAT3 activation assay
HEK-Blue IL-6 cells are specifically designed to monitor the activation of the JAK–STAT pathway induced by IL-6. HEK-Blue IL-6 cells were seeded in a flat-bottom 96-well plate by a density of 50,000 cells per well and treated with CTS at serial concentrations (0, 0.1, 0.3, 1, 3, 6 and 9 μmol/L), followed by the addition of recombinant human IL-6 at 1 ng/mL in one well. After 16 hours of treatment, the supernatant (20 μL) was incubated with 180 mL of resuspended QUANTI-Blue per well for 1 hour, then the secreted embryonic alkaline phosphatase (SEAP) levels, reflecting STAT3 signaling activation, were determined using a spectrophotometer at 620–655 nm.
Cell apoptosis analysis
Apoptosis was analyzed using the Staralow annexin V–FITC/PI apoptosis analysis kit (Genstar, Beijing, People’s Republic of China) according to the manufacturer’s instructions. Cells were seeded in six-well plates treated without or with CTS (1, 2.5, 5, 10 and 20 μM) for 24 hours or 48 hours, then harvested, washed twice in cold PBS and resuspended in 500 μL binding buffer containing 5 μL annexin V–FITC staining solution. The cells were incubated in the dark for 30 minutes, then 10 μL PI staining solution was added to the cell suspension. The cells were gently mixed and incubated for 15 minutes in the dark at room temperature, then stained cells were immediately analyzed. The number of apoptotic cells was quantified using a flow cytometer that collected 10,000 events for analysis, and apoptosis was detected using a BD FACS Calibur flow cytometer (BD Biosciences, San Jose, CA, USA). All samples were assayed in triplicate.
Cell cycle analysis
Cells treated without or with CTS (5, 10 and 20 μM) for 24 hours, then harvested by trypsinization (without EDTA), washed twice in PBS and fixed in cold ethanol (70%) at −20°C overnight. After fixation, the cells were washed and resuspended in cold PBS, then treated with staining buffer (PBS containing 1 mg/mL PI and 10 mg/mL RNase A; Sigma-Aldrich, St. Louis, MO, USA) at 37°C in the dark for 30 minutes. The cells were then analyzed by a BD FACS Calibur flow cytometer (BD Biosciences, San Jose, CA, USA), and the percentages of cells in G1, S and G2/M phase were calculated. All samples were assayed in triplicate.
Xenografts and animal experiments
Athymic nude mice (BALB/c-nu males, 6–8 weeks old, 18–20 g) were purchased from HFK Bioscience Co. (Beijing, People’s Republic of China) and housed at a temperature of 23.1°C with a 12-hour light–dark cycle with free access to food and water. A human esophageal cancer EC109 tumor from a donor mouse was cut into small pieces with surgical scissors and implanted subcutaneously in the flank of nude mice with a gauge trocar.21 When the tumor had grown to approximately 100 mm3 in volume, the 18 nude mice were divided into three groups with almost equal mean tumor volume (six mice per group) and weighed. Then, mice were treated with intratumoral injection of vehicle (normal saline) or CTS 25 mg/kg and CTS 50 mg/kg once every 2 days for 3 weeks. Nude mice were monitored twice a week and tumor volume was estimated by measuring tumor size and using the following formula: Tumor volume =0.5×L×W2, where L and W represent the largest diameter and the smallest diameter, respectively. After the final treatment, animals were killed, and tumors were removed for further immunohistochemical and Western blot analysis. All animal experiments were approved by the Ethics Committee for Animal Experiments of the Institute of Materia Medica, Chinese Academy of Medical Sciences & Peking Union Medical College, and conducted in accordance with the Guidelines for Animal Experiments of Peking Union Medical College.
Immunohistochemical staining
Tumors were fixed in 4% paraformaldehyde and then processed and embedded in paraffin. Dewaxed and hydrated sections were exposed to 3% H2O2 to eliminate endogenous peroxidase activity. Then, heat-mediated antigen retrieval was performed using a microwave. After 5% goat serum blocking, monoclonal anti-KI67 antibody (Cell Signaling Technology, Danvers, MA, USA) was used at a dilution of 1:400 at 4°C overnight. Detection was performed by standard avidin–biotin complex methods. Sections for all experimental conditions were reacted simultaneously to reduce processing variability. Finally, slides were examined using an upright microscope (Nikon, Tokyo, Japan).
Results
CTS inhibits STAT3 (Tyr705) phosphorylation
First, we explored the expression levels of the JAK2/STAT3 signaling pathway in CAES17 and EC109 esophageal cancer cells. As shown in Figure 1A, compared with pancreatic cancer cell lines SW1990 and BXPC3, which are reported as STAT3 signaling overactivated cells,22 esophageal carcinoma cells exhibited significantly higher levels of p-STAT3 at Tyr705, especially in EC109 cells. Then, we treated esophageal carcinoma cells with increasing doses of CTS for 24 hours. Western blot analysis showed that CTS significantly reduced p-STAT3 (Tyr705) and p-JAK2 in a dose-dependent manner without affecting total STAT3 and JAK2 expression in both CAES17 and EC109 cells (Figure 1B). Furthermore, after treatment with 5 μmol/L of CTS, the protein levels of p-STAT3 (Tyr705) and p-JAK2 decreased time-dependently in both EC109 and CAES17 cells (Figure 1C).
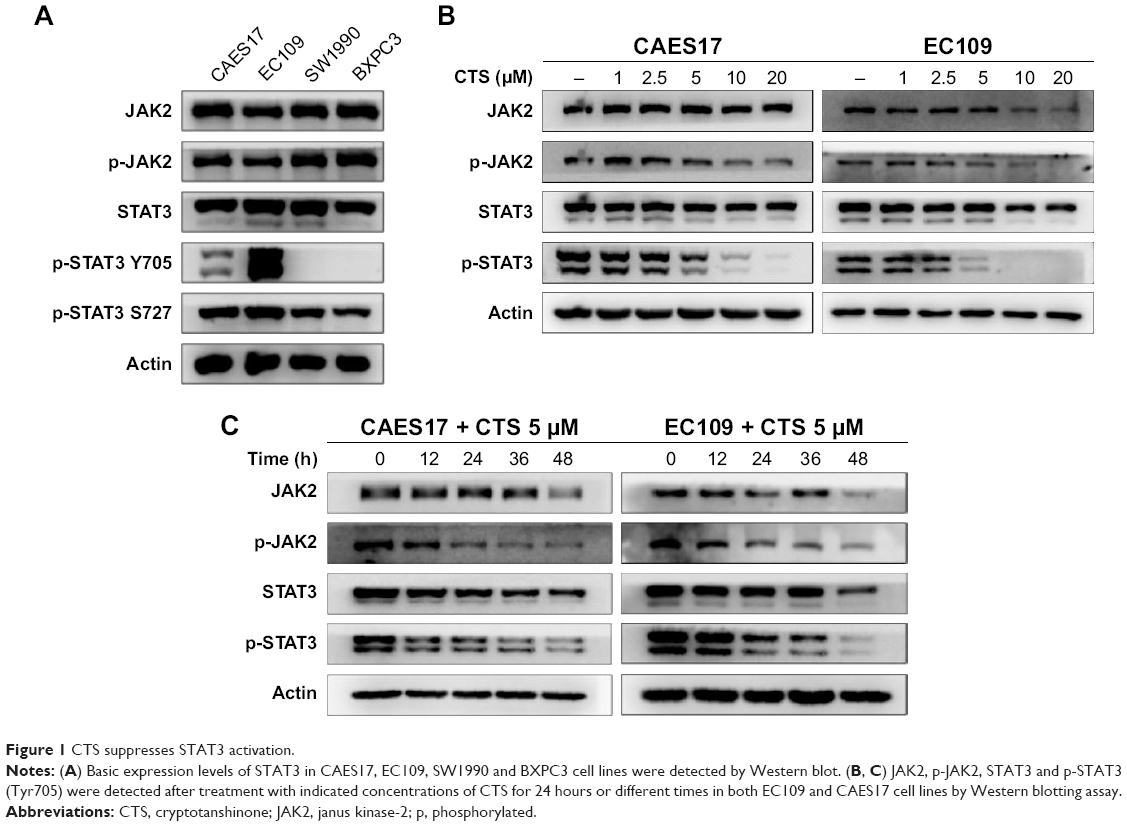 | Figure 1 CTS suppresses STAT3 activation. |
CTS inhibits IL-6-mediated STAT3 activation
IL-6 is a representative stimulant of STAT3 signaling. To further investigate the inhibitory molecular mechanism of CTS on STAT3 activation, we stimulated esophageal carcinoma cells with IL-6 to detect the effect of CTS on the STAT3 signaling pathway. We found that administration of IL-6 (25 ng/mL) for 30 minutes could effectively increase p-STAT3 expression in EC109 and CAES17 cells (Figures 2A and S1A). As shown in Figure 2B, CTS significantly restrained the IL-6-stimulated expression of p-STAT3 (Tyr705) and p-JAK2 of EC109 cells in a dose-dependent manner. The same results could be observed in CAES17 cells (Figure S1B). From all of these results, it seems reasonable to conclude that the inhibition of STAT3 phosphorylation by CTS is associated with the IL-6-induced JAK2/STAT3 signaling pathway. We used HEK-Blue IL-6 cells to verify our conclusion. The SEAP level is taken as an index of STAT3 activation. The SEAP content was significantly increased by IL-6 (1 ng/mL) stimulation (Figure S1C). As shown in Figure 2C, CTS significantly reduced SEAP levels in HEK-Blue IL-6 cells, confirming that CTS inhibited the IL-6-induced STAT3 activation.
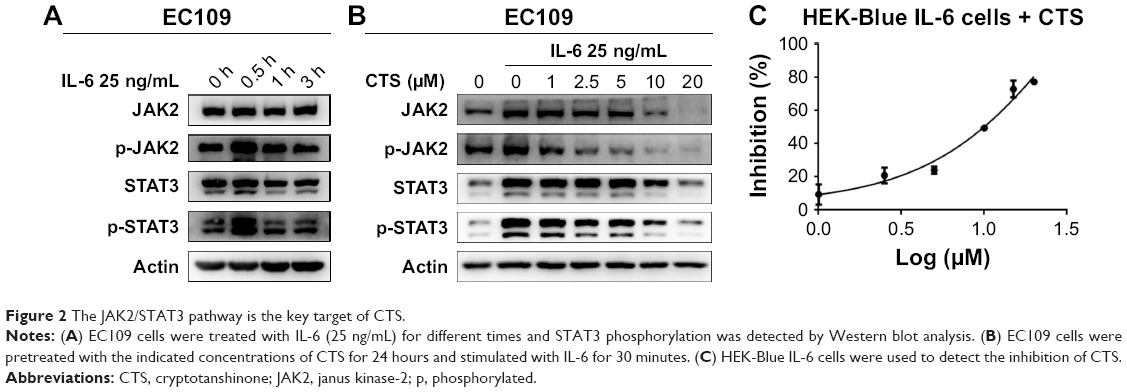 | Figure 2 The JAK2/STAT3 pathway is the key target of CTS. |
CTS inhibits the proliferation of EC109 and CAES17 cells
According to the inhibition of the JAK/STAT3 pathway by CTS, the MTT assay was performed to evaluate the anti-proliferative effects of CTS in EC109 and CAES17 cells. As shown in Figure 3, CTS significantly diminishes proliferative activities of both esophageal cancer cell lines in a time- and dose-dependent manner. The IC50 values (the concentration of drug inhibiting 50% of the cells) of EC109 and CAES17 cells at 72 hours were 2.57 and 10.07 μmol/L, respectively. These results indicate that CTS not only inhibits cell proliferation but also has a better therapeutic selectivity towards STAT3-overexpressed ESCC cells.
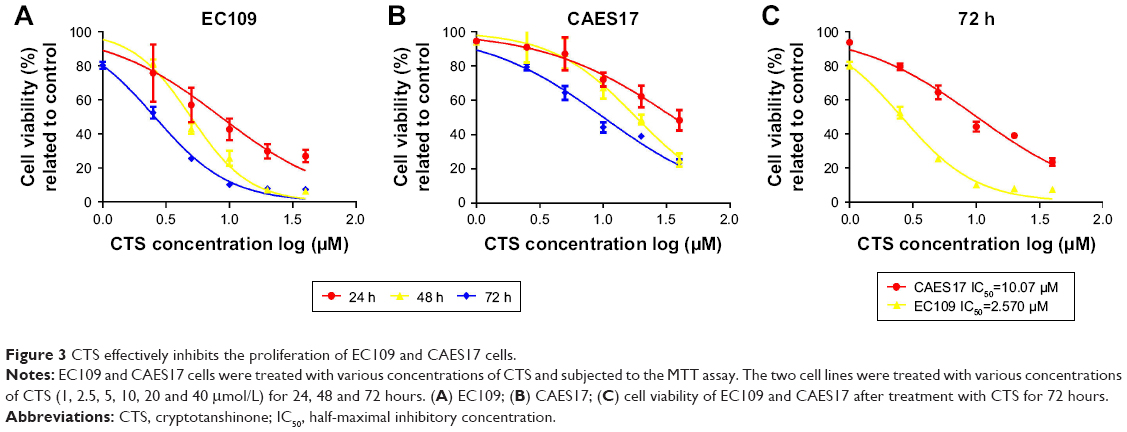 | Figure 3 CTS effectively inhibits the proliferation of EC109 and CAES17 cells. |
CTS induces apoptosis in CAES17 and EC109 cells
To determine whether the CTS-induced growth inhibition was associated with apoptosis, flow cytometric analysis was performed using annexin V/PI dual staining after treatment with CTS on esophageal cancer cells for 24 and 48 hours. As described in Figure S2A, CTS dose-dependently increased the proportion of apoptotic cells, which increased from 14.38% in the untreated group to 54.10% in the treated group (20 μmol/L) in EC109 cells, and from 7.33% to 18.14% in CAES17 cells over 24 hours. After exposure to CTS for 48 hours, the proportions of apoptotic cells increased from 18.78% to 76.54% in EC109 cells and from 8.46% to 37.4% in CAES17 cells (Figure 4A). As shown in Figures 4B and S2B, CTS significantly promoted apoptosis of esophageal cancer cells. These results suggest that induction of apoptosis is involved in the inhibition of proliferation by CTS in esophageal cancer cells.
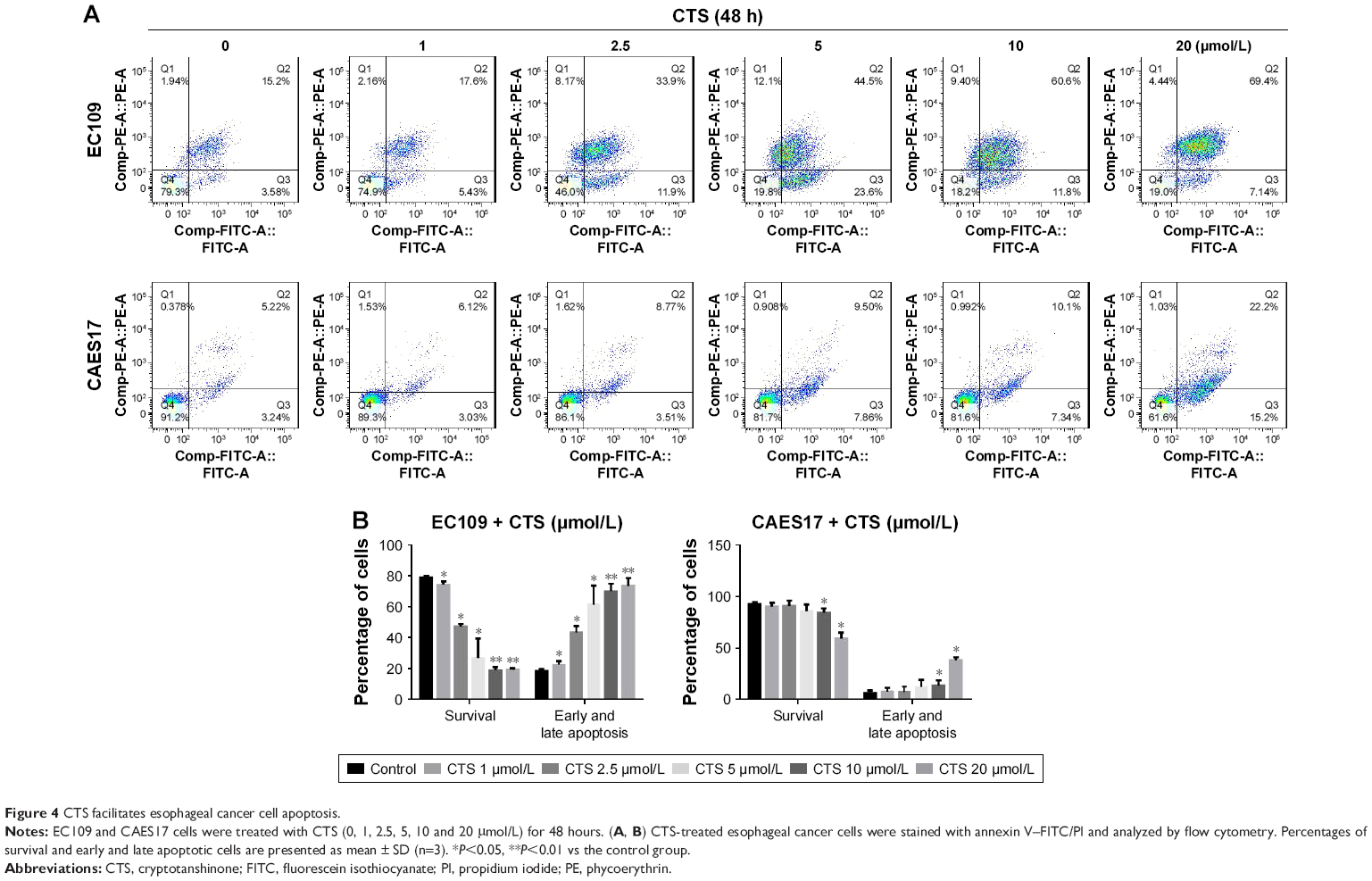 | Figure 4 CTS facilitates esophageal cancer cell apoptosis. |
CTS has no effect on the cell cycle of CAES17 and EC109 cells
Furthermore, we investigated whether the cell cycle arrest was affected by CTS using flow cytometric analysis. Figure 5 shows that the distribution of G0/1, S and G2/M phases did not change after treatment with CTS in CAES17 and EC109 cells for 24 hours. Taken together, the results suggest that CTS-induced growth inhibition is a result of apoptosis rather than cell cycle arrest.
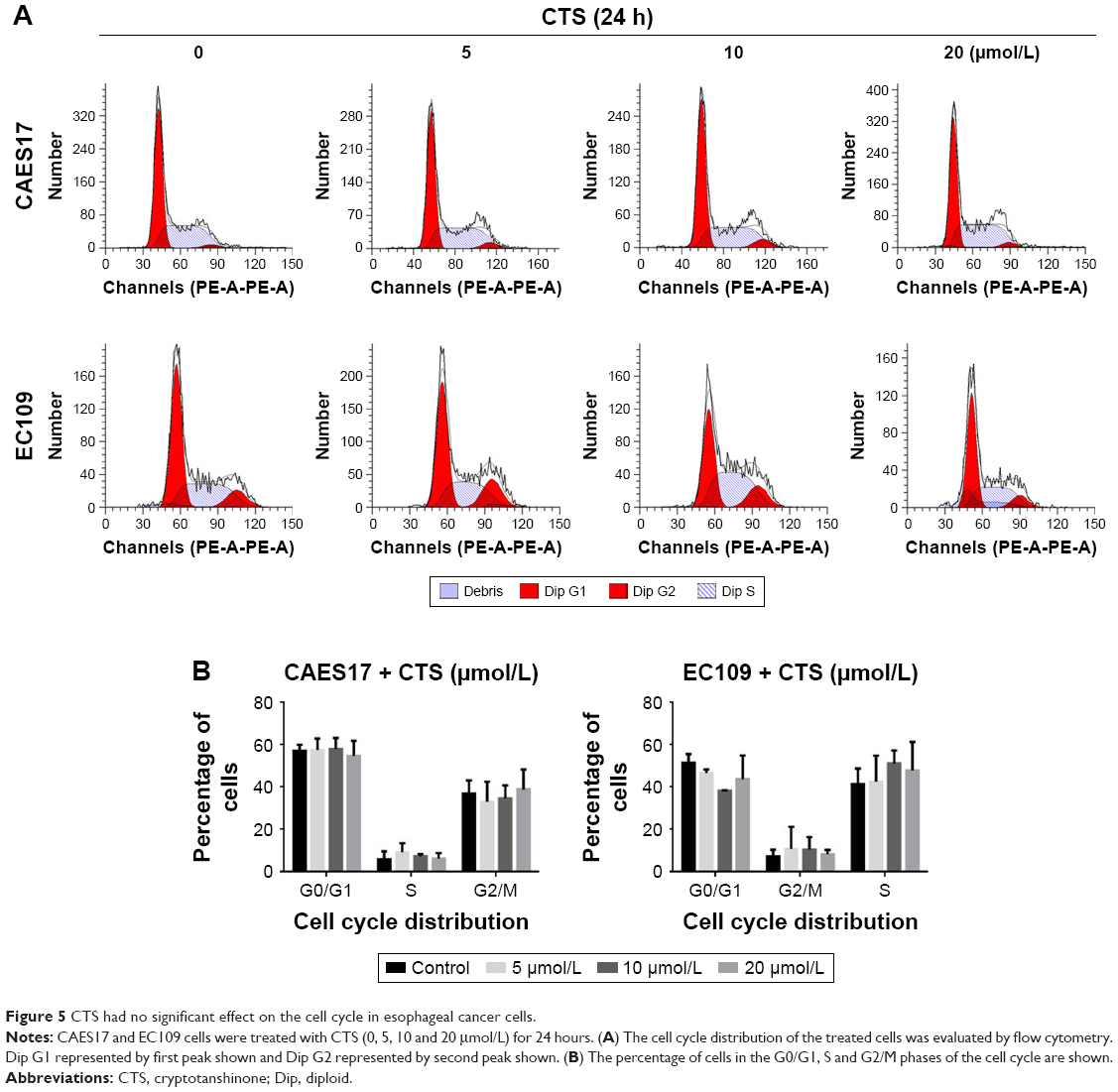 | Figure 5 CTS had no significant effect on the cell cycle in esophageal cancer cells. |
CTS reduces the migration ability of EC109 cells
Next, we investigated the influence of CTS on the migratory capacity of EC109 cells using a scratch wound assay. After scrapping by a sterile pipette tip, the EC109 cells were treated with CTS (0, 2.5 and 10 μmol/L) for 20 hours. Cell migration into the wound was measured according to the proportion of distances between the wound edges before and after CTS treatment. CTS significantly inhibited EC109 cell migration in a dose-dependent manner. As shown in Figure 6, after treatment of EC109 cells for 10 hours, the scratch proportion in the treatment groups was smaller than in the control group. After 20 hours, the wound of the control had almost healed and the closure rates of CTS-treated cells were obviously slower. These results suggest that CTS could suppress the migration of ESCC.
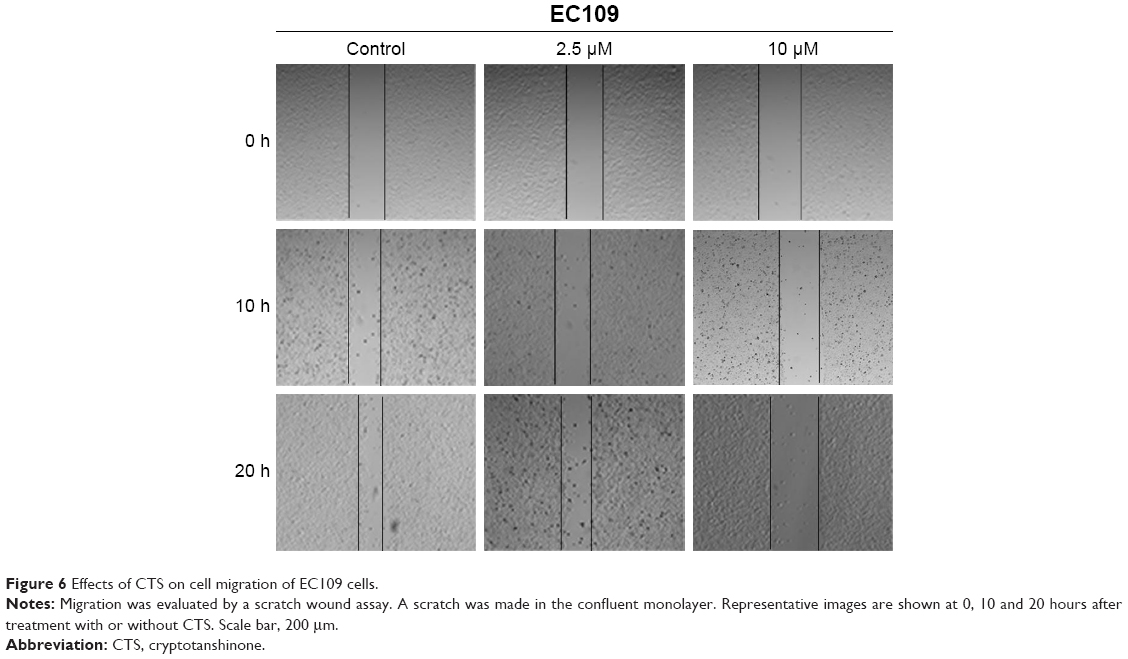 | Figure 6 Effects of CTS on cell migration of EC109 cells. |
CTS inhibits tumor growth in EC109 cell xenograft mice
We examined the anti-tumor activity of CTS in a xenograft model of EC109 cells. As shown in Figure 7A and B, compared to the control group, CTS treatment significantly decreased the tumor volume and tumor weight. In addition, there was no significant influence of CTS on the body weight of the nude mice (Figure 7C).
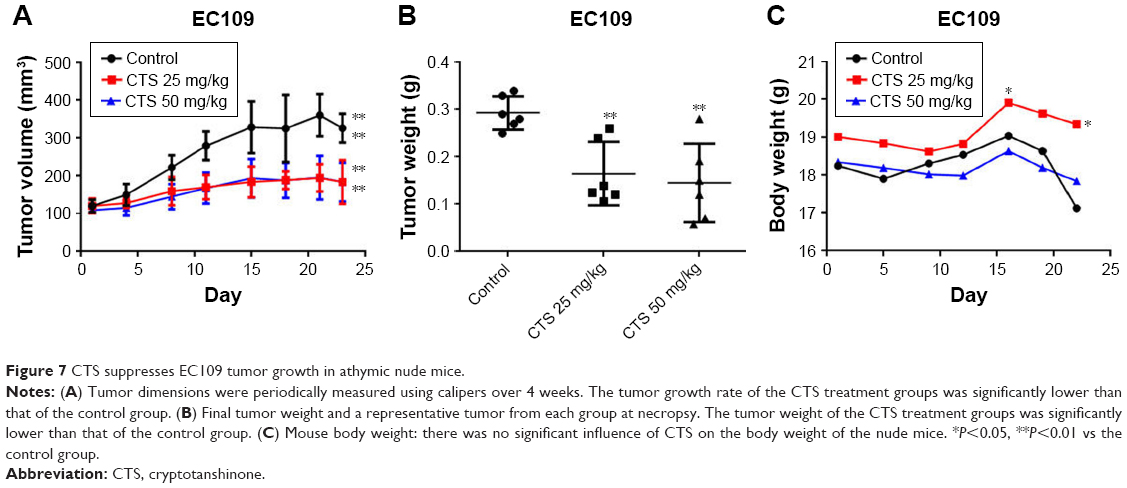 | Figure 7 CTS suppresses EC109 tumor growth in athymic nude mice. |
Immunohistochemical analysis of tumor tissues revealed that CTS obviously decreased the proportion of KI-67-positive cells in tumor tissue (Figure 8A–D). In the cell experiments, we found that CTS promoted the apoptosis of EC109 cells. Immunohistochemical analysis revealed that CTS enhanced the expression of cleaved caspase3 in tumor tissue (Figure S3). Western blot analysis showed that CTS treatment could suppress the protein expression of JAK2, p-JAK2, STAT3 and p-STAT3 (Tyr705) in tumor tissue (Figure 8E and F). Together, these results suggest that CTS showed anti-cancer properties via restriction of the JAK2/STAT3 signaling pathway in vivo, as well as in cell culture systems.
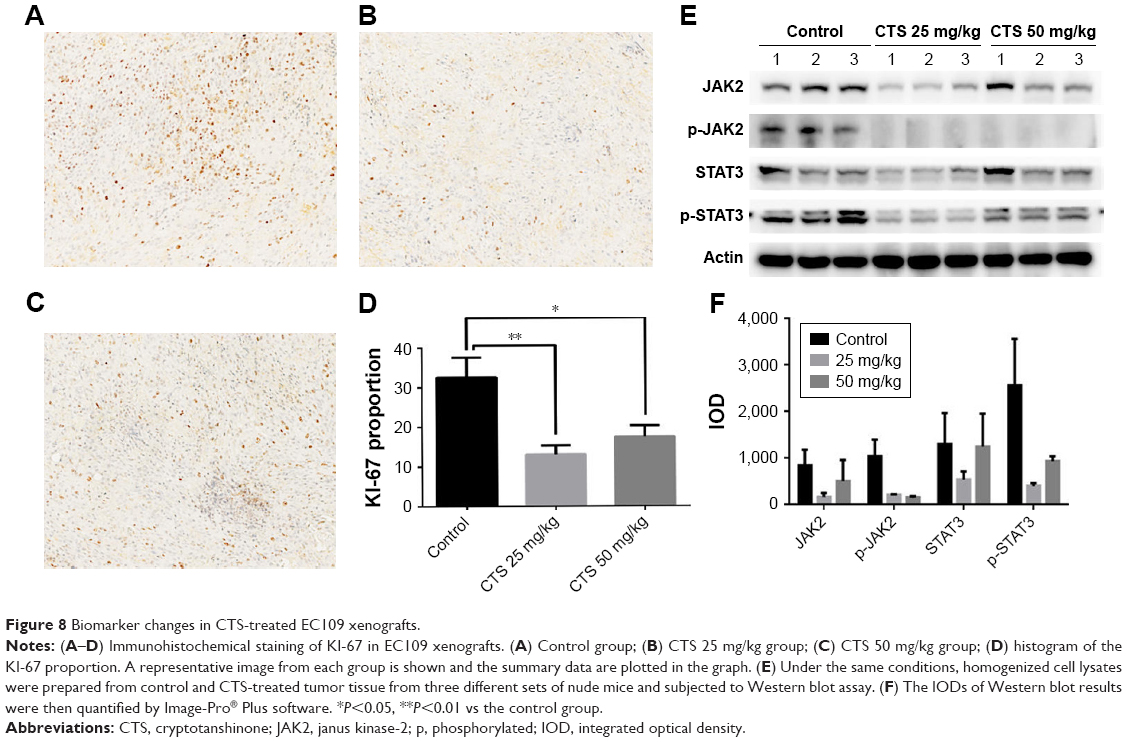 | Figure 8 Biomarker changes in CTS-treated EC109 xenografts. |
Discussion
Esophageal cancer is the eighth most common type of cancer worldwide, affecting more than 450,000 people.23,24 ESCC is the predominant histological type of esophageal carcinoma worldwide. The incidence of esophageal cancer varies greatly by region. The “Asian belt”, which encompasses Turkey, north-eastern Iran, Kazakhstan and northern and central China, has a very high incidence of ESCC, with more than 100 cases per 100,000 population annually. Classic treatments for ESCC include surgical excision and radiotherapy.25 However, owing to invasive growth, incomplete surgical resection and loss of sensitivity to radiation therapy, the 5-year overall survival rate ranges from 30% to 50%, without significant improvement during the past few years and with more than half of patients relapsing within 2–3 years after surgery.5,25 The exploration of more effective therapeutic approaches is therefore necessary for improving the outcome of ESCC treatment. In our research, CTS inhibited the growth of ESCC in vitro and in vivo, suggesting that CTS is a potential compound for the treatment of ESCC.
CTS, the major active constituent isolated from the root of S. miltiorrhiza Bunge, has exhibited significant anti-cancer activity in multiple cancer cells through different mechanisms, such as inhibiting DNA topoisomerase 2 to suppress the growth of PC3 cells,26 inhibiting mTORC1 signaling in Rh30 cells27 and inducing JNK signaling in A549 cells.28 There is growing evidence that CTS inhibits proliferation and growth via the STAT3 signaling pathway in human glioma cells,29 cholangiocarcinoma cells30 and gastric cancer cells,31 among others. Activated STAT3 has been observed in malignant cells and is capable of inducing the expression of a large number of genes involved in tumorigenesis.32 In our research, STAT3 was significantly activated in esophageal cancer cells (Figure 1), which is consistent with research based on clinical samples. This indicates that esophageal cancer may be sensitive to STAT3 inhibitors. Thus, we suggest that CTS could exhibit anti-tumor effects on esophageal cancer by regulating STAT3 activation. Phosphorylation of STAT3 at Tyr705 is a main representative of STAT3 overactivation. We found that CTS dose- and time-dependently inhibits p-STAT3 (Tyr705) protein levels in both CAES17 and EC109 esophageal cancer cell lines.
Activation of STAT3 is associated with a variety of upstream molecules, such as the JAK family of protein tyrosine kinases,6 and various growth factors, such as EGFRs, HGFRs and VEGFRs.33 IL-6 binds to IL-6Rα and the co-receptor gp130 to activate JAK and the phosphorylation of STAT3, which consequently activates inflammatory cascades and oncogenic pathways.34 Our study showed that CTS not only directly inhibits the p-STAT3 level, but also inhibits JAK2-mediated STAT3 phosphorylation during IL-6 stimulation. Regarding the mechanism by which CTS inhibits STAT3 activity, Ge et al found no relation to the activation of JAK235 and Lu et al found that CTS can inhibit IL-6-stimulated STAT3 activation.29 We believe that whether the inhibition of CTS is related to the activation of JAK2 depends on the degree of JAK2 activation in different cells, and further confirmation is needed. CTS has been reported to possess a great anti-proliferation effect on different cancer cells by suppressing STAT3 activation.18,35,36 In the present study, we confirmed that CTS could significantly inhibit the proliferation of CAES17 and EC109 cells in vitro and was more sensitive in EC109 cells, meaning that CTS has better selectivity. The proliferation of cancer cells is related to apoptosis and cell cycle arrest. In HepG2 cells, CTS inhibits cell proliferation by inducing G1 cell arrest.37 CTS inhibits proliferation by inducing apoptosis in renal cell carcinoma.18 Our study suggests that in ESCC, CTS inhibits cell proliferation by inducing apoptosis and shows no effects on the cell cycle.
Xenotransplantation is an effective model for evaluating the effect of compounds on tumor formation and growth, and is widely used in preclinical studies. Tumor size and tumor weight are the most important data.38 Although the in vitro anti-tumor effect of CTS has been reported in various types of tumor, few papers have reported its anti-tumor efficacy in vivo.39 In particular, its in vivo efficacy on esophageal cancer has not been reported. Our results showed that CTS markedly inhibited tumor growth of EC109 cell xenograft mice without affecting the body weight. KI-67 is a meaningful marker for proliferation, and CTS significantly reduced the proportion of KI-67-positive cells in EC109 cell tumors. The results provide further evidence for the proliferative inhibition of CTS. The Western blot results show that CTS could also reduce the phosphorylation levels of JAK2 and STAT3. These results were in accordance with the data from cultured cells.
In summary, our study showed that CTS is a potent growth inhibitor of esophageal carcinoma cells in vitro and in vivo. In future studies, we will select a known STAT3 inhibitor as a comparison to further investigate the inhibitory effect of CTS and structurally similar compounds on the STAT3 pathway. Taking into account the wide acceptance and low toxicity of this plant extract, the results of the present study suggest that it may offer an effective method to treat esophageal cancer by blocking the JAK2/STAT3 signaling pathway, demonstrating the possibility of further developing CTS as an alternative treatment option or an adjuvant chemotherapeutic agent in esophageal cancer treatment.
Conclusion
Our experiments have demonstrated that CTS can inhibit ESCC by inhibiting the JAK/STAT3 signaling pathway, which indicates that CTS can be used as a potential therapeutic agent for esophageal cancer and provides a possibility to target the JAK/STAT3 signaling pathway for the treatment of esophageal cancer.
Acknowledgment
This work was financially supported by the CAMS Innovation Fund for Medical Sciences (grant no 2017-I2M-1-010).
Disclosure
The authors report no conflicts of interest in this work.
References
Fichter CD, Gudernatsch V, Przypadlo CM, et al. ErbB targeting inhibitors repress cell migration of esophageal squamous cell carcinoma and adenocarcinoma cells by distinct signaling pathways. J Mol Med. 2014;92(11):1209–1223. | ||
Torre LA, Bray F, Siegel RL, Ferlay J, Lortet-Tieulent J, Jemal A. Global Cancer statistics, 2012. CA Cancer J Clin. 2015;65(2):87–108. | ||
Li T, Lu ZM, Chen KN, et al. Human papillomavirus type 16 is an important infectious factor in the high incidence of esophageal cancer in Anyang area of China. Carcinogenesis. 2001;22(6):929–934. | ||
Lu CL, Ji Y, Ge D, Guo J, Ding JY. The expression of CXCR4 and its relationship with matrix metalloproteinase-9/vascular endothelial growth factor in esophageal squamous cell cancer. Dis Esophagus. 2011;24(4):283–290. | ||
Pennathur A, Gibson MK, Jobe BA, Luketich JD. Oesophageal carcinoma. The Lancet. 2013;381(9864):400–412. | ||
Yu H, Lee H, Herrmann A, Buettner R, Jove R. Revisiting STAT3 signalling in cancer: new and unexpected biological functions. Nat Rev Cancer. 2014;14(11):736–746. | ||
Li W, Saud SM, Young MR, Colburn NH, Hua B. Cryptotanshinone, a STAT3 inhibitor, suppresses colorectal cancer proliferation and growth in vitro. Mol Cell Biochem. 2015;406(1–2):63–73. | ||
Feng T, Cao W, Shen W, et al. Arctigenin inhibits STAT3 and exhibits anticancer potential in human triple-negative breast cancer therapy. Oncotarget. 2017;8(1):329–344. | ||
Yan H, Guo BY, Zhang S. Cancer-associated fibroblasts attenuate Cisplatin-induced apoptosis in ovarian cancer cells by promoting STAT3 signaling. Biochem Biophys Res Commun. 2016;470(4):947–954. | ||
Alvarez JV, Greulich H, Sellers WR, Meyerson M, Frank DA. Signal transducer and activator of transcription 3 is required for the oncogenic effects of non-small-cell lung cancer-associated mutations of the epidermal growth factor receptor. Cancer Res. 2006;66(6):3162–3168. | ||
Zamo A, Chiarle R, Piva R, et al. Anaplastic lymphoma kinase (ALK) activates STAT3 and protects hematopoietic cells from cell death. Oncogene. 2002;21(7):1038–1047. | ||
Bhat AA, Lu H, Soutto M, et al. Exposure of Barrett’s and esophageal adenocarcinoma cells to bile acids activates EGFR–STAT3 signaling axis via induction of APE1. Oncogene. 2018;37(46):6011–6024. | ||
Zhang Y, Du X-L, Wang C-J, et al. Reciprocal activation between Plk1 and STAT3 contributes to survival and proliferation of esophageal cancer cells. Gastroenterology. 2012;142(3):521–530. | ||
Liu C, Sun HN, Luo YH, et al. Cryptotanshinone induces ROS-mediated apoptosis in human gastric cancer cells. Oncotarget. 2017;8(70):115398–115412. | ||
Tang S, Shen XY, Huang HQ, et al. Cryptotanshinone suppressed inflammatory cytokines secretion in RAW264.7 macrophages through inhibition of the NF-κB and MAPK signaling pathways. Inflammation. 2011;34(2):111–118. | ||
Ma Y, Li H, Yue Z, et al. Cryptotanshinone attenuates cardiac fibrosis via downregulation of COX-2, NOX-2, and Nox-4. J Cardiovasc Pharmacol. 2014;64(1):28–37. | ||
Yu XY, Lin SG, Chen X, et al. Transport of cryptotanshinone, a major active triterpenoid in Salvia miltiorrhiza Bunge widely used in the treatment of stroke and Alzheimer’s disease, across the blood-brain barrier. Curr Drug Metab. 2007;8(4):365–378. | ||
Chen Z, Zhu R, Zheng J, et al. Cryptotanshinone inhibits proliferation yet induces apoptosis by suppressing STAT3 signals in renal cell carcinoma. Oncotarget. 2017;8(30):50023–50033. | ||
Lv G, Sun D, Zhang J, et al. Lx2-32c, a novel semi-synthetic taxane, exerts antitumor activity against prostate cancer cells in vitro and in vivo. Acta Pharm Sin B. 2017;7(1):52–58. | ||
Yang Y, Guan D, Lei L, et al. H6, a novel hederagenin derivative, reverses multidrug resistance in vitro and in vivo. Toxicol Appl Pharmacol. 2018;341:98–105. | ||
Ma YT, Yang Y, Cai P, et al. A series of Enthalpically optimized docetaxel analogues exhibiting enhanced antitumor activity and water solubility. J Nat Prod. 2018;81(3):524–533. | ||
Huang X, Dai S, Dai J, et al. Luteolin decreases invasiveness, deactivates STAT3 signaling, and reverses interleukin-6 induced epithelial-mesenchymal transition and matrix metalloproteinase secretion of pancreatic cancer cells. Onco Targets Ther. 2015;8:2989–3001. | ||
Ferlay J, Shin HR, Bray F, Forman D, Mathers C, Parkin DM. Estimates of worldwide burden of cancer in 2008: GLOBOCAN 2008. Int J Cancer. 2010;127(12):2893–2917. | ||
Lepage C, Rachet B, Jooste V, Faivre J, Coleman MP. Continuing rapid increase in esophageal adenocarcinoma in England and Wales. Am J Gastroenterol. 2008;103(11):2694–2699. | ||
Naveed M, Kubiliun N. Endoscopic treatment of early-stage esophageal cancer. Curr Oncol Rep. 2018;20(9):71. | ||
Kim EJ, Kim SY, Kim SM, Lee M. A novel topoisomerase 2A inhibitor, cryptotanshinone, suppresses the growth of PC3 cells without apparent cytotoxicity. Toxicol Appl Pharmacol. 2017;330:84–92. | ||
Chen W, Pan Y, Wang S, et al. Cryptotanshinone activates AMPK-TSC2 axis leading to inhibition of mTORC1 signaling in cancer cells. BMC Cancer. 2017;17(1):34. | ||
Hao W, Zhang X, Zhao W, et al. Cryptotanshinone induces pro-death autophagy through JNK signaling mediated by reactive oxygen species generation in lung cancer cells. Anticancer Agents Med Chem. 2016;16(5):593–600. | ||
Lu L, Li C, Li D, et al. Cryptotanshinone inhibits human glioma cell proliferation by suppressing STAT3 signaling. Mol Cell Biochem. 2013;381(1–2):273–282. | ||
Ke F, Wang Z, Song X, et al. Cryptotanshinone induces cell cycle arrest and apoptosis through the JAK2/STAT3 and PI3K/Akt/NFκB pathways in cholangiocarcinoma cells. Drug Des Devel Ther. 2017;11:1753–1766. | ||
Wang J, Zhang G, Dai C, et al. Cryptotanshinone potentiates the antitumor effects of doxorubicin on gastric cancer cells via inhibition of STAT3 activity. J Int Med Res. 2017;45(1):220–230. | ||
Morikawa T, Baba Y, Yamauchi M, et al. STAT3 expression, molecular features, inflammation patterns, and prognosis in a database of 724 colorectal cancers. Clin Cancer Res. 2011;17(6):1452–1462. | ||
Debnath B, Xu S, Neamati N. Small molecule inhibitors of signal transducer and activator of transcription 3 (STAT3) protein. J Med Chem. 2012;55(15):6645–6668. | ||
Yin Z, Ma T, Lin Y, et al. IL-6/STAT3 pathway intermediates M1/M2 macrophage polarization during the development of hepatocellular carcinoma. J Cell Biochem. 2018;119(11):9419–9432. | ||
Ge Y, Yang B, Chen Z, Cheng R. Cryptotanshinone suppresses the proliferation and induces the apoptosis of pancreatic cancer cells via the STAT3 signaling pathway. Mol Med Rep. 2015;12(5):7782–7788. | ||
Chen W, Luo Y, Liu L, et al. Cryptotanshinone inhibits cancer cell proliferation by suppressing mammalian target of rapamycin-mediated cyclin D1 expression and RB phosphorylation. Cancer Prev Res. 2010;3(8):1015–1025. | ||
Park IJ, Yang WK, Nam SH, et al. Cryptotanshinone induces G1 cell cycle arrest and autophagic cell death by activating the AMP-activated protein kinase signal pathway in HepG2 hepatoma. Apoptosis. 2014;19(4):615–628. | ||
Chen L, Wang HJ, Xie W, Yao Y, Zhang YS, Wang H. Cryptotanshinone inhibits lung tumorigenesis and induces apoptosis in cancer cells in vitro and in vivo. Mol Med Rep. 2014;9(6):2447–2452. | ||
Li S, Wang H, Hong L, et al. Cryptotanshinone inhibits breast cancer cell growth by suppressing estrogen receptor signaling. Cancer Biol Ther. 2015;16(1):176–184. |
Supplementary materials
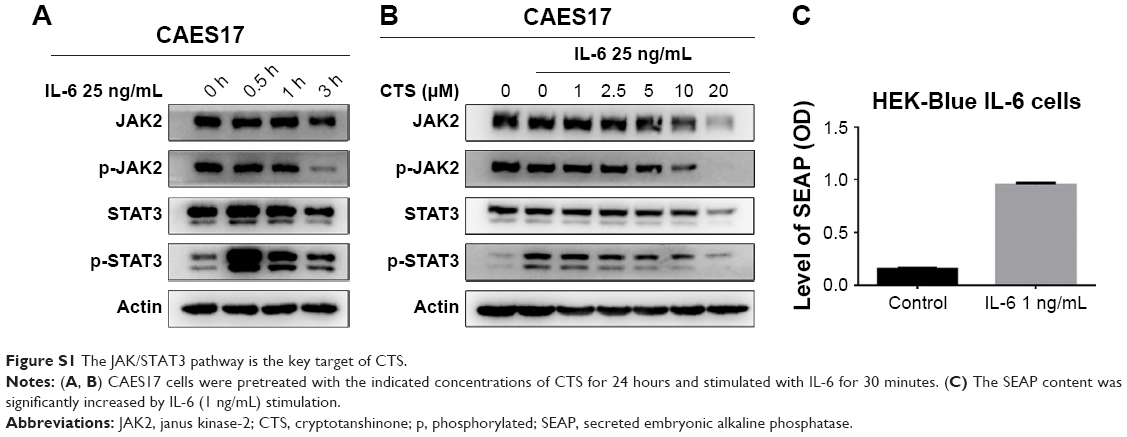 | Figure S1 The JAK/STAT3 pathway is the key target of CTS. |
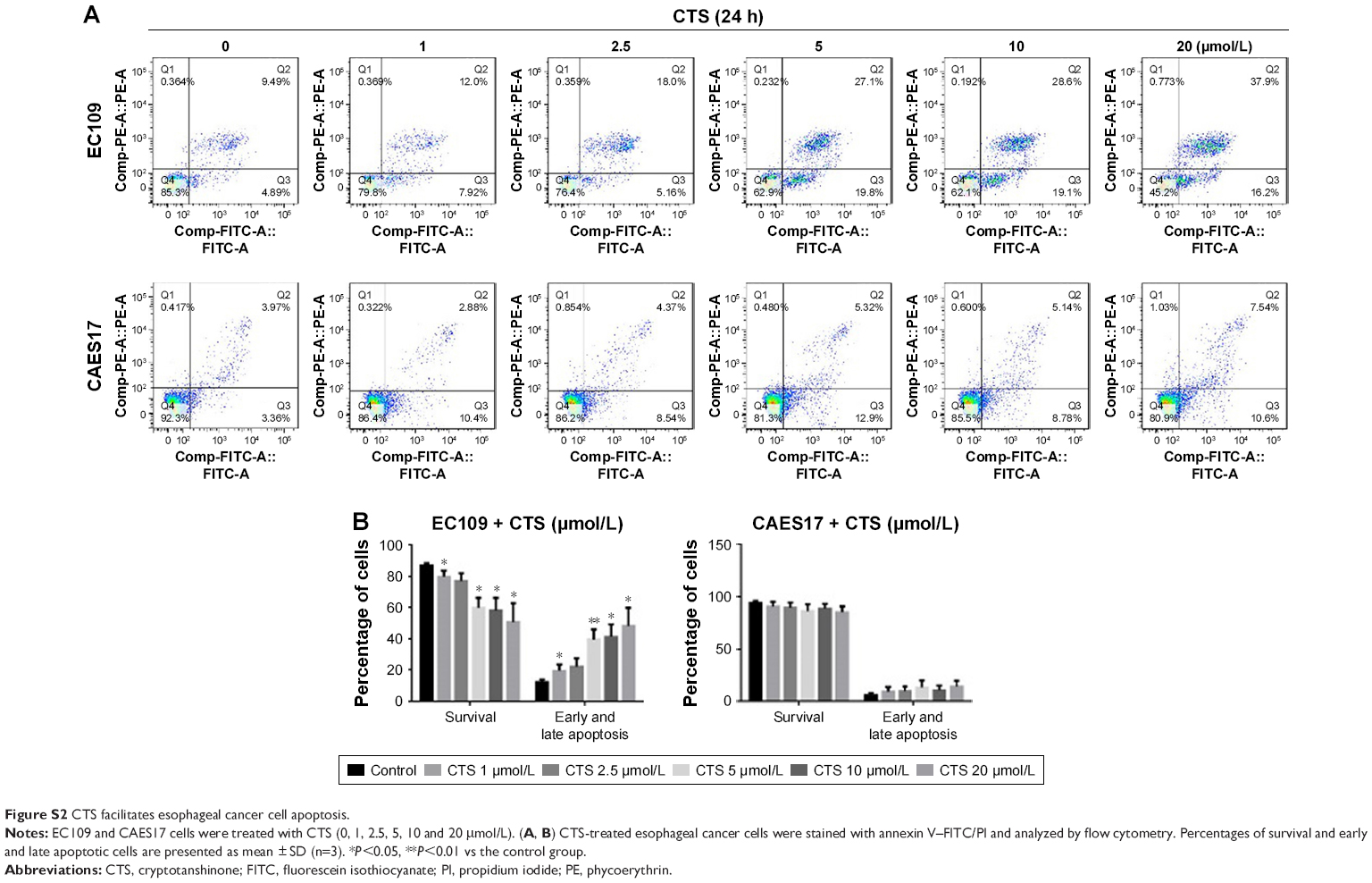 | Figure S2 CTS facilitates esophageal cancer cell apoptosis. |
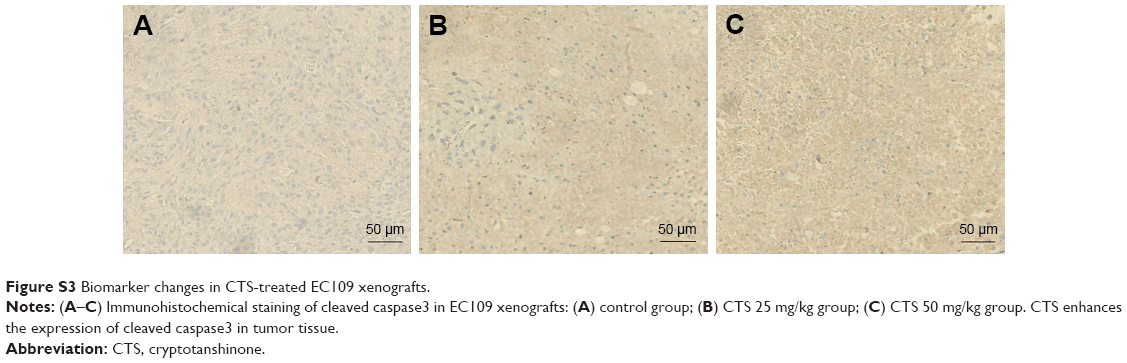 | Figure S3 Biomarker changes in CTS-treated EC109 xenografts. |
 © 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.