Back to Journals » Journal of Inflammation Research » Volume 18
Crosstalk Between Keratinocytes and T Cells in Ulcerative Oral Mucosal Diseases: Mechanisms of Epithelial Dysfunction and Therapeutic Perspectives
Authors Hao Y ![]() , Yuan Y, Yu Y, Liu C, Wang Z, Li Y, Chen Q
, Yuan Y, Yu Y, Liu C, Wang Z, Li Y, Chen Q
Received 27 May 2025
Accepted for publication 30 September 2025
Published 16 October 2025 Volume 2025:18 Pages 14405—14421
DOI https://doi.org/10.2147/JIR.S543082
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Professor Ning Quan
Yilong Hao,1,* Yao Yuan,2,* Yang Yu,1 Chuanxia Liu,1 Zhiyong Wang,1 Yining Li,1 Qianming Chen1
1Stomatology Hospital, School of Stomatology, Zhejiang University School of Medicine, Zhejiang Provincial Clinical Research Center for Oral Diseases, Zhejiang Key Laboratory of Oral Biomedical, Hangzhou, Zhejiang, 310000, People’s Republic of China; 2State Key Laboratory of Oral Diseases, National Clinical Research Center for Oral Diseases, Chinese Academy of Medical Sciences Research Unit of Oral Carcinogenesis and Management, West China Hospital of Stomatology, Sichuan University, Chengdu, Sichuan, 610041, People’s Republic of China
*These authors contributed equally to this work
Correspondence: Yining Li, Email [email protected] Qianming Chen, Email [email protected]
Abstract: Erosive and ulcerative oral mucosal diseases (OMDs) are characterized by persistent inflammation, epithelial barrier disruption, and impaired tissue repair, including oral lichen planus (OLP), discoid lupus erythematosus (DLE), pemphigus vulgaris (PV), mucous membrane pemphigoid (MMP), and recurrent aphthous ulcers (RAU). Aberrant activation of T cells induces cytotoxic and cytokine-mediated injury to the oral mucosa, impairing epithelial stem cell (EpSC) function, damaging the basement membrane, and compromising epithelial regeneration, which eventually results in sustained barrier failure. Under physiological conditions, EpSCs maintain mucosal resilience through continuous self-renewal and rapid turnover. In ulcerative OMDs, however, T cells drive inflammatory signals disrupt these processes. To systematically understand these mechanisms, this review summarizes current evidence on disease specific T cell subsets, cytokine networks, and keratinocyte responses that drive oral epithelial dysfunction. It also highlights emerging therapeutic strategies aimed at restoring epithelial homeostasis by targeting T cell and keratinocyte interactions.
Keywords: epithelial stemness, keratinocytes, oral mucosal diseases, mucosa immunity, T cell
Introduction
Erosive and ulcerative oral mucosal diseases (ulcerative OMDs) are chronic, recurrent disorders. They are characterized by epithelial barrier breakdown, persistent inflammation and pain, which together lead to substantial reduction in quality of life.1 Representative conditions include oral lichen planus (OLP), discoid lupus erythematosus (DLE), pemphigus vulgaris (PV), mucous membrane pemphigoid (MMP), and recurrent aphthous ulcers (RAU). Although their etiologies and effector mechanisms differ, these disorders share clinical features such as painful erosions or ulcerations, repeated relapses, and therapeutic challenges.2–4 A common pathological feature is impaired epithelial regeneration associated with excessive immune activation.1
A central concept of this review is epithelial stemness maintenance. In oral epithelium, the basal keratinocytes function as oral epithelial stem cells (EpSCs). They maintain the capacity to self-renew, generate differentiated progeny, and preserve epithelial architecture and barrier function during both homeostasis and repair.5,6 Stemness is essential for the rapid turnover and effective wound healing that characterize healthy oral mucosa. Recent single-cell transcriptomic and lineage-tracing studies have revealed a discrete stem like compartment within the basal layer that drives mucosal regeneration.7–10 Disruption of this compartment has been linked to impaired healing, chronic ulceration, and a higher risk of malignant transformation in some cases.6,10
Under physiological conditions, EpSCs respond rapidly to injury through proliferation and migration, restoring epithelial stratification. In ulcerative OMDs, immune perturbations impair these regenerative programs, particularly T cell dominated inflammation. Activated T cells and their cytokines promote basal keratinocyte apoptosis, liquefactive degeneration, and adhesion loss (Figure 1). These processes deplete stem like keratinocyte pools and impair re-epithelialization, leading to barrier failure and chronic non-healing lesions.11 For example, in OLP, cytotoxic CD8⁺ T cells directly mediate basal keratinocyte apoptosis, whereas in PV, autoreactive T cells provide B cell help to drive pathogenic autoantibody production, further aggravating epithelial damage. While basal keratinocyte biology in epithelium homeostasis has been described in detail, the ways in which T cell rich inflammatory microenvironments disrupt EpSCs function remain poorly understood.1,2,6,12
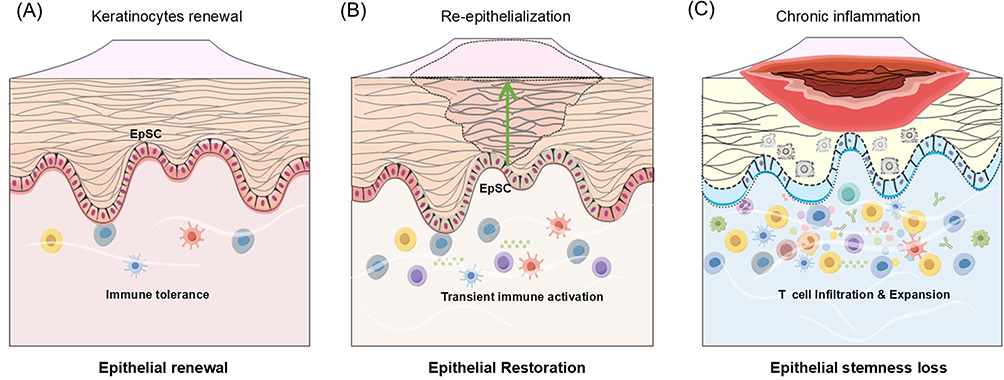 |
Figure 1 Oral epithelial homeostasis, repair, and stemness loss in ulcerative OMDs. (A) Under physiological conditions, EpSCs continuously self-renew and differentiate to sustain epithelial renewal, while resident immune cells maintain immune tolerance. (B) Following injury, EpSCs proliferate and migrate to restore epithelial integrity, supported by transient immune activation that facilitates wound closure and resolution. (C) In erosive/ulcerative oral mucosal diseases, however, persistent T-cell infiltration and chronic inflammation disrupt EpSCs stemness, impair epithelial regeneration, and drive recurrent erosions and barrier breakdown.. |
This review therefore focuses on the role of epithelial stemness in oral mucosal pathology. We discuss how interactions between T cells and keratinocytes disrupt this process and contribute to barrier dysfunction. By drawing on evidence from OLP, DLE, PV, MMP and RAU, we highlight mechanistic insights and discuss therapeutic opportunities aimed at restoring epithelial homeostasis and mucosal immune balance. In particular, targeting immune microenvironmental signals to preserve or restore epithelial stemness emerges as a promising strategy for improving barrier repair and preventing chronic disease progression.
Ulcerative OMDs Show Epithelial Stemness Disruption and Immune Activation
Ulcerative OMDs are associated with impaired epithelial regeneration and excessive immune activation. A key pathological process involves the targeting of basal keratinocytes by infiltrating T cells, disrupting the epithelial progenitor pool that sustains mucosal renewal. These cells serve as the main epithelial progenitors responsible for epithelial renewal, and their destruction is especially prominent in OLP.13,14 This immune mediated injury leads to loss of epithelial stemness, reflected in diminished self-renewal and restricted differentiation capacity of basal keratinocytes. Meanwhile, keratinocyte dysfunction, including apoptosis, abnormal proliferation, premature differentiation, and impaired adhesion, further compromises mucosal barrier integrity.15–17 Loss of stemness contributes to keratinocyte dysfunction, in part through downregulation of p63 and disruption of Wnt/β-catenin signaling. Keratinocyte dysfunction may also arise independently through inflammatory or autoimmune pathways, underscoring the complex interplay between epithelial and immune dysregulation.
Under physiological conditions, EpSCs rapidly proliferate and migrate to injury sites to restore epithelial integrity and rebuild the stratified epithelium.18,19 Compared with skin, the oral mucosa exhibits faster wound healing, which enhances protection against infection. This advantage is supported by oral epithelium specific transcriptional programs that promote rapid repair.6,14,19 Transient immune cell recruitment during normal wound healing can also stimulate keratinocyte proliferation and differentiation, thereby accelerating tissue repair.3,9,17 In ulcerative OMDs, however, chronic immune activation disrupts this regenerative process. Persistent infiltration alters keratinocyte behavior. In OLP, for example, basal cell degeneration and abnormal differentiation impair progenitor function and weaken epithelial protection.2,15,20,21
Dysregulated immune signaling is a major driver of tissue injury in ulcerative OMDs.22,23 Persistent T cell dominated responses and a pathologic cytokine signal directly compromise EpSCs function and reshape cell fate within the inflammatory microenvironment.13,14 In health, the oral mucosa displays minimal inflammation despite constant exposure to environmental stimuli, reflecting a tightly regulated state of immune tolerance.2,17 This controlled equilibrium relies on specialized immune mechanisms that are now being elucidated, offering new insights into mucosal homeostasis and its breakdown in disease.24 Once tolerance is lost, chronic inflammation disrupts epithelial stemness, impairs barrier function, and disturbs keratinocyte differentiation.2,21 This high sensitivity to immune imbalance also helps explain why the oral mucosa is often the first site affected in systemic immune mediated disorders.25 Among the immune populations involved, T cells constitute the dominant subset in both homeostasis and disease, and they play a pivotal role in driving ulcerative OMDs.6,15,17,21,26 Consequently, immunomodulation emerges as a promising therapeutic strategy to rebalance the immune microenvironment and preserve epithelial regenerative capacity.17,25,27 This review focuses on the specific contributions of T cells and their effector molecules to the pathogenesis of ulcerative OMDs and highlights the emerging importance of immune regulation in managing these conditions.
T Cell Activation Disrupts EpSCs Function and Mucosal Homeostasis
T cells are a critical resident immune population in the oral mucosa and represent a major expanded population in various diseased states.6,17 Conventional αβ T cells consist of CD4⁺ Th cells (recognizing peptide MHC II) and CD8⁺ cytotoxic T cells(CTLs) (MHC I restricted). These cells are typically primed in lymphoid organs and then traffic to peripheral tissues such as the oral mucosa to exert effector functions.28 By contrast, γδ T cells form a distinct, tissue-resident lineage at barrier sites, such as oral mucosa, skin. They act in an MHC-unrestricted, innate like manner and provide rapid local defense.29,30
Innate lymphoid cells (ILCs) cooperate with T cells to maintain homeostasis and to regulate inflammation.31,32 ILC1s are located near the basement membrane of the oral mucosa.33–35 Meanwhile, ILC2s are implicated in tissue repair, as seen in skin36 and ILC3s have been found to increase skin thickness in psoriasis and may also have an impact on the oral mucosa.37 Dysregulated ILCs responses may foster chronic oral inflammation by sustaining cytokine production and impairing healing.38
Infiltration and local expansion of T cells are hallmarks of ulcerative OMDs and can act as both initiators and perpetuators of tissue injury. By shaping a pro-inflammatory microenvironment, T cells disrupt epithelial homeostasis, induce keratinocyte death or dysfunction, and deplete basal progenitor pools, which ultimately leading to mucosal breakdown.15,39 This review systematically summarizes the roles of T-cell subsets and associated cytokines in OLP, DLE, PV, MMP, and RAU, and explores the common immunopathological mechanisms shared across these disorders as well as the exploration of clinically therapeutic prospects (Table 1).
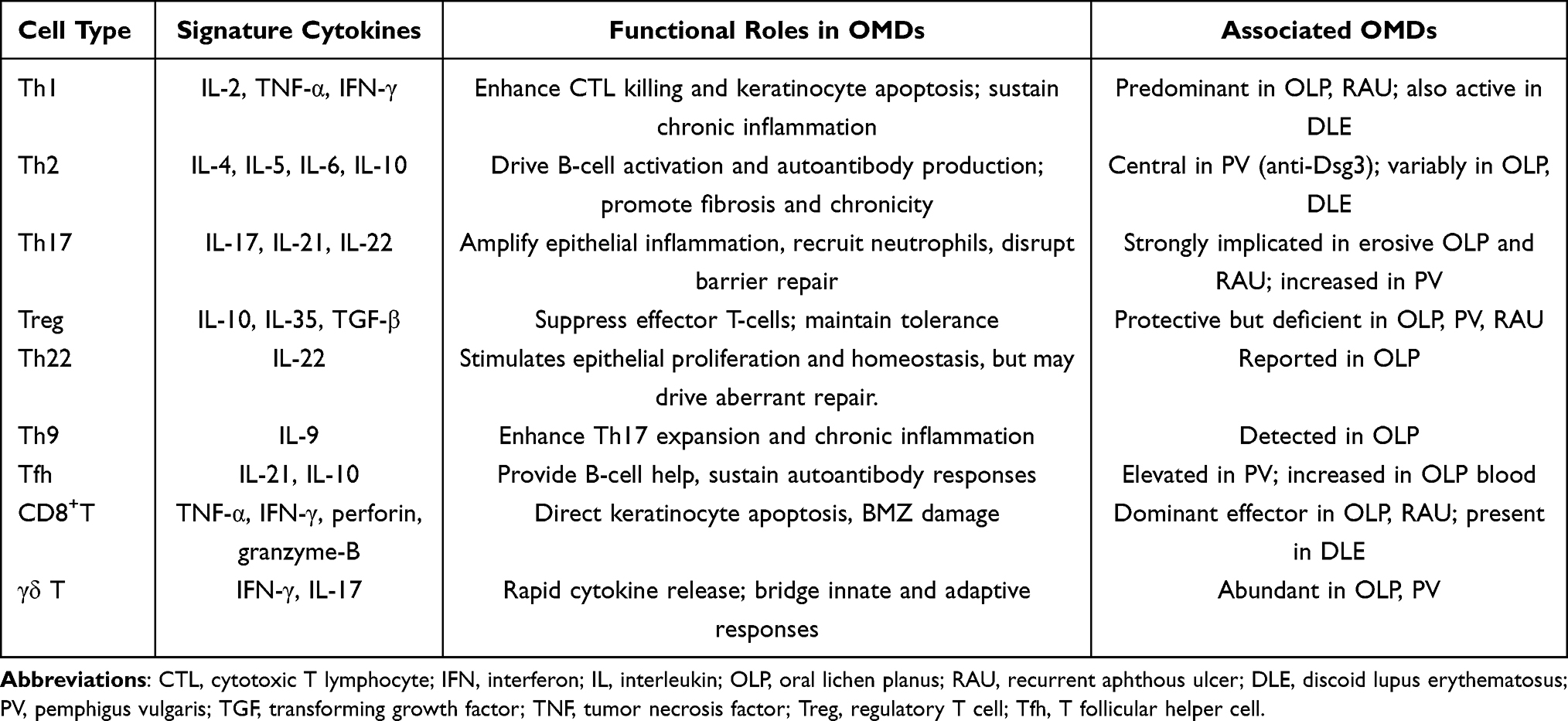 |
Table 1 T Cell Subset Differentiation and Their Lineage-Signature Cytokines in OMD |
T Cell Induced Basal Keratinocyte Liquefaction Degeneration and Disruption of Epithelial Integrity
EpSCs are anchored to the basement membrane zone (BMZ) by hemidesmosomal α6β4 integrin and by β1 integrin mediated adhesion to collagen IV and laminins. These adhesion mechanisms are essential for maintaining stemness, polarity and cell survival.5,40 Basal keratinocytes proliferate within the basal layer and initiate terminal differentiation as they migrate suprabasally.5 In ulcerative OMDs, T cell dominated inflammation damages the BMZ and intercellular junctions, such as desmosomes and adherens junctions. CD8⁺ cytotoxic effectors together with Th1/Th17 cytokines such as IFN‑γ, TNF‑α, and IL‑17 directly stress basal keratinocytes. This stress precipitates vacuolar (liquefactive) degeneration and apoptosis, depleting the basal EpSCs pool and compromising re-epithelialization.15,21,41
Here we distinguish loss of epithelial stemness from general keratinocyte dysfunction across the epithelial lineage. T cell mediated vacuolar degeneration primarily targets the basal compartment. This initiates stemness loss, which then propagates adhesion failure, premature differentiation and impaired re-epithelialization. The following subsections summarize disease specific evidence from OLP and DLE, two interface pattern disorders in which BMZ injury and EpSCs stress are prominent.
Oral Lichen Planus
OLP is a chronic T cell mediated inflammatory disorder recognized by the WHO as an oral potentially malignant disorder (OPMD).42 Clinically, mucosal lesions manifest as reticular or erosive forms that may fluctuate with inflammatory activity.21,43 Histopathology shows basal keratinocyte degeneration, BMZ disruption, and band like subepithelial T cell infiltrates.44 These changes directly compromise EpSCs homeostasis, resulting in impaired regeneration and aberrant differentiation. Basal keratinocytes in OLP exhibit abnormal expression of lineage and adhesion markers, including downregulation of K15 and K19 and upregulation of β1 and α6 integrins.8,45 In active lesions, E-cadherin loss and vimentin upregulation further reduce epithelial cohesion and barrier function, facilitating T cell infiltration across the epithelial stromal interface.13,14,46 These alterations reflect EpSCs stress and contribute to a blurred boundary between the epithelium and lamina propria.
Effector T Cells Drive Epithelial Damage
CD8⁺ T cells dominate the intraepithelial compartment, whereas CD4⁺ T cells are more abundant in the lamina propria.15 Activated CD8⁺ T cells induce keratinocyte apoptosis via Fas/FasL, perforin/ granzyme, and TNF-α pathways,16,43,47 and their infiltration intensifies upon BMZ degradation.15,48 Concurrently, extracellular matrix remodeling in the immune microenvironment leads to the BMZ degradation and epithelial exposure. This loss of anchoring disrupts mechanosensory signaling and niche support, ultimately resulting in the loss of stemness in EpSCs.15,49
CD4⁺ Th cells orchestrate the inflammatory milieu of OLP.50 The classic Th1/Th2 paradigm illustrates how distinct cytokine profiles influence mucosal immunity.47,51 A shift toward Th1 dominance in OLP is associated with chronicity and treatment resistance.52–54 Th1 polarization enhances CD8⁺ T cell cytotoxic activity primarily via IFN-γ and TNF-α, both highly expressed in erosive and ulcerative lesions.15,55–57 IFN-γ not only activates CD8⁺ T cells,43 but also directly modulates keratinocyte behavior by suppressing proliferation and altering EpSCs properties specifically upregulating β1 and α6 integrins, and Nestin, while downregulating E-cadherin thereby weakening epithelial integrity. TNF-α synergistically amplifies cytotoxic signaling, and therapeutic inhibition of TNF-α has shown promise in OLP.58,59 In contrast, the Th2 subset produces IL-4, IL-5, and IL-13. Notably, higher IL-4 levels frequently induced by IL-25 are more common in reticular OLP than in erosive forms, suggesting a role in sustained inflammation and chronicity.53
Beyond the Th1/Th2 paradigm, increasing evidence highlights the Th17 axis as another critical contributor to epithelial injury in OLP. Th17 cytokines, particularly IL-17, stimulate keratinocytes to secrete chemokines and proinflammatory mediators, such as IL-8, TNF-α, and β-defensins, thereby amplifying local inflammation.60,61 IL-23 derived from keratinocytes may further sustain Th17 responses.52,60–62 Elevated IL-17/IL-23 in erosive OLP suggests a predominant Th17 role in ulcerative forms,43,63 whereas Th2 responses appears more closely associated with the reticular form of OLP.
Dysregulated Immune Regulation
Under physiological conditions, Treg cells maintain immune tolerance by suppressing excessive Th1 and Th17 responses. In OLP, however, although Treg cells are numerically increased within lesions,64–66 their immunosuppressive function appears compromised. Defects in TGF-β signaling, which modulates the Th1/Th2 balance by inhibiting IFN-γ and TNF-α, along with reduced production of IL-10 (inhibiting IL-2, IFN-γ, IL-4, and IL-5) and IL-35 (which suppresses Th17 and Th1 development), collectively contribute to a failure in immune regulation and promote disease chronicity.58,67–69 Importantly, Treg cell numbers are generally lower in erosive OLP than in the reticular form,65,66 suggesting that both qualitative and quantitative deficiencies in Treg function may correlate with disease severity.
Immune Complexity and Network Effects
In addition to the classical Th1, Th2, Th17, and Treg subsets, several other T cell populations contribute to OLP pathogenesis. Th22 cells and their cytokine IL-22 are associated with epithelial hyperplasia in OLP,70,71 while Th9 cells and IL-9 may exacerbate inflammation by amplifying Th17 responses.71,72 T follicular helper (Tfh) cells are elevated in OLP and may contribute to pathogenesis through aberrant B-cell activation.73 Nonconventional γδ T cells are also enriched at mucosal sites, where they rapidly produce IFN-γ and IL-17 to amplify local inflammation.74–76 Beyond T cells, ILCs show subset imbalance, increased ILC1s and ILC3s with reduced ILC2s, which may impair tissue repair and perpetuate inflammation.34,35 Collectively, these additional lymphocyte subsets extend the complexity of the immune microenvironment and further compromise epithelial integrity.
In summary, basal EpSCs are central targets of OLP associated inflammation. Liquefactive degeneration reflects both direct cytotoxic injury and EpSCs impairment, leading to compromised epithelial regeneration. Chronic epithelial disruption arises from the combined effects of CD8⁺ cytotoxicity, dysregulated CD4⁺ helper responses, and defective immune regulation, ultimately perpetuating barrier breakdown and lesion persistence (Figure 2).
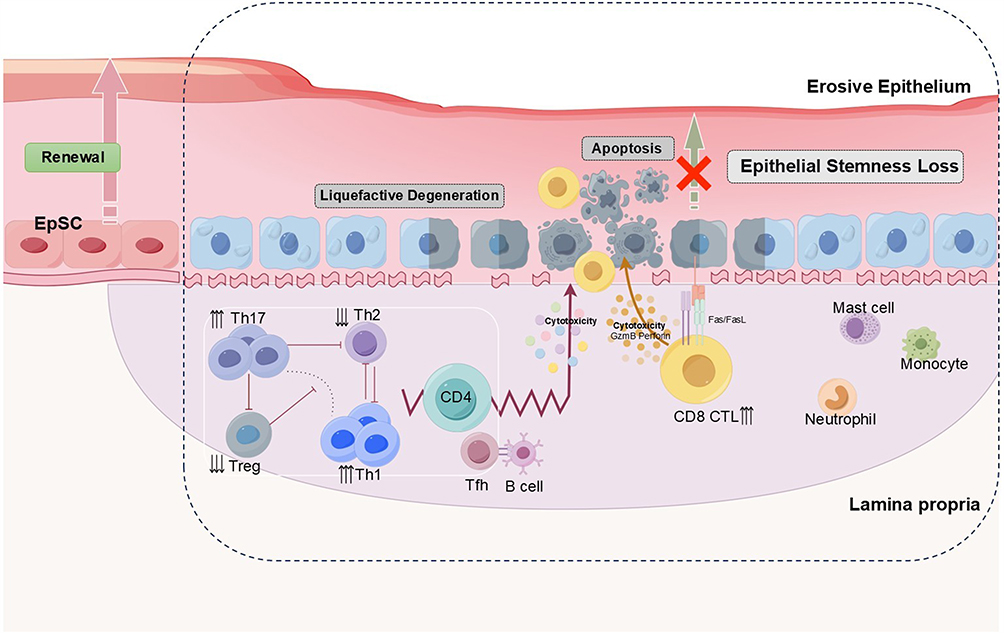 |
Figure 2 Epithelial stemness loss and T cell mediated pathology in OLP. In OLP, infiltrating CD8⁺ CTLs and CD4⁺ subsets, particularly Th1 and Th17, are markedly increased, while Treg and Th2 populations are reduced. CTLs mediate basal keratinocyte death via perforin/granzyme release and Fas–FasL interaction, while Th1/Th17 cytokines (TNF-α, IFN-γ, IL-17, IL-22) further impair keratinocyte function. These processes lead to basal apoptosis, liquefactive degeneration, and progressive epithelial stemness loss, culminating in barrier breakdown and erosive lesions characteristic of OLP. |
Discoid Lupus Erythematosus
DLE is a chronic autoimmune mucocutaneous disease commonly involves sun exposed maxillofacial skin and oral mucosa.77 Oral lesions may accompany cutaneous involvement or present independently. Compared with cutaneous sites, oral DLE tends to be more severe and persistent, leading to significant symptoms, and has been classified by the WHO as an OPMD.25,26,78 Clinically, oral DLE lesions typically present as central erythema or atrophy with radiating white keratotic striae and a raised keratotic rim, often accompanied by peripheral hyperkeratosis and telangiectasia, closely resembling OLP.79 Histopathology shows basal keratinocyte degeneration, epithelial atrophy, BMZ disruption, and chronic inflammatory infiltrates in the lamina propria, together with lupus-specific immunofluorescence findings such as granular deposition of immunoglobulins and complement along the BMZ (“lupus band”).79,80 In some cases, additional features such as follicular plugging and telangiectasia can be observed.81
Keratinocyte Apoptosis and Effector T Cell Attack
Ultraviolet (UV) light is a well-established trigger of lupus flares.82–84 Keratinocytes in DLE exhibit heightened UVB sensitivity, which promotes local inflammation and microvascular changes.85,86 UV exposure also upregulates heat shock proteins, enhancing keratinocyte apoptosis and release of autoantigens.87,88 These apoptotic bodies increase the local antigenic load, fueling immune complex deposition and T cell activation.89–91 T cells represent the predominant infiltrating population in DLE lesions.91,92 While some studies identify CD8⁺ CTLs as the major effector subset,84,93 others report balanced or increased CD4⁺ T cell proportions.81,94 These discrepancies suggest heterogeneity in immune clustering across patients and disease stages. Activated CD8⁺ T cells contribute to basal keratinocyte apoptosis through Fas/FasL, perforin/granzyme, and cytokine mediated mechanisms. CD4⁺ T helper subsets play a central role in modulating the immune response in DLE.95 A Th1 dominant milieu is typically observed in active lesions, characterized by elevated IL-2 and IFN-γ, which reinforce CD8⁺ T cell cytotoxic activity and promote keratinocyte apoptosis.82,84,92 As lesions evolve, a shift toward Th2 responses is observed, marked by IL-4, IL-10, IL-13, and TGF-β, which support B cell activation and plasma cell differentiation.70,92,95 Among these, IL-10 is of particular interest, which is constitutively produced by keratinocytes and further upregulated following UV exposure,96,97 suggesting a dual role in both immune regulation and disease chronicity.
Dysregulated Immune Regulation and Chronic Inflammation
Additional T cell subsets contribute to disease heterogeneity. Reports on Th17 cells are inconsistent: some studies describe increased infiltration compared with normal tissue,98,99 while others show minimal involvement.81,95 Treg cells are more consistently reported to be expanded and persistently recruited within DLE lesions. However, despite their increased numbers, their regulatory function appears inadequate to control Th1 driven inflammation,81,82 indicating a state of functional impairment rather than mere numerical deficiency. The mechanisms underlying chronic T cell recruitment to lesions and the crosstalk among different subsets that lead to loss of immune tolerance remain important topics for future research.
In summary, UV-induced keratinocyte apoptosis and autoantigen release initiate a self-perpetuating immune cycle. Effector T cells, including CD8⁺ cytotoxic and Th1/Th2 polarized CD4⁺ helper cells, disrupt basal EpSCs integrity and adhesion, while defective Treg-mediated regulation allows inflammation to persist. Together, these processes impair epithelial stemness, promote epithelial atrophy, compromise repair, and contribute to the malignant potential of oral DLE lesions.
Loss of Keratinocytes Adhesion Leads to Epithelial Structural Damage
Epithelial integrity relies on robust keratinocyte adhesion. Desmosomes, specialized intercellular junctions, provide strong cell-cell adhesion and act as mechanical “spot welds” between keratinocytes, enabling the epithelium to withstand physical stress and preserve tissue architecture.100,101 Desmogleins (Dsgs) are critical for keratinocyte cohesion and also function as extra desmosomal adhesion receptors in stratified epithelia.102 The expression patterns of Dsg differ by tissue, in oral epithelium, Dsg3 predominates across multiple epithelial layers, whereas in skin, both Dsg1 and Dsg3 are expressed, with Dsg1 largely confined to the suprabasal compartment.103
In addition to cell-cell adhesion, epithelial anchorage to the underlying connective tissue is essential for homeostasis. This attachment is mediated by hemidesmosomes, which connect intermediate filaments of basal keratinocytes to the BMZ. Within hemidesmosomes, plaque proteins such as BP230 link to intracellular keratin filaments, while transmembrane proteins including BP180 (type XVII collagen) and the α6β4 integrin extend across the plasma membrane to bind laminins within the lamina lucida. These laminins further interact with type IV collagen and fibronectin in the lamina densa. The entire structure is ultimately anchored to the subepithelial connective tissue through anchoring fibrils composed of type VII collagen.104 Thus, epithelial structural integrity depends on both desmosome mediated cell-cell adhesion and hemidesmosome mediated epithelial stromal adhesion at the BMZ. Disruption of either system can compromise the barrier function and regenerative capacity of the oral mucosa.
Pemphigus Vulgaris: Impaired Intraepithelial Cohesion
PV is a chronic and potentially life threatening autoimmune blistering disorder that affects the epidermis and oral mucosa. Oral lesions often precede cutaneous involvement and appear at trauma prone sites such as the buccal mucosa, tongue, and palate. The blisters rupture easily, leading to painful erosions or chronic ulcerations. Histologically, PV is characterized by acantholysis, intraepithelial clefting, and discontinuous epithelial layers due to loss of cell-cell adhesion.103,105
Autoantibody Mediated Loss of Keratinocyte Adhesion
The major pathogenic mechanism in PV involves IgG autoantibodies-most notably against Dsg-3 (and Dsg-1 in some cases), which disrupt desmosomal cadherins and impair intercellular adhesion. In mucosal dominant PV, anti-Dsg3 IgG is predominant, mostly of the IgG4 subclass, which does not efficiently fix complement but directly interferes with desmosomal cohesion. This leads to autoantibody-mediated acantholysis and blistering, largely independent of innate inflammation.106 Importantly, basal EpSCs loss in PV is generally secondary to mechanical disruption rather than a primary stemness defect, distinguishing it from interface diseases like OLP and DLE. Barrier breakdown and microbial exposure can trigger secondary inflammation.
Genetic Susceptibility and T Cell Contributions
Genetic factors, particularly HLA class II alleles (eg, DRβ104:02, DQβ105:03), play a crucial role in PV susceptibility.107–109 Antigen presenting cells display Dsg peptides via these alleles, activating autoreactive CD4⁺ T cells, which in turn help B cells produce pathogenic anti-Dsg antibodies. Dendritic cells and autoreactive CD4⁺ T cells therefore bridge genetic risk and humoral autoimmunity.110 Although PV is antibody mediated, T cells are indispensable for disease initiation and maintenance. Evidence from patient studies and murine models shows that Dsg3 specific CD4⁺ T cells support autoantibody production and modulate downstream immune pathways. These cells produce cytokines such as IL-10 and IFN-γ; while IL-10 promotes IgG4 class switching and tolerance, it may also indirectly facilitate autoantibody production, underscoring its dual role.106 CD8⁺ T cells are present at lower frequencies in PV compared with OLP,111 but they can secrete IL-2 and IFN-γ when stimulated with Dsg3, contributing to keratinocyte stress and apoptosis in some contexts.109
Effector T Cell Polarization and Regulatory Imbalance
Active PV typically displays a Th2 biased cytokine environment, with reduced Th1 signals (IL-2, IFN-γ) and elevated Th2 cytokines (IL-4, IL-5, IL-6, IL-10), promoting B cell activation and pathogenic autoantibody production.112–114 Th2 activity correlates with disease severity, and IL-6 often remains elevated in glucocorticoid-refractory patients.108,115 Rituximab efficacy partly reflects depletion of autoreactive Th2 helper signals.102,116 While systemic Th1 signals are comparatively subdued, Dsg3 reactive Th1 cells can induce interface dermatitis in mice,117 indicating context dependent tissue damaging potential. PV also features a Th17/Treg disequilibrium. Increased frequencies of Th17 cells and elevated levels of IL-17 and IL-21 enhance local inflammation and B cell assistance.107,118,119 Conversely, Tregs are consistently reduced in number and exhibit functional impairment,102 including diminished CD28 expression limiting suppressive activity.120–122 Animal studies show that restoring Treg function suppresses anti-Dsg3 antibody formation and ameliorates disease.123,124 Additional subsets such as expanded circulating Tfh and γδ T cells further support IL-21 dependent B cell responses in a subset of patients.125
In summary, PV arises from a dysregulated T cell response, featuring a Th2 and Th17 cytokine response and compromised Treg function. This immune environment promotes sustained autoantibody production by B cells, leading to acantholysis and intraepithelial blistering (Figure 3). Genetic susceptibility enables Dsg3 specific CD4⁺ T cells even in healthy carriers, but clinical disease requires additional breaks in immune regulation. Thus, autoreactive T cells are necessary but insufficient alone; they act in concert with autoantibodies and impaired immune tolerance to drive overt PV.
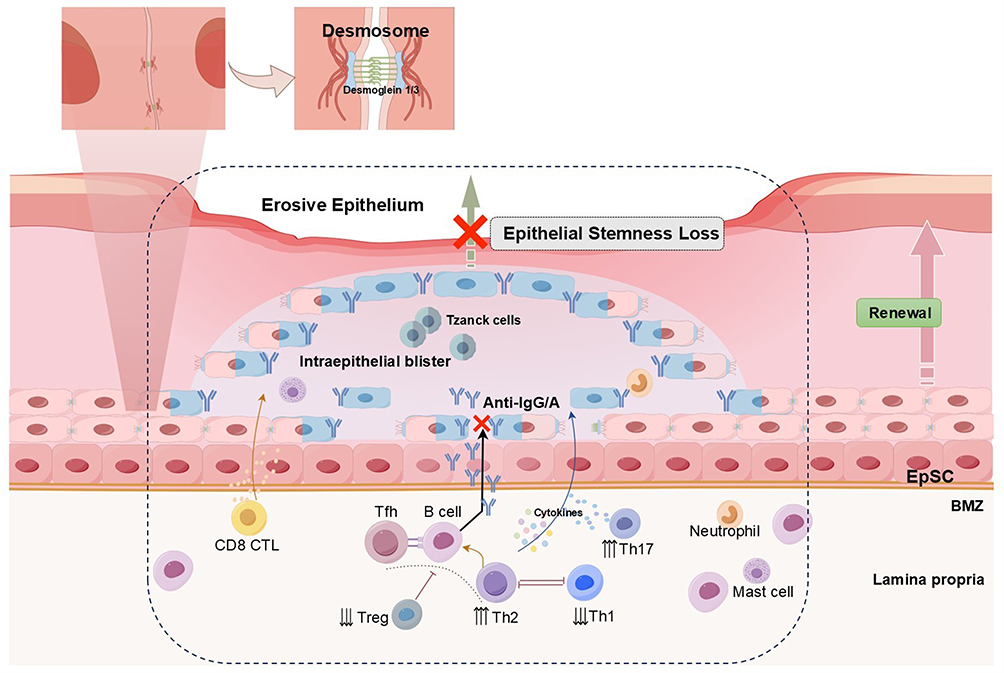 |
Figure 3 Immune dysregulation and epithelial stemness loss in PV. In PV, autoantibodies against Dsg1 and Dsg3 disrupt desmosomes, causing acantholysis, intraepithelial blister formation, and the appearance of Tzanck cells. Th2 associated cytokines (IL-4, IL-10) promote B cell activation and plasma cell differentiation, leading to autoantibody production, while reduced Treg activity impairs immune tolerance. Th1 cytokines (TNF-α, IFN-γ) and CD8⁺ CTLs further drive keratinocyte apoptosis. These processes converge to impair EpSCs renewal and induce epithelial stemness loss, resulting in erosive epithelial pathology. |
Mucous Membrane Pemphigoid: Subepithelial Loss of Adhesion
MMP is a chronic autoimmune blistering disorder that predominantly affects mucosal surfaces, especially the oral cavity, where it often presents as desquamative gingivitis or fragile vesicles that rupture into painful erosions.126 Oral involvement occurs in up to 85% of cases, and gingiva is the most common site.127 Histologically, MMP is characterized by subepithelial blister formation, resulting from autoantibodies targeting the BMZ, accompanied by inflammatory infiltrates in the underlying connective tissue.107
Autoantibody Mediated BMZ Disruption
The central pathogenic mechanism of MMP involves IgG and/or IgA autoantibodies against BMZ components, including BP180, BP230, laminin-332, type VII collagen, and α6β4 integrin.126 BP180 is the main target antigen in MMP and is recognized in about 75% of patients,127 and is essential for hemidesmosome mediated epithelial stromal cohesion.107 Binding of autoantibodies triggers complement activation and deposition along the BMZ, followed by recruitment of neutrophils, eosinophils, and lymphocytes. The inflammatory response in MMP is believed to involve autoantibodies (IgG and/or IgA) attacking antigen sites that connect the epithelium to the lamina propria, preventing the linkage of hemidesmosomes and BMZ. The ensuing inflammation damages hemidesmosomal complexes and anchoring fibrils, leading to epithelial detachment and subepithelial blistering. Disease severity correlates with autoantibody titers and the involvement of specific Ig classes, particularly IgG and IgA.
T Cell Contributions and Immune Amplification
Although MMP is primarily antibody mediated, T cells also play a supportive and regulatory role in disease initiation and progression. Patients with MMP harbor autoreactive CD4⁺ T cells that recognize BP180 epitopes and secrete IFN-γ when stimulated with the NC16A domain.128 These autoreactive T cells provide help to B cells for autoantibody production and likely shape cytokine networks in the local immune microenvironment. Peripheral blood and lesion infiltrates show both CD4⁺ and CD8⁺ T cells, consistent with their contribution to inflammatory amplification at the BMZ. Subsets of Th cells contribute variably: Th1 cytokines (IFN-γ, IL-2) sustain inflammation, while Th2 cytokines (IL-4, IL-5) promote B cell activation and antibody production.129 Evidence for Th17 cells in MMP remains limited, but IL-17 driven neutrophil recruitment may further compromise epithelial integrity in severe cases. HLA associations (eg, DQB1*0301) suggest a genetic predisposition to T cell mediated antigen presentation in MMP, linking adaptive immunity to disease severity.
In summary, MMP exemplifies a subepithelial adhesion disorder, in contrast to PV and interface diseases like OLP/DLE. Autoantibodies against BMZ proteins directly mediate epithelial detachment, while autoreactive T cells support B cell responses and modulate local cytokine networks, sustaining chronic inflammation (Figure 4). The interplay between antibody mediated adhesion loss and T cell driven immune dysregulation underlies the persistence and severity of oral lesions.
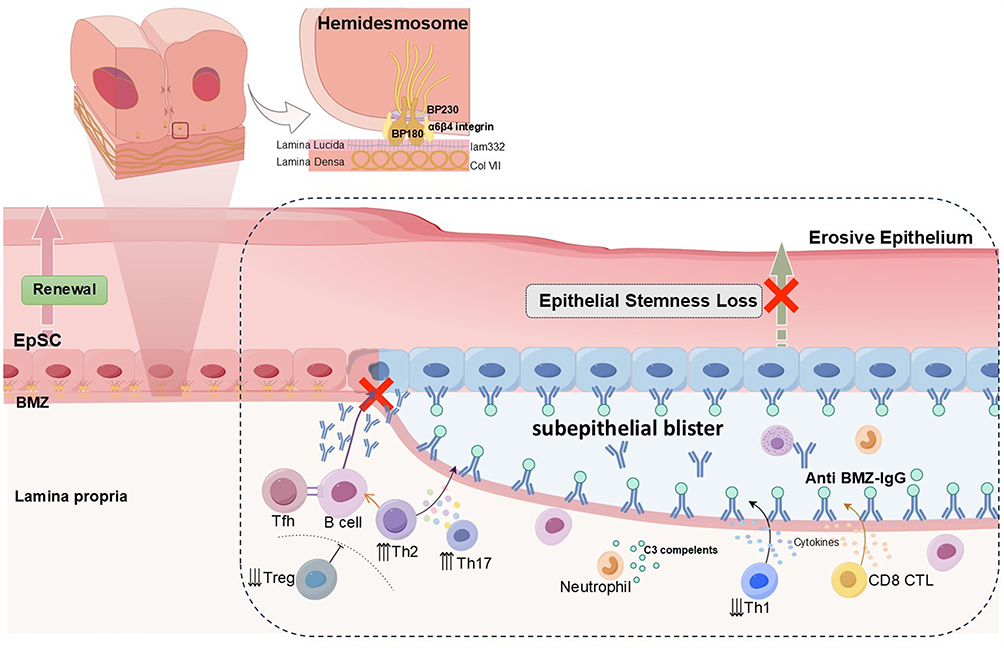 |
Figure 4 Autoantibody-driven basement membrane disruption and epithelial stemness loss in MMP. In MMP, autoantibodies against BMZ proteins, including BP180, BP230, α6β4 integrin, laminin-332, and type VII collagen, disrupt hemidesmosomes and trigger subepithelial blister formation. Th2 and Th17 associated cytokines (IL-4, IL-10, IL-17, IL-22) and Tfh driven B cell activation promote autoantibody production, while reduced Treg and Th1 activity weaken immune regulation. C3 complements activation recruit neutrophils, and CD8⁺ CTL further contribute to basal keratinocyte injury. These immune-mediated processes impair EpSCs renewal, leading to epithelial stemness loss and persistent erosive pathology. |
Non-Specific Epithelial Structural Disruption
Oral epithelial homeostasis depends on slow cycling basal keratinocyte EpSCs that proliferate, differentiate, and migrate upward to replenish supra basal layers. This renewal process ensures barrier continuity, with oral mucosa turnover spanning 14–24 days.5 Persistent immune infiltration disrupts EpSCs renewal, leading to premature apoptosis or aberrant differentiation, which manifests as epithelial thinning, barrier fragility, and impaired wound healing.
Recurrent Aphthous Ulcer
RAU is the most common chronic inflammatory disorder of the oral mucosa, affecting up to 25% of certain populations.130 It is characterized by recurrent, painful, shallow, round or oval ulcers that occur predominantly on non-keratinized oral mucosa, including the buccal mucosa, ventral tongue, labial mucosa, and soft palate.131 Lesions are typically covered by a yellowish fibrinopurulent pseudomembrane, surrounded by an erythematous halo, and heal within 7–14 days, though recurrence is frequent. Histology shows epithelial ulceration with a subjacent mixed inflammatory infiltrate dominated by T lymphocytes.132,133
Immune Dysregulation and Epithelial Damage
Although RAU etiology involves multiple factors-including genetic predisposition, nutritional deficiencies, microbial antigens, and local trauma. A central pathogenic feature is dysregulated cell mediated immunity directed against the oral epithelium. In susceptible individuals, focal infiltration by monocytes and lymphocytes precedes epithelial damage and delayed wound healing.134–136 An imbalance between effector and regulatory immune mechanisms promotes a pro-inflammatory mucosal environment, which disrupts epithelial integrity and impairs regeneration.
T Cell Subsets and Cytokine Profiles
Active RAU lesions are enriched with CD8⁺ CTLs, accompanied by a relative reduction in CD4⁺ T cells.137 CD8⁺ CTLs contribute directly to keratinocyte apoptosis and ulcer formation. This process is reinforced by a Th1 polarized cytokine environment, with elevated levels of IFN-γ, IL-2, and TNF-α enhancing T cell cytotoxicity and epithelial damage. Concurrently, levels of Th2-associated cytokines (IL-4, IL-10, TGF-β) are frequently reduced, further compromising anti-inflammatory regulation.133,138–140 Emerging evidence also supports a role for Th17/Treg imbalance. Elevated IL-17 levels promote neutrophil infiltration and epithelial activation, perpetuating local inflammation.132,135 Meanwhile, Treg cells are often decreased in number or functionally impaired both systemically and within lesions, resulting in inadequate immune suppression and failure to control effector T cell activity.133 Collectively, these alterations sustain a pro-inflammatory state and delay mucosal repair.
In summary, RAU is a nonspecific ulcerative disorder driven primarily by localized immune dysregulation rather than defined autoantibody mediated attack. In contrast to disorders such as OLP and DLE (which feature interface inflammation with basal liquefaction) or PV and MMP (characterized by autoantibody induced loss of epithelial adhesion), RAU is immune pathologically defined by CD8⁺ T cell mediated cytotoxicity and exaggerated Th1/Th17 responses against a background of inadequate Treg mediated regulation141 (Figure 5). It exemplifies immune hyperreactivity that disrupts oral mucosal tolerance and compromises epithelial barrier function.
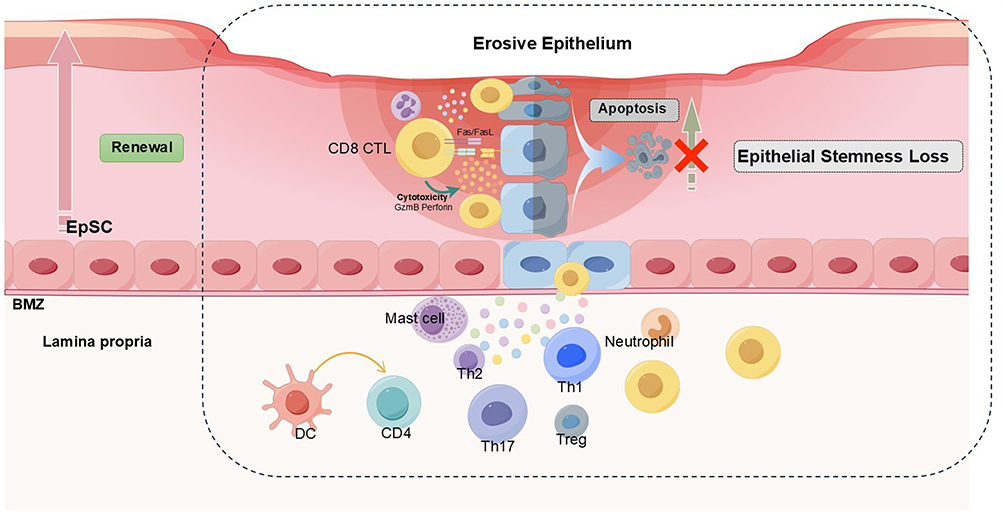 |
Figure 5 Non-specific epithelium structure destruction and epithelial stemness loss in RAU. The pathogenic process in RAU is dominated by CD8⁺ CTLs, which induce basal keratinocyte apoptosis via cytotoxic granules and Fas–FasL signaling. Th1 and Th17 derived cytokines (TNF-α, IFN-γ, IL-17, IL-22) further amplify tissue injury, whereas reduced Treg and Th2 activity weakens immune regulation. Neutrophils and mast cells enhance local inflammation. These combined immune responses impair EpSCs renewal, resulting in stemness loss, barrier breakdown, and recurrent erosions. |
Conclusion
The oral mucosa functions as a critical immune and structural barrier, maintained by basal keratinocyte progenitor/stem cells that drive continuous self-renewal and regeneration. However, its high turnover also makes it vulnerable to immune-mediated injury. In ulcerative OMDs, persistent T cell activation disrupts epithelial integrity through direct cytotoxicity, impaired adhesion, and dysregulated cytokine signaling. These mechanisms compromise basal keratinocyte function, diminish regenerative capacity, and perpetuate barrier failure.
Current therapeutic approaches for ulcerative oral mucosal diseases primarily involve topical or systemic corticosteroids, calcineurin inhibitors, and other broad spectrum immunomodulatory agents. Although these treatments provide symptomatic relief for many patients, their efficacy is often constrained by high relapse rates, adverse effects, and limited specificity for the underlying pathogenic pathways. Recently, targeted biologic agents, such as cytokine inhibitors and JAK–STAT signaling blockers, have shown promising results in early clinical studies, demonstrating potential for improved outcomes in selected patient cohorts (Table 2). A more profound mechanistic understanding of T cell mediated disruption of epithelial progenitor function remains essential for the development of more precise and effective therapeutics.
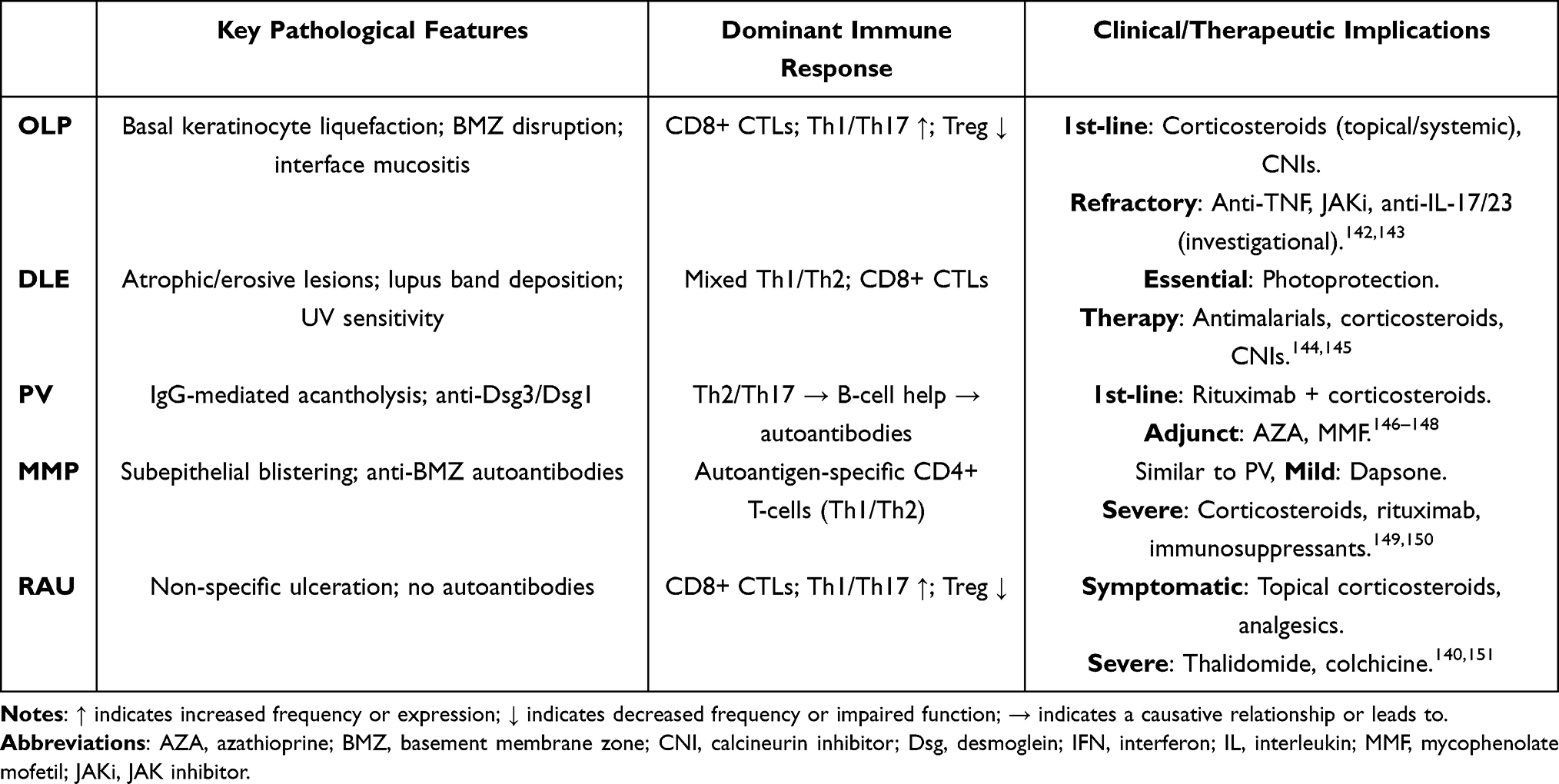 |
Table 2 Clinical and Therapeutic Implications of Ulcerative Oral Mucosal Diseases |
The need for this therapeutic transition is reinforced by mechanistic insights highlighted in this review: chronic T cell activation, skewed Th1/Th17 responses, insufficient Treg function, and downstream disruption of basal keratinocyte stemness are central to epithelial barrier failure. These findings provide a strong biological rationale for expanding the clinical application of targeted therapies that directly modulate T cell epithelial interactions. By integrating mechanistic understanding with therapeutic innovation, future strategies should aim not only to dampen inflammation but also to restore epithelial stemness and regenerative capacity. This approach may hold great promise for achieving durable remission and improving long term outcomes in patients with refractory ulcerative OMDs.
Author Contributions
All authors made a significant contribution to the work reported, whether that is in the conception, study design, execution, acquisition of data, analysis and interpretation, or in all these areas; took part in drafting, revising or critically reviewing the article; gave final approval of the version to be published; have agreed on the journal to which the article has been submitted; and agree to be accountable for all aspects of the work.
Funding
The National Natural Science Foundation of China (NSFC) (Grant Nos 82001047; 32200577; 81902755; 82101007), Zhejiang Provincial Key Research and Development Program (2021C03074) and Zhejiang Provincial Medical Health 360 Major Science Program (WKJ-ZJ-2212), Zhejiang University Global Partnership Fund (No. 188170 and 194452307/004), Department of Science and Technology of Zhejiang Province (No.2024C03193), and the Fundamental Research Funds for the Central Universities (No. 2023QZJH58).
Disclosure
The authors declare that they have no competing interests.
References
1. Lin D, Yang L, Wen L, Lu H, Chen Q, Wang Z. Crosstalk between the oral microbiota, mucosal immunity, and the epithelial barrier regulates oral mucosal disease pathogenesis. Mucosal Immunol. 2021;14(6):1247–1258. doi:10.1038/s41385-021-00413-7
2. Moutsopoulos NM, Moutsopoulos HM. The oral mucosa: a barrier site participating in tissue-specific and systemic immunity. Oral Dis. 2018;24(1–2):22–25. doi:10.1111/odi.12729
3. Moutsopoulos NM, Konkel JE. Tissue-specific immunity at the oral mucosal barrier. Trends Immunol. 2018;39(4):276–287. doi:10.1016/j.it.2017.08.005
4. Akdis CA. Does the epithelial barrier hypothesis explain the increase in allergy, autoimmunity and other chronic conditions? Nat Rev Immunol. 2021;21(11):739–751. doi:10.1038/s41577-021-00538-7
5. Calenic B, Greabu M, Caruntu C, Tanase C, Battino M. Oral keratinocyte stem/progenitor cells: specific markers, molecular signaling pathways and potential uses. Periodontol. 2015;69(1):68–82. doi:10.1111/prd.12097
6. Williams DW, Greenwell-Wild T, Brenchley L, et al. Human oral mucosa cell atlas reveals a stromal-neutrophil axis regulating tissue immunity. Cell. 2021;184(15):4090–4104.e15. doi:10.1016/j.cell.2021.05.013
7. Alonso L, Fuchs E. Stem cells of the skin epithelium. Proc Natl Acad Sci U S A. 2003;100(Suppl 1):11830–11835. doi:10.1073/pnas.1734203100
8. Bose A, Teh MT, Hutchison IL, Wan H, Leigh IM, Waseem A. Two mechanisms regulate keratin K15 expression in keratinocytes: role of PKC/AP-1 and FOXM1 mediated signalling. PLoS One. 2012;7(6):e38599. doi:10.1371/journal.pone.0038599
9. Groeger S, Meyle J. Oral mucosal epithelial cells. Front Immunol. 2019;10:208. doi:10.3389/fimmu.2019.00208
10. Jones KB, Furukawa S, Marangoni P, et al. Quantitative clonal analysis and single-cell transcriptomics reveal division kinetics, hierarchy, and fate of oral epithelial progenitor cells. Cell Stem Cell. 2019;24(1):183–192.e8. doi:10.1016/j.stem.2018.10.015
11. Yuan Y, Park J, Feng A, et al. YAP1/TAZ-TEAD transcriptional networks maintain skin homeostasis by regulating cell proliferation and limiting KLF4 activity. Nat Commun. 2020;11(1):1472. doi:10.1038/s41467-020-15301-0
12. Bouaoud J, Foy JP, Tortereau A, et al. Early changes in the immune microenvironment of oral potentially malignant disorders reveal an unexpected association of M2 macrophages with oral cancer free survival. Oncoimmunology. 2021;10(1):1944554. doi:10.1080/2162402x.2021.1944554
13. Köse O, Lalli A, Kutulola AO, Odell EW, Waseem A. Changes in the expression of stem cell markers in oral lichen planus and hyperkeratotic lesions. J Oral Sci. 2007;49(2):133–139. doi:10.2334/josnusd.49.133
14. Boccellino M, Di Stasio D, Romano A, et al. Lichen planus: molecular pathway and clinical implications in oral disorders. J Biol Regul Homeost Agents. 2018;32(2 Suppl. 1):135–138.
15. El-Howati A, Thornhill MH, Colley HE, Murdoch C. Immune mechanisms in oral lichen planus. Oral Dis. 2023;29(4):1400–1415. doi:10.1111/odi.14142
16. Wang F, Zhang J, Zhou G. 2-Deoxy-D-glucose impedes T cell-induced apoptosis of keratinocytes in oral lichen planus. J Cell Mol Med. 2021;25(21):10257–10267. doi:10.1111/jcmm.16964
17. Gomez-Casado C, Sanchez-Solares J, Izquierdo E, Díaz-Perales A, Barber D, Escribese MM. Oral mucosa as a potential site for diagnosis and treatment of allergic and autoimmune diseases. Foods. 2021;10(5):970. doi:10.3390/foods10050970
18. Castilho RM, Squarize CH, Leelahavanichkul K, Zheng Y, Bugge T, Gutkind JS. Rac1 is required for epithelial stem cell function during dermal and oral mucosal wound healing but not for tissue homeostasis in mice. PLoS One. 2010;5(5):e10503. doi:10.1371/journal.pone.0010503
19. Iglesias-Bartolome R, Uchiyama A, Molinolo AA, et al. Transcriptional signature primes human oral mucosa for rapid wound healing. Sci Transl Med. 2018;10(451). doi:10.1126/scitranslmed.aap8798
20. Schreurs O, Karatsaidis A, Balta MG, Grung B, Hals EKB, Schenck K. Expression of keratins 8, 18, and 19 in epithelia of atrophic oral lichen planus. Eur J Oral Sci. 2020;128(1):7–17. doi:10.1111/eos.12666
21. Schreurs O, Balta MG, Karatsaidis A, Schenck K. Composition of hemidesmosomes in basal keratinocytes of normal buccal mucosa and oral lichen planus. Eur J Oral Sci. 2020;128(5):369–378. doi:10.1111/eos.12732
22. Peskoller M, Bhosale A, Göbel K, et al. How to build and regenerate a functional skin barrier: the adhesive and cell shaping travels of a keratinocyte. J Invest Dermatol. 2022;142(4):1020–1025. doi:10.1016/j.jid.2021.12.034
23. Margadant C, Charafeddine RA, Sonnenberg A. Unique and redundant functions of integrins in the epidermis. FASEB J. 2010;24(11):4133–4152. doi:10.1096/fj.09-151449
24. Silva LM, Doyle AD, Greenwell-Wild T, et al. Fibrin is a critical regulator of neutrophil effector function at the oral mucosal barrier. Science. 2021;374(6575):eabl5450. doi:10.1126/science.abl5450
25. Saccucci M, Di Carlo G, Bossù M, Giovarruscio F, Salucci A, Polimeni A. Autoimmune diseases and their manifestations on oral cavity: diagnosis and clinical management. J Immunol Res. 2018;2018:6061825. doi:10.1155/2018/6061825
26. Salah E. Clinical and dermoscopic spectrum of discoid lupus erythematosus: novel observations from lips and oral mucosa. Int J Dermatol. 2018;57(7):830–836. doi:10.1111/ijd.14015
27. Pelaez-Prestel HF, Sanchez-Trincado JL, Lafuente EM, Reche PA. Immune tolerance in the oral mucosa. Int J Mol Sci. 2021;22(22):12149. doi:10.3390/ijms222212149
28. Wu RQ, Zhang DF, Tu E, Chen QM, Chen W. The mucosal immune system in the oral cavity-an orchestra of T cell diversity. Int J Oral Sci. 2014;6(3):125–132. doi:10.1038/ijos.2014.48
29. Hovav AH, Wilharm A, Barel O, Prinz I. Development and function of γδT cells in the oral mucosa. J Dent Res. 2020;99(5):498–505. doi:10.1177/0022034520908839
30. Nielsen MM, Witherden DA, Havran WL. γδ T cells in homeostasis and host defence of epithelial barrier tissues. Nat Rev Immunol. 2017;17(12):733–745. doi:10.1038/nri.2017.101
31. Zhou S, Li Q, Wu H, Lu Q. The pathogenic role of innate lymphoid cells in autoimmune-related and inflammatory skin diseases. Cell Mol Immunol. 2020;17(4):335–346. doi:10.1038/s41423-020-0399-6
32. Brown JL, Campbell L, Malcolm J, Adrados Planell A, Butcher JP, Culshaw S. Enrichment of innate lymphoid cell populations in gingival tissue. J Dent Res. 2018;97(12):1399–1405. doi:10.1177/0022034518782141
33. Nabekura T, Shibuya A. ILC1: guardians of the oral mucosa against enemy viruses. Immunity. 2021;54(2):196–198. doi:10.1016/j.immuni.2021.01.002
34. Shannon JP, Vrba SM, Reynoso GV, et al. Group 1 innate lymphoid-cell-derived interferon-γ maintains anti-viral vigilance in the mucosal epithelium. Immunity. 2021;54(2):276–290.e5. doi:10.1016/j.immuni.2020.12.004
35. Pan L, Feng M, Chen J, et al. Group 1 and 3 innate lymphoid cells are increased in oral lichen planus and oral lichenoid lesions. Oral Dis. 2023;29(8):3372–3380. doi:10.1111/odi.14384
36. Ebbo M, Crinier A, Vély F, Vivier E. Innate lymphoid cells: major players in inflammatory diseases. Nat Rev Immunol. 2017;17(11):665–678. doi:10.1038/nri.2017.86
37. Teunissen MBM, Munneke JM, Bernink JH, et al. Composition of innate lymphoid cell subsets in the human skin: enrichment of NCR(+) ILC3 in lesional skin and blood of psoriasis patients. J Invest Dermatol. 2014;134(9):2351–2360. doi:10.1038/jid.2014.146
38. Wang ZM, Zhang J, Wang F, Zhou G. The tipped balance of ILC1/ILC2 in peripheral blood of oral lichen planus is related to inflammatory cytokines. Front Cell Dev Biol. 2021;9:725169. doi:10.3389/fcell.2021.725169
39. Şenel S. An overview of physical, microbiological and immune barriers of oral mucosa. Int J Mol Sci. 2021;22(15):7821. doi:10.3390/ijms22157821
40. Ning W, Muroyama A, Li H, Lechler T. Differentiated daughter cells regulate stem cell proliferation and fate through intra-tissue tension. Cell Stem Cell. 2021;28(3):436–452.e5. doi:10.1016/j.stem.2020.11.002
41. Xing C, Duan T, Li L, et al. T(H)17 cells regulate chemokine expression in epithelial cells through C/EBPβ and dictate host sensitivity to colitis and cancer immunity. Sci Adv. 2025;11(31):eads3530. doi:10.1126/sciadv.ads3530
42. Arduino PG, Magliano A, Gambino A, et al. Risk of malignant transformation in 3173 subjects with histopathologically confirmed oral lichen planus: a 33-year cohort study in Northern Italy. Cancers. 2021;13(22):5740. doi:10.3390/cancers13225740
43. Piccinni MP, Lombardelli L, Logiodice F, et al. Potential pathogenetic role of Th17, Th0, and Th2 cells in erosive and reticular oral lichen planus. Oral Dis. 2014;20(2):212–218. doi:10.1111/odi.12094
44. Qing M, Yang D, Shang Q, et al. CD8(+) tissue-resident memory T cells induce oral lichen planus erosion via cytokine network. Elife. 2023;12. doi:10.7554/eLife.83981
45. Bose A, Teh MT, Mackenzie IC, Waseem A. Keratin k15 as a biomarker of epidermal stem cells. Int J Mol Sci. 2013;14(10):19385–19398. doi:10.3390/ijms141019385
46. Liu Y, Liu G, Liu Q, et al. The cellular character of liquefaction degeneration in oral lichen planus and the role of interferon gamma. J Oral Pathol Med. 2017;46(10):1015–1022. doi:10.1111/jop.12595
47. Ding M, Zeng J, Sroussi H, et al. Interactions between Golli-MBP and Th1/Th2 cytokines in patients with oral lichen planus. Oral Dis. 2014;20(2):205–211. doi:10.1111/odi.12090
48. Neppelberg E, Loro LL, Oijordsbakken G, Johannessen AC. Altered CD40 and E-cadherin expression--putative role in oral lichen planus. J Oral Pathol Med. 2007;36(3):153–160. doi:10.1111/j.1600-0714.2007.00511.x
49. Grossmann J. Molecular mechanisms of “detachment-induced apoptosis--Anoikis”. Apoptosis. 2002;7(3):247–260. doi:10.1023/a:1015312119693
50. Van Dyke TE, Serhan CN. Resolution of inflammation: a new paradigm for the pathogenesis of periodontal diseases. J Dent Res. 2003;82(2):82–90. doi:10.1177/154405910308200202
51. Rhodus NL, Cheng B, Ondrey F. Th1/Th2 cytokine ratio in tissue transudates from patients with oral lichen planus. Mediators Inflamm. 2007;2007:19854. doi:10.1155/2007/19854
52. Zhao Z, Wang L, Zhang M, et al. Reveals of quercetin’s therapeutic effects on oral lichen planus based on network pharmacology approach and experimental validation. Sci Rep. 2022;12(1):1162. doi:10.1038/s41598-022-04769-z
53. Wang H, Jiang Y, Wang H, Luo Z, Wang Y, Guan X. IL-25 promotes Th2-type reactions and correlates with disease severity in the pathogenesis of oral lichen planus. Arch Oral Biol. 2019;98:115–121. doi:10.1016/j.archoralbio.2018.11.015
54. Wang L, Wu W, Chen J, Li Y, Xu M, Cai Y. MicroRNA microarray-based identification of involvement of miR-155 and miR-19a in development of Oral Lichen Planus (OLP) by modulating Th1/Th2 balance via targeting eNOS and Toll-Like Receptor 2 (TLR2). Med Sci Monit. 2018;24:3591–3603. doi:10.12659/msm.907497
55. Wang Y, Zhou J, Fu S, Wang C, Zhou B. A study of association between oral lichen planus and immune balance of Th1/Th2 cells. Inflammation. 2015;38(5):1874–1879. doi:10.1007/s10753-015-0167-4
56. Hu JY, Zhang J, Ma JZ, et al. MicroRNA-155-IFN-γ feedback loop in CD4(+)T cells of erosive type oral lichen planus. Sci Rep. 2015;5:16935. doi:10.1038/srep16935
57. Wei W, Sun Q, Deng Y, et al. Mixed and inhomogeneous expression profile of Th1/Th2 related cytokines detected by cytometric bead array in the saliva of patients with oral lichen planus. Oral Surg Oral Med Oral Pathol Oral Radiol. 2018;126(2):142–151. doi:10.1016/j.oooo.2018.02.013
58. Wang H, Zhang D, Han Q, et al. Role of distinct CD4(+) T helper subset in pathogenesis of oral lichen planus. J Oral Pathol Med. 2016;45(6):385–393. doi:10.1111/jop.12405
59. Yarom N. Etanercept for the management of oral lichen planus. Am J Clin Dermatol. 2007;8(2):121. doi:10.2165/00128071-200708020-00010
60. Tan ZY, Bealgey KW, Fang Y, Gong YM, Bao S. Interleukin-23: immunological roles and clinical implications. Int J Biochem Cell Biol. 2009;41(4):733–735. doi:10.1016/j.biocel.2008.04.027
61. Korn T, Bettelli E, Oukka M, Kuchroo VK. IL-17 and Th17 Cells. Annu Rev Immunol. 2009;27:485–517. doi:10.1146/annurev.immunol.021908.132710
62. Lu R, Zeng X, Han Q, et al. Overexpression and selectively regulatory roles of IL-23/IL-17 axis in the lesions of oral lichen planus. Mediators Inflamm. 2014;2014:701094. doi:10.1155/2014/701094
63. Monteiro BV, Pereira Jdos S, Nonaka CF, Godoy GP, da Silveira ÉJ, Miguel MC. Immunoexpression of Th17-related cytokines in oral lichen planus. Appl Immunohistochem Mol Morphol. 2015;23(6):409–415. doi:10.1097/pai.0000000000000096
64. Zhu Y, Li J, Bai Y, et al. Hydroxychloroquine decreases the upregulated frequencies of Tregs in patients with oral lichen planus. Clin Oral Investig. 2014;18(8):1903–1911. doi:10.1007/s00784-013-1176-z
65. Tao XA, Xia J, Chen XB, et al. FOXP3 T regulatory cells in lesions of oral lichen planus correlated with disease activity. Oral Dis. 2010;16(1):76–82. doi:10.1111/j.1601-0825.2009.01608.x
66. Zhang D, Wang J, Li Z, et al. The activation of NF-κB in infiltrated mononuclear cells negatively correlates with treg cell frequency in oral lichen planus. Inflammation. 2015;38(4):1683–1689. doi:10.1007/s10753-015-0145-x
67. Mucida D, Pino-Lagos K, Kim G, et al. Retinoic acid can directly promote TGF-beta-mediated Foxp3(+) Treg cell conversion of naive T cells. Immunity. 2009;30(4):471–472; author reply 472–473. doi:10.1016/j.immuni.2009.03.008
68. Josefowicz SZ, Lu LF, Rudensky AY. Regulatory T cells: mechanisms of differentiation and function. Annu Rev Immunol. 2012;30:531–564. doi:10.1146/annurev.immunol.25.022106.141623
69. Amend A, Wickli N, Schäfer AL, et al. Dual role of interleukin-10 in murine NZB/W F1 lupus. Int J Mol Sci. 2021;22(3):1347. doi:10.3390/ijms22031347
70. Raphael I, Nalawade S, Eagar TN, Forsthuber TG. T cell subsets and their signature cytokines in autoimmune and inflammatory diseases. Cytokine. 2015;74(1):5–17. doi:10.1016/j.cyto.2014.09.011
71. Deng X, Wang Y, Jiang L, Li J, Chen Q. Updates on immunological mechanistic insights and targeting of the oral lichen planus microenvironment. Front Immunol. 2022;13:1023213. doi:10.3389/fimmu.2022.1023213
72. Wang H, Bai J, Luo Z, Fu J, Wang H, Sun Z. Overexpression and varied clinical significance of Th9 versus Th17 cells in distinct subtypes of oral lichen planus. Arch Oral Biol. 2017;80:110–116. doi:10.1016/j.archoralbio.2017.04.003
73. Tan YQ, Li Q, Zhang J, Du GF, Lu R, Zhou G. Increased circulating CXCR5(+) CD4(+) T follicular helper-like cells in oral lichen planus. J Oral Pathol Med. 2017;46(9):803–809. doi:10.1111/jop.12550
74. Fan X, Rudensky AY. Hallmarks of Tissue-Resident Lymphocytes. Cell. 2016;164(6):1198–1211. doi:10.1016/j.cell.2016.02.048
75. Yang JY, Wang F, Zhou G. Characterization and function of circulating mucosal-associated invariant T cells and γδT cells in oral lichen planus. J Oral Pathol Med. 2022;51(1):74–85. doi:10.1111/jop.13250
76. Vičić M, Hlača N, Kaštelan M, Brajac I, Sotošek V, Prpić Massari L. Comprehensive Insight into Lichen Planus Immunopathogenesis. Int J Mol Sci. 2023;24(3):3038. doi:10.3390/ijms24033038
77. Gao X, Ma L, Zhou Z, Jian X, Liu W. Podoplanin expression is correlated with the progression of chronic discoid lupus erythematosus to lip squamous cell carcinoma. Int J Surg Pathol. 2016;24(7):595–599. doi:10.1177/1066896916652220
78. Nico MM, Bologna SB, Lourenço SV. The lip in lupus erythematosus. Clin Exp Dermatol. 2014;39(5):563–569. doi:10.1111/ced.12368
79. Simões DM, Fava M, Figueiredo MA, Salum FG, Cherubini K. Oral manifestations of lupus erythematosus - report of two cases. Gerodontology. 2013;30(4):303–308. doi:10.1111/j.1741-2358.2012.00686.x
80. Andreadis D, Pavlou A, Vakirlis E, et al. Actinic cheilitis may resemble oral lichenoid-type lesions or discoid lupus erythematosus. Arch Dermatol Res. 2021;313(10):891–892. doi:10.1007/s00403-021-02194-2
81. Solé C, Gimenez-Barcons M, Ferrer B, Ordi-Ros J, Cortés-Hernández J. Microarray study reveals a transforming growth factor-β-dependent mechanism of fibrosis in discoid lupus erythematosus. Br J Dermatol. 2016;175(2):302–313. doi:10.1111/bjd.14539
82. Chen AC, Halliday GM, Damian DL. Non-melanoma skin cancer: carcinogenesis and chemoprevention. Pathology. 2013;45(3):331–341. doi:10.1097/PAT.0b013e32835f515c
83. Zaalberg A, Moradi Tuchayi S, Ameri AH, et al. Chronic inflammation promotes skin carcinogenesis in cancer-prone discoid lupus erythematosus. J Invest Dermatol. 2019;139(1):62–70. doi:10.1016/j.jid.2018.06.185
84. Xie Y, Jinnin M, Zhang X, et al. Immunohistochemical characterization of the cellular infiltrate in discoid lupus erythematosus. Biosci Trends. 2011;5(2):83–88. doi:10.5582/bst.2011.v5.2.83
85. Warnakulasuriya S. Clinical features and presentation of oral potentially malignant disorders. Oral Surg Oral Med Oral Pathol Oral Radiol. 2018;125(6):582–590. doi:10.1016/j.oooo.2018.03.011
86. Furukawa F, Itoh T, Wakita H, et al. Keratinocytes from patients with lupus erythematosus show enhanced cytotoxicity to ultraviolet radiation and to antibody-mediated cytotoxicity. Clin Exp Immunol. 1999;118(1):164–170. doi:10.1046/j.1365-2249.1999.01026.x
87. Domingo S, Solé C, Moliné T, Ferrer B, Ordi-Ros J, Cortés-Hernández J. Efficacy of thalidomide in discoid lupus erythematosus: insights into the molecular mechanisms. Dermatology. 2020;236(5):467–476. doi:10.1159/000508672
88. Donnelly AM, Halbert AR, Rohr JB. Discoid lupus erythematosus. Australas J Dermatol. 1995;36(1):3–10; quiz 11–2. doi:10.1111/j.1440-0960.1995.tb00916.x
89. Garelli CJ, Refat MA, Nanaware PP, Ramirez-Ortiz ZG, Rashighi M, Richmond JM. Current insights in cutaneous lupus erythematosus immunopathogenesis. Front Immunol. 2020;11:1353. doi:10.3389/fimmu.2020.01353
90. Kuhn A, Wenzel J, Weyd H. Photosensitivity, apoptosis, and cytokines in the pathogenesis of lupus erythematosus: a critical review. Clin Rev Allergy Immunol. 2014;47(2):148–162. doi:10.1007/s12016-013-8403-x
91. O’Brien JC, Hosler GA, Chong BF. Changes in T cell and B cell composition in discoid lupus erythematosus skin at different stages. J Dermatol Sci. 2017;85(3):247–249. doi:10.1016/j.jdermsci.2016.12.004
92. Coias J, Marzuka A, Hosler GA, Chong BF. T-cell polarization differs in various stages of discoid lupus erythematosus skin. Br J Dermatol. 2020;182(5):1291–1293. doi:10.1111/bjd.18704
93. Thorpe RB, Gray A, Kumar KR, Susa JS, Chong BF. Site-specific analysis of inflammatory markers in discoid lupus erythematosus skin. Sci World J. 2014;2014:925805. doi:10.1155/2014/925805
94. Wenzel J, Uerlich M, Wörrenkämper E, Freutel S, Bieber T, Tüting T. Scarring skin lesions of discoid lupus erythematosus are characterized by high numbers of skin-homing cytotoxic lymphocytes associated with strong expression of the type I interferon-induced protein MxA. Br J Dermatol. 2005;153(5):1011–1015. doi:10.1111/j.1365-2133.2005.06784.x
95. Jabbari A, Suárez-Fariñas M, Fuentes-Duculan J, et al. Dominant Th1 and minimal Th17 skewing in discoid lupus revealed by transcriptomic comparison with psoriasis. J Invest Dermatol. 2014;134(1):87–95. doi:10.1038/jid.2013.269
96. Wang R, Zhang X, Wang S. Differential genotypes of TNF-α and IL-10 for immunological diagnosis in discoid lupus erythematosus and oral lichen planus: a narrative review. Front Immunol. 2022;13:967281. doi:10.3389/fimmu.2022.967281
97. Grewe M, Gyufko K, Krutmann J. Interleukin-10 production by cultured human keratinocytes: regulation by ultraviolet B and ultraviolet A1 radiation. J Invest Dermatol. 1995;104(1):3–6. doi:10.1111/1523-1747.ep12613446
98. Tanasescu C, Balanescu E, Balanescu P, et al. IL-17 in cutaneous lupus erythematosus. Eur J Intern Med. 2010;21(3):202–207. doi:10.1016/j.ejim.2010.03.004
99. Shahidi Dadras M, Rakhshan A, Dadkhahfar S, Barat T. The role of interleukin-17 (IL-17) in the pathogenesis of discoid lupus erythematosus and lichen planopilaris: is immunohistochemistry for IL-17 a promising way to differentiate these entities? Int J Dermatol. 2022;61(6):647–652. doi:10.1111/ijd.15885
100. Schmidt E, Kasperkiewicz M, Joly P. Pemphigus. Lancet. 2019;394(10201):882–894. doi:10.1016/s0140-6736(19)31778-7
101. Ebert LM, Tan LY, Johan MZ, et al. A non-canonical role for desmoglein-2 in endothelial cells: implications for neoangiogenesis. Angiogenesis. 2016;19(4):463–486. doi:10.1007/s10456-016-9520-y
102. Di Zenzo G, Amber KT, Sayar BS, Müller EJ, Borradori L. Immune response in pemphigus and beyond: progresses and emerging concepts. Semin Immunopathol. 2016;38(1):57–74. doi:10.1007/s00281-015-0541-1
103. Bystryn JC, Rudolph JL. Pemphigus. Lancet. 2005;366(9479):61–73. doi:10.1016/s0140-6736(05)66829-8
104. Opelka B, Schmidt E, Goletz S. Type XVII collagen: relevance of distinct epitopes, complement-independent effects, and association with neurological disorders in pemphigoid disorders. Front Immunol. 2022;13:948108. doi:10.3389/fimmu.2022.948108
105. Egu DT, Sigmund AM, Schmidt E, Spindler V, Walter E, Waschke J. A new ex vivo human oral mucosa model reveals that p38MAPK inhibition is not effective in preventing autoantibody-induced mucosal blistering in pemphigus. Br J Dermatol. 2020;182(4):987–994. doi:10.1111/bjd.18237
106. Kasperkiewicz M, Ellebrecht CT, Takahashi H, et al. Pemphigus. Nat Rev Dis Primers. 2017;3:17026. doi:10.1038/nrdp.2017.26
107. Fang H, Li Q, Wang G. The role of T cells in pemphigus vulgaris and bullous pemphigoid. Autoimmun Rev. 2020;19(11):102661. doi:10.1016/j.autrev.2020.102661
108. Amber KT, Staropoli P, Shiman MI, Elgart GW, Hertl M. Autoreactive T cells in the immune pathogenesis of pemphigus vulgaris. Exp Dermatol. 2013;22(11):699–704. doi:10.1111/exd.12229
109. Hertl M, Veldman C. T-cellular autoimmunity against desmogleins in pemphigus, an autoantibody-mediated bullous disorder of the skin. Autoimmun Rev. 2003;2(5):278–283. doi:10.1016/s1568-9972(03)00035-1
110. Lee AY, Kim T, Kim JH. Understanding CD4(+) T cells in autoimmune bullous diseases. Front Immunol. 2023;14:1161927. doi:10.3389/fimmu.2023.1161927
111. Schinner J, Cunha T, Mayer JU, et al. Skin-infiltrating T cells display distinct inflammatory signatures in lichen planus, bullous pemphigoid and pemphigus vulgaris. Front Immunol. 2023;14:1203776. doi:10.3389/fimmu.2023.1203776
112. Takahashi H, Amagai M, Tanikawa A, et al. T helper type 2-biased natural killer cell phenotype in patients with pemphigus vulgaris. J Invest Dermatol. 2007;127(2):324–330. doi:10.1038/sj.jid.5700527
113. Holstein J, Solimani F, Baum C, et al. Immunophenotyping in pemphigus reveals a T(H)17/T(FH)17 cell-dominated immune response promoting desmoglein1/3-specific autoantibody production. J Allergy Clin Immunol. 2021;147(6):2358–2369. doi:10.1016/j.jaci.2020.11.008
114. Chriguer RS, Roselino AM, de Castro M. Glucocorticoid sensitivity and proinflammatory cytokines pattern in pemphigus. J Clin Immunol. 2012;32(4):786–793. doi:10.1007/s10875-012-9679-y
115. Mortazavi H, Babaeijandaghi F, Akbarzadeh M, et al. The influence of systemic therapy on the serum levels of IL-6 and IL-8 in pemphigus vulgaris. J Eur Acad Dermatol Venereol. 2013;27(3):387–390. doi:10.1111/j.1468-3083.2011.04319.x
116. Zhu H, Chen Y, Zhou Y, Wang Y, Zheng J, Pan M. Cognate Th2-B cell interaction is essential for the autoantibody production in pemphigus vulgaris. J Clin Immunol. 2012;32(1):114–123. doi:10.1007/s10875-011-9597-4
117. Takahashi H, Kouno M, Nagao K, et al. Desmoglein 3-specific CD4+ T cells induce pemphigus vulgaris and interface dermatitis in mice. J Clin Invest. 2011;121(9):3677–3688. doi:10.1172/jci57379
118. Timoteo RP, da Silva MV, Miguel CB, et al. Th1/Th17-related cytokines and chemokines and their implications in the pathogenesis of pemphigus vulgaris. Mediators Inflamm. 2017;2017:7151285. doi:10.1155/2017/7151285
119. Hennerici T, Pollmann R, Schmidt T, et al. Increased frequency of T follicular helper cells and elevated interleukin-27 plasma levels in patients with pemphigus. PLoS One. 2016;11(2):e0148919. doi:10.1371/journal.pone.0148919
120. Asothai R, Anand V, Das D, et al. Distinctive Treg associated CCR4-CCL22 expression profile with altered frequency of Th17/Treg cell in the immunopathogenesis of Pemphigus Vulgaris. Immunobiology. 2015;220(10):1129–1135. doi:10.1016/j.imbio.2015.06.008
121. Xu RC, Zhu HQ, Li WP, et al. The imbalance of Th17 and regulatory T cells in pemphigus patients. Eur J Dermatol. 2013;23(6):795–802. doi:10.1684/ejd.2013.2177
122. Alecu M, Ursaciuc C, Surcel M, Coman G, Ciotaru D, Dobre M. CD28 T-cell costimulatory molecule expression in pemphigus vulgaris. J Eur Acad Dermatol Venereol. 2009;23(3):288–291. doi:10.1111/j.1468-3083.2008.03035.x
123. Yokoyama T, Matsuda S, Takae Y, et al. Antigen-independent development of Foxp3+ regulatory T cells suppressing autoantibody production in experimental pemphigus vulgaris. Int Immunol. 2011;23(6):365–373. doi:10.1093/intimm/dxr020
124. Schmidt T, Willenborg S, Hünig T, et al. Induction of T regulatory cells by the superagonistic anti-CD28 antibody D665 leads to decreased pathogenic IgG autoantibodies against desmoglein 3 in a HLA-transgenic mouse model of pemphigus vulgaris. Exp Dermatol. 2016;25(4):293–298. doi:10.1111/exd.12919
125. Das D, Anand V, Khandpur S, Sharma VK, Sharma A. T helper type 1 polarizing γδ T cells and Scavenger receptors contribute to the pathogenesis of Pemphigus vulgaris. Immunology. 2018;153(1):97–104. doi:10.1111/imm.12814
126. Schmidt E, Zillikens D. Pemphigoid diseases. Lancet. 2013;381(9863):320–332. doi:10.1016/s0140-6736(12)61140-4
127. Du G, Patzelt S, van Beek N, Schmidt E. Mucous membrane pemphigoid. Autoimmun Rev. 2022;21(4):103036. doi:10.1016/j.autrev.2022.103036
128. Black AP, Seneviratne SL, Jones L, et al. Rapid effector function of circulating NC16A-specific T cells in individuals with mucous membrane pemphigoid. Br J Dermatol. 2004;151(6):1160–1164. doi:10.1111/j.1365-2133.2004.06219.x
129. Black AP, Wojnarowska F, Ogg GS. Role of T cells in the pathogenesis of mucous membrane pemphigoid. Expert Rev Dermatol. 2006;1:25–30. doi:10.1586/17469872.1.1.25
130. Deng Y, Yao Y, Du G, Liu W. Changes in Th1/Th2-related cytokine expression in the saliva of patients with recurrent aphthous stomatitis before and after prednisone treatment. Clin Oral Investig. 2022;26(1):1089–1093. doi:10.1007/s00784-021-04349-x
131. Lewkowicz N, Lewkowicz P, Dzitko K, et al. Dysfunction of CD4+CD25high T regulatory cells in patients with recurrent aphthous stomatitis. J Oral Pathol Med. 2008;37(8):454–461. doi:10.1111/j.1600-0714.2008.00661.x
132. Lewkowicz N, Kur B, Kurnatowska A, Tchorzewski H, Lewkowicz P. Expression of Th1/Th2/Th3/Th17-related genes in recurrent aphthous ulcers. Arch Immunol Ther Exp. 2011;59(5):399–406. doi:10.1007/s00005-011-0134-1
133. Lewkowicz N, Lewkowicz P, Banasik M, Kurnatowska A, Tchórzewski H. Predominance of Type 1 cytokines and decreased number of CD4(+)CD25(+high) T regulatory cells in peripheral blood of patients with recurrent aphthous ulcerations. Immunol Lett. 2005;99(1):57–62. doi:10.1016/j.imlet.2005.01.002
134. Slebioda Z, Szponar E, Kowalska A. Etiopathogenesis of recurrent aphthous stomatitis and the role of immunologic aspects: literature review. Arch Immunol Ther Exp. 2014;62(3):205–215. doi:10.1007/s00005-013-0261-y
135. Ozyurt K, Celik A, Sayarlıoglu M, et al. Serum Th1, Th2 and Th17 cytokine profiles and alpha-enolase levels in recurrent aphthous stomatitis. J Oral Pathol Med. 2014;43(9):691–695. doi:10.1111/jop.12182
136. Dudding T, Haworth S, Lind PA, et al. Genome wide analysis for mouth ulcers identifies associations at immune regulatory loci. Nat Commun. 2019;10(1):1052. doi:10.1038/s41467-019-08923-6
137. Ruan HH, Li GY, Duan N, et al. Frequencies of abnormal humoral and cellular immune component levels in peripheral blood of patients with recurrent aphthous ulceration. J Dent Sci. 2018;13(2):124–130. doi:10.1016/j.jds.2017.09.003
138. Wardhana DEA, Datau EA. Recurrent aphthous stomatitis caused by food allergy. Acta Med Indones. 2010;42(4):236–240.
139. Borra RC, Andrade PM, Silva ID, et al. The Th1 /Th2 immune-type response of the recurrent aphthous ulceration analyzed by cDNA microarray. J Oral Pathol Med. 2004;33(3):140–146. doi:10.1111/j.0904-2512.2004.00089.x
140. Lau CB, Smith GP. Recurrent aphthous stomatitis: a comprehensive review and recommendations on therapeutic options. Dermatol Ther. 2022;35(6):e15500. doi:10.1111/dth.15500
141. Milia E, Sotgiu MA, Spano G, Filigheddu E, Gallusi G, Campanella V. Recurrent aphthous stomatitis (RAS): guideline for differential diagnosis and management. Eur J Paediatr Dent. 2022;23(1):73–78. doi:10.23804/ejpd.2022.23.01.14
142. Louisy A, Humbert E, Samimi M. Oral lichen planus: an update on diagnosis and management. Am J Clin Dermatol. 2024;25(1):35–53. doi:10.1007/s40257-023-00814-3
143. Balestri R, Bortolotti R, Rech G, Girardelli CR, Zorzi MG, Magnano M. Treatment of oral erosive lichen planus with upadacitinib. JAMA Dermatol. 2022;158(4):457–458. doi:10.1001/jamadermatol.2022.0147
144. Scherlinger M, Kolios AGA, Kyttaris VC, Tsokos GC. Advances in the treatment of systemic lupus erythematosus. Nat Rev Drug Discov. 2025. doi:10.1038/s41573-025-01242-0
145. Lazar S, Kahlenberg JM. Systemic lupus erythematosus: new diagnostic and therapeutic approaches. Annu Rev Med. 2023;74:339–352. doi:10.1146/annurev-med-043021-032611
146. Lin X, Li X, Zhai Z, Zhang M. JAK-STAT pathway, type I/II cytokines, and new potential therapeutic strategy for autoimmune bullous diseases: update on pemphigus vulgaris and bullous pemphigoid. Front Immunol. 2025;16:1563286. doi:10.3389/fimmu.2025.1563286
147. Melchionda V, Harman KE. Pemphigus vulgaris and pemphigus foliaceus: an overview of the clinical presentation, investigations and management. Clin Exp Dermatol. 2019;44(7):740–746. doi:10.1111/ced.14041
148. Cao S, Yang B, Wang Z, et al. Efficacy, safety, and B-cell depletion capacity of 3 rituximab dosing regimens in the treatment of moderate-to-severe pemphigus vulgaris and pemphigus foliaceus: a 52-week clinical trial. J Am Acad Dermatol. 2025;93(3):634–643. doi:10.1016/j.jaad.2025.05.1374
149. Lytvyn Y, Rahat S, Mufti A, et al. Biologic treatment outcomes in mucous membrane pemphigoid: a systematic review. J Am Acad Dermatol. 2022;87(1):110–120. doi:10.1016/j.jaad.2020.12.056
150. Bohelay G, Alexandre M, Le Roux-Villet C, et al. Rituximab therapy for mucous membrane pemphigoid: a retrospective monocentric study with long-term follow-up in 109 patients. Front Immunol. 2022;13:915205. doi:10.3389/fimmu.2022.915205
151. Altenburg A, El-Haj N, Micheli C, Puttkammer M, Abdel-Naser MB, Zouboulis CC. The treatment of chronic recurrent oral aphthous ulcers. Dtsch Arztebl Int. 2014;111(40):665–673. doi:10.3238/arztebl.2014.0665
 © 2025 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 4.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2025 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 4.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.