Back to Journals » Drug Design, Development and Therapy » Volume 8
Cross-sectional and longitudinal analysis of cancer vaccination trials registered on the US Clinical Trials Database demonstrates paucity of immunological trial endpoints and decline in registration since 2008
Authors Lu L, Yan H, Shyam-Sundar V, Janowitz T
Received 11 April 2014
Accepted for publication 17 June 2014
Published 27 September 2014 Volume 2014:8 Pages 1539—1553
DOI https://doi.org/10.2147/DDDT.S65963
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Liangjian Lu,1 Haixi Yan,1 Vijay Shyam-Sundar,1 Tobias Janowitz2
1School of Clinical Medicine, 2Cancer Research UK, Cambridge Institute, Li Ka Shing Centre, University of Cambridge, Cambridge, UK
Introduction: Cancer vaccination has been researched as a means of treating and preventing cancer, but successful translational efforts yielding clinical therapeutics have been limited. Numerous reasons have been offered in explanation, pertaining both to the vaccine formulation, and the clinical trial methodology used. This study aims to characterize the tumor vaccine clinical trial landscape quantitatively, and explore the possible validity of the offered explanations including the translational obstacles posed by the current common endpoints.
Methods: We performed a detailed cross-sectional and longitudinal analysis of tumor vaccine trials (n=955) registered in the US Clinical Trials database.
Results: The number of tumor vaccine trials initiated per annum has declined 30% since a peak in 2008. In terms of vaccine formulation, 25% of trials use tumor cell/lysate preparations; whereas, 73% of trials vaccinate subjects against defined protein/peptide antigens. Also, 68% of trials do not use vectors for antigen delivery. Both these characteristics of tumor vaccines have remained unchanged since 1996. The top five types of cancer studied are: melanoma (22.6%); cervical cancer (13.0%); breast cancer (11.3%); lung cancer (9.5%); and prostate cancer (9.4%). In addition, 86% of the trials are performed where there is established disease rather than prophylactically, of which 67% are performed exclusively in the adjuvant setting. Also, 42% of Phase II trials do not measure any survival-related endpoint, and only 23% of Phase III trials assess the immune response to vaccination.
Conclusion: The clinical trial effort in tumor vaccination is declining, necessitating a greater urgency in identifying and removing the obstacles to clinical translation. These obstacles may include: 1) vaccination against a small range of antigens; 2) naked delivery of antigen; 3) investigation of less immunogenic cancer types; and 4) investigation in the setting of established disease. In addition, the prevalence of late phase failure may be due to inadequate assessment of survival-related endpoints in Phase II trials. The clinical trial development of tumor vaccines should include mechanism-based translational endpoints, as well as the discovery of immune biomarkers with which to stratify, monitor, and prognosticate patients.
Keywords: cancer vaccination, cancer prevention, clinical trials, translational trial endpoints, immunotherapy
Introduction
Numerous lines of evidence suggest that tumor cells can be recognized and eliminated by the immune system.1 On April 29, 2010, sipuleucel-T (Dendreon Corporation, Seattle, WA, USA) received US Food and Drug Administration approval for use in minimally symptomatic castration-resistant prostate cancer.2,3 This was the first therapeutic cancer vaccine to obtain the US Food and Drug Administration approval, a landmark success in the field of cancer vaccines, and – at the same time – a reminder of the hitherto low therapeutic yield4 of the field. In the context of the recent successes of immunomodulatory strategies in Phase II and Phase III trials,5,6 it is questionable whether cancer vaccines represent an optimal approach for inducing greater immunological control of established disease. It can be argued that a cancer vaccine will have greater efficacy if antigen presentation and the afferent arm of the immune system are impaired, while the immunomodulatory strategies directed at the T-cell checkpoints and signaling will be more effective if the efferent arm of the immune system, especially T-cell function, is impaired.1,7 A definitive answer that instructs clinical translational efforts8 is some years away.
Over the years, numerous reviews have examined the obstacles in translating vaccinations that show promise in preclinical research into clinical practice. To understand to what extent these obstacles are reflected in the clinical trial effort as a whole, we characterized the trial landscape by analyzing the registered trials on the US trial database (http://www.clinicaltrials.gov). We also investigate if the current trial methodology includes the translational research endpoints that can inform vaccine development in the future.
These aforementioned obstacles can be broadly divided into two areas: 1) the nature of the vaccination approach; and 2) the existing clinical trial methodology.9
The therapeutic intervention in cancer vaccine trials consists of a particular formulation of tumor-associated antigens (TAAs) delivered together with adjuvant(s). There remains considerable uncertainty as to the optimal formulation of TAAs. Irradiated tumor cells or tumor cell lysates have been suggested to be superior antigen formulations, as they stimulate an immune response against diverse tumor antigens making immune escape less likely, compared to the formulations containing one or a few recombinant TAAs.10
It is also important to consider the growing evidence for the necessity of stimulating cytotoxic T-lymphocytes (CTLs) to produce a tumor response.11 CTLs express the cluster of differentiation 8 (CD8) T-cell receptor (TCR) coreceptor and are major histocompatibility complex class I-restricted.12 Consequently, this suggests that TAAs need to be delivered packaged – eg, in viral vectors – as opposed to naked, or alternatively pulsed into antigen presenting cells (APCs) ex vivo before infusion.
Besides the antigen formulation, there is similarly little clarity as to the optimal adjuvants.4,13 Classically, adjuvants stimulate APC maturation, inducing the expression of costimulatory molecules and proinflammatory cytokines, which are necessary for the complete activation of T-cells.10 Increasingly, with the characterization of immune checkpoints that prevent a spontaneously effective antitumor immune response in cancer patients, eg, the engagement of T-cell cytotoxic T-lymphocyte antigen 4 and programmed death 1 (PD-1) in the immunosuppressive tumor microenvironment, adjuvants may more broadly include biologicals that relieve these checkpoints, such as ipilimumab and nivolumab, respectively.14,15
Besides the actual vaccination approach, another broad area of criticism is the existing clinical trial methodology. Both the suitability of patient enrollment criteria used in cancer vaccine trials – as well as the methods of assessing the therapeutic effects of the vaccine – are thought to be implicated in low translational yields from cancer vaccine trials.
Some tumors – for instance, melanomas and clear cell renal cell carcinomas – are considered more immunogenic than others, as evidenced by the tumors being frequently infiltrated by the CD8+ T-lymphocytes, which correlates with favorable prognosis, and occasionally undergoing spontaneous regression.14,16–21 These tumor types may be more amenable to immunotherapy, with tumor vaccination at least serving to trigger the generation of an antitumor immune response in the proportion of patients who fail to do so spontaneously.22 Thus, the clinical trial efforts focused on other tumor types less tractable to immunotherapy could explain, in part, the failures of translational efforts.4,14
In advanced disease, multiple redundant mechanisms of immune escape are present in the tumor microenvironment, and there may be a global systemic dysfunction of T-cells as well, perhaps due to chronic nonproductive antigenic stimulation.16,23 This may explain poor vaccine efficacy in advanced cancers. Maybe cancer vaccines should be mainly trialled in the prophylactic or, at the very least adjuvant, setting.8,23,24 Cancers with well-established precursor lesions – eg, colonic adenomas and pancreatic intraepithelial neoplasia – may be good candidates for prophylactic vaccination, as they allow a high-risk group to be targeted for vaccination.25,26
Another area of intense discussion concerns the suitability of trial endpoints, which were originally formulated to assess the efficacy of cytotoxic chemotherapies. Immune responses have different kinetics from cytotoxic chemotherapies. They may also appear differently in the conventional trial assessment methods, for instance, the infiltration of the tumor by lymphocytes leading to apparent radiological progression.9,27 This calls into question the validity of survival measures, such as the radiologically assessed progression free survival (PFS) being used as the primary endpoint of clinical trials, in spite of its acceptance by regulatory authorities.28
Many of the aforementioned reasons proffered to explain the poor therapeutic yield of tumor vaccines are based on a careful consideration of preclinical data as well as the published results of certain clinical trials. It remains an open question, whether such analyses are valid across the entire spectrum of cancer vaccine interventional trials, so as to have sufficient explanatory power to account for the limited translational success of the field in general. It is also still uncertain whether these analyses have encouraged, or at least correspond with, changes across the field. Consequently, these analyses will be greatly complemented by a cross-sectional and longitudinal study of the tumor vaccine clinical trial landscape, which we have undertaken and report here.
Materials and methods
Database creation and analysis
This study is a cross-sectional and longitudinal study of interventional cancer vaccine trials registered on the Clinical Trials Database (http:// www.clinicaltrials.gov). It has been conducted and reported according to the Strengthening the Reporting of Observational Studies in Epidemiology (STROBE) criteria.29 On June 19, 2013, using the advanced search function, trials with the terms “cancer” in their list of conditions, and “vaccine” in their list of interventions, as well as registered as being of an “interventional” study type, were selected. Interventional studies refer to those in which an intervention of any type, including drugs, procedures, and rehabilitation strategies, was investigated, as opposed to purely observational studies.30 Study details were downloaded as datasets for review using Microsoft Excel (Microsoft Corporation, Redmond, WA, USA).
All studies were manually checked to ensure that they were trials of cancer vaccines in the prevention or therapy of cancer. This led to the exclusion of 43 trials in which the primary condition being investigated was not cancer, eg, influenza vaccinations in children with cancer (NCT00022035), as well as the exclusion of a further 78 trials in which the intervention was not a cancer vaccine per se, eg, the Bacillus Calmette–Guérin vaccine for bladder cancer (NCT00427570). All suitable trials registered before 2013 were included, yielding a total of 955 trials for analysis (Figure 1).
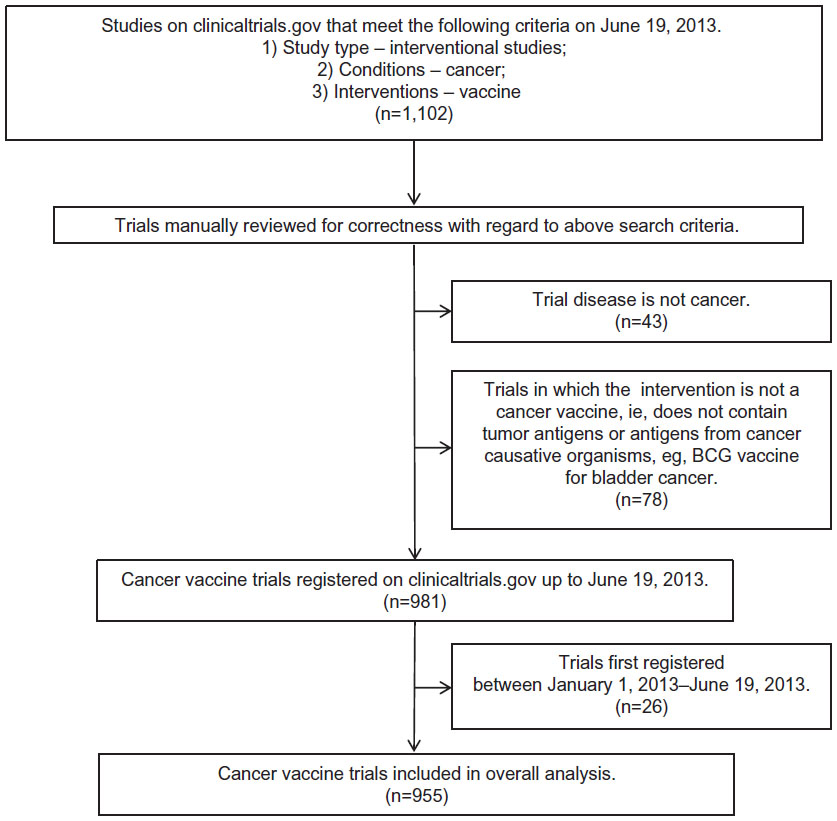 | Figure 1 Flow diagram of database creation and manual review of trials. |
The data that were downloaded into Microsoft Excel were not edited. As a result, a proportion of the trials was missing certain data fields. Incomplete registration details are a known limitation of trial database entries.31 The respective trials were excluded from the relevant parts of the analysis,31 leading to a total trial count of <955 for most subsections. Total trial counts are always specified for each analysis performed. For certain analyses, such as the clinical setting of vaccination, the downloaded datasets provided insufficient information. In these cases, the online registration entry of the trial on the Clinical Trials Database was referred to for further details. For the analysis of enrollment numbers, comparison with interventional cancer trials in general, ie, all interventions, was needed. The relevant trial data were downloaded from the Clinical Trials Database on June 19, 2013 with the following criteria: 1) study type – interventional studies; and 2) conditions – cancer. This yielded a total of 30,859 trials. These data were not subject to manual review.
Statistical analysis
All averages are given as mean ± standard error of mean, unless otherwise stated. For selected longitudinal series, linear regression analysis was performed. For comparison of means and medians, the Student’s t-test and the Wilcoxon rank-sum test were performed, respectively. Also, P<0.05 was taken to indicate statistical significance.
Results
Overall trial characteristics
Of the 955 trials included in the overall analysis, data for the trial start date and the trial primary completion date were available for 935 trials and 776 trials, respectively. A longitudinal analysis of trial start dates (Figure 2A) reveals a decline in the number of clinical trials initiated, with the count declining 30% from a peak of 87 in 2008 to 61 in 2012. On a per annum basis, the number of vaccine trials decreased on average by 6.5 each year from 2008 onward (P<0.01). The corresponding peak in trial completion is in 2013 (Figure 2A). This is broadly in line with a calculated mean trial duration of 4.0±0.1 years (n=776). In contrast, the counts of trial start dates for the nonvaccine interventional cancer vaccine trials have stayed approximately constant from 2008–2012 (P=0.68). Relative to 2008, the trial counts for subsequent years are significantly different (P=0.02), when comparing between vaccine and nonvaccine trials.
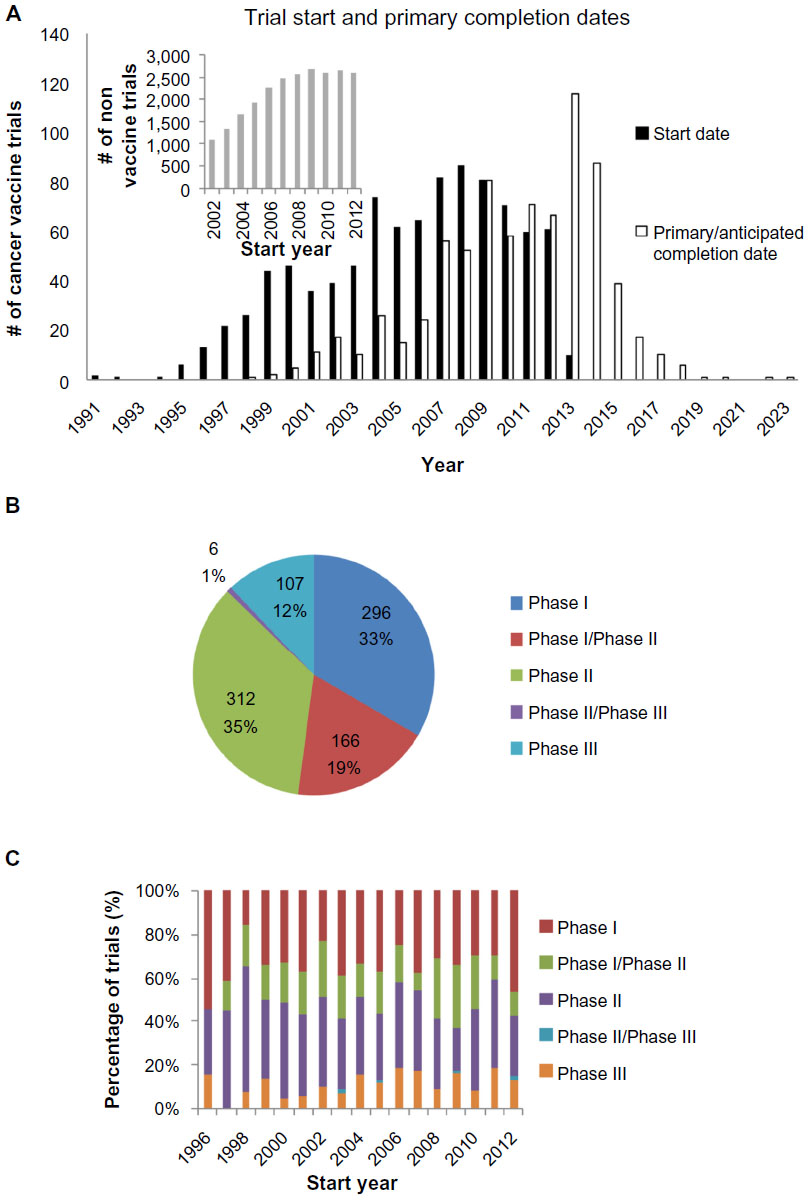 | Figure 2 Overall trial characteristics. |
Phase data were available for 887 Phase I–Phase III trials. There are similar numbers of Phase I and Phase II trials, which together account for 88% of trials conducted (Figure 2B). This relative proportion of clinical trials has stayed fairly constant since 1996 (Figure 2C).
Nature of vaccination approach
A cross-sectional analysis of the antigen formulation used in the 955 trials was performed as shown in Figure 3. In 75% of the trials, the tumor antigens vaccinated against were a relatively small number of specified protein or carbohydrate antigens; whereas, in 25% of the trials, patients were exposed to a wide range of tumor-associated antigens via administration of tumor cells or their lysates (Figure 3A). This proportion has remained relatively stable across time, with an annual mean of 24.2%±2.0% from 1996–2012 (Figure 3B).
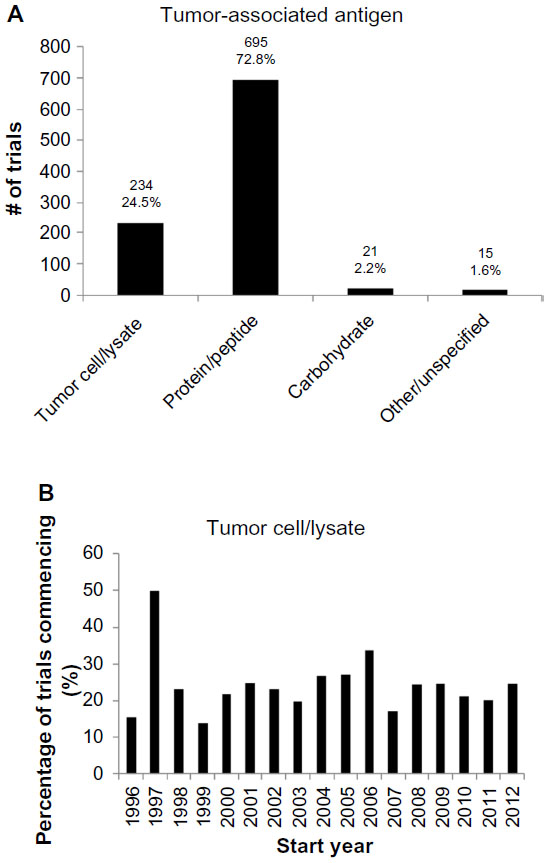 | Figure 3 Types of antigen in vaccine formulation. |
The majority (68%) of the trials involved the administration of naked antigen (Figure 4A). This proportion has stayed relatively constant across the time, with an annual mean of 68.9%±1.8% from 1996–2012 (Figure 4B). The remaining 32% of the trials adopted vectors that comprised: dendritic cells (20%); viral vectors (9%), especially poxviruses and adenoviruses; and naked nucleic acids (4%), including unpackaged deoxyribonucleic acid, ribonucleic acid, and plasmids. Notably, even though only 1% of all the trials employed anti-idiotype vaccines, 33% of the trials in which carbohydrate antigens were vaccinated against used anti-idiotype vaccines. This reveals the utility of the anti-idiotype vaccines in specifying carbohydrate antigens, which are generally less immunogenic than protein antigens.
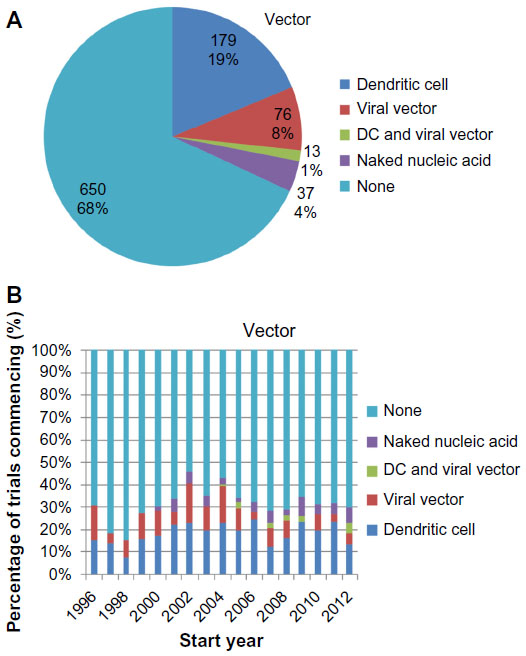 | Figure 4 Use of vectors in vaccine formulation. |
By an inspection of the data, we observed that almost all trials included the administration of adjuvants in the intervention under study. However, a wide range of adjuvants was used, rendering systematic categorization impossible and inappropriate. To demonstrate this spectrum of adjuvants, we chose to examine those employed in Phase III trials initiated within the last 5 years (n=40; Table S1), as these experimental vaccines are likely to have the greatest near-term clinical significance. As shown in Table 1, a total of 15 vaccines were studied in 40 trials. These vaccines utilized eight different adjuvants. There was, thus, no apparent preference for any adjuvant. Even among the four human papillomavirus (HPV) vaccines analyzed, all of which comprised viruslike particles that were made up of L1 capsid protein of various HPV strains, different adjuvants were used. For example, one used alum (aluminum hydroxide); another two, amorphous aluminum hydroxyphosphate sulfate; and a final one, a combination of alum and monophosphoryl lipid A.
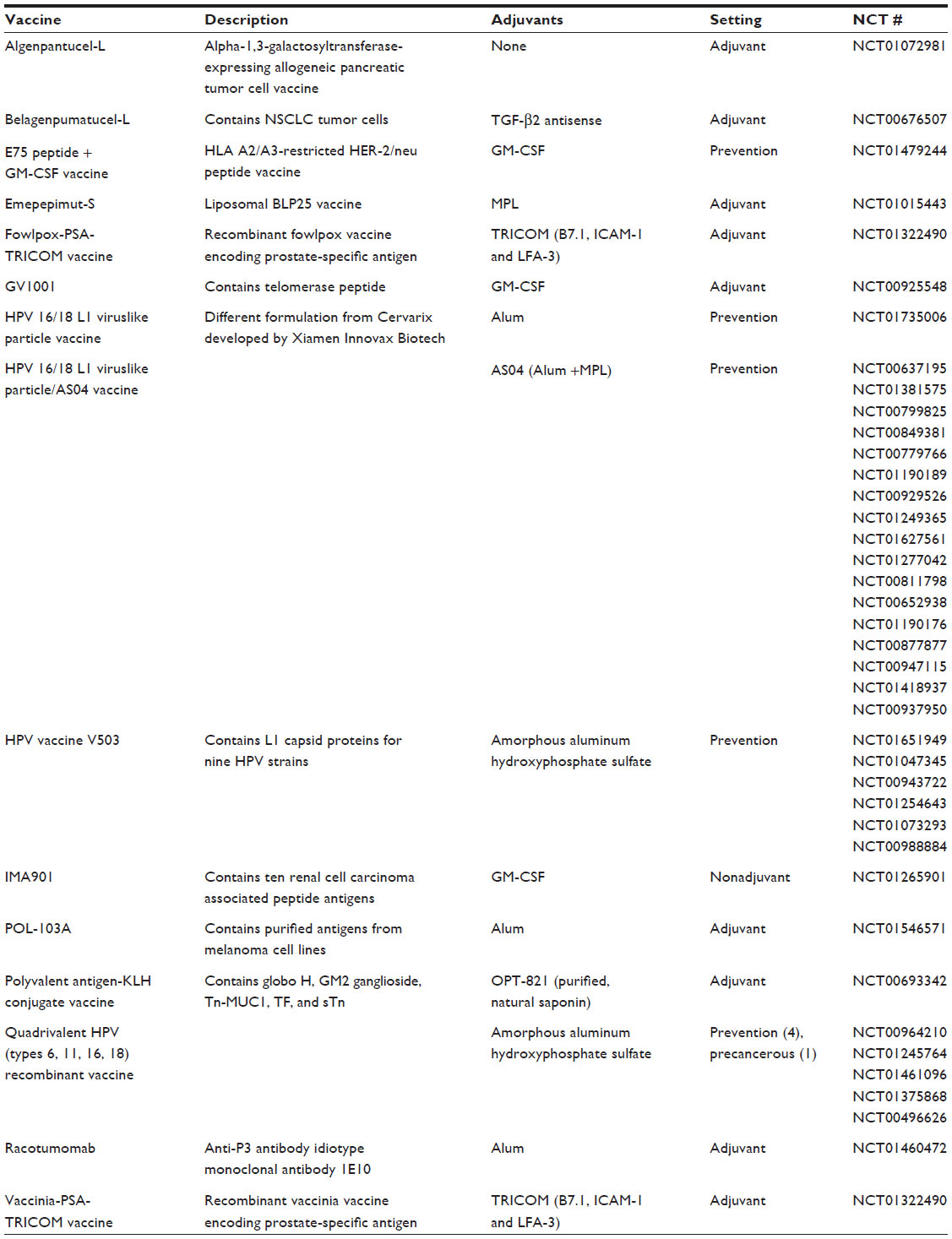 | Table 1 Vaccines investigated in Phase III trials commencing between 2008 and 2012, (n=40) |
Existing trial methodology: patient characteristics
In addition, 1,178 instances of various cancers were studied across 955 trials; 28 trials did not specify, or incompletely specified, the type of cancer studied. Of the remaining 927 trials, 860 (93%) studied one cancer type. Many of the cancers that did not adequately specify cancer type had – instead – inclusion criteria based on the tumor antigen expression, eg, overexpression of vaccine antigen.
The distribution of cancer types across trials is shown in Figure 5A, with the nine cancer types studied in 5% or more of trials being reflected. The top five cancer types were: melanoma (22.6%); cervical cancer (13.0%); breast cancer (11.3%); lung cancer (9.5%); and prostate cancer (9.4%). The other cancer type thought to be immunogenic, renal cancer, was studied in only 36 (3.8%) trials. While the predominance of melanoma is striking, there is a statistically significant decrease in the percentage of trials in which it is studied (Figure 5B).
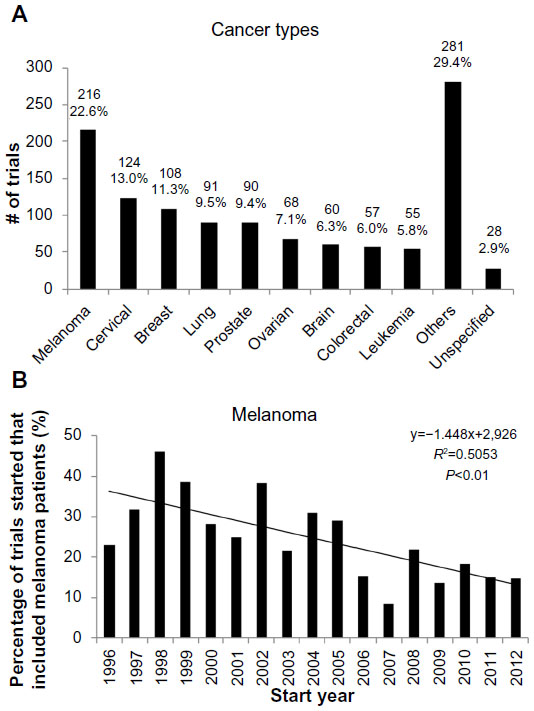 | Figure 5 Cancer types in cancer vaccine trials. |
Data on the primary purpose were available for 933 of the 955 trials included. Overall, the majority of trials investigated therapeutic cancer vaccines, although a minority of the trials (13%) was performed in the preventive setting. Most of the preventive trials involved cancer types with a suggested or proven infectious etiology, especially HPV in cervical cancer (Figure 6B). Since 1996, there is a trend toward a higher proportion of preventive cancer vaccine trials (Figure 6B).
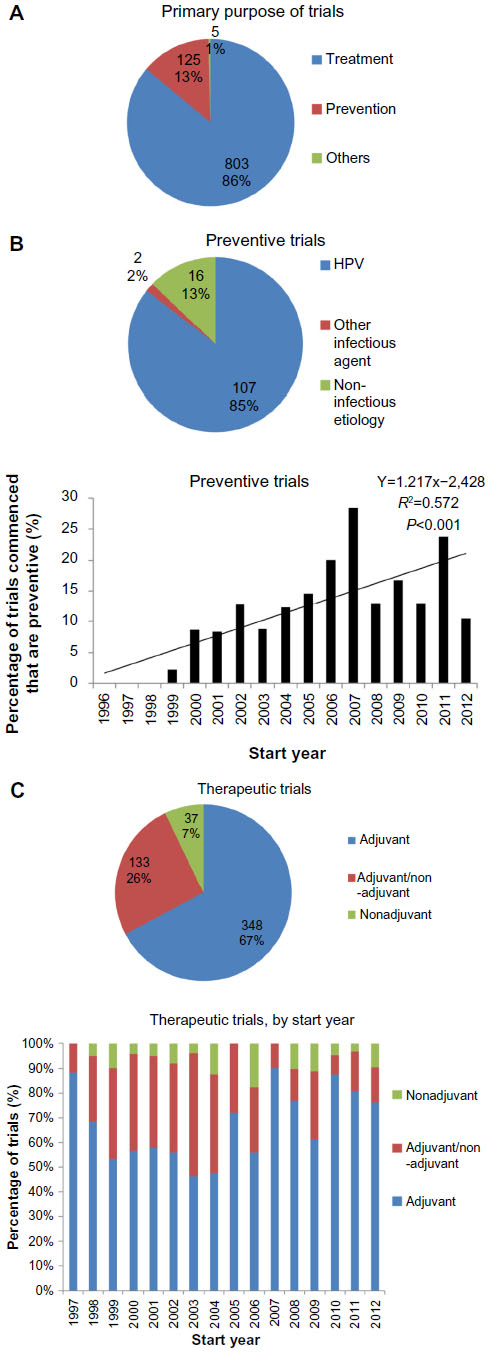 | Figure 6 Clinical setting of cancer vaccine trials. |
Of the 803 therapeutic trials performed, we sought to determine whether they were performed in the adjuvant setting, or in the clinical context of unresected, and likely advanced and thus unresectable, disease. We excluded trials that considered premalignant conditions (14) or hematological malignancies (110), as well as trials that provided insufficient information to determine the clinical context of vaccination (161). This left 518 trials for analysis. This analysis revealed that in 67% of trials, the vaccine was administered exclusively in the adjuvant setting, a proportion that has stayed relatively constant over time (Figure 6C).
Existing trial methodology: endpoints
Considering only therapeutic (as oppose to preventive) trials, endpoint data were available for 637 of the 803 therapeutic trials. There is a decrease in the proportion of the trials assessing the immune response, eg, through antibody titers and T-cell enzyme-linked immunosorbent spot assays, declining from 78% of Phase I trials to 23% of Phase III trials (Figure 7A).
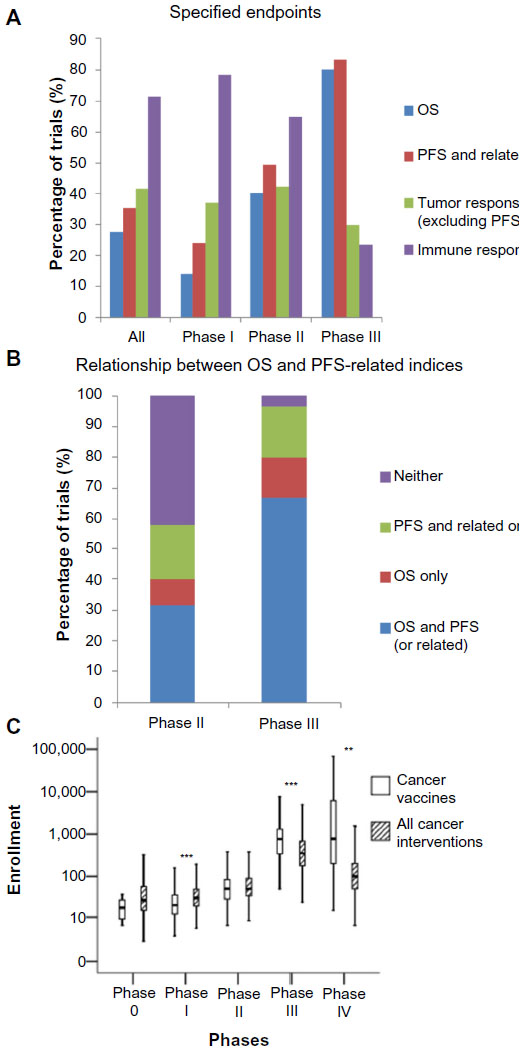 | Figure 7 Trial methodology. |
Examining the use of survival-related endpoints, such as overall survival (OS) and PFS, more closely (Figure 7B), 42% of the Phase II trials did not include any of these endpoints in the assessment of therapeutic efficacy. In the Phase III trials, 17% of trials did not measure OS. Instead, they relied on other survival-related endpoints, such as PFS (Figure 7B).
Trial size was also assessed in terms of enrollment numbers. Enrollment numbers were available for 838 of the 955 trials, but suspended (nine), withdrawn (61), and terminated (20) trials were further excluded due to incomplete enrollment, thus yielding 748 trials in this analysis. Data were analyzed by phase, with data from mixed-phase trials discarded. As expected, trial enrollment numbers increase with the trial phase (Figure 7C). Comparing trial enrollment numbers of the cancer vaccine trials with those of cancer trials in general (n=25,638), trial size is significantly smaller in Phase I cancer vaccine trials, but significantly larger in Phase III and Phase IV cancer vaccine trials, compared to all cancer trials. The former may be due to the reduced toxicity of cancer vaccines compared to cytotoxic chemotherapies or even targeted molecular inhibitors,3 while the latter is likely to reflect the delayed effect of the cancer vaccines mentioned earlier, which necessitates larger patient populations to achieve adequate statistical power.
Discussion
In this study, we have analyzed the characteristics of all the cancer vaccine trials registered in the US Clinical Trials Database before 2013, corresponding to 955 trials. The chosen database is the most comprehensive trial database available.32 Such a methodology has been previously applied by ourselves and others to characterize the trial landscape in traumatic brain injury,30 as well as nephrology33 and oncology in general.34
Overall trial characteristics
A longitudinal analysis of cancer vaccine trials revealed a peak in 2008 followed by a decline (Figure 2A). While this could be due to the global reasons across the cancer research sector, or perhaps the entire clinical trial landscape, this is made unlikely by the fact that a similar peak in trial count could not be observed when nonvaccine interventional cancer trials were analyzed. Instead, this finding suggests that there is indeed a real and significant decline in the translational effort for cancer vaccines; this is probably a reflection of the poor therapeutic yield of the field as earlier discussed and suggests that a greater urgency is necessary in identifying and addressing the underlying causes.
An analysis of the trials by phase obtained a distribution that was not dissimilar to that obtained by others who have analyzed cancer interventional trials.35 These data are in line with previous data that had shown the response rate in early phase cancer vaccine trials to be similar to the lower end of the response rates observed in early phase interventional cancer trials.4 This suggests that, in their current design, Phase II cancer vaccination trials excessively overestimate therapeutic efficacy. Consequently, although it has been argued that the selection of trial endpoints underestimates the benefit of the cancer vaccines,4,27 the precise effects of trial endpoints on response rates are likely to be more complex.
Nature of vaccination approach
Our analyses of antigen formulation revealed that 25% of the trials included a wide range of tumor-associated antigens (Figure 3), and 32% of the trials included a delivery vector (Figure 4). This lends quantitative support to earlier suggestions that the contributing factors to the poor efficacy of the cancer vaccines include (i) vaccination against too few antigens, thus making tumor escape possible,10 as well as (ii) the failure to deliver antigens into the cytosol of antigen-presenting cells so as to enable major histocompatibility complex class I presentation and thus a cytotoxic T-lymphocyte response.11
Interestingly, the previously mentioned proportions have remained approximately constant over time. This suggests that – despite calls to modify the antigen formulation – the approach to clinical trials remains conservative on these questions. This may be due to significant logistical difficulties in the production of such vaccines, in spite of their theoretical advantages, most notably for autologous vaccine formulations that require the preparation of patient-specific vaccines from their tumor material and/or peripheral blood APCs.11
We have observed that a wide spectrum of adjuvants was used, indicating that adjuvants account for a sizable proportion of variation between vaccine formulations (Table 1). This observation implies that the optimal adjuvant is still uncertain, and it also supports earlier observations of a nontargeted approach across the field toward adjuvant selection, which may be hindering the identification of the optimal adjuvant.36 Indeed, the necessity of carefully testing the effect of adjuvants was highlighted by a recent trial, which showed that the addition of granulocyte macrophage colony-stimulating factor to an melanoma tumor cell vaccine reduced T-cell responses and OS at 2 years.36 Thus, the lack of a systematic approach to adjuvant testing may contribute to the poor efficacy of the cancer vaccines in general.
Existing trial methodology
The most frequently studied cancers are melanoma and cancers of the cervix, breast, lung, and prostate (Figure 5A). These data are broadly in line with that of Dayoub and Davis, who performed a cross-sectional analysis of the therapeutic tumor vaccine trials registered from January–May 2011.37
The frequency with which breast, lung, and prostate cancers are studied is likely to reflect their high incidence and contribution to annual mortality,37 while the study of melanoma and cervical cancers is likely to be driven – at least in part – by their perceived tractability to vaccination strategies, due to the immunogenicity of melanomas and the infectious etiology of cervical cancers. While renal cancers have been considered immunogenic as well, the low frequency with which they have been studied may be due to disappointing results from a number of adjuvant clinical trials of autologous renal cell carcinoma vaccines in the late 1990s to the early 2000s.8
This analysis of the cancer types studied provides some support for the argument that the translational failures of cancer vaccines are due to efforts being directed at the common cancers less amenable to vaccine therapy. Further support can be obtained from the observation that – apart from sipuleucel-T – the other cancer vaccines receiving approval are targeted at cervical cancer, malignant melanoma, or renal carcinoma.
Interestingly, however, the proportion of trials involving melanoma patients is on the decrease. This may reflect a more nuanced and less empirical understanding of cancer immunogenicity, for instance through the identification of more TAAs38 across different cancer types. An additional explanation for this relative decline of melanoma vaccine trials could be the recent success of immune checkpoint inhibition, eg, ipilimumab,6 in treating metastatic melanoma. Both these developments may have resulted in translational efforts being refocused away from melanoma, which may subsequently lead to a further dilution and decline of therapeutic success in the field.
Also, this study has revealed that a relatively small proportion of the translational effort has been directed at preventive cancer vaccines. Most of these vaccines are directed against HPV (Figure 6B). Although the proportion of cancer vaccine trials conducted in the preventive setting has been increasing, it is doubtful whether this overall trend will continue, given the presence of a noticeable peak in trials initiated in 2007, and the fact that two vaccines against HPV strains, Cervarix® (GlaxoSmithKline plc, London, UK) and Gardasil® (Merck & Co, Inc., Whitehouse Station, NJ, USA), have already received regulatory approval.
Further efforts in the field may be driven by the development of vaccines against other infectious agents, for instance, Helicobacter pylori (NCT00613665) in the context of gastric carcinoma or gastric mucosa-associated lymphoid tissue lymphoma. However, the economic drivers of these translational efforts may be weaker, given the fact that some, if not many, of the infectious causes of cancers have a higher incidence in poorer countries, eg, H. pylori and Schistosoma haematobium.21
For the remaining trials that were conducted with the aim of treatment rather than prevention, we have shown that 67% of the trials assessed vaccine efficacy exclusively in the adjuvant setting, with a further 26% including at least some patients free of macroscopic disease (Figure 6C). This suggests that the lack of therapeutic efficacy of candidate vaccines cannot be attributed to the immunosuppressive effects of the tumor in situ. The local immunosuppressive effect of the stroma in micrometastases, however, is potentially causative and – in the preclinical setting – is currently actively researched. Also, these data cannot exclude the possibility that vaccine efficacy is attenuated by immunosuppressive effects of the tumor that persist even after resection, as has been demonstrated in a mice melanoma model.39 Indeed, poor vaccine responses in the adjuvant setting may even perhaps be attributable to the long-term immunosuppressive effects of adjuvant cytotoxic chemotherapy,2,40 a valuable question that can be addressed in future clinical trial analyses.
With regard to trial endpoints, our data suggest some further explanations regarding the use of existing endpoints that may contribute to the high rate of late phase failures. First, a minority of Phase III trials do not assess OS, but depend instead on endpoints, such as PFS and disease-free survival, even though several immunotherapies, (eg, sipuleucel-T and ipilimumab), have demonstrated increases in OS without attendant increases in the PFS or related measures.3 Second, in 42% of Phase II trials, no survival-related endpoint was measured, suggesting that a significant proportion of candidate vaccines enter late phase trials on the basis of radiological evidence of tumor response, a surrogate measure that is known to be problematic for immunotherapies.41
Most importantly from a translational research point of view, this study has also revealed that only a small proportion of late phase trials assess the immune response to the tumor vaccine under investigation (Figure 7A). We suggest that Phase II and especially Phase III trials should include objective immune response analyses more frequently to facilitate translational science and a better understanding of positive and negative trial outcomes, especially in the context of a high proportion of failed late stage clinical trials. Moreover, immunological data from the late phase trials will be vital to enabling the optimal use of cancer vaccines in clinical practice postlicensing, as given the aforementioned problems with assessing vaccine response radiologically, some surrogate of the clinical benefit is required to prognosticate and plan for additional therapies, which may be therapeutic or palliative.3
Trial design can be adjusted to take into account the logistical demands of immunological endpoint assessment, including assessing these endpoints in a subgroup of patients to reduce costs or only in selected participating tertiary referral centers, where the necessary technical expertise is available. Regardless, beginning to assess immunological endpoints now will enable the development of the necessary clinical trial infrastructure to do so more reliably and cost effectively in the future.
Conclusion
By characterizing the landscape of interventional clinical vaccine trials, this study has revealed declining numbers of trials initiated since 2008; there is a need for greater urgency in removing the obstacles to the clinical translation of experimental vaccines.
Our data have demonstrated that only in a minority of trials are vaccines that incorporate a wide range of tumor antigens or utilize vectors for antigen delivery assessed, providing quantitative support for the hypotheses that these characteristics of experimental vaccines are impeding clinical translation. We have also observed a significant lack of consistency in terms of the adjuvants employed in the various trials, including Phase III trials – suggesting that cancer vaccines, in general, still lack effective adjuvants. In terms of the clinical trial methodology, our data reveal that while melanoma is the most common cancer studied, significant clinical efforts are being directed at common cancers not regarded as particularly immunogenic. Also, we have confirmed the observation that only a minority of cancer vaccines is used prophylactically, predominantly HPV vaccines. We have demonstrated that, in addition to this, the majority of therapeutic cancer vaccines are trialed in the adjuvant setting, suggesting that disease volume has little impact on vaccine efficacy. Longitudinally, the overall picture is generally one of stasis, with minimal evolution of the trial landscape in spite of various calls to the contrary. Finally, considering both the finding of a surprisingly high proportion of Phase III trials and the observation that 42% of Phase II trials did not utilize any survival-related endpoints, we suggest that a failure to adequately estimate therapeutic efficacy in Phase II trials is contributing, at least in part, to the high rate of translational failure in the late phase trials. We also note the relative paucity of mechanistic immunological endpoints in the Phase III trials, which – if not rectified – is likely to hinder translational efforts in the field as well as the optimal clinical use of approved vaccines.
Acknowledgments
The authors thank Ehsan Ghorani for critical review of the manuscript. TJ is funded by the Wellcome Trust Translational Medicine and Therapeutics program.
Disclosure
The authors report no conflicts of interest in this work.
References
Kim R, Emi M, Tanabe K. Cancer immunoediting from immune surveillance to immune escape. Immunology. 2007;121(1):1–14. | |
Bilusic M, Gulley JL. Endpoints, patient selection, and biomarkers in the design of clinical trials for cancer vaccines. Cancer Immunol Immunother. 2012;61(1):109–117. | |
Bilusic M, Madan RA. Therapeutic cancer vaccines: the latest advancement in targeted therapy. Am J Ther. 2012;19(6):e172–e181. | |
Klebanoff CA, Acquavella N, Yu Z, Restifo NP. Therapeutic cancer vaccines: are we there yet? Immunol Rev. 2011;239(1):27–44. | |
Topalian SL, Hodi FS, Brahmer JR, et al. Safety, activity, and immune correlates of anti-PD-1 antibody in cancer. N Engl J Med. 2012; 366(26):2443–2454. | |
Hodi FS, O’Day SJ, McDermott DF, et al. Improved survival with ipilimumab in patients with metastatic melanoma. N Engl J Med. 2010;363(8):711–723. | |
Feig C, Jones JO, Kraman M, et al. Targeting CXCL12 from FAP-expressing carcinoma-associated fibroblasts synergizes with anti-PD-L1 immunotherapy in pancreatic cancer. Proc Natl Acad Sci U S A. 2013;110(50):20212–20217. | |
Janowitz T, Welsh SJ, Zaki K, Mulders P, Eisen T. Adjuvant therapy in renal cell carcinoma-past, present, and future. Semin Oncol. 2013;40(4):482–491. | |
Hoos A. Evolution of end points for cancer immunotherapy trials. Ann Oncol. 2012;23 Suppl 8(Suppl 8):viii47–viii52. | |
Andersen BM, Ohlfest JR. Increasing the efficacy of tumor cell vaccines by enhancing cross priming. Cancer Lett. 2012;325(2):155–164. | |
Schlom J. Therapeutic cancer vaccines: current status and moving forward. J Natl Cancer Inst. 2012;104(8):599–613. | |
Balwit JM, Hwu P, Urba WJ, Marincola FM. The iSBTc/SITC primer on tumor immunology and biological therapy of cancer: a summary of the 2010 program. J Transl Med. 2011;9:18. | |
Butt AQ, Mills KH. Immunosuppressive networks and checkpoints controlling antitumor immunity and their blockade in the development of cancer immunotherapeutics and vaccines. Oncogene. Epub October 21, 2013. | |
Drake CG, Lipson EJ, Brahmer JR. Breathing new life into immunotherapy: review of melanoma, lung and kidney cancer. Nat Rev Clin Oncol. 2014;11(1):24–37. | |
Riley JL. Combination checkpoint blockade – taking melanoma immunotherapy to the next level. N Engl J Med. 2013:369(2):187–189. | |
Koos D, Josephs SF, Alexandrescu DT, et al. Tumor vaccines in 2010: need for integration. Cell Immunol. 2010;263(2):138–147. | |
Nakano O, Sato M, Naito Y, et al. Proliferative activity of intratumoral CD8(+) T-lymphocytes as a prognostic factor in human renal cell carcinoma: clinicopathologic demonstration of antitumor immunity. Cancer Res. 2001;61(13):5132–5136. | |
Dougan M, Dranoff G. Immune therapy for cancer. Annu Rev Immunol. 2009;27:83–117. | |
Marzbani E, Inatsuka C, Lu H, Disis ML. The invisible arm of immunity in common cancer chemoprevention agents. Cancer Prev Res (Phila). 2013;6(8):764–773. | |
Kalialis LV, Drzewiecki KT, Klyver H. Spontaneous regression of metastases from melanoma: review of the literature. Melanoma Res. 2009;19(5):275–282. | |
Liu MA. Cancer vaccines. Philos Trans R Soc Lond B Biol Sci. 2011;366(1579):2823–2826. | |
Clemente CG, Mihm MC Jr, Bufalino R, Zurrida S, Collini P, Cascinelli N. Prognostic value of tumor infiltrating lymphocytes in the vertical growth phase of primary cutaneous melanoma. Cancer. 1996;77(7):1303–1310. | |
Gray A, Raff AB, Chiriva-Internati M, Chen SY, Kast WM. A paradigm shift in therapeutic vaccination of cancer patients: the need to apply therapeutic vaccination strategies in the preventive setting. Immunol Rev. 2008;222:316–327. | |
Sobol RE. The rationale for prophylactic cancer vaccines and need for a paradigm shift. Cancer Gene Ther. 2006;13(8):725–731. | |
Kimura T, McKolanis JR, Dzubinski LA, et al. MUC1 vaccine for individuals with advanced adenoma of the colon: a cancer immunoprevention feasibility study. Cancer Prev Res (Phila). 2013;6(1):18–26. | |
Finn OJ. Cancer vaccines: between the idea and the reality. Nat Rev Immunol. 2003;3(8):630–641. | |
Hoos A, Eggermont AM, Janetzki S, et al. Improved endpoints for cancer immunotherapy trials. J Natl Cancer Inst. 2010;102(18):1388–1397. | |
Begg CB. Justifying the choice of endpoints for clinical trials. J Natl Cancer Inst. 2013;105(21):1594–1595. | |
von Elm E, Altman DG, Egger M, et al; Strobe Initiative. The Strengthening the Reporting of Observational Studies in Epidemiology (STROBE) statement: guidelines for reporting observational studies. Lancet. 2007;370(9596):1453–1457. | |
Li LM, Menon DK, Janowitz T. Cross-sectional analysis of data from the US clinical trials database reveals poor translational clinical trial effort for traumatic brain injury, compared with stroke. PLoS One. 2014;9(1):e84336. | |
Janowitz T. Biopharmaceuticals and monoclonal antibodies in oncology trials – a cross-sectional analysis. Protein Eng Des Sel. 2011;24(1–2):105–111. | |
Zarin D. Newsmaker interview: Debora Zarin. Unseen world of clinical trials emerges from US database. Interview by Eliot Marshall. Science. 2011;333(6039):145. | |
Inrig JK, Califf RM, Tasneem A, et al. The landscape of clinical trials in nephrology: a systematic review of Clinicaltrials.gov. Am J Kidney Dis. 2014;63(5):771–780. | |
Hirsch BR, Califf RM, Cheng SK, et al. Characteristics of oncology clinical trials: insights from a systematic analysis of Clinicaltrials.gov. JAMA Intern Med. 2013;173(11):972–979. | |
Califf RM, Zarin DA, Kramer JM, Sherman RE, Aberle LH, Tasneem A. Characteristics of clinical trials registered in Clinicaltrials.gov, 2007–2010. JAMA. 2012;307(17):1838–1847. | |
Faries MB, Hsueh EC, Ye X, Hoban M, Morton DL. Effect of granulocyte/macrophage colony-stimulating factor on vaccination with an allogeneic whole-cell melanoma vaccine. Clin Cancer Res. 2009;15(22):7029–7035. | |
Dayoub EJ, Davis MM. Relationship of therapeutic cancer vaccine development to population disease burden and five-year survival. Hum Vaccin. 2011;7(11):1124–1129. | |
Buonaguro L, Petrizzo A, Tagliamonte M, Tornesello ML, Buonaguro FM. Challenges in cancer vaccine development for hepatocellular carcinoma. J Hepatol. 2013;59(4):897–903. | |
Russ AJ, Wentworth L, Xu K, et al. Suppression of T-cell expansion by melanoma is exerted on resting cells. Ann Surg Oncol. 2011;18(13):3848–3857. | |
von Mehren M, Arlen P, Gulley J, et al. The influence of granulocyte macrophage colony-stimulating factor and prior chemotherapy on the immunological response to a vaccine (ALVAC-CEA B7.1) in patients with metastatic carcinoma. Clin Cancer Res. 2001;7(5):1181–1191. | |
Wolchok JD, Hoos A, O’Day S, et al. Guidelines for the evaluation of immune therapy activity in solid tumors: immune-related response criteria. Clin Cancer Res. 2009;15(23):7412–7420. |
Supplementary material
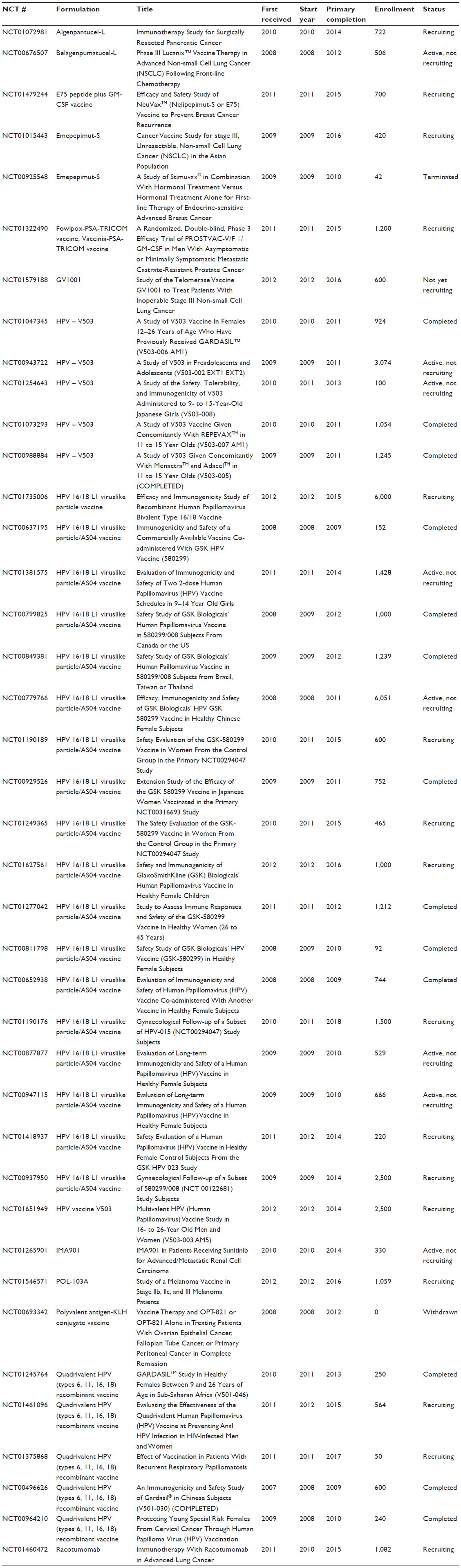 | Table S1 Details of Phase III trials commencing between 2008 and 2012 |
 © 2014 The Author(s). This work is published by Dove Medical Press Limited, and licensed under a
Creative Commons Attribution License.
The full terms of the License are available at http://creativecommons.org/licenses/by/4.0/.
The license permits unrestricted use, distribution, and reproduction in any medium, provided the
original author and source are credited.
© 2014 The Author(s). This work is published by Dove Medical Press Limited, and licensed under a
Creative Commons Attribution License.
The full terms of the License are available at http://creativecommons.org/licenses/by/4.0/.
The license permits unrestricted use, distribution, and reproduction in any medium, provided the
original author and source are credited.